

Metody usuwania grupy karbonylowej z ketonów i aldehydów
Metody usuwania grupy karbonylowej z ketonów i aldehydów
Metody usuwania grupy karbonylowej z ketonów i aldehydów

You also want an ePaper? Increase the reach of your titles
YUMPU automatically turns print PDFs into web optimized ePapers that Google loves.
Z W I Ą Z K I K A R B O N Y L O W E 10.2006<br />
Aleksander Kołodziejczyk<br />
Grupa karbonylowa -C=O występuje w wielu związkach organicznych, ale tylko aldehydy i<br />
ketony nazywane są związkami karbonylowymi. W jedynie nich karbonylowy atom węgla jest<br />
związany wyłącznie z atomami wodoru lub/i węgla. W pozostałych związkach zawierających<br />
grupę -C=O, tj. w kwasach karboksylowych i ich pochodnych do karbonylowego atomu węgla<br />
dołączona jeszcze inna grupa funkcyjna, np. -OH, -OR, -COR czy -X.<br />
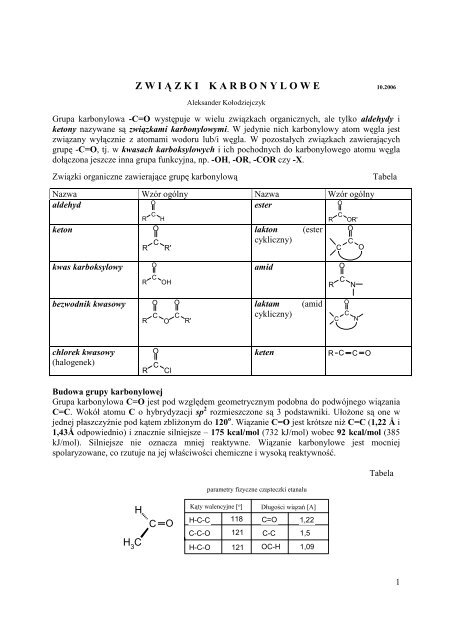
Związki organiczne zawierające grupę karbonylową Tabela<br />
Nazwa Wzór ogólny Nazwa Wzór ogólny<br />
aldehyd<br />
O<br />
ester<br />
O<br />
keton<br />
kwas karboksylowy<br />
bezwodnik kwasowy<br />
chlorek kwasowy<br />
(halogenek)<br />
R<br />
C<br />
O<br />
H<br />
C<br />
R R'<br />
O<br />
C<br />
R OH<br />
O<br />
C C<br />
R O<br />
O<br />
C<br />
R Cl<br />
O<br />
R'<br />
lakton (ester<br />
cykliczny)<br />
amid<br />
laktam (amid<br />
cykliczny)<br />
R<br />
C<br />
OR'<br />
O<br />
C<br />
O<br />
C<br />
C<br />
R N<br />
keten R C C O<br />
Budowa <strong>grupy</strong> <strong>karbonylowej</strong><br />
Grupa karbonylowa C=O jest pod względem geometrycznym podobna do podwójnego wiązania<br />
C=C. Wokół atomu C o hybrydyzacji sp 2 rozmieszczone są 3 podstawniki. UłoŜone są one w<br />
jednej płaszczyźnie pod kątem zbliŜonym do 120 o . Wiązanie C=O jest krótsze niŜ C=C (1,22 Å i<br />
1,43Å odpowiednio) i znacznie silniejsze – 175 kcal/mol (732 kJ/mol) wobec 92 kcal/mol (385<br />
kJ/mol). Silniejsze nie oznacza mniej reaktywne. Wiązanie karbonylowe jest mocniej<br />
spolaryzowane, co rzutuje na jej właściwości chemiczne i wysoką reaktywność.<br />
H<br />
C<br />
H 3<br />
C O<br />
parametry fizyczne cząsteczki etanalu<br />
Kąty walencyjne [ o ] Długości wiązań [A]<br />
H-C-C 118 C=O 1,22<br />
C-C-O<br />
121<br />
H-C-O 121<br />
C-C 1,5<br />
OC-H 1,09<br />
C<br />
O<br />
C<br />
N<br />
O<br />
Tabela<br />
1
Karbonylowy atom węgla o hybrydyzacji sp 2 tworzy 3 wiązania typu σ i jedno π.<br />
.<br />
.<br />
C O<br />
orbitale p<br />
..<br />
.<br />
..<br />
C O .<br />
elektrony na niezhybrydyzowanych<br />
orbitalach p po nałoŜeniu<br />
tworzą wiązanie typu π<br />
Związki zawierające grupę karbonylową są polarne, poniewaŜ wiązanie C=O jest<br />
spolaryzowane. Łatwo się domyślić, Ŝe częściowy ładunek dodatni jest zlokalizowany na atomie<br />
węgla, zaś ujemny na atomie tlenu.<br />
+δ -δ ...<br />
C<br />
Grupę karbonylową moŜna przedstawić w postaci wzorów mezomerycznych, przy czym na<br />
jednym z nich są całkowicie rozdzielone ładunki. Prawdopodobieństwo występowania <strong>grupy</strong><br />
<strong>karbonylowej</strong> z rozdzielonymi ładunkami jest niskie, ale wyjaśnia on podatność tego typu<br />
związków na atak odczynników nukleofilowych i kwasów:<br />
+δ -δ<br />
C<br />
..<br />
O.<br />
.<br />
O.<br />
+ .. -<br />
C O..<br />
:<br />
Dodatkowe podstawniki i <strong>grupy</strong> funkcyjne mogą zwiększać lub zmniejszać polarność tego<br />
wiązania, a tym samym wpływać na wielkość momentu dipolowego cząsteczki i reaktywność<br />
<strong>grupy</strong>. W tabeli przedstawiono wartości momentów dipolowych wybranych związków<br />
zawierających grupę karbonylową.<br />
Wartości momentu dipolowego wybranych związków zawierających Tabela<br />
grupę karbonylową<br />
Nazwa Wzór Grupa związku Moment<br />
dipolowy [D]<br />
metanal H2C=O aldehyd 2,33<br />
etanal CH3HC=O aldehyd 2,72<br />
aceton (CH3)2C=O keton 2,88<br />
acetofenon PhCOCH3 keton 3,02<br />
cyklobutanon<br />
O<br />
keton 2,99<br />
kwas octowy CH3COOH kwas karboksylowy 1,74<br />
chlorek acetylu CH3COCl chlorek kwasowy 2,72<br />
octan metylu CH3COOCH3 ester 1,72<br />
acetamid CH3CONH2 amid 3,72<br />
N,N-dimetyloacetamid<br />
CH3CON(CH3)2 amid 3,81<br />
2
Reaktywność związków karbonylowych<br />
Reaktywność związków zawierających grupę karbonylowa jest związana z właściwościami<br />
elektronowymi atomów wchodzących w jej skład, przede wszystkim z dodatnio naładowanym<br />
karbonylowym atomem węgla C +δ i ujemnie naładowanym atomem tlenu O -δ .<br />
Karbonylowy atom węgla C +δ jest podatny na atak odczynników nukleofilowych (:Nu), a atom<br />
tlenu O -δ na działanie kwasów (A). Powinowactwo karbonylowego atomu węgla do nukleofila<br />
:Nu będzie modyfikowane właściwościami elektronowymi związanej z nim <strong>grupy</strong> funkcyjnej Y.<br />
Grupy elektronoakceptorowe będą powinowactwo zwiększały, a eletronodonorowe –<br />
zmniejszały. Reaktywne są równieŜ atomy wodoru przy Cα. Mają one podwyŜszoną ruchliwość.<br />
Sąsiedztwo <strong>grupy</strong> <strong>karbonylowej</strong> zwiększa ich kwasowość kwaśne w porównaniu z typowym<br />
alifatycznym atomem wodoru, a przez to łatwiej ulegają oderwaniu pod wpływem zasad (B:).<br />
:B<br />
-δ .. A<br />
O:<br />
+δ<br />
C C<br />
Y<br />
H<br />
:Nu<br />
A kwas<br />
:B<br />
zasada<br />
:Nu nukleofil<br />
-δ .. + A<br />
O:<br />
+δ<br />
C C<br />
Y<br />
H<br />
: O<br />
C C<br />
Y<br />
H<br />
.. + A<br />
A – kwasy Lewisa (np.: + H, + NH2, BF3, AlCl3, itp.) wykazując duŜe powinowactwo do<br />
karbonylowego atomu tlenu O tworzą z nim kompleksy.<br />
:B – zasady Lewisa (np.: - H, - NH2, - OH, - OR, NR3, itp.) mając powinowactwo do ruchliwych<br />
protonów odrywają H przy Cα w stosunku do <strong>grupy</strong> <strong>karbonylowej</strong>:<br />
:B<br />
-δ ..<br />
O:<br />
+δ<br />
C C<br />
Y<br />
H<br />
- + BH<br />
..<br />
O:<br />
C C<br />
-<br />
Y<br />
.. -<br />
: O:<br />
C C<br />
Y<br />
↑<br />
Powstający po oderwaniu protonu karboanion jest podatny na atak odczynnika elektrofilowego –<br />
E + ; w ten sposób biegną reakcje kondensacji <strong>karbonylowej</strong>.<br />
:Nu – wykazuje powinowactwo do dodatnio naładowanego węgla <strong>grupy</strong> <strong>karbonylowej</strong> i reakcja z<br />
nim zaczyna się od addycji do karbonylowego atomu węgla. Dalszy bieg reakcji zaleŜy od<br />
właściwości podstawnika Y:<br />
– moŜe nastąpić protonowanie atomu O lub<br />
– eliminacja podstawnika - Y, jeŜeli jest dobrą grupą odchodzącą.<br />
+ H<br />
.. -<br />
: O:<br />
C C<br />
Y<br />
Nu<br />
protonowanie<br />
atomu O<br />
albo<br />
eliminacja<br />
<strong>grupy</strong> Y<br />
3
Do najpopularniejszych reakcji jakim ulegają związki zawierające grupę karbonylową naleŜą:<br />
addycja nukleofilowa<br />
acylowa substytucja nukleofilowa<br />
substytucja α α α α i<br />
kondensacja karbonylowa<br />
Addycja nukleofilowa do <strong>grupy</strong> <strong>karbonylowej</strong><br />
JeŜeli podstawnik <strong>grupy</strong> <strong>karbonylowej</strong> -Y jest złą grupą odchodzącą (-H lub -R) to w reakcji z<br />
nukleofilem dochodzi do addycji nukleofilowej, przy czym zmienia się hybrydyzacja obu<br />
atomów tworzących grupę karbonylową (z sp 2 na sp 3 ), a po sprotonowaniu produktu addycji<br />
(produktu pośredniego) powstaje alkohol. Tak reagują aldehydy i ketony z nukleofili.<br />
.. -<br />
: O:<br />
: O:<br />
: O H<br />
..<br />
C<br />
R Y<br />
:Nu<br />
C<br />
R<br />
Y<br />
Csp2 Csp3<br />
Nu<br />
+ H<br />
R<br />
Y<br />
C<br />
Csp 3<br />
aldehyd lub keton produkt addycji alkohol<br />
R i Y: H, alkil lub aryl Nu: -H (jon wodorkowy) lub związki Grignarda (R ’ MgX)<br />
Substytucja na karbonylowym atomie węgla<br />
Reakcja substytucji na grupie C=O moŜe biec dwoma sposobami (A i B). Wg A w końcowym<br />
produkcie zostaje zachowana grupa C=O, a wg B tworzy się ugrupowanie C=Nu.<br />
Sposób A<br />
Związki karbonylowe zawierające łatwo odchodząca grupę Y, np. -OH, -OR, -SR, -C(O)OR,<br />
lub -halogen po przyłączeniu nukleofila :Nu tworzą produkt pośredni (addukt), z którego<br />
odszczepia się grupa Y i powstaje inny związek zawierający grupę karbonylową, np. kwas<br />
karboksylowy zostaje przekształcony w ester, chlorek kwasy w amid itp. Są to reakcje<br />
charakterystyczne dla kwasów karboksylowych i ich pochodnych. Nie wszystkie przedstawione<br />
powyŜej podstawniki Y naleŜą do grup łatwo odchodzących, ale w odpowiednich warunkach<br />
mogą zostać w takie <strong>grupy</strong> przekształcone, np. -OH czy -OR po protonowaniu do H + OH lub<br />
H + OR stają się grupami łatwo odchodzącymi.<br />
-δ ..<br />
-<br />
: O:<br />
: O:<br />
:Nu<br />
C<br />
C<br />
R Y<br />
R Nu<br />
+ δ<br />
Y<br />
Csp2 Csp3<br />
- Y -<br />
: O:<br />
C<br />
R Nu<br />
Csp 2<br />
R: H, alkil lub aryl Y: halogen; -OR, -OH, -SR, -C(O)OR<br />
Według sposobu A z nukleofilami reagują kwasy karboksylowe i ich pochodne.<br />
- δ<br />
+ δ<br />
Nu<br />
4
Sposób B<br />
Aldehydy i ketony reagują z niektórymi związkami zawierającymi grupę aminową -NH2 w ten<br />
sposób, Ŝe po ich addycji do karbonylowego atomu węgla, następuje eliminacja atomu tlenu w<br />
postaci cząsteczki wody, jako najlepiej odchodzącej <strong>grupy</strong> w powstałym związku przejściowym.<br />
Produktem takiej reakcji jest imina.<br />
-δ ..<br />
..<br />
-<br />
-<br />
δ<br />
: O:<br />
: O:<br />
: O H : N R'<br />
:NH2R' - HOH<br />
C<br />
C + C<br />
R Y<br />
R NH2R' R NHR' C<br />
+ δ<br />
..<br />
Y<br />
Y<br />
R + δ Y<br />
Csp2 Csp3<br />
Csp 3<br />
Csp 2<br />
aldehydy lub ketony iminy<br />
Y: H, alkil lub aryl :NH2R’: aminy 1 o , hydrazyna i inne związki zawierające grupę -NH2<br />
Substytucja α<br />
W reakcji substytucji αααα dochodzi do podstawienia uaktywnionego atomu wodoru przy Cα w<br />
stosunku do <strong>grupy</strong> <strong>karbonylowej</strong>. Miejsce Hα moŜe zająć elektrofil, np. halogen lub alkil.<br />
Reakcja biegnie poprzez formę enolanową.<br />
H<br />
C<br />
: O:<br />
C<br />
E<br />
Y<br />
związek karbonylowy<br />
C<br />
: O:<br />
C<br />
Y<br />
C<br />
..<br />
: O<br />
C<br />
- H +<br />
α-podstawiony związek karbonylowy<br />
..<br />
H : O H<br />
Y<br />
+ E<br />
E<br />
E<br />
C<br />
C + Y<br />
C<br />
+<br />
: O H<br />
Przykładem takiej reakcji moŜe być otrzymywanie α-bromoacetofenonu. Związek ten jest silnym<br />
lakrymatorem (draŜni oczy).<br />
O<br />
C<br />
CH 3<br />
+ Br 2<br />
AcOH<br />
O<br />
C<br />
C<br />
CH 3<br />
Y<br />
+ HBr<br />
acetofenon α-bromoacetofenon (72%)<br />
Reakcje kondensacji związków karbonylowych<br />
W środowisku zasadowym aldehydy i ketony zawierające Hα ulegają dimeryzują.<br />
O O - OH O<br />
OH<br />
CH3CH + CH3CH CH3CHCH2CH etanal etanal 3-hydroksybutanal<br />
5
Mechanizm<br />
Reakcja zaczyna się od oderwania protonu Hα i utworzenia karboanionu, który jako nukleofil<br />
przyłącza się do karbonylowego atomu węgla wg uprzednio poznanych reguł. Powstający enolan<br />
po zakwaszeniu tworzy aldol. Ta nazwa pochodzi od skrótu sporządzone ze słów<br />
aldehydoalkohol.<br />
..<br />
: O<br />
C<br />
H2C H<br />
H<br />
etanal<br />
- HB<br />
:B -<br />
: O<br />
..<br />
- C<br />
H2C.. H<br />
aldol<br />
(3-hydroksybutanal)<br />
O<br />
C H3C H3C H H<br />
C<br />
H 3<br />
.. -<br />
: O :<br />
H<br />
C<br />
CH 2<br />
..<br />
: O H<br />
C<br />
CH 2<br />
O<br />
C<br />
+ H/HOH<br />
Do tego typu reakcji naleŜy kondensacja aldolowa <strong>aldehydów</strong> i <strong>ketonów</strong>, kondensacja Claisena<br />
estrów i szereg innych podobnych reakcji.<br />
A L D E H Y D Y I K E T O N Y<br />
Do <strong>aldehydów</strong> zalicza się związki zawierające przy karbonylowym atomie węgla resztę<br />
organiczną i atom wodoru. Szczególnym przypadkiem jest metanal (aldehyd mrówkowy inaczej<br />
formaldehyd), poniewaŜ z karbonylowym atomem węgla związane są dwa atomy wodoru.<br />
W ketonach do <strong>grupy</strong> <strong>karbonylowej</strong> przyłączone są dwie reszty organiczne.<br />
R<br />
O<br />
C<br />
H<br />
aldehydy, R: alkil, aryl lub H ketony, R, R’: alkil lub aryl<br />
Występowanie<br />
Aldehydy są związkami nietrwałymi, poniewaŜ łatwo ulegają utlenieniu. Dlatego chociaŜ<br />
występują często w naturze, rzadko spotyka się je w duŜych stęŜeniach. Jako składniki niektórych<br />
olejków eterycznych znaleziono: cytronellal, neral (cytral b), geranial (cytral a), aldehyd<br />
benzoesowy (benzaldehyd) czy aldehyd anyŜowy. Znane są teŜ pochodne aldehydu 3,4dihydroksybenzoesowego.<br />
Aldehydy stanowią waŜne substraty w biosyntezie.<br />
CHO CHO CHO<br />
α β<br />
cytranellal neral geranial<br />
H<br />
R<br />
H<br />
O<br />
C<br />
R'<br />
CHO<br />
O<br />
C<br />
H<br />
H<br />
CHO<br />
aldehyd<br />
benzoesowy<br />
6
Do <strong>aldehydów</strong> naleŜy aldosteron, przedstawiciel hormonów kortykosterydowych (hormonów<br />
nadnercza). Obecność <strong>grupy</strong> aldehydowej wyraŜona jest w nazwie związku – aldosteron.<br />
O<br />
HO<br />
OHC<br />
H<br />
H H<br />
COCH 2 OH<br />
aldosteron<br />
Popularnymi aldehydami są cukry – aldozy, np. D-glukoza. W odróŜnieniu od innych <strong>aldehydów</strong><br />
są to związki trwałe, poniewaŜ jako polihydroksyaldehydy tworzą hemiacetale – odporne na<br />
utleniające działanie tlenu z powietrza.<br />
H<br />
HO<br />
H<br />
H<br />
CHO<br />
C<br />
C<br />
C<br />
C<br />
OH<br />
H<br />
OH<br />
OH<br />
CH 2 OH<br />
HO<br />
HO<br />
OH<br />
O<br />
HO<br />
H<br />
OH<br />
D-glukoza hemiacetal<br />
Niezwykle interesującymi (ze względu na moŜliwość praktycznego zastosowania do zwalczania<br />
owadów) są substancje utrudniające Ŝerowanie owadów - IAF (ang. insect antifeedants). Są<br />
pośród nich aldehydy naleŜące do terpenoidów, np. poligodial (tadeonal), ajugaryny (I-V) lub<br />
ajugomaryna. IAF wydzielają niektóre rośliny, Ŝeby chronić się przed Ŝerującymi na nich<br />
owadami, np. przed szarańczom. Jak dotychczas naturalne IAF nie znalazły praktycznego<br />
zastosowania (są zbyt skomplikowane, Ŝeby je syntezować, a w naturze występują w małym<br />
stęŜeniu). Jednak na ich podstawie zaprojektowano prostsze związki o zbliŜonej aktywności.<br />
H 3 C<br />
H<br />
CHO<br />
CHO<br />
H3C H<br />
OCOCH 3<br />
O<br />
CH 3<br />
O CH2 OCOCH 3<br />
poligolial (tadeonal) ajugaryna I<br />
Naturalne ketony są znacznie popularniejsze od <strong>aldehydów</strong>. Nietrudno zauwaŜyć, Ŝe aldosteron<br />
jest takŜe ketonem. Pozostałe kortykosterydy teŜ zawierają grupę lub <strong>grupy</strong> ketonowe. Do<br />
<strong>ketonów</strong> naleŜy większość hormonów płciowych: estron, progesteron, testosteron i androsteron.<br />
H<br />
O<br />
O<br />
H H<br />
O<br />
H H<br />
progesteron testosteron<br />
H<br />
O<br />
OH<br />
7
Pośród terpenoidów spotyka się wiele związków zawierających grupę ketonową; jako przykłady<br />
moŜna podać menton, karwon, tujon czy kamforę:<br />
O<br />
O O<br />
O<br />
(-)-menton (+)-karwon (-)-α-tujon kamfora<br />
Ketonami są substancje zapachowe pochodzenia zwierzęcego, takie jak cybeton i muskon.<br />
O<br />
cybeton muskon<br />
Grupę ketonową zawierają cukry – ketozy, czyli polihydroksyketony, a najpopularniejszą pośród<br />
nich jest D-fruktoza. Ketozy, tak jak i aldozy występują w formie hemiacetalowej.<br />
Nomenklatura<br />
Aldehydów<br />
1.Systematyczna<br />
Nazwy <strong>aldehydów</strong> tworzy się przez dodanie końcówki -al do nazwy węglowodoru<br />
macierzystego.<br />
O<br />
O C<br />
H C H<br />
3<br />
C<br />
H H etanal CH CH 3 2<br />
O<br />
C<br />
H<br />
O<br />
CH CH 2 3<br />
CH3CHCH2CHCHO CH 3<br />
metanal propanal 4-etylo-2-metylopentanal<br />
2. Karboaldehydowa<br />
Inny systematyczny sposób nazywania <strong>aldehydów</strong> polega na dodaniu do nazwy węglowodoru<br />
związanego z grupą -CHO przyrostku karboaldehyd wraz z łącznikiem „o”. Stosowany<br />
najczęściej w nazywaniu skomplikowanych związków.<br />
CHO<br />
H<br />
H<br />
Et H<br />
CHO<br />
cykloheksanokarboaldehyd (1R,2R)-2-etylocyklo-heksano-1-karboaldehyd<br />
3. Podobną rolę pełni przedrostek formylo:<br />
CH 2 CHO<br />
OCHCH 2 CH 2 CHCHCHO<br />
CH 3<br />
OHC COOH<br />
3-(formylometylo)-2-metyloheksanodial kwas 4-formylobenzoesowy<br />
8
4. Odkwasowa<br />
Popularny, półsystematyczny sposób nadawania nazw aldehydom wywodzi się od kwasów<br />
karboksylowych. Te dwie <strong>grupy</strong> związków są z sobą spokrewnione, poniewaŜ utlenienie<br />
<strong>aldehydów</strong> prowadzi do kwasów zawierających tę samą resztę organiczną. Mamy więc aldehyd<br />
mrówkowy (metanal, zwany takŜe formaldehydem) wywodzący się od kwasu mrówkowego,<br />
podobnie aldehyd octowy (etanal), propionowy (propanal), masłowy (butanal), izomasłowy (2metylopropanal)<br />
benzoesowy czy salicylowy.<br />
5. Nazwy zwyczajowe<br />
Niektóre aldehydy znane są prawie wyłącznie pod nazwami zwyczajowymi, np. aldehyd<br />
salicylowy czy anyŜowy.<br />
CHO<br />
(CH CHO<br />
3 ) 2CHCHO H<br />
OH 3CO aldehyd izomasłowy aldehyd anyŜowy<br />
(2-metylopropanal) aldehyd salicylowy (p-metoksybenzoesowy)<br />
(o-hydroksybenzoesowy)<br />
Nomenklatura <strong>ketonów</strong><br />
1. Systematyczna<br />
Zgodnie z zasadami IUPAC nazwy <strong>ketonów</strong> tworzy się przez dodanie do nazwy węglowodoru<br />
macierzystego końcówki „on”:<br />
O O<br />
O CH3 O<br />
C CH<br />
CH3CH=CCH2CCH3 3CCH2CH2CH3 H3C CH3 pentan-2-on<br />
4-metyloheks-4-en-2-on<br />
propanon (aceton) cykloheksanon<br />
2. Grupowo-funkcjonalna<br />
Powszechnie stosowany sposób nazywania <strong>ketonów</strong>, tzw. grupowo-funkcyjny polega na<br />
wymienieniu po słowie keton w porządku alfabetycznym, rozdzielone myślnikiem nazwy reszt<br />
alkilowych (arylowych) związanych z grupą karbonylową,. NaleŜy pamiętać o tym, Ŝeby uŜywać<br />
nazw reszt organicznych w formie przymiotnikowej, tj. np. keton metylowo-propylowy, a nie<br />
metylo-propylowy, gdyŜ w wymowie ta druga forma jest równoznaczna z ketonem<br />
metylopropylowym, a to oznacza inny związek.<br />
C<br />
H 3<br />
O<br />
C<br />
CH 2 CH 2 CH 3<br />
CH 3 CHCH 2<br />
CH 3<br />
O<br />
CH 2 CHCH 3<br />
CH 3<br />
keton metylowo-propylowy (keton metylopropylowy)<br />
keton bis(2-metylopropylowy)<br />
3. Podstawnikowa<br />
Tlen <strong>grupy</strong> ketonowej =O, jako podstawnik nosi nazwę okso i moŜna tego przedrostka uŜywać w<br />
tworzeniu nazw podstawnikowych. Zastępuje ona dawniej uŜywany przedrostek „keto”.<br />
C<br />
9
O CH 2 CH 2 CH 2 COOH<br />
kwas 4(4-oksocykloheksylo)butanowy<br />
O<br />
8<br />
6<br />
9<br />
O<br />
10<br />
5<br />
O<br />
1<br />
4<br />
3<br />
COOH<br />
H<br />
O<br />
kwas 1,3,6,8-tetraokso-1,2,3,6,7,8,-<br />
-heksahydropireno-2-karboksylowy<br />
4. Zwyczajowa<br />
Wiele <strong>ketonów</strong> ma nazwy zwyczajowe. Oprócz wymienionych uprzednio <strong>ketonów</strong> naturalnych<br />
naleŜy znać takie związki jak aceton, acetofenon czy benzofenon:<br />
C<br />
H 3<br />
O<br />
C aceton<br />
CH 3<br />
O<br />
O<br />
acetofenon<br />
C C<br />
CH 3<br />
benzofenon<br />
Właściwości fizykochemiczne<br />
Metanal jest gazem (tw. -19 o C), etanal zaś bardzo lotna cieczą, lotniejszą niŜ eter etylowy (tw.<br />
20,8 o C), a propanal wrze w temp. 48 o C. Metanal i etanal mieszają się z wodą w kaŜdym<br />
stosunku; rozpuszczalność w wodzie wyŜszych <strong>aldehydów</strong> maleje wraz ze wzrostem ich masy<br />
cząsteczkowej; przykładowo propanal, n-butanal i aldehyd benzoesowy rozpuszczają się w<br />
wodzie odpowiednio: 28, 4 i 3 g/100 ml w 20 o C. Aldehydy i ketony rozpuszczają się dobrze w<br />
większości rozpuszczalników organicznych. Wodny roztwór metanalu (25-37%) znany pod<br />
nazwą formaliny zawiera często metanol jako stabilizator. Formalina ma silne działanie<br />
antyseptyczne i słuŜy do przechowywania preparatów organicznych.<br />
Metanal ma bardzo silny, draŜniący i duszący zapach, etanal nieco słabiej, ale teŜ bardzo<br />
intensywnie oddziałuje na zmysł powonienia. Zapach wyŜszych <strong>aldehydów</strong> alifatycznych jest<br />
bardzo nieprzyjemny („smrodliwy”). Benzaldehyd i wiele innych <strong>aldehydów</strong> aromatycznych<br />
charakteryzują się specyficznym zapachem gorzkich migdałów.<br />
Aceton wrze w temperaturze 56 o C i miesza się z wodą w kaŜdym stosunku. Ma<br />
charakterystyczny rozpuszczalnikowy zapach. Aceton jest popularnym rozpuszczalnikiem<br />
stosowanym do rozpuszczania związków organicznych (w tym farb i lakierów).<br />
Otrzymywanie <strong>aldehydów</strong><br />
1. Utlenianie<br />
Alkohole 1 o moŜna przeprowadzić w aldehydy pod wpływem odpowiednio dobranych<br />
utleniaczy, naleŜy jednak zapewnić warunki zabezpieczające przed dalszym utlenieniem do<br />
kwasów, poniewaŜ aldehydy łatwiej się utleniają do kwasów niŜ alkohole do <strong>aldehydów</strong>.<br />
Rekomendowanym utleniaczem alkoholi 1 o do <strong>aldehydów</strong> jest chlorochromian pirydyny – w<br />
skrócie PCC (ang. pyridium chlorochromate). Reakcja biegnie w dichlorometanie w<br />
temperaturze pokojowej (RT – ang. room temperature).<br />
N + CrO3Cl- H<br />
PCC, chlorochromian pirydyny<br />
10
PCC, RT<br />
CH2OH CHO<br />
CH2Cl2 cytronellol cytronellal (82%)<br />
Aldehydy lotniejsze od wody otrzymuje się z odpowiednich alkoholi poprzez ich utlenienie za<br />
pomocą dichromianu sodu we wrzącej wodzie. W tych warunkach powstający aldehyd usuwany<br />
jest z utleniającego środowiska przez oddestylowanie. Przykładem moŜe być przekształcanie nbutanolu<br />
(tw. 118 o C) w n-butanal (tw. 75 o C). Wydajności są jednak duŜo niŜsze niŜ w utlenianiu<br />
za pomocą PCC.<br />
Na 2 Cr 2 O 7 /H 2 SO 4<br />
CH2OH HOH/tw<br />
CHO<br />
n-butanolu n-butanal (32%)<br />
Jedna z przemysłowych metod otrzymywania aldehydu octowego polega na utlenianiu etanolu w<br />
fazie gazowej w obecności katalizatora srebrowego.<br />
500 o C<br />
CH3CH2OH + 0,5 O2 CH<br />
Ag<br />
3CHO + HOH<br />
etanol etanal (90%)<br />
2. Odwodornianie<br />
Alkohole 1 o moŜna odwodornić katalitycznie w obecności, np. katalizatora złoŜonego z miedzi i<br />
tlenku chromu osadzonego na pumeksie. Jest to metoda preferowana w przemyśle.<br />
CH 2 OH<br />
Cu-CrO 3 /pumeks<br />
CHO<br />
- H 2 , 330 o C<br />
n-heksanol n-hesanal (30%)<br />
3.Ozonoliza<br />
Z alkenów zawierających winylowy atom wodoru powstają aldehydy w reakcji ozonolizy.<br />
1. O 3<br />
2. Zn, AcOH 2<br />
dec-5-en n-pentanal<br />
4. Redukcja pochodnych kwasów karboksylowych<br />
Dobrymi substratami w syntezie <strong>aldehydów</strong> są pochodne kwasów karboksylowych, głównie estry<br />
i chlorki kwasowe. Większość reduktorów przeprowadza kwasy karboksylowe i ich pochodne je<br />
w alkohole 1 o . Do otrzymania <strong>aldehydów</strong> potrzebne są reduktory specjalne, umoŜliwiające<br />
selektywną redukcję. NaleŜy do nich hydroglinian diizobutylowy (wodorek didizobutyloglinowy<br />
– DIBAH, ang. diisobutylaluminium hydride).<br />
CH 3 (CH 2 ) 10 COOCH 3<br />
1. DIBAH, toluen,-78 o C<br />
2. H + /HOH<br />
CHO<br />
CH 3 (CH 2 ) 10 CHO<br />
dodekanian metylu dodekanal (88%)<br />
DIBAH: (CH 3 ) 2 CHCH 2 -Al(H)-CH 2 CH(CH 3 ) 2<br />
H<br />
Al<br />
11
5. Redukcja Rosenmunda<br />
Dawniej aldehydy otrzymywało się z chlorków kwasowych w reakcji uwodornienia wobec<br />
zdezaktywowanego katalizatora palladynowego. Metoda ta nazywa się redukcja Rosenmunda.<br />
6. Hydroliza geminalnych dihalogenopochodnych<br />
Zarówno aldehydy, jak i ketony moŜna otrzymać poprzez hydrolizę geminalnych<br />
dihalogenopochodnych. Jest to sposób przydatny wówczas, kiedy takie pochodne są łatwo<br />
dostępne. Do popularnych naleŜy chlorek benzylidenu, otrzymywany w reakcji chlorowania<br />
toluenu. W trakcie jego hydrolizy łatwo powstaje benzaldehyd:<br />
CH 3<br />
Cl 2 , hν<br />
CHCl 2<br />
- OH/HOH<br />
CHO<br />
toluen chlorek benzylidenu aldehyd benzoesowy<br />
7. Hydratacja alkinów<br />
Aldehydy i ketony tworzą się w reakcji addycji wody do alkinów. Reakcja jest katalizowana<br />
przez sole rtęci. Z etynu powstaje etanal, a z pozostałych alkinów ketony. W ten sposób<br />
otrzymywano aldehyd octowy przemysłowo, obecnie dominuje utlenianie etenu tlenem<br />
gazowym.<br />
HgSO 4<br />
HC CH + HOH<br />
CH3CHO etyn H2SO4 /HOH etanal<br />
Otrzymywanie <strong>ketonów</strong><br />
120<br />
CH2 =CH2 + 0,5 O2 oC CH3CHO Pd/CuCl2 eten etanal (94%)<br />
1. Utlenianie<br />
Ketony najłatwiej otrzymać z alkoholi 2 o , nie ma w tym przypadku obawy o ich dalsze<br />
utlenienie. W ten sposób powstaje cykloheksanon z cykloheksanolu.<br />
cykloheksanol<br />
OH<br />
Na 2 Cr 2 O 7 /H 2 SO 4<br />
HOH<br />
Utleniaczem alkoholi 2 o , tak jak 1 o moŜe być PCC.<br />
O<br />
cykloheksanon<br />
W przemyśle cykloheksanon produkuje się przez utlenienie cykloheksanu powietrzem. Ten<br />
sposób otrzymywania <strong>ketonów</strong> jest szczególnie przydatny w syntezie <strong>ketonów</strong> alifatycznoaromatycznych<br />
poprzez utlenianie arenów zawierające odpowiednie łańcuchy boczne.<br />
CH 2<br />
powietrze<br />
O<br />
(79%)<br />
CH C<br />
3 CH3<br />
100 o C<br />
etylobenzen benzofenon<br />
12
2. Ozonoliza<br />
Ozonoliza odpowiednich alkenów prowadzi do <strong>ketonów</strong>:<br />
1. O 3<br />
2. Zn, AcOH<br />
1-metylocykloheksen 6-oksoheptanal (86%)<br />
3. Acylowanie<br />
Niezwykle wygodnym sposobem otrzymywania <strong>ketonów</strong> alifatyczno-aromatycznych lub<br />
aromatycznych jest reakcja acylowania arenów Friedla-Craftsa. Polega ona na działaniu<br />
chlorkami kwasowymi na związki zawierające pierścień aromatyczny.<br />
+<br />
Cl<br />
O<br />
C<br />
AlCl 3<br />
benzen chlorek benzoilu benzofenon (85%)<br />
benzen<br />
+<br />
O<br />
O<br />
O<br />
AlCl 3<br />
bezwodnik<br />
bursztynowy<br />
Δ<br />
O<br />
O<br />
O<br />
O<br />
C<br />
H<br />
C-CH 2 CH 2 COOH<br />
kwas 3-benzoilopropanowy<br />
(94%)<br />
Za pomocą reakcji Friedla-Craftsa moŜna otrzymywać związki cykliczne; w ten sposób z kwasu<br />
fenylobutanowego powstaje tetralon:<br />
Właściwości chemiczne<br />
O (CH ) COOH<br />
2 3<br />
C-CH2CH2COOH NaBH4 kwas 4-fenylo-<br />
kwas 3-benzoilopropanowy<br />
tetralon (79%) O<br />
AlCl 3<br />
butanowy<br />
SOCl 2<br />
ClC=O<br />
(89%)<br />
chlorek<br />
kwasowy<br />
1. Utlenianie<br />
Aldehydy są podatne na działanie utleniaczy bardziej niŜ ketony; nawet pod wpływem słabych<br />
utleniaczy zostają przekształcane w kwasy karboksylowe.<br />
aldehyd<br />
R<br />
C<br />
O<br />
H<br />
[O]<br />
R<br />
C<br />
O<br />
OH<br />
kwas<br />
karboksylowy<br />
Utleniacze: HNO3, KMnO4, O2, Na2CrO7, odczynniki Jonesa, Tollensa, Fehlinga i inne.<br />
13
Odczynnik Jonesa – CrO3/H2SO4/HOH – naleŜy do najłagodniejszch utleniaczy<br />
przekształcających aldehydy w kwasy.<br />
Odczynniki Tollensa – (AgNO3/NH3/HOH) i Fehlinga – (CuSO4/H2SO4/HOH-<br />
NaOH/KNaC4H4O6/HOH) są stosowane w testach do wykrywania <strong>aldehydów</strong>.<br />
O<br />
O<br />
R C<br />
H<br />
+ 2 Ag + R C<br />
OH<br />
+ 2 Ag<br />
aldehyd kwas karboksylowy lustro srebrowe<br />
Odczynnik Tolensa w obecności reduktora typu aldehydu ulega redukcji do srebra metalicznego,<br />
które tworzy lustro, jako Ŝe w postaci cienkiej warstwy osadza się na dnie naczynia szklanego.<br />
Tworzenie się takiego lustra jest testem na obecność aldehydu. Ta reakcja wykorzystywana jest<br />
do produkcji luster.<br />
W próbie Fehlinga obserwuje się pomarańczowy osad wytrącającego się tlenku miedzi (I) pod<br />
wpływem aldehydu.<br />
O<br />
O<br />
R C<br />
H<br />
+ 2 Cu R<br />
OH<br />
2+ aldehyd<br />
C<br />
-OH kwas karboksylowy<br />
+ Cu2O pomarańczowy<br />
osad<br />
Ketony teŜ ulegają utlenieniu, ale jedynie pod wpływem silniejszych utleniaczy, następuje przy<br />
tym rozerwanie wiązania pomiędzy atomem Cα a grupą karbonylową. Z <strong>ketonów</strong> alifatycznych<br />
powstaje mieszanina kwasów karboksylowych.<br />
O<br />
R-CH 2 -C-CH 2 -R'<br />
KMnO 4<br />
- OH/HOH,Δ<br />
RCOOH + RCH 2 COOH +<br />
HOOCR' + HOOCCH 2 R'<br />
keton alifatyczny mieszanina kwasów karboksylowych<br />
Z <strong>ketonów</strong> cyklicznych otrzymuje się jednorodne kwasy dikarboksylowe.<br />
O<br />
1. KMnO4 /NaOH/HOH, Δ<br />
2. H<br />
O<br />
HO<br />
O<br />
OH<br />
+ /HOH<br />
cykloheksanon kwas adypinowy (kwas heksano-1,6-dionowy) (65%)<br />
2. Addycja nukleofilowa<br />
Addycja nukleofilowa do <strong>grupy</strong> <strong>karbonylowej</strong> jest charakterystyczną reakcją <strong>aldehydów</strong> i<br />
<strong>ketonów</strong>. Na atomie węgla <strong>grupy</strong> C=O zlokalizowany jest częściowy ładunek dodatni, a przez to<br />
karbonylowy atom węgla jest podatny na atak nukleofilowy.<br />
O<br />
δ -<br />
C<br />
R R(H)<br />
δ+<br />
:Nu<br />
14
Nukleofilami (Nu: - ) mogą być atomy lub <strong>grupy</strong> obdarzone ładunkiem ujemnym, np.:<br />
RO: -<br />
..<br />
..<br />
HO: -<br />
..<br />
N C:<br />
..<br />
-<br />
R3C: - H: -<br />
aniony: cyjankowy karboanion wodorkowy alkoksylowy hydroksylowy<br />
lub bez formalnego ładunku (cząsteczki obojętne – :Nu-H), ale zawierające elektroujemne atomy<br />
z wolną para elektronową, np.:<br />
RNH2 R2NH ROH<br />
..<br />
.. ..<br />
.. HOH<br />
..<br />
H3N: ..<br />
amoniak amina 1 o amina 2 o alkohol woda<br />
Addycja nukleofilowa do <strong>grupy</strong> <strong>karbonylowej</strong> biegnie jednym z dwóch wariantów: A jedynie<br />
addycja lub B addycja i eliminacja. Kierunek reakcji zaleŜny jest głównie od charakteru<br />
odczynnika nukleofilowego. W obu przypadkach pierwszym etapem jest atak nukleofila na<br />
karbonylowy atom węgla i utworzenie adduktu, przy czym atom ten zmienia hybrydyzację na<br />
sp 3 . W wariancie A addukt stabilizuje się przez protonowanie atomu tlenu, a w drugim<br />
przypadku – B, następuje odszczepione cząsteczki wody (eliminacja) i atom węgla powraca do<br />
poprzedniej hybrydyzacji sp 2 . Odszczepienie cząsteczki wody jest moŜliwe dzięki temu, Ŝe<br />
nukleofil zawiera 2 ruchliwe atomy wodoru.<br />
A<br />
..<br />
: O:<br />
-<br />
C<br />
R Nu<br />
R'<br />
..<br />
: O<br />
H +<br />
H<br />
C<br />
R Nu<br />
R'<br />
: O:<br />
C<br />
R R'<br />
:Nu - :NuNH 2<br />
Rys. Mechanizm addycji nukleofilowej do karbonylowego atomu węgla<br />
aldehyd<br />
lub keton<br />
..<br />
: O:<br />
-<br />
C +<br />
R NuH2 R'<br />
Nu<br />
B<br />
- HOH<br />
C<br />
R R'<br />
15
Addycja nukleofilowa do <strong>grupy</strong> <strong>karbonylowej</strong> <strong>aldehydów</strong> i <strong>ketonów</strong> jest szeroko<br />
wykorzystywana w syntezie chemicznej, moŜna za jej pomocą otrzymywać wiele cennych<br />
produktów. PoniŜszy schemat obrazuje te moŜliwości:<br />
alkohole<br />
OH<br />
R<br />
C C<br />
R<br />
alkeny<br />
C<br />
OR<br />
C<br />
acetale<br />
H<br />
OR<br />
NaBH 4<br />
(Ph) 3 P=C(R) 2<br />
ylidy<br />
:H -<br />
OH<br />
C<br />
alkohole<br />
O<br />
C<br />
H<br />
R<br />
RMgX<br />
H 2 N-Y<br />
aldehydy lub ketony<br />
ROH<br />
C<br />
H<br />
alkany<br />
HCN<br />
Y<br />
N<br />
OH<br />
C<br />
CN<br />
cyjanohydryny<br />
C<br />
1. HSCH 2 CH 2 SH<br />
2. Ni Ra<br />
iminy<br />
oksymy<br />
hydrazony<br />
itp<br />
pochodne azotowe<br />
R 2 NH<br />
C C<br />
enaminy<br />
2.1 Addycja wody – tworzenie geminalnych dioli<br />
W wyniku addycji cząsteczki wody do <strong>grupy</strong> <strong>karbonylowej</strong> powstają geminalne diole, zwane w<br />
skrócie gemdiolami lub gemglikolami (grec. geminalny czyli bliźniaczy; w tym przypadku<br />
odnosi się do dwóch grup na tym samym atomie węgla). Hydratacja <strong>grupy</strong> <strong>karbonylowej</strong> jest<br />
reakcją odwracalną i najczęściej jej równowaga jest przesunięta na korzyść formy <strong>karbonylowej</strong>.<br />
Zdarzają się jednak trwałe gemglikole (hydraty).<br />
O<br />
OH<br />
C + HOH C<br />
R<br />
R R'<br />
R'<br />
OH<br />
R 2 N<br />
forma karbonylowa forma uwodniona (hydrat)<br />
Trwałymi gemdiolami są hydrat metanalu i chloralu. W większości wodnych roztworów<br />
<strong>aldehydów</strong> i <strong>ketonów</strong> udział formy uwodnionej nie przekracza 0,1%, podczas gdy metanal i<br />
chloral występują głównie w formie uwodnionej (99,9%). Hydrat chloralu jest silnym lekiem<br />
usypiającym, o działaniu zbliŜonym do barbituranów. Jego działanie uboczne polega na<br />
draŜniącym działaniu na skórę i błony śluzowe. Stosowany takŜe jako surowiec do produkcji<br />
DDT.<br />
H2C(OH) 2<br />
O<br />
ciecz wrząca<br />
Cl3CC w temp. 97<br />
H<br />
Cl3CC(OH) 2<br />
oC t.t. = 57oC hydrat metanalu chloral hydrat chloralu<br />
16
Hydrat metanalu występuje jedynie w roztworze wodnym, podczas gdy hydrat chloralu jest<br />
trwałym, krystalicznym związkiem.<br />
Występowanie hydratów związków karbonylowych udowodniono na podstawie widm<br />
spektroskopowych, a takŜe za pomocą reakcji izotopowej z wodą zawierającą 18 O. Po pewnym<br />
czasie od rozpuszczenia aldehydu w takiej wodzie izolowano aldehyd zawierający w grupie<br />
C=O tlen 18 O.<br />
RCHO + H 18 OH RCH(OH)( 18 OH) RCH 18 O + HOH<br />
Reakcja hydratacji <strong>aldehydów</strong> i <strong>ketonów</strong> jest katalizowana zarówno zasadowo, jak i kwasowo. W<br />
środowisku obojętnym biegnie wolno.<br />
Mechanizm hydratacji <strong>grupy</strong> <strong>karbonylowej</strong><br />
kataliza zasadowa kataliza kwasowa<br />
..<br />
..<br />
O:<br />
O:<br />
C<br />
C<br />
..<br />
: O:<br />
C ..<br />
OH ..<br />
..<br />
: O<br />
C<br />
- ..<br />
:OH ..<br />
..<br />
HO..<br />
H<br />
H - ..<br />
.. + :OH<br />
..<br />
OH ..<br />
H 3 O +<br />
+<br />
.. +<br />
O H<br />
C<br />
..<br />
: O<br />
C<br />
H<br />
..<br />
O<br />
H<br />
.. +<br />
H H 2O.. : O H<br />
C<br />
..<br />
+<br />
..<br />
: O<br />
C<br />
..<br />
H2O.. ..<br />
H2O.. H<br />
.. H<br />
O<br />
+<br />
H<br />
2.2 Addycja cząsteczki alkoholu - tworzenie hemiacetali, acetali i tioacetali<br />
Podobnie jak cząsteczka wody do <strong>grupy</strong> <strong>karbonylowej</strong> przyłącza się cząsteczka alkoholu, a<br />
produkt takiej addycji nosi nazwę hemiacetalu (grec. hemi = w połowie). Dawniej tego typu<br />
związki nazywano półacetalami, dla odróŜnienia od acetali (pełnych acetali). Reakcja jest<br />
katalizowana kwasami.<br />
17
..<br />
O:<br />
C<br />
aldehyd<br />
lub keton<br />
H +<br />
.. +<br />
O H<br />
C<br />
hemiacetal<br />
: O H<br />
C<br />
..<br />
+<br />
..<br />
: OH<br />
C ..<br />
OR ..<br />
..<br />
ROH ..<br />
- H +<br />
..<br />
: O H<br />
C .. H<br />
O<br />
+<br />
R<br />
Zwykle hemiacetale są nietrwałe i występują jedynie w roztworze alkoholowym w stanie<br />
równowagi z formą karbonylową. W środowisku kwaśnym reagują dalej z alkoholem w kierunku<br />
acetali. Acetale pochodzące od <strong>ketonów</strong> nazywa się często ketalami.<br />
Mechanizm<br />
Reakcja tworzenia acetali zaczyna się od protonowania <strong>grupy</strong> -OH, eliminacji cząsteczki wody,<br />
przyłączenia cząsteczki alkoholu do atomu węgla i kończy się stabilizacją przez odszczepienie<br />
protonu:<br />
..<br />
: OH<br />
C ..<br />
OR ..<br />
hemiacetal<br />
..<br />
: O<br />
R<br />
O R C<br />
..<br />
acetal<br />
H +<br />
- H +<br />
H<br />
H<br />
..<br />
O<br />
H<br />
+<br />
C<br />
O..<br />
R<br />
-HOH<br />
O<br />
O R<br />
..<br />
..<br />
R ROH ..<br />
+<br />
C<br />
..<br />
O R<br />
C +<br />
..<br />
..<br />
O R<br />
C +<br />
..<br />
ChociaŜ większość hemiacetali występuje jedynie w roztworach alkoholowych w równowadze<br />
ze związkami karbonylowymi, to hemiacetale wewnątrzcząsteczkowe, do których naleŜą cukry<br />
są trwałe. W cukrach równowaga przesunięta jest na korzyść hemiacetali.<br />
H<br />
HO<br />
H<br />
H<br />
CHO<br />
C<br />
C<br />
C<br />
C<br />
OH<br />
H<br />
OH<br />
OH<br />
CH 2 OH<br />
H<br />
HO<br />
H<br />
H<br />
OH<br />
HC<br />
C<br />
C<br />
C<br />
C<br />
OH<br />
H<br />
OH<br />
CH 2 OH<br />
O<br />
HO<br />
HO<br />
OH<br />
O<br />
HO<br />
D-glukoza<br />
Wiązanie hemiacetalowe w D-glukozie tworzy się pomiędzy grupą -OH przy C5 i grupą<br />
aldehydową. Cząsteczka glukozy przyjmuje konformację krzesłową. Acetale utworzone z cukrów<br />
nazywają się glikozydami. Występują powszechnie w przyrodzie.<br />
Synteza acetali polega na ogrzewaniu <strong>aldehydów</strong> lub <strong>ketonów</strong> z bezwodnym alkoholem w<br />
kwaśnym środowisku. Ulegają one łatwo hydrolizie w środowisku kwaśnym, a są odporne na<br />
hydrolizę zasadową. Stosuje się je do czasowej osłony <strong>grupy</strong> <strong>karbonylowej</strong>, tak Ŝeby moŜna<br />
H<br />
OH<br />
18
yło przeprowadzić reakcję w innej części cząsteczki, chroniąc grupę karbonylową, np. przed<br />
utlenieniem lub redukcją. Po zaplanowanych przekształceniach łatwo odzyskuje się ją w reakcji<br />
hydrolizy kwaśnej. Często dla ochrony funkcji <strong>karbonylowej</strong> uŜywa się glikolu etylowego,<br />
poniewaŜ z niego wyjątkowo łatwo tworzy się acetal cykliczny.<br />
O<br />
O<br />
O<br />
O<br />
4-oksopentanian etylu<br />
OH<br />
4-oksopent-1-ol<br />
-<br />
HO OH<br />
H +<br />
+ H/HOH<br />
HO OH<br />
O<br />
O<br />
O<br />
O<br />
1. LiAlH 4<br />
O<br />
O<br />
O<br />
2. + H/HOH<br />
OH<br />
+ EtOH<br />
Selektywna redukcja przedstawiona na powyŜszym schemacie byłaby niemoŜliwa do<br />
przeprowadzenia bez czasowej ochrony <strong>grupy</strong> ketonowej.<br />
Podobną rolę pełnią tioacetale, które tworzą się jeszcze łatwiej, a ulegają usunięciu, np. podczas<br />
katalitycznej redukcji. Warto wiedzieć, Ŝe związki siarki dezaktywują katalizatory typu Pt czy<br />
Pd, ale nie nikiel Renaya (NiRa). W ten sposób moŜna całkowicie zredukować grupę<br />
karbonylową, czyli do węglowodoru.<br />
O 2 RSH RS SR<br />
C<br />
C<br />
+ H<br />
Ni Ra<br />
EtOH<br />
CH 2<br />
aldehyd lub keton tioacetal węglowodór<br />
Do osłony <strong>grupy</strong> <strong>karbonylowej</strong> moŜna teŜ uŜyć tioglikolu etylenowego.<br />
O<br />
HS SH<br />
BF 3<br />
S<br />
S<br />
Ni Ra<br />
+ NiS<br />
4-metylocykloheksanon tioacetal 4-metylocykloheksan<br />
2.3 Reakcje <strong>aldehydów</strong> i <strong>ketonów</strong> ze związkami Grignarda<br />
Związki Grignarda reagują z grupą karbonylową w ten sposób, Ŝe ujemnie naładowana reszta<br />
organiczna R przyłącza się do karbonylowego atomu węgla, a -MgX tworzy wiązanie<br />
alkoholanowe z atomem tlenu. Po hydrolizie alkoholanu otrzymuje się alkohole.<br />
O<br />
: O:<br />
MgX<br />
R<br />
.. - +<br />
-δ : :<br />
MgX +H/HOH<br />
+δ C + R C<br />
-δ +δ<br />
: OH<br />
..<br />
związek Grignarda<br />
aldehyd lub keton alkoholan alkohol<br />
C<br />
R<br />
19
W reakcji związków Grignarda z metanalem powstają alkohole 1 o , z innymi aldehydami –<br />
alkohole 2 o , a z ketonami – alkohole 3 o .<br />
C<br />
H 3<br />
bromek<br />
cyklopentylu<br />
H<br />
C=O<br />
Br<br />
Mg<br />
eter<br />
H<br />
MgBr<br />
bromek cyklopentylowomagnezowy<br />
cyklopentylometanol (48%)<br />
(alkohol 1 o )<br />
H<br />
C<br />
H<br />
- +<br />
O MgBr<br />
+ H/HOH<br />
CH 2 OH<br />
CH3 CH<br />
O<br />
3 OH<br />
C MgCl + C H3C C C<br />
H<br />
H<br />
CH 3<br />
CH 3<br />
bromek-t-butylo - benzaldehyd 1-fenylo-2,2-dimetylo-<br />
magnezowy propanol (alkohol 2 o )<br />
CH 3<br />
MgI + O<br />
IMgO<br />
C<br />
H 3<br />
+H/HOH<br />
HO<br />
C<br />
H 3<br />
jodek metylo- cyklopentanon 1-metylocyklopentanol<br />
magnezowy (alkohol 3 o )<br />
2.4 Addycja nukleofilowa HCN; otrzymywanie cyjanohydryn<br />
Cyjanowodór przyłącza się do <strong>grupy</strong> <strong>karbonylowej</strong> <strong>aldehydów</strong> i <strong>ketonów</strong> jeŜeli nie ma zawady<br />
przestrzennej wokół tej <strong>grupy</strong>. Produktem reakcji są cyjanohydryny (inaczej nitryle ααααhydroksykwasów),<br />
czyli związki zawierające funkcję -CN i -OH przy tym samym atomie węgla.<br />
W wyniku addycji cyjanowodoru do benzaldehydu powstaje nitryl kwasu migdałowego:<br />
O<br />
OH<br />
C<br />
H<br />
HC N<br />
C<br />
H<br />
CN<br />
benzaldehyd nitryl kwasu migdałowego (cyjanohydryna) (88%)<br />
Addycja cyjanowodoru do <strong>aldehydów</strong> lub <strong>ketonów</strong> jest reakcją odwracalną, zwykle z równowagą<br />
przesuniętą na korzyść produktu. Reakcję ułatwia kataliza zasadowa, gdyŜ jon - CN przyłącza się<br />
łatwiej niŜ HCN. Jon cyjankowy jest silnym nukleofilem i dlatego równowaga jego addycji do<br />
<strong>grupy</strong> <strong>karbonylowej</strong> jest przesunięta w stronę produktów. Inne kwasy, np. halogenowodory,<br />
kwas siarkowy czy kwasy karboksylowe są słabymi nukleofilami i nie tworzą adduktów ze<br />
związkami karbonylowymi.<br />
20
W reakcji addycji cyjanowodoru do <strong>grupy</strong> <strong>karbonylowej</strong> wystarczają śladowe ilości zasady,<br />
poniewaŜ w warunkach równowagi jon cyjankowy odtwarza się:<br />
..<br />
.. -<br />
O:<br />
: O:<br />
C<br />
H<br />
: C -<br />
N<br />
C<br />
H<br />
CN<br />
HCN<br />
OH<br />
C<br />
+<br />
H<br />
CN<br />
aldehyd produkt przejściowy cyjanohydryna<br />
Cyjanohydryny stanowią dogodny półprodukt w syntezie organicznej. Poprzez ich redukcję<br />
moŜna otrzymać ββββ-aminoalkohole, a w wyniku hydrolizy αααα-hydroksykwasy.<br />
OH<br />
H CN<br />
C<br />
cyjanohydryna<br />
benzaldehydu<br />
1. LiAlH 4 ,THF<br />
2. HOH<br />
+ H/HOH<br />
lub - Δ<br />
OH/HOH<br />
OH<br />
C<br />
:C -<br />
H CH2NH2 2-amino-1-fenyloetanol<br />
OH<br />
N<br />
C<br />
COOH<br />
H kwas migdałowy<br />
2.5 Addycja jonu wodorkowego – redukcja wodorkami<br />
Wodorki typu NaBH4 lub LiAlH4 redukują aldehydy i ketony do odpowiednich alkoholi, przy<br />
czym czynnikiem redukującym jest anion wodorkowy – H - .<br />
Na + BH 4 - Na + BH 3 + :H -<br />
tetrahydroboran sodu anion wodorkowy<br />
Szczególnie przydatnym okazał się tetrahydroboran sodu, poniewaŜ jest trwalszy (odporniejszy<br />
na hydrolizę) od LiAlH4, działa selektywnie (nie redukuje kwasów karboksylowych i ich<br />
pochodnych), a reakcję moŜna prowadzić w metanolu, nawet uwodnionym.<br />
: O:<br />
:H- C<br />
R R'<br />
NaBH 4 lub<br />
LiAlH 4<br />
aldehyd lub keton<br />
R, R': H, alkil lub aryl<br />
R R'<br />
.. -<br />
: O:<br />
C<br />
H<br />
R<br />
.. -<br />
: O:<br />
+ C<br />
H R<br />
R'<br />
OH<br />
C<br />
R'<br />
+ H/HOH<br />
H<br />
alkohol<br />
3. Reakcja Cannizzaro<br />
Jest to reakcja dysproporcjonowania, w wyniku której z aldehydu tworzy się alkohol (redukcja) i<br />
kwas karboksylowy (utlenienie). Zachodzi ona w środowisku alkalicznym, a ulegają jej aldehydy<br />
nie zawierające atomów wodoru przy Cα, np. metanal i aldehydy aromatyczne.<br />
21
CHO 1. CH2OH COOH<br />
2 +<br />
-OH/HOH 2. H + /HOH (86%) (79%)<br />
benzaldehyd alkohol benzylowy kwas benzoesowy<br />
Charakterystyczną cechą tej reakcji jest to, Ŝe bierze w niej udział anion wodorkowy. W<br />
pierwszym etapie reakcji grupa hydroksylowa ulega addycji do karbonylowego atomu węgla, po<br />
czym anion wodorkowy, jako nukleofil atakuje grupę karbonylową drugiej cząsteczki aldehydu.<br />
Następnie proton z kwasu karboksylowego (silniejszy kwas) przechodzi do alkoholanowego<br />
atomu tlenu:<br />
..<br />
O:<br />
C<br />
H<br />
- ..<br />
: OH ..<br />
- ..<br />
: O:<br />
C H<br />
OH<br />
benzaldehyd<br />
O<br />
C .. -<br />
O..<br />
:<br />
..<br />
O:<br />
C<br />
H<br />
CH 2 OH<br />
O<br />
C<br />
+<br />
- ..<br />
: O:<br />
C H<br />
H<br />
anion benzoesanowy alkohol benzylowy<br />
+<br />
ChociaŜ produkt reakcji Cannizzaro jest złoŜony (kwas i alkohol), to łatwo udaje się go<br />
rozdzielić, poniewaŜ oba składniki róŜnią się właściwościami chemicznymi i fizycznymi. Z<br />
zasadowego, wodnego środowiska alkohol wyekstrahuje się rozpuszczalnikiem organicznym, nie<br />
mieszającym się z wodą, a kwas wytrąca z wodnego roztworu jego soli przez zakwaszenie. Kwas<br />
moŜna izolować teŜ za pomocą jonitów.<br />
Pomimo tych ułatwień nie jest to szeroko stosowana metoda otrzymywania ani kwasu<br />
benzoesowego, ani alkoholu benzylowego, poniewaŜ znane są lepsze sposoby syntezy tych<br />
związków. Większe znaczenie preparatywne ma tzw. krzyŜowa reakcja Cannizzaro, w której<br />
wykorzystuje się fakt, Ŝe w obecności <strong>aldehydów</strong> aromatycznych metanal ulega głównie<br />
utlenieniu do mrówczanu, natomiast większa część aldehydu aromatycznego ulega redukcji<br />
alkoholu aromatycznego. Ta róŜnica w reaktywności spowodowana jest większą<br />
elektrofilowością karbonylowego atomu węgla metanalu – jest on bardziej podatny na atak reszty<br />
hydroksylowej niŜ karbonylowy atom aldehydu aromatycznego.<br />
CHO<br />
CH 3<br />
+<br />
HCHO<br />
KOH<br />
CH 2 OH<br />
CH 3<br />
OH<br />
+ HCOO - K +<br />
metanal mrówczan potasu<br />
aldehyd p-toluilowy alkohol p-metybenzylowy (72%)<br />
Z aldehydu ftalowego w reakcji Cannizzaro powstaje kwas (hydroksymetylo)benzoesowy.<br />
22
CHO<br />
1. KOH<br />
2. + H/HOH<br />
COOH<br />
CHO CH 2 OH<br />
aldehyd o-ftalowy kwas o-(hydroksymetylo)benzoesowy<br />
W najwaŜniejszym procesie redukcji biochemicznej, z udziałem zredukowanego dinukleotyd<br />
nikotynoamidoadeninowy (NADH), widoczny jest mechanizm reakcji Cannizzaro:<br />
N<br />
H 2<br />
R''<br />
N<br />
R<br />
O<br />
R' H2N R''<br />
N<br />
R R'<br />
O H H<br />
O H H<br />
H<br />
C<br />
..<br />
+<br />
..<br />
:<br />
C<br />
C<br />
+<br />
+<br />
OH<br />
C<br />
zw. karbonylowy<br />
hydroksyzwiązek<br />
NADH NAD + R’’: reszta cukrowo-fosforanowa<br />
4. Reakcje kondensacji<br />
4.1 Kondensacja aldolowa<br />
Aldehydy i ketony zawierające w atom wodoru przy Cα ulegają kondensacji katalizowanej przez<br />
zasady i kwasy. W reakcji tej powstają dimery – aldehydoalkohole, dlatego została ona nazwana<br />
kondensacją aldolową a jej najprostszy produkt dimeryzacji etanalu zwany jest aldolem.<br />
2 CH 3 CH<br />
O OH O<br />
NaOEt<br />
CH 3 CH-CH 2 CH<br />
EtOH<br />
acetaldehyd aldol (3-hydroksybutanal)<br />
W odracalnej reakcji kondensacji aldolowej dla monopodstawionych <strong>aldehydów</strong> (RCH2CHO)<br />
równowaga jest przesunięta na prawo, zaś dla rozgałęzionych <strong>aldehydów</strong> i większości <strong>ketonów</strong><br />
na lewo. Z aldehydami biegnie ona z większą szybkością. Zwykle w warunkach reakcji tworzą<br />
się nienasycone aldehydy lub ketony, poniewaŜ w produktach dochodzi do eliminacji cząsteczki<br />
wody. ββββ-podstawione aldehydy i ketony są bardzo podatne na eliminację. Dehydratacja<br />
powoduje przesunięcie równowagi reakcji w kierunku dimerów.<br />
Z podatności produktów do dehydratacji reakcja ta powszechnie nazywana jest kondensacją, a<br />
nie jak powinna – dimeryzacją. Kondensacja bowiem to reakcja dimeryzacji (oligomeryzacji lub<br />
polimeryzacji), w której obok głównego produktu wydzielają się małe cząsteczki – w tym<br />
wypadku wody.<br />
Kondensacja aldolowa polega na addycji nukleofilowej do karbonylowego atomu węgla anionu<br />
powstałego w wyniku oderwania protonu z Cα.<br />
23
.. -<br />
HO:<br />
+<br />
O<br />
H-CH2-CH : O<br />
-<br />
H2C-CH ..<br />
..<br />
: O:<br />
H2C=CH -<br />
acetaldehyd<br />
- HOH<br />
:<br />
.. -<br />
: O:<br />
OH<br />
O<br />
O<br />
CH 3 -CH-CH 2 -CH<br />
..<br />
HOH ..<br />
- HO -<br />
aldol CH3-CH-CH2-CH - HOH<br />
O<br />
CH3CH aldehyd krotonowy<br />
(but-2-enal)<br />
O<br />
CH 3 CH=CH-CH<br />
Podobnie wygląda kondensacja aldolowa acetonu, jednak wydajność tej reakcji jest niewielka:<br />
Mechanizm:<br />
O OH<br />
H 3 C-C-CH 3<br />
aceton<br />
- OH<br />
CH 3<br />
O<br />
H 3 C-C-CH 2 -C-CH 3<br />
4-hydroksy-<br />
- 4-metylopent-2-on<br />
(5%)<br />
O<br />
H-H2C-C-CH3 aceton<br />
..-<br />
: OH ..<br />
O<br />
-<br />
H2C-C-CH .. 3<br />
- ..<br />
: O:<br />
H2C=C-CH3 OH<br />
H 3 C-C-CH 2 -C-CH 3<br />
CH 3<br />
C<br />
H 3<br />
4-hydroksy-4-metylopent-2-on<br />
O<br />
C<br />
CH 3<br />
O : O:<br />
.. -<br />
HOH<br />
- - OH<br />
CH 3<br />
O<br />
H 3 C-C-CH 2 -C-CH 3<br />
Wydajność kondensacji aldolowej acetonu moŜna zwiększyć przez usuwanie produktu ze<br />
środowiska reakcji, tzn. poprzez odizolowanie go od alkalicznego katalizatora. Tę reakcję moŜna<br />
prowadzić w kolumnie wypełnionej, np. kawałkami Ba(OH)2. W kolumnie aceton spływający z<br />
chłodnicy zwrotnej ulega dimeryzacji w zetknięciu z Ba(OH)2 i spływa do kolby zawierając<br />
kilka procent 4-hydroksy-4-metylopentan-2-onu. Aceton w kolbie wrze, a jego pary skraplając<br />
się w chłodnicy zwrotnej ponownie przepływają przez kolumnę z Ba(OH)2, podczas czego<br />
kolejna porcja acetonu ulegnie dimeryzacji. Ten proces trwa aŜ do wyczerpania acetonu, a w<br />
kolbie pozostaje prawie czysty produkt.<br />
W cykloheksanonie zawada przestrzenna jest mniejsza niŜ w acetonie (sztywny układ) i dzięki<br />
temu wydajność jego dimeryzacji jest kilka razy większa (22%):<br />
2<br />
O<br />
NaOH<br />
OH<br />
O<br />
cykloheksanon 1-hydroksy-2’-oksodicykloheksyl<br />
24
Kondensację aldolową stosuje się często w syntezie róŜnorodnych związków organicznych,<br />
zarówno w laboratorium, jak i w skali technicznej. Powstające w reakcji aldole moŜna poddawać<br />
dehydratacji, dalszej kondensacji aldolowej lub selektywnej redukcji.<br />
O OH O OH<br />
RCH2CH NaOH<br />
RCH2CHCHCH NaBH4 2<br />
RCH2CHCHCH2OH aldehyd aldol<br />
R<br />
R<br />
- HOH<br />
RCH2CH=CCH H2 /Ni<br />
R<br />
aldehyd<br />
αααα,ββββ-nienasycony<br />
RCH2CH2CHCH2OH alkohol<br />
O<br />
R<br />
O<br />
1,3-diol<br />
R<br />
RCH 2 CH=CCH 2 OH<br />
H R<br />
2 /Pd/C alkohol allilowy<br />
RCH 2 CH 2 CHCH<br />
aldehyd<br />
Kondensacja aldolowa jest szeroko stosowana w przemyśle, szczególnie do otrzymywania<br />
alkoholi. Tą drogą produkuje się butan-1-ol.<br />
NaBH 4<br />
Zadanie: zaproponuj schemat reakcji prowadzących do butan-1-olu.<br />
Dehydratacja ββββ-hydroksy<strong>aldehydów</strong> i <strong>ketonów</strong> jest charakterystyczną reakcją tych związków.<br />
Często juŜ w warunkach kondensacji aldolowej dochodzi do dehydratacji, jeŜeli nie zachowuje<br />
się specjalnego reŜimu (niskiej temperatury). Powstają przy tym αααα,ββββ-nienasycone aldehydy lub<br />
ketony, zwane inaczej sprzęŜonymi enonami. Reakcja dehydratacji ββββ-hydroksy<strong>aldehydów</strong> i<br />
<strong>ketonów</strong> jest katalizowana zarówno przez kwasy, jak i zasady. Usunięcie cząsteczki wody z<br />
produktu kondensacji aldolowej przesuwa równowagę reakcji na prawo i w ten sposób otrzymuje<br />
się sprzęŜone enony z wysoką wydajnością, pomimo tego Ŝe równowaga samej reakcji nie<br />
sprzyja dimeryzacji. W warunkach równowagi z cykloheksanonu powstaje jedynie 22% produktu<br />
dimeryzacji, ale w wyniku dehydratacji tworzy się ponad 90% sprzęŜonego enonu, co świadczy<br />
o prawie całkowitym zuŜyciu substratu (ilościowej dimeryzacji wyjściowego ketonu).<br />
2<br />
O<br />
cykloheksanon<br />
NaOH<br />
OH<br />
O<br />
O<br />
+ HOH<br />
1-hydroksy-2’oksodicykloheksyl cykloheksylideno-cykloheksan-2-on (90%)<br />
Jak uprzednio wspomniano produkt kondensacji aldolowej acetonu – 4-hydroksy-4-metylopenta-<br />
2-on powstaje w reakcji równowagowej jedynie z wydajnością 5%. JeŜeli jednak reakcja będzie<br />
prowadzona w temperaturze powyŜej 50 o C, to z dobrą wydajnością powstanie sprzęŜony enon,<br />
zwany tlenkiem mezytylu.<br />
25
O OH<br />
H 3 C-C-CH 3<br />
aceton<br />
Ba(OH) 2<br />
CH 3<br />
O<br />
H 3 C-C-CH 2 -C-CH 3<br />
50o C<br />
C<br />
H 3<br />
C<br />
H 3<br />
O<br />
C=C C<br />
H<br />
CH 3<br />
4-hydroksy-4-metylopenta-2-on (5%) tlenek mezytylu (95%)<br />
Reakcja podobna do kondensacji aldolowej zachodzi równieŜ w środowisku kwaśnym. Aceton<br />
po nasyceniu chlorowodorem ulega przekształceniu w tlenek mezytylu; równocześnie tworzy się<br />
produkt trimeryzacji i dehydratacji, nazywany foronem.<br />
C<br />
H 3<br />
C<br />
H 3<br />
O<br />
CH 3<br />
C=C<br />
H H CH3 C C=C foron<br />
Zadanie: napisz etapy powstawania foronu i jego cyklizacji do mezytylenu, który tworzy się z<br />
acetonu pod wpływem stęŜonego kwasu siarkowego.<br />
4.2 Mieszane (krzyŜowe) reakcje kondensacji aldolowej<br />
W reakcji kondensacji aldolowej dwóch róŜnych <strong>aldehydów</strong>, np. etanalu i propanalu powstaną 4<br />
produkty dimeryzacji: dwa symetryczne w wyniku połączenia się wzajemnego 2 cząsteczek<br />
etanalu i 2 cząsteczek propanalu oraz dwa jako efekt dimeryzacji mieszanej: (etanal + propanal) i<br />
(propanal + etanal).<br />
CH 3 CHO<br />
etanal<br />
+<br />
CH 3 CH 2 CHO<br />
propanal<br />
:B -<br />
OH<br />
CH 3 CHCH 2 CHO<br />
3-hydroksybutanal<br />
OH<br />
CH3CH2CHCHCHO CH 3<br />
3-hydroksy-2-metylopentanal<br />
OH<br />
CH3CHCHCHO CH 3<br />
3-hydroksy-2-metylobutanal<br />
OH<br />
CH 3 CH 2 CHCH 2 CHO<br />
3-hydroksypentanal<br />
symetryczne<br />
niesymetryczne<br />
Reakcja, w której powstają 4 róŜne produkty o podobnych właściwościach fizycznych i<br />
chemicznych jest mało przydatna, nie tylko dlatego, Ŝe wydajności poszczególnych związków są<br />
niskie, ale równieŜ z tego powodu, Ŝe trudno je rozdzielić. Syntezę chemiczną naleŜy tak<br />
prowadzić, Ŝeby z maksymalnie wysoką wydajnością otrzymywać jeden czysty produkt.<br />
Wobec tego wydawać się moŜe, Ŝe mieszane kondensacje aldolowe są praktycznie nieprzydatne.<br />
JeŜeli jednak w reakcji będzie brał udział jeden z substratów, który nie zawiera atomu wodoru<br />
26
przy Cα, to teoretycznie powstaną dwa produkty: symetryczny i mieszany. Jest to sytuacja<br />
znacznie korzystniejsza. Ponadto tę reakcję moŜna tak prowadzić, Ŝeby zmniejszyć wydajność<br />
dimeru symetrycznego, który w takiej reakcji jest produktem ubocznym (niepoŜądanym).<br />
ZałoŜony cel osiągną się przez wkraplanie substratu zawierającego Hα (A) do alkalicznego<br />
środowiska reakcji, w którym znajduje się składnik bez Hα (B). W ten sposób substrat A dopiero<br />
w środowisku zasadowym moŜe autokondesować, spotyka jest jednak ze znacznym nadmiarem<br />
składnika B i wobec tego powstaje głównie produkt mieszany. Tworzeniu się produktu<br />
mieszanego sprzyja równieŜ to, Ŝe grupa karbonylowa składnika B jest często silniejszym<br />
elektrofilem niŜ reagenta A, np. <strong>grupy</strong> karbonylowe benzaldehydu czy metanalu są silniejszymi<br />
elektrofilami niŜ większość innych <strong>aldehydów</strong>, nie wspominając o ketonach. Substrat A<br />
nazywany jest często donorem (eletronów), a B akceptorem.<br />
Z benzaldehydu i etanalu otrzymuje się w ten sposób aldehyd cynamonowy (główny składnik<br />
aromatu cynamonowego), a z benzaldehydu i propanalu powstaje aldehyd αmetylocynamonowy.<br />
O O<br />
CH<br />
+ CH 3 CH 2 CH<br />
NaOH<br />
10 o C<br />
CH<br />
O<br />
CCH<br />
CH 3<br />
benzaldehyd B propanal A aldehyd α-metylocynamonowy (80%)<br />
(akceptor) (donor)<br />
Zadanie: wyjaśnij dlaczego naleŜy roztwór propanalu wkraplać do intensywnie mieszanego roztworu benzaldehydu<br />
w środowisku zasadowym, a nie odwrotnie.<br />
Jako inny przykład krzyŜowej kondensacji aldolowej moŜe słuŜyć reakcja benzaldehydu z<br />
ketonem, np. metylocykloheksanonem.<br />
CHO O<br />
+<br />
CH 3<br />
NaOEt<br />
- HOH<br />
benzaldehyd 2-metylocyloheksanon 2-benzylideno-6-metylocykloheksanon (78%)<br />
W reakcji benzaldehydu z acetonem powstaje benzylidenoaceton.<br />
CHO<br />
O<br />
+ H3C C<br />
beznaldehyd aceton<br />
- HOH<br />
CH3 O<br />
CH CH<br />
O<br />
CH 3<br />
NaOH CCH 3<br />
benzylidenoaceton<br />
(77%)<br />
Benzylidenoaceton moŜna przekształcić w dibenzylidenoaceton przez traktowanie go kolejną<br />
porcją benzaldehydu.<br />
27
CH CH<br />
O<br />
CCH 3<br />
benzylidenoaceton<br />
NaOH<br />
+<br />
O<br />
O<br />
HC<br />
- HOH<br />
CH CHCCH=CH benzaldehyd<br />
dibenzylidenoaceton<br />
W krzyŜowej kondensacji aldolowej często wykorzystywanym akceptorem jest metanal. W<br />
reakcji z etanalem powstaje produkt addycji trzech cząsteczek metanalu do etanalu, po czym<br />
następuje spontaniczna redukcja (nadmiarem metanalu) do polialkoholu zwanego<br />
pentaerytrytolem; preparat uŜywany w leczeniu zaparć, a jego ester z kwasem azotowym –<br />
tetraazotan pentaerytrytolu – podobnie jak nitrogliceryna rozszerza naczynia wieńcowe i<br />
stosowany jest leczniczo oraz zapobiegawczo w przypadku dusznicy bolesnej. Działa on dłuŜej<br />
niŜ nitrogliceryna. Tetraazotan pentaerytrytolu równieŜ jak nitrogliceryna jest silnym materiałem<br />
wybuchowym.<br />
O<br />
3 HCH + C<br />
H<br />
H<br />
CHO<br />
H<br />
metanal etanal<br />
Ca(OH) 2<br />
krzyŜowa reakcja<br />
Cannizzaro<br />
pentaerytrytol<br />
(55%)<br />
HOCH 2<br />
- HCOOH<br />
HO<br />
HO<br />
(77%)<br />
CHO<br />
C<br />
CH 2 OH<br />
CH 2 OH<br />
(trihydroksymetylo)etanal<br />
HCHO<br />
Wysoką wydajność jednego tylko produktu w krzyŜowych kondensacjach aldolowych zapewnia<br />
uŜycie jako donora związku posiadającego bardziej kwaśne atomy wodoru niŜ Hα<br />
<strong>aldehydów</strong> i <strong>ketonów</strong>. Do takich związków naleŜy, np. acetylooctan etylu. Dwie <strong>grupy</strong><br />
karbonylowe mocniej uaktywniają atomy wodoru i przez to łatwiej ulegają oderwaniu tworząc<br />
karboanion, który przyłącza się do <strong>grupy</strong> <strong>karbonylowej</strong> akceptora.<br />
O<br />
O O<br />
+ CH 3 CCH 2 COEt<br />
acetylooctan etylu<br />
NaOEt<br />
- HOH<br />
OH<br />
OH<br />
O O<br />
cykloheksan (donor) cykloheksylidenoacetylooctan etylu (80%)<br />
(akceptor)<br />
4.3 Wewnątrzcząsteczkowa kondensacja aldolowa – reakcje cyklizacji<br />
Cząsteczki zawierające grupę akceptorową i donorową mogą ulegać wewnątrzcząsteczkowej<br />
kondensacji aldolowej w wyniku czego dochodzi do cyklizacji. W tego typu reakcjach biorą<br />
udział dialdehydy i diketony (takŜe oksoaldehydy), najłatwiej kondensuję te, w których <strong>grupy</strong><br />
karbonylowe oddalone są od siebie tak, Ŝeby powstawały pięcio- lub sześcioczłonowe<br />
pierścienie. Z 1,4-diketonu powstaje pochodna cyklopentanonu.<br />
OEt<br />
28
heksano-<br />
-2,5-dion<br />
O<br />
CH 3<br />
O CH 3<br />
NaOH<br />
- HOH<br />
Z 1,5-ketonu powstanie pochodna cykloheksanonu.<br />
heptano-<br />
2,6-dion<br />
O<br />
CH3 CH3 O<br />
NaOH<br />
- HOH<br />
O<br />
O<br />
3-metylocyklopent-2-enon<br />
(40%)<br />
3-metylocykloheks-2-enon<br />
(44%)<br />
Natomiast z 6-oksoheptanalu tworzy się głównie pochodna cyklopetanonu pomimo tego, Ŝe<br />
odległość pomiędzy grupami karbonylowymi jest taka sama jak w hepta-2,6-dionie. W tym<br />
przypadku lepszym akceptorem jest grupa aldehydowa, zaś atomy wodoru przy C5 są bardziej<br />
kwaśne niŜ przy C7.<br />
Mechanizm reakcji:<br />
O O<br />
CH 3 CCH 2 CH 2 CH 2 CH 2 CH<br />
NaOH<br />
- HOH<br />
O<br />
CCH 3<br />
6-oksoheptanal keton 1-cyklopentenylowometylowy (73%)<br />
O<br />
H<br />
O<br />
CCH 3<br />
O<br />
CH 3 CCHCH 2 CH 2 CH 2 CH -OH<br />
6-oksoheptanal<br />
- OH<br />
keton 1-cyklopentenylowo-metylowy<br />
..<br />
O<br />
: O<br />
-<br />
CH3CCHCH .. 2CH2CH2CH ..<br />
.. -<br />
: OH<br />
: O:<br />
O HOH O<br />
Właściwości elektronoakceptorowe ketonowej <strong>grupy</strong> <strong>karbonylowej</strong> są zmniejszone jako efekt<br />
elektrodonorowego oddziaływania dwóch reszt alkilowych:<br />
H<br />
CCH 3<br />
O O<br />
R C R' R C H<br />
w ketonach w aldehydach<br />
4.4 Addycja nukleofilowa do α,β-nienasyconych <strong>aldehydów</strong> i <strong>ketonów</strong><br />
Przyłączenie nukleofila do karbonylowego atomu węgla sprzęŜonych enonów nie zawsze<br />
prowadzi do addycji 1,2. MoŜe nastąpić addycja 1,4. Kierunek reakcji w duŜej mierze zaleŜy od<br />
właściwości nukleofilowo-zasadowych odczynnika nukleofilowego.<br />
Grupa karbonylowa jest spolaryzowana, przy czym częściowy ładunek dodatni jest<br />
zlokalizowany przy atomie C i w to miejsce skierowany jest atak nukleofila – :Nu - . W enonach<br />
znajduje się dodatkowo podwójne wiązanie C=C, które ma powinowactwo do elektrofila E + .<br />
CCH 3<br />
29
δ-<br />
δ+<br />
- ..<br />
: O: : O:<br />
C<br />
+ C C<br />
C<br />
:Nu- E+<br />
W enonach sprzęŜonych na skutek oddziaływania elektronów π obu podwójnych wiązań (C=C i<br />
C=O)dochodzi do polaryzacji cząsteczki i utworzenia częściowego ładunku dodatniego na<br />
atomie węgla C4, który tym samym staje się podatny na oddziaływanie odczynnika<br />
nukleofilowego :Nu - .<br />
Mechanizm addycji 1,4:<br />
: O:<br />
C<br />
C C<br />
alken<br />
C C<br />
δ-<br />
Nu<br />
Nu<br />
δ+<br />
: O:<br />
C<br />
addycja<br />
1,2<br />
1<br />
- ..<br />
: O:<br />
C<br />
+ H/HOH<br />
.. 1<br />
: OH<br />
- ..<br />
: O:<br />
C<br />
sprzęŜony enon<br />
C C<br />
2 3<br />
4<br />
:Nu -<br />
E +<br />
C C +<br />
1 ..<br />
C<br />
2<br />
2<br />
3<br />
C C<br />
4<br />
:Nu -<br />
brak reakcji<br />
:Nu -<br />
: O:<br />
C<br />
2<br />
..<br />
- : O:<br />
C<br />
2<br />
: O:<br />
3<br />
4<br />
C<br />
2<br />
C C<br />
+<br />
C C<br />
3<br />
E<br />
.. -<br />
: O:<br />
C<br />
C C<br />
3<br />
+<br />
4<br />
C<br />
Nu<br />
C<br />
C<br />
H Nu<br />
C<br />
C C<br />
addycja<br />
1,4<br />
4<br />
+ H/HOH<br />
nienasycony alkohol aldehyd lub keton<br />
1<br />
1<br />
3<br />
4<br />
Nu<br />
30
Silnie zasadowe odczynniki nukleofilowe, np. związki Grignarda przyłączają się głównie w<br />
sposób 1,2.<br />
Mechanizm reakcji addycji 1,2:<br />
O<br />
CH 3 CH=CHCCH 3 + CH 3 MgBr<br />
OH<br />
CH 3<br />
THF<br />
pent-3-en-2-on bromek metylomagnezowy<br />
CH 3<br />
+ H/HOH<br />
CH 3 CH=CHCCH 3 + CH 3 CHCH 2 CCH 3<br />
3-metylopent-2-en-3-ol (72%) 4-metylopent-2-on (20%)<br />
addycja 1,2 addycja 1,4<br />
δ-<br />
: O<br />
..<br />
C C C<br />
δ+<br />
Mechanizm reakcji addycji 1,4:<br />
..<br />
O:<br />
C<br />
C C<br />
δ+<br />
sprzęŜony enon<br />
MgX<br />
R δ-<br />
δ+<br />
MgX<br />
R δ-<br />
aldehyd<br />
lub keton<br />
C<br />
C<br />
H<br />
..<br />
: O<br />
C C C<br />
: O<br />
C<br />
..<br />
R<br />
C C<br />
R<br />
R<br />
O<br />
..<br />
MgX : OH<br />
HX<br />
- MgX 2<br />
C C C<br />
R<br />
addycja 1,2<br />
MgX : O<br />
HX C<br />
..<br />
- MgX 2<br />
C C<br />
.. enol<br />
O:<br />
C<br />
addycja 1,4<br />
Mniej zasadowe nukleofile, np. odczynniki Gilmana przyłączają się prawie wyłącznie w<br />
sposób 1,4.<br />
Otrzymywanie odczynników Gilmana:<br />
R<br />
X<br />
+ 2 Li<br />
pentan<br />
- LiX<br />
R<br />
Li<br />
CuI<br />
eter<br />
H<br />
R<br />
Li + ( - R-Cu-R) + LiI<br />
halogenek alkilu alkilolit dialkilomiedzian litu (odczynnik Gilmana)<br />
Za pomocą dimetylomiedzianu litu moŜna wprowadzić grupę metylową do łańcucha węglowego<br />
sprzęŜonego enonu:<br />
O<br />
CH 3 CCH=CH 2 + Li(CH 3 ) 2 Cu<br />
+H/HOH<br />
O<br />
CH 3 CCH 2 CH 2 CH 3<br />
but-1-en-3-on dimetylomiedzian litu pentan-2-on (97%)<br />
31
Tym sposobem moŜna równieŜ przyłączyć reszty nienasycone lub aromatyczne:<br />
O<br />
+<br />
Li(H 2 C=CH) 2 Cu<br />
+H/HOH<br />
O<br />
(65%)<br />
CH=CH 2<br />
cykloheks-2-enon diwinylomiedzian litu 3-winylocykloheksanon<br />
O O<br />
+<br />
Li(C 6 H 5 ) 2 Cu<br />
+H/HOH<br />
(70%)<br />
cykloheks-2-enon difenylomiedzian litu 3-fenylocykloheksanon<br />
Odczynniki Gilmana stanowią wyjątek pośród związków metaloorganicznych, np. butylolit i<br />
inne alkilolity jako silnie zasadowe ulegają addycji 1,2.<br />
1. CH 3 MgBr 1. CH 3 Li<br />
2. + H/HOH<br />
CH 3<br />
2. + H/HOH<br />
O<br />
HO HO<br />
CH3 cykloheks-2-enon<br />
1. Li(CH 3 ) 2 Cu<br />
2. + H/HOH<br />
O<br />
CH 3<br />
1-metylocykloheks-2-enon 3-metylocykloheksanon<br />
Do <strong>grupy</strong> nukleofili, które do sprzęŜonych enonów przyłączają się w sposób 1,4 naleŜy jon<br />
cyjankowy. Jest on silnym nukleofilem o umiarkowanej zasadowości.<br />
CH=CH<br />
O<br />
C<br />
1. CN -<br />
1. H +<br />
CHCH 2<br />
keton fenylowo-(2-fenylowinylowy) 3-cyjano-1,3-difenylo-3-oksopropan (95%)<br />
TakŜe karboaniony powstałe z C-kwasów są silnymi nukleofilami i przyłączają się zwykle w<br />
sposób 1,4. Za ich pomocą moŜna wykonywać skomplikowane syntezy.<br />
CN<br />
O<br />
C<br />
32
O CH3<br />
H<br />
O<br />
- OH<br />
MeOH<br />
O<br />
- CH3<br />
..<br />
O<br />
karboanion<br />
2-metylocykloheksa-1,3-dion<br />
5,6-didehydro-<br />
1,7-diokso-10metylodekalina<br />
(65%)<br />
O<br />
O<br />
O<br />
CH 2 =CHCCH 3<br />
- OH/MeOH<br />
- HOH<br />
- OH/MeOH<br />
O<br />
O CH3<br />
CH 2<br />
C<br />
H 3<br />
OH<br />
O CH 2<br />
5. Azotowe pochodne <strong>aldehydów</strong> i <strong>ketonów</strong><br />
Aldehydy i ketony reagują z aminami i ich analogami tworząc pochodne, których budowa zaleŜy<br />
od rzędowości amin. Z aminami 1 o powstają iminy (aldiminy lub ketiminy) zwane zasadami<br />
Schiffa, podczas gdy z aminami 2 o tworzą się enaminy (grupa aminowa przyłączona do C=C).<br />
Reakcja jest katalizowana kwasami, jednak środowisko silnie kwaśnie nie sprzyja reakcji;<br />
optymalne pH wynosi ~ 4.<br />
Mechanizm addycji amin 1 o<br />
.. - ..<br />
..<br />
: O .. : O:<br />
:<br />
H OH<br />
2N-Y C<br />
C + C ..<br />
NH2 NH<br />
aldehyd<br />
lub keton<br />
Y<br />
Y aminoalkohol<br />
(karbinoloamina)<br />
+ H<br />
N Y<br />
:<br />
C<br />
- H +<br />
H<br />
N Y +<br />
C<br />
- HOH<br />
H<br />
.. + H<br />
O<br />
C ..<br />
NH<br />
Y<br />
C<br />
O<br />
O<br />
33
W zaleŜności od reszty Y przyłączonej do <strong>grupy</strong> aminowe otrzymuje się szereg azotowych<br />
pochodnych <strong>aldehydów</strong> i <strong>ketonów</strong>.<br />
:NH2-Y<br />
Y: wzór nazwa<br />
N H<br />
-H C ..<br />
N R<br />
-R (alkil, aryl) C ..<br />
-OH<br />
N OH<br />
N NH -NH2 2 C ..<br />
-NH-<br />
-NH-<br />
O 2 N<br />
NO 2<br />
C<br />
N NH<br />
C ..<br />
aldimina lub ketimina<br />
zasada Schiff<br />
(imina)<br />
oksym<br />
hydrazon<br />
fenylohydrazon<br />
N NH NO2 C ..<br />
O2N 2,4-dinitrofenylohydrazon<br />
O<br />
N NH NH O<br />
-NH-C-NH2 C ..<br />
C 2<br />
semikarbazon<br />
Jedną z najbardziej znanych imin jest benzylidenoanilina, preparat, który prawie kaŜdy student<br />
wydziału chemicznego otrzymuje w trakcie zajęć laboratoryjnych z chemii organicznej,<br />
poniewaŜ jest on niezwykle łatwy w wykonaniu – miesza się ze sobą oba substraty i krystalizuje<br />
produkt.<br />
CHO + H2N - HOH<br />
CH N<br />
benzaldehyd anilina benzylidenoanilina (84%)<br />
DuŜe znaczenie pośród azotowych pochodnych <strong>ketonów</strong> ma oksym cykloheksanonu. Z niego po<br />
przeształceniu w kaprolaktam (przegrupowanie Hofmanna) wytwarzana jest lizyny, słuŜy takŜe<br />
jako monomer w przemyśle włókien syntetycznych. Oksym cykloheksanonu otrzymuje się w<br />
reakcji cykloheksanonu z chloroworkiem hydroksyloaminy w obecności octanu sodu (buforu<br />
obniŜającego pH).<br />
34
O<br />
+ H 2 NOH . HCl<br />
NaAc<br />
..<br />
N<br />
OH<br />
cykloheksanon chlorowodorek hydroksyloaminy oksym cykloheksanonu (87%)<br />
Oksymy i innego podobne związki zawierające układ =N- tworzą stereoizomery typu cis-/transzwane<br />
syn- i anti-.<br />
syn<br />
.. OH<br />
N<br />
H O . .<br />
N<br />
anti<br />
C<br />
H<br />
C<br />
H<br />
stereoizomery oksymu benzaldehydu<br />
Takie pochodne <strong>aldehydów</strong> i <strong>ketonów</strong>, jak oksymy, fenylohydrazony, 2,4dinitrofenylohydrazony<br />
i semikarbazony wykorzystuje się do charakteryzowania związków<br />
karbonylowych. Są one zwykle krystaliczne i dzięki temu po specyficznych temperaturach<br />
topnienia moŜna zidentyfikować aldehydy i ketony, z których powstały. Ten sposób<br />
identyfikacji stracił obecnie na znaczeniu, z uwagi na powszechne stosowanie metod<br />
spektralnych, szczególnie uŜytecznego do tego celu NMR.<br />
Przykłady krystalicznych pochodnych ciekłych związków karbonylowych:<br />
C<br />
H 3<br />
N<br />
C<br />
OH<br />
H<br />
H H<br />
C<br />
N N<br />
oksym etanalu fenylohydrazon benzaldehydu 2,4-dinitrofenylohydrazon semikarbazon<br />
(tt. 47 o C) (tt. 158 o C) acetonu (tt. 126 o C) benzaldehydu (tt. 222 o C)<br />
C<br />
H 3<br />
Mechanizm addycji 2 o amin do związków karbonylowych<br />
enamina<br />
..<br />
R2NH +<br />
R ..<br />
N<br />
R<br />
C C<br />
: O:<br />
C<br />
H<br />
C<br />
- + H/HOH<br />
: O:<br />
.. -<br />
C<br />
+<br />
R N 2<br />
H<br />
R + R<br />
N<br />
C C<br />
H<br />
N<br />
C<br />
H<br />
C<br />
NH<br />
CH 3<br />
R N 2 ..<br />
NO 2<br />
: OH<br />
..<br />
NO 2<br />
C H<br />
C<br />
+ H<br />
+<br />
: OH2 C H<br />
C<br />
R2N ..<br />
- HOH<br />
..<br />
HOH ..<br />
..<br />
Często do otrzymywania enamin uŜywa się 2 o amin cyklicznych, np. pirolidyny.<br />
N<br />
C<br />
NH<br />
H<br />
C<br />
O<br />
NH 2<br />
35
O + HN:<br />
- HOH<br />
cykloheksanon pirolidyna pirolidynowa enamina cykloheksanonu<br />
6. Redukcja Wolffa-KiŜnera<br />
Grupę karbonylową moŜna zredukować do CH2. SłuŜy do tego redukcja Wolffa-KiŜnera.<br />
Ludwig Wolff (1857-1919); ur. w Neustadt/Hardt (Niemcy), doktorat w Strasburgu, Francja, u Fittiga; prof. Uniw.<br />
w Jenie.<br />
N. M. KiŜner (1867-1935); ur. w Moskwie; doktorat w Moskwie u Markownikowa; prof. w Tomsku i w Moskwie.<br />
Reakcja addycji hydrazyny do <strong>grupy</strong> <strong>karbonylowej</strong> jest wykorzystywana do redukcji tej <strong>grupy</strong> do<br />
-CH2-, czyli przekształcania związku karbonylowego w węglowodór; nosi ona nazwę redukcji<br />
Wolffa-KiŜnera. Polega ona na ogrzewaniu aldehydu lub ketonu z hydrazyną w obecności<br />
KOH w temperaturze 240 o C. Pośrednio powstaje fenylohydrazon. Późniejsza modyfikacja<br />
polegająca na zastosowaniu DMSO jako rozpuszczalnika pozwoliła na znaczne obniŜenie<br />
temperatury reakcji. Redukcja Wolffa-Kiznera jest alternatywnym redukcji <strong>grupy</strong> <strong>karbonylowej</strong><br />
do metody Clemmensena (za pomocą amalgamatu rtęci).<br />
O<br />
C<br />
CH 2 CH 3<br />
H 2 NNH 2 /KOH<br />
DMSO<br />
N:<br />
CH2CH2CH3 + HOH + N2 propiofenon propylobenzen (keton etylowo-fenylowy) (82%)<br />
7. Reakcja haloformowa<br />
Ketony metylowe i acetaldehyd rozpadają się w środowisku zasadowym pod wpływem<br />
halogenów; wydziela się przy tym haloform (CHX3), a z reszty tworzy się sól kwasu<br />
karboksylowego.<br />
O<br />
C<br />
CH 3<br />
X 2<br />
- OH<br />
O<br />
C<br />
CX 3<br />
- OH<br />
-<br />
COO<br />
+ CHX3 acetofenon trihalogenoacetofenon benzoesan haloform<br />
W reakcji metylowego ketonu z halogenem w środowisku zasadowym reakcja nie zatrzymuje się<br />
na monohalogenowaniu, ale biegnie dalej aŜ do podstawienia wszystkich atomów wodoru <strong>grupy</strong><br />
metylowej. Taki trihalogenometylowy keton jest nietrwały w obecności zasad i rozpada się na<br />
haloform i jon karboksylanowy:<br />
36
O H<br />
O<br />
O<br />
:B<br />
CH2 -<br />
..<br />
: :<br />
: : : :<br />
-<br />
R C<br />
R C<br />
R C<br />
-<br />
CH 2<br />
: O:<br />
R C<br />
:B -<br />
CBr 3<br />
Br 2<br />
:B -<br />
jon enolanowy Br 2<br />
Br 2<br />
:B -<br />
: O:<br />
-<br />
R C COO<br />
: O:<br />
Br<br />
R C<br />
CH 2<br />
CH 2<br />
+ CHBr 3<br />
+ Br -<br />
jon karboksylanowy bromoform<br />
Podobnej reakcji ulegają 2 o alkohole zawierające grupę CH3 przy hydroksylowanym atomie<br />
węgla. Tego typu alkohole łatwo utleniają się pod wpływem halogenu do metylowego ketonu:<br />
OH<br />
RCHCH 3<br />
X 2<br />
- HX<br />
O<br />
RCCH 3<br />
2 o alkohol zawierający grupę keton metylowy<br />
CH3 przy hydroksylowanym atomie C<br />
Działanie jodem na ketony metylowe, acetaldehyd i 2 o alkohole zawierające grupę CH3 przy<br />
hydroksylowanym atomie C, słuŜy jako test do wykrywania tego typu ugrupowań:<br />
O OH<br />
i<br />
-CCH 3<br />
-CHCH 3<br />
Po dodaniu jodu do roztworu takich związków wypada jodoform w formie Ŝółtych kłaczków.<br />
Test ten nosi nazwę reakcji jodoformowej.<br />
O<br />
-CCH 3<br />
I 2<br />
- OH<br />
-COO -<br />
+ CHI 3<br />
jodoform<br />
8. Kondensacja pinakolinowa<br />
Pinakolanami nazywane są wicynalne glikole zawierające <strong>grupy</strong> hydroksylowe przy 3 o atomach<br />
węgla, przy czym najprostszy z nich czyli 2,3-dimetylo-2,3-dihydroksybutan nosi nazwę<br />
pinakolu. Pinakole powstają w wyniku dimeryzacji <strong>ketonów</strong> pod wpływem reaktywnych metali<br />
w środowisku aprotonowym. W środowisku protonowym metale te redukują ketony do alkoholi.<br />
Pierwszy etapem reakcji w obu przypadkach jest utworzenie anionorodnika (ketylu) w procesie<br />
przeniesienia pojedynczego elektronu (SET, ang. single electron transfer).<br />
: O:<br />
R C R' + Mo Dalsze etapy reakcji zaleŜą od środowiska:<br />
SET<br />
.. -<br />
: O:<br />
R C.<br />
R'<br />
anionorodnik<br />
(ketyl)<br />
37
A. W środowisku protonowym anionorodnik przyłącza proton i przekształca się w rodnik,<br />
który ponownie w reakcji SET pobiera elektron od atomu metalu przechodząc w anion.<br />
Z anionu po kolejnym protonowaniu tworzy się alkohol.<br />
B. Zaś w środowisku aprotonowym anionorodnik dimeryzuje tworząc dianion<br />
(alkoholan), z którego po zakwaszeniu powstaje pinakol.<br />
..<br />
: O:<br />
anionorodnik<br />
A<br />
R C . R' (ketyl)<br />
B<br />
-<br />
H +<br />
..<br />
: OH<br />
R C.<br />
R'<br />
SET M o<br />
..<br />
: OH<br />
-<br />
R C..<br />
R'<br />
: OH<br />
..<br />
R C<br />
H<br />
środowisko<br />
protonowe<br />
H +<br />
alkohol<br />
R'<br />
rodnik<br />
anion<br />
anionorodnik<br />
anionorodnik<br />
środowisko<br />
aprotonowe<br />
alkoholan<br />
pinakol<br />
R<br />
R<br />
R'<br />
C .<br />
+<br />
.<br />
C<br />
R'<br />
.. -<br />
O..<br />
:<br />
.. -<br />
O..<br />
:<br />
R<br />
R<br />
R'<br />
C<br />
C<br />
R'<br />
.. -<br />
O..<br />
:<br />
..<br />
O..<br />
:<br />
-<br />
H +<br />
R' R'<br />
R C C<br />
Dowodem na to, Ŝe reakcja kondensacji pinakolinowej biegnie przez anionorodnik jest zmiana<br />
barwy roztworu, w którym zachodzi reakcja. Zarówno substrat – keton, jak i produkt – pinakol<br />
są bezbarwne, natomiast w trakcie reakcji pojawia się intensywnie niebieskie (lub fioletowe)<br />
zabarwienie, typowe dla anionorodników.<br />
HO<br />
.. +<br />
: O: : O:<br />
- K<br />
C<br />
benzofenon<br />
bezbarwny<br />
HO<br />
C C<br />
K<br />
THF<br />
OH<br />
+ H<br />
C.<br />
ketyl benzofenonu<br />
intensywnie<br />
niebieski<br />
benzopinakol<br />
bezbarwny<br />
OH<br />
R<br />
38
Pinakol, czyli 2,3-dimetylobutano-2,3-diol jest najczęściej otrzymywanym pinakolem.<br />
Otrzymuje się z bardzo dobrą wydajnością w reakcji acetonu z magnezem; krystalizuje w postaci<br />
hydratu.<br />
..<br />
: O: : O:<br />
-<br />
2 H3C C CH3 C<br />
6 HOH .<br />
aceton<br />
C<br />
H 3<br />
CH3 CH3 C C<br />
HO<br />
OH<br />
Mg<br />
benzen<br />
CH 3<br />
hydrat pinakolu (95%)<br />
+ H<br />
HOH<br />
H 3<br />
C<br />
H 3<br />
C<br />
H 3<br />
C.<br />
.<br />
C<br />
: O:<br />
.. -<br />
CH 3<br />
CH 3<br />
CH3 CH3 C C<br />
: O O<br />
- ..<br />
: :<br />
..<br />
:<br />
-<br />
2+<br />
Mg<br />
9. Addycja wodorosiarczanu (IV) – połączenia bisulfitowe<br />
Wodorosiarczany (IV) przyłączają się do <strong>grupy</strong> <strong>karbonylowej</strong> <strong>aldehydów</strong> i niektórych <strong>ketonów</strong><br />
(nie zawierających objętościowo duŜych podstawników) tworząc krystaliczne, trudno<br />
rozpuszczalne w wodzie sole kwasów αααα-hydroksysulfonowych, zwane połączeniami<br />
bisulfitowymi.<br />
O<br />
OH<br />
C + Na + - C SO HSO3 3 Na +<br />
CH 3<br />
aldehyd lub keton połączenie bisulfitowe (αααα-hydroksysulfonian sodu)<br />
Z połączeń bisulfitowych łatwo odtwarza się wyjściowe związki karbonylowe. Ulegają one<br />
rozkładowi pod wpływem kwasów lub zasad. Często słuŜą do wyodrębniania związków<br />
karbonylowych z mieszanin (ze środowiska reakcji) lub do <strong>usuwania</strong> ich resztek, kiedy słuŜyły<br />
jako substraty.<br />
10. Reakcja Wittiga<br />
OH<br />
C<br />
-<br />
SO 3<br />
+ H<br />
- OH<br />
O<br />
C + SO 2 + HOH<br />
O<br />
C<br />
-<br />
+ SO 3 -2 + HOH<br />
Georg Wittig (1897-1987), ur. w Berlinie; doktorat w Marburgu u von Auwersa; prof. uniwersytetów we Freiburgu,<br />
Tybindze i Heiderbergu; laureat nagrody Nobla w 1979 r.<br />
Reakcja Wittiga słuŜy do otrzymywania alkenów z <strong>aldehydów</strong> lub <strong>ketonów</strong>. Kluczowym<br />
reagentem w tej reakcji są ylidy – specyficzne pochodne trifenylofosfiny.<br />
39
..<br />
P<br />
trifenylofosfina<br />
Trifenylofosfina ulega łatwo alkilowaniu do 4 o soli fosfoniowej.<br />
Ph 3 P: + CH 3<br />
trifenylofosfina<br />
bromek<br />
metylu<br />
SN2 Br Ph3P CH3 Br - +<br />
bromek trifenylometylofosfoniowy<br />
4 o sole fosfoniowe, np. bromek trifenylometylofosfoniowy pod wpływem silnych zasad<br />
przekształca się w betainę, będącą izomeryczną formą ylidu. Zasada odrywa z halogeneku<br />
trifenyloalkilofosfoniowego kwaśny atom wodoru znajdujący się przy Cα w stosunku do atomu<br />
fosforu.<br />
+<br />
BuLi + ..<br />
Ph3P CH2 H Ph3P CH<br />
- 2<br />
Br -<br />
betaina<br />
bromek<br />
trifenylometylofosfoniowy<br />
Betaina tym róŜni się od ylidu, Ŝe w betainie rozdzielone są ładunki. Obie te formy są<br />
odmianami mezomerycznymi tego samego związku.<br />
Ylid reaguje ze związkami karbonylowymi tworząc nietrwały addukt, który się rozpada na alken i<br />
tlenek trifenylofosfiny.<br />
Ph 3 P<br />
ylid<br />
H C C 2<br />
alken<br />
CH 2<br />
- Ph 3 P=O<br />
+<br />
Ph3P -<br />
..<br />
C O:<br />
+<br />
Ph3P ..<br />
: O<br />
-<br />
..<br />
tlenek trifenylofosfiny<br />
Reakcja Wittiga, pośród wielu metod otrzymywania alkenów, naleŜy do najbardziej<br />
selektywnych, szczególnie gdy produktem ma być związek o rozbudowanym szkielecie<br />
węglowym.<br />
Z <strong>aldehydów</strong> lub <strong>ketonów</strong> moŜna trzymać alkeny równieŜ innym sposobem: poprzez ich reakcję<br />
ze związkami Grignarda, a następnie dehydratację otrzymanych alkoholi pod wpływem POCl3,<br />
jednak produktem jest zwykle mieszanina izomerów.<br />
CH 2<br />
CH 2<br />
C<br />
Ph 3 P<br />
ylid<br />
CH 2<br />
40
O<br />
1. CH 3 MgBr<br />
2. POCl 3<br />
Ph 3 P=CH 2<br />
CH 3<br />
1-metylocykloheksen<br />
CH 2<br />
+<br />
CH 2<br />
metylenocykloheksen<br />
metylenocykloheksen<br />
O efektywności reakcji Wittiga świadczy to, Ŝe znalazła zastosowanie przemysłowe, np. do<br />
wytwarzania β-karotenu (prowitaminy A).<br />
retinal<br />
O<br />
CH<br />
reakcja<br />
Wittiga<br />
+<br />
HC<br />
β-karoten<br />
PPh 3<br />
(84%)<br />
ylid retinylidenotrifenylofosfoniowy<br />
11. Tautomeria keto-enolowa<br />
Związki karbonylowe zawierające atom wodoru w połoŜeniu α ulegają tautomerii, polegającej na<br />
tym, Ŝe Hαααα przemieszcza się pomiędzy atomami O↔C. W większości przypadków równowaga<br />
jest przesunięta w kierunku <strong>grupy</strong> <strong>karbonylowej</strong> (na lewo):<br />
H<br />
..<br />
O:<br />
R<br />
..<br />
: O H<br />
R<br />
R'<br />
C C<br />
R'' R'<br />
C C<br />
R''<br />
Zjawisko przejścia atomu wodoru (przegrupowania) w obrębie tej samej cząsteczki nazywane<br />
jest tautomerią (grec. tauto – takie samo, meros – część). Poszczególne izomery tautomeryczne<br />
nazywane są tautomerami. Tautomeria jest związana z mezomerią. Etapem pośrednim<br />
tautomeryzacji jest jon enolanowy, a powstający tautomer jest enolem.<br />
Nazwa enol pochodzi od połączenia końcówek dwóch grup funkcyjnych „en” (od alkenu) i „ol”<br />
(od alkoholu) i oznacza, Ŝe grupa -OH jest przyłączona do atomu węgla tworzącego podwójne<br />
wiązanie (tzn. o hybrydyzacji sp 2 ).<br />
41
..... ..<br />
.. -<br />
R : O:<br />
C C jon enolanowy<br />
R' R''<br />
- H + + H+ - H + + H+<br />
..<br />
..<br />
R O:<br />
R : O H<br />
R' C C C C<br />
H R''<br />
R' R''<br />
forma ketonowa (karbonylowa) forma enolowa<br />
W środowisku obojętnym tautomeria jest reakcją wolną, natomiast w obecności zarówno<br />
kwasów jak i zasad jej szybkość znacznie wzrasta.<br />
Kataliza kwasowa:<br />
H<br />
.. +<br />
O H<br />
C C<br />
H<br />
..<br />
O:<br />
+H<br />
O H<br />
C C<br />
-<br />
H<br />
C<br />
..<br />
: O<br />
C<br />
+<br />
H<br />
+ ..<br />
:<br />
H C C<br />
forma ketonowa forma enolowa<br />
Kataliza zasadowa:<br />
..<br />
H O:<br />
-OH C C<br />
- HOH<br />
forma ketonowa<br />
C<br />
-<br />
C<br />
..<br />
O:<br />
C<br />
.. -<br />
: O:<br />
C<br />
+ H<br />
..<br />
: O H<br />
C C<br />
forma enolowa<br />
Aldehydy i ketony chociaŜ występuję w obu formach (ketonowej i enolowej) to najczęściej<br />
równowaga jest zdecydowanie przesunięta w kierunku <strong>grupy</strong> <strong>karbonylowej</strong>. Znane są jednak<br />
związki, w których forma enolowa przewaŜa, naleŜy do nich fenol, a takŜe 1,3-diony.<br />
42
Równowaga tautomeryczna przykładowych związków<br />
aldehyd<br />
octowy<br />
forma ketonowa forma enolowa<br />
O<br />
O<br />
aceton CH 3 CCH 3<br />
cykloheksanon<br />
fenol<br />
O<br />
O<br />
C<br />
H 2<br />
OH<br />
CH3CH CH2 CH<br />
99,99%<br />
Przyczyną tej racemizacji jest przejściowo tworząca się, pozbawiona chiralnego atomu węgla<br />
forma enolowa. W trakcie odtworzenie <strong>grupy</strong> <strong>karbonylowej</strong> -C=O dochodzi do powstanie takiej<br />
samej ilości izomeru (+) jak i (-), czyli racematu.<br />
Dzięki tautomerii moŜna wprowadzać izotopy wodoru w połoŜenie α do <strong>grupy</strong> <strong>karbonylowej</strong>.<br />
H3C O<br />
CH 3 CH 2 CC<br />
H<br />
+ D/DOD<br />
lub - OD/DOD<br />
H3C O<br />
CH 3 CH 2 CC<br />
1-fenylo-2-metylobutan-1-on 2-deutero-1-fenylo-2-metylobutan-1-on<br />
Zadanie: przedstaw mechanizm α deuterowania związków karbonylowych<br />
Stosunkowo łatwa wymiana protonu przy Cα na deuteron (lub tryton) jest moŜliwa dzięki<br />
stosunkowo duŜej wartości pKa (kwasowości) atomu wodoru w tej pozycji – pKa przy Cα<br />
wynosi 19-20, podczas gdy przy Cβ jedynie 40-50.<br />
R<br />
O<br />
C C C<br />
H H<br />
α β<br />
19-20 40-50<br />
pKa ↑ ↑<br />
kwasowy atom H niekwasowy alifatyczny atom H<br />
1 o i 2 o nitrozwiązki teŜ ulegają tautomerii, tworząc przy tym stosunkowo silne kwasy.<br />
Nitrozwiązki w formie tautomerycznej noszą nazwę aci-nitropochodnych. Stała kwasowa Hα dla<br />
nitrometanu wynosi 6,1.10 -11 , podczas gdy jego formy aci – 5,6.10 -4 . Roztwór nitrometanu nie<br />
daje odczynu kwaśnego wobec papierka uniwersalnego, natomiast nitrometan w formie aci<br />
zabarwi ten papierek podobnie jak kwas octowy. W nitrozwiązkach w środowisku obojętnym<br />
równowaga przesunięta jest w kierunku formy nitrowej; w nitrometanie formy aci jest jedynie<br />
około 0,00001%, w nitroetanie 0,009%, a w 2-nitropropanie 0,3%.<br />
H<br />
..<br />
O:<br />
..<br />
: O H<br />
R C N ..<br />
R N<br />
+<br />
-<br />
C ..<br />
O..<br />
:<br />
+<br />
-<br />
O..<br />
:<br />
forma nitro forma aci<br />
W obecności zasad nitrozwiązki wolno tworzą rozpuszczalne w wodzie sole formy aci. Sole te<br />
pod wpływem mocnych kwasów zostają przekształcane w nietrwałe kwasy – aci-nitrozwiązki. Po<br />
ponownym zalkalizowaniu świeŜo otrzymanych aci-nitrozwiązków powstają z nich sole tak<br />
szybko, jak z kwasów mineralnych. Aci-nitrozwiązki z ciągu kilku godzin samorzutnie<br />
przechodzą w formę nitro.<br />
D<br />
44
H ..<br />
O:<br />
CH N ..<br />
+<br />
-<br />
O..<br />
:<br />
fenylonitrometan<br />
forma nitro<br />
ciecz, tw. 226 o C<br />
forma aci<br />
NaOH<br />
wolno<br />
kilka<br />
godzin<br />
fenylonitrometan<br />
ciało stałe, tt. 84 o C<br />
NaOH<br />
szybko<br />
..<br />
: O Na<br />
CH N ..<br />
+<br />
-<br />
O..<br />
:<br />
+ -<br />
H +<br />
szybko<br />
..<br />
: O H<br />
CH N ..<br />
+<br />
-<br />
O..<br />
:<br />
Forma aci większości nitrozwiązków jest tak nietrwała, Ŝe trudno je wyizolować w postaci<br />
indywiduów chemicznych; fenylonitrometan naleŜy do tych nitrozwiązków, które trwają przez<br />
kilka godzin, moŜna więc je wyizolować i scharakteryzować.<br />
Aci-nitropochodne są silniejszymi kwasami niŜ kwas węglowy i rozpuszczają się w roztworach<br />
węglanu sodu czy potasu, natomiast nitrozwiązki w formie nitro nie rozpuszczają się w<br />
roztworach węglanów. Aci-nitrozwiązków pod wpływem roztworu chlorku Ŝelaza (III) przyjmują<br />
czerwone zabarwienie, podobnie jak enole i fenole.<br />
Fenole są przykładem trwałych enoli. Do twałych enoli naleŜą równieŜ 1,3-diony, ββββoksyaldehydy,<br />
ββββ-oksynitryle czy ββββ-oksyestry.<br />
12. Polimeryzacja <strong>aldehydów</strong><br />
Aldehydy niskocząsteczowe (metanal i etanal) ulegają cyklicznej oligomeryzacji lub liniowej<br />
polimeryzacji. Tworzą się przy tym produkty zawierające wiązania acetalowe. Aldehydy<br />
aromatyczne zawierające grupę karbonylową przy pierścieniu nie ulegają polimeryzacji.<br />
Tendencja do polimeryzacji <strong>aldehydów</strong> alifatycznych zmniejsza się wraz ze wzrostem ich masy<br />
cząsteczkowej.<br />
Metanal jest trwały w stanie gazowym. Ślady wody i wiele innych zanieczyszczeń powodują jego<br />
szybką polimeryzację. W roztworach wodnych polimeryzacja jest wolniejsza, ale widoczny osad<br />
(polimer metanalu) na dnie formaliny (wodnego roztworu metanalu) świadczy, Ŝe polimeryzacja<br />
ma miejsce. MoŜna ją przyspieszyć. Katalitycznie działają zarówno kwasy, jak i zasady. Z<br />
wodnego roztworu metanalu pod wpływem wodorotlenków alkalicznych powstaje αpolioksymetylen.<br />
(n + 2) HCHO + HOH<br />
+ H lub -OH HOCH2 (OCH2 ) nOCH2OH metanal polioksymetylen<br />
Pod wpływem stęŜonego kwasu siarkowego wytrąca się β-polioksymetylen, a paraformaldehyd<br />
tworzy się w trakcie odparowywania pod obniŜonym ciśnieniem wodnych roztworów metanalu.<br />
Wszystkie te trzy formy są liniowo spolimeryzowanym metanalem. RóŜnica polega na tym, Ŝe<br />
stopień polimeryzacji n paraformaldehydu mieści się w granicach 10-50, zaś stopień<br />
polimeryzacji polioksymetylenów jest większy. β-Polioksymetylen zawiera małe ilości kwasu<br />
siarkowego związanego estrowo. Obecnie paraformaldehyd jest równieŜ zaliczany do<br />
polioksymetylenów.<br />
45
Paraformaldehyd jest produkowany na skalę przemysłową, stanowi bowiem dogodną formę<br />
przechowywania i transportowania metanalu. Paraformaldehyd łatwo przekształcić w wolny<br />
aldehyd, poniewaŜ depolimeryzuje on w temperaturze 140-160 o C bezpośrednio do gazowego<br />
produktu. RównieŜ w niektórych reakcjach zachowuje się jak wolny aldehyd.<br />
Trioksan (trioksymetylen), cykliczny trimer metanalu powstaje w trakcie ogrzewania<br />
paraformaldehydu (polioksymetylenu) z katalityczną ilością kwasu siarkowego w zamkniętym<br />
naczyniu. Jest to w odróŜnieniu od polimerów liniowych związek jednorodny i w odróŜnieniu od<br />
metanalu ma przyjemny zapach.<br />
HOCH 2 (OCH 2 ) n OCH 2 OH<br />
polioksymetylen<br />
H 2 SO 4<br />
O<br />
O O<br />
trioksan<br />
tt. 64 o C<br />
Etanal i wyŜsze aldehydy przechowywane w stanie czystym, w szczelnie zamkniętym naczyniu są<br />
trwałe – nie ulegają samorzutnej polimeryzacji. Jednak pod wpływem katalitycznych ilości<br />
silnych kwasów etanal szybko trimeryzuje. Ten trimer nazywany jest paraacetaldehydem lub<br />
krócej paraaldehydem (paraldehydem). Równocześnie powstaje tetramer zwany metaaldehydem<br />
(metaldehydem).<br />
3 CH 3 CHO<br />
+ H<br />
C<br />
H 3<br />
O CH 3<br />
O O<br />
CH 3<br />
H C 3<br />
O<br />
C<br />
H 3<br />
O<br />
O<br />
O<br />
CH 3<br />
CH 3<br />
etanal paraaldehyd metaaldehyd<br />
(tw. 21 o C, tt. –125 o C) (tw. 124 o C, tt. 10 o C) (tt. 190 o C, subl.)<br />
Reakcja oligomeryzacji etanalu jest reakcją odwracalną, przy czym stan równowagi zaleŜy od<br />
temperatury; w temp. 50 o C 40% aldehydu ulega oligomeryzacji. W niŜszych temperaturach<br />
równowaga przesuwa się w kierunku oligomerów. Czysty paraaldehyd otrzymuje się przez<br />
oddestylowanie go po zobojętnieniu mieszaniny równowagowej. Z pozostałości wydziela się<br />
metaaldehyd. Dawniej metaaldehyd w formie kostek słuŜył jako bezpieczne paliwo turystyczne.<br />
Nie stwarzało ono tyle problemów co, np. benzyna w szklanych butelkach. Wadą metaaldehydu<br />
jako paliwa jest jego niska wartość energetyczna, poniewaŜ jest w znacznym stopniu utleniony.<br />
Obecnie metalowe butle z ciekłym gazem (propan/butan) są najpopularniejszym paliwem<br />
turystycznym.<br />
Z paraaldehydu po zakwaszeniu wydziela się etanal. Jest to jedna z metod wprowadzania etanalu<br />
jako reagenta do wielu syntez.<br />
Wielocząsteczkowy, liniowy polimer etanalu powstaje poprzez polimeryzację etanalu wobec<br />
tlenku glinu. MoŜna go równieŜ otrzymać z zamroŜonego do -120 o C monomeru. Po jego<br />
podgrzaniu do temperatury pokojowej otrzymuje się gęstą ciecz, która samorzutnie przekształca<br />
się w stały produkt.<br />
+<br />
46

