Role of Intestinal Microbiota in Ulcerative Colitis
Role of Intestinal Microbiota in Ulcerative Colitis Role of Intestinal Microbiota in Ulcerative Colitis
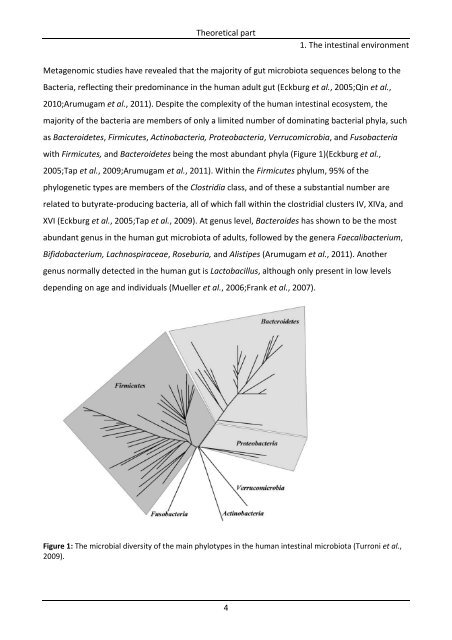
Theoretical part 4 1. The intestinal environment Metagenomic studies have revealed that the majority of gut microbiota sequences belong to the Bacteria, reflecting their predominance in the human adult gut (Eckburg et al., 2005;Qin et al., 2010;Arumugam et al., 2011). Despite the complexity of the human intestinal ecosystem, the majority of the bacteria are members of only a limited number of dominating bacterial phyla, such as Bacteroidetes, Firmicutes, Actinobacteria, Proteobacteria, Verrucomicrobia, and Fusobacteria with Firmicutes, and Bacteroidetes being the most abundant phyla (Figure 1)(Eckburg et al., 2005;Tap et al., 2009;Arumugam et al., 2011). Within the Firmicutes phylum, 95% of the phylogenetic types are members of the Clostridia class, and of these a substantial number are related to butyrate‐producing bacteria, all of which fall within the clostridial clusters IV, XIVa, and XVI (Eckburg et al., 2005;Tap et al., 2009). At genus level, Bacteroides has shown to be the most abundant genus in the human gut microbiota of adults, followed by the genera Faecalibacterium, Bifidobacterium, Lachnospiraceae, Roseburia, and Alistipes (Arumugam et al., 2011). Another genus normally detected in the human gut is Lactobacillus, although only present in low levels depending on age and individuals (Mueller et al., 2006;Frank et al., 2007). Figure 1: The microbial diversity of the main phylotypes in the human intestinal microbiota (Turroni et al., 2009).
Theoretical part 5 1. The intestinal environment 1.2. Function and physiology of the large intestine The main functions of the large intestine are food storage, absorption of water and electrolytes, and digestion of indigestible carbohydrates by the colonic microbiota (Vander et al., 1998). Anatomically, the large intestine consists of the cecum, ascending colon, transverse colon, descending colon, sigmoid colon, and rectum (Guarner and Malagelada, 2003). The ascending colon is a saccharolytic environment where most bacterial metabolic activity and carbohydrate fermentation occur. The pH of the ascending colon is generally lower (approximately 5‐6) than that of the distal colon. The reduced pH is considered to be the result of carbohydrate fermentation, which gives rise to the production of Short‐Chain Fatty Acids (SCFAs) (Macfarlane et al., 1992). Consequently, the carbohydrate availability decreases in the descending colon, which leads to a pH close to neutral. The rate of bacterial metabolism is lower, and protein and amino acids become a more dominant metabolic energy source for bacteria (Figure 2A) (Guarner and Malagelada, 2003;Vernazza et al., 2006). Anaerobic fermentation of proteins by the microbiota produces branched SCFAs, however, it also generates a series of potentially toxic compounds such as ammonia, amines, and phenolic compounds (Vernazza et al., 2006). The wall of the colon consists of four tissue compartments (Figure 2B): mucosa, submucosa, muscularis externa, and serosa. The mucosa consists of a mucus layer, single layer of epithelium, the lamina propria, and a thin muscle layer (muscularis mucosae). The epithelium covering the mucosa consists of different types of cells, namely goblet cells (mucin secreting cells), enterocytes (absorptive cells), and endocrine cells (hormone secreting cells). These cells are linked together along the edges of their luminal surface by tight junctions. The submucosa is a connective tissue supporting the mucosa with blood vessels, lymphatic vessels, and nerves. The muscularis externa consists of two muscle layers: the longitudinal and the smooth muscle layer. Between the two muscle layers is a network of nerves (the myenteric nerve plexus). The serosa is the outer connective tissue layer, which connects the colon to the abdominal cavity (Vander et al., 1998).
- Page 1: Role of Intestinal Microbiota in Ul
- Page 4 and 5: Role of Intestinal Microbiota in Ul
- Page 6 and 7: Preface Preface This thesis present
- Page 8 and 9: Summary Summary The microbiota of t
- Page 10 and 11: Dansk sammendrag Dansk sammendrag M
- Page 12 and 13: Introduction and objectives Introdu
- Page 14 and 15: List of Manuscripts Not included in
- Page 16 and 17: List of contents List of Centents P
- Page 18 and 19: List of Centents Methodology append
- Page 21: 1. The intestinal environment Theor
- Page 25 and 26: 2. The colonic environment Theoreti
- Page 27 and 28: Theoretical part 9 2. The colonic e
- Page 29 and 30: Table 1: The presence of glycoside
- Page 31 and 32: Theoretical part Figure 3: The colo
- Page 33 and 34: 3. Inflammatory Bowel disease Theor
- Page 35 and 36: Theoretical part 17 3. Inflammatory
- Page 37 and 38: Theoretical part 19 4. Modulation o
- Page 39 and 40: Theoretical part 21 4. Modulation o
- Page 41 and 42: Theoretical part 23 4. Modulation o
- Page 43 and 44: Table 4: Clinical trials on the pre
- Page 45 and 46: Theoretical part 5. Production of p
- Page 47 and 48: Theoretical part 5. Production of p
- Page 49 and 50: Theoretical part 5. Production of p
- Page 51: Methodology part
- Page 54 and 55: Methodology part 6. Methodology, co
- Page 56 and 57: Methodology part 6. Methodology, co
- Page 58 and 59: Methodology part 6. Methodology, co
- Page 60 and 61: Introduction Methodology part 42 Pa
- Page 62 and 63: Abstract Background Detailed knowle
- Page 64 and 65: depending the level of disease acti
- Page 66 and 67: in 1 x TAE at 60 °C for 16 h at 36
- Page 68 and 69: Statistics PCA were generated by SA
- Page 70 and 71: The PCA of the Gram‐positive bact
Theoretical part<br />
4<br />
1. The <strong>in</strong>test<strong>in</strong>al environment<br />
Metagenomic studies have revealed that the majority <strong>of</strong> gut microbiota sequences belong to the<br />
Bacteria, reflect<strong>in</strong>g their predom<strong>in</strong>ance <strong>in</strong> the human adult gut (Eckburg et al., 2005;Q<strong>in</strong> et al.,<br />
2010;Arumugam et al., 2011). Despite the complexity <strong>of</strong> the human <strong>in</strong>test<strong>in</strong>al ecosystem, the<br />
majority <strong>of</strong> the bacteria are members <strong>of</strong> only a limited number <strong>of</strong> dom<strong>in</strong>at<strong>in</strong>g bacterial phyla, such<br />
as Bacteroidetes, Firmicutes, Act<strong>in</strong>obacteria, Proteobacteria, Verrucomicrobia, and Fusobacteria<br />
with Firmicutes, and Bacteroidetes be<strong>in</strong>g the most abundant phyla (Figure 1)(Eckburg et al.,<br />
2005;Tap et al., 2009;Arumugam et al., 2011). With<strong>in</strong> the Firmicutes phylum, 95% <strong>of</strong> the<br />
phylogenetic types are members <strong>of</strong> the Clostridia class, and <strong>of</strong> these a substantial number are<br />
related to butyrate‐produc<strong>in</strong>g bacteria, all <strong>of</strong> which fall with<strong>in</strong> the clostridial clusters IV, XIVa, and<br />
XVI (Eckburg et al., 2005;Tap et al., 2009). At genus level, Bacteroides has shown to be the most<br />
abundant genus <strong>in</strong> the human gut microbiota <strong>of</strong> adults, followed by the genera Faecalibacterium,<br />
Bifidobacterium, Lachnospiraceae, Roseburia, and Alistipes (Arumugam et al., 2011). Another<br />
genus normally detected <strong>in</strong> the human gut is Lactobacillus, although only present <strong>in</strong> low levels<br />
depend<strong>in</strong>g on age and <strong>in</strong>dividuals (Mueller et al., 2006;Frank et al., 2007).<br />
Figure 1: The microbial diversity <strong>of</strong> the ma<strong>in</strong> phylotypes <strong>in</strong> the human <strong>in</strong>test<strong>in</strong>al microbiota (Turroni et al.,<br />
2009).


