Alkyl & Aryl Ligands - Chemistry
Alkyl & Aryl Ligands - Chemistry
Alkyl & Aryl Ligands - Chemistry

You also want an ePaper? Increase the reach of your titles
YUMPU automatically turns print PDFs into web optimized ePapers that Google loves.
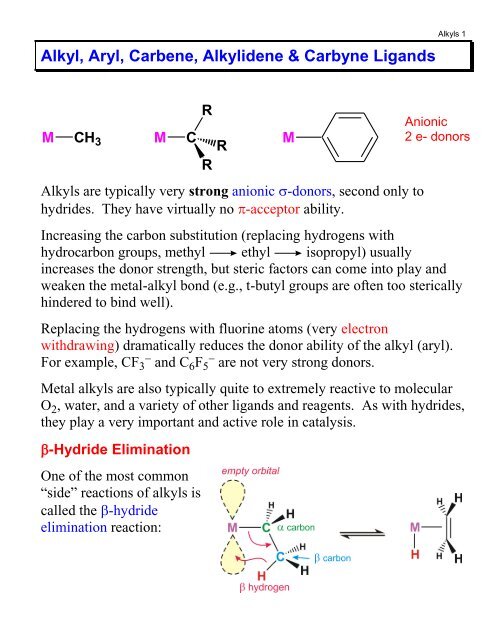
<strong>Alkyl</strong>, <strong>Aryl</strong>, Carbene, <strong>Alkyl</strong>idene & Carbyne <strong>Ligands</strong><strong>Alkyl</strong>s 1M CH 3 M CRRR<strong>Alkyl</strong>s are typically very strong anionic -donors, second only tohydrides. They have virtually no -acceptor ability.Increasing the carbon substitution (replacing hydrogens withhydrocarbon groups, methyl ethyl isopropyl) usuallyincreases the donor strength, but steric factors can come into play andweaken the metal-alkyl bond (e.g., t-butyl groups are often too stericallyhindered to bind well).Replacing the hydrogens with fluorine atoms (very electronwithdrawing) dramatically reduces the donor ability of the alkyl (aryl).For example, CF 3and C 6 F 5are not very strong donors.Metal alkyls are also typically quite to extremely reactive to molecularO 2 , water, and a variety of other ligands and reagents. As with hydrides,they play a very important and active role in catalysis.-Hydride EliminationOne of the most common“side” reactions of alkyls iscalled the -hydrideelimination reaction:MAnionic2 e- donors
<strong>Alkyl</strong>s 2The main driving force for -hydride elimination is the formation of astronger M-H bond (almost always stronger than M-alkyl) and thegeneration of an alkene ligand that reduces the unsaturation of the metalcomplex. The reverse reaction, however, also can occur and is called amigratory insertion. This is very important in transition metal reactionchemistry and catalysis, as we will see in later chapters.Note that in order to have a -hydride elimination you MUST havea empty orbital on the metal cisoidal (next) to the alkyl ligand. Youalso must have -hydrogens present on the alkyl.In order to prepare stable M-alkyl complexes one, therefore, often needsto stay away from alkyls with -hydrogens (or avoid metals with emptycoordination sites). Some common ligands used to avoid -hydrideelimination reactions are shown below.M CH 3MMeMMSiMeMeMeMeMemethyl neopentyl benzyl trimethylsilylmethylProblems:a) Why doesn’t a 16e- M-phenyl do a -hydride elimination?HHMeMHMMeHHMeb) Would a 16 e- M-(t-butyl) complex be stable or not? Why?
-Hydride EliminationA less common reaction with metal alkyls is the -hydride elimination,where a hydrogen atom on the -carbon is added to the metal togenerate a M=CR 2 (carbene or alkylidene) group and a hydride:<strong>Alkyl</strong>s 3MeMePPMeTaClHHCl-hydrideelminationcould alsobe consideredan oxidativeadditionMeMeClPPMeHHTaClmigratoryinsertionMeMePPMeTaClPractice your electron counting!!ClNote that just as with a -hydride elimination, it is important to havean empty orbital cis to the -hydrogen in order to have the -hydride elimination occur. In the next section on carbene/alkylideneligands, we will see that depending on how you electron count, an -hydride elimination can also be considered to be a C-H bond oxidativeaddition (see that chapter as well).Synthesis:The most common way of making metal alkyls is to do what is called atransmetallation, that is react a transition metal halide with a alkali ormain group metal alkyl, which is typically far more ionic and reactive:M–X + LiR M–R + LiXOther reactive alkyl reagents: RMgX (Gignard), R 2 Zn, R 2 Hg, R 2 Cu, AlR 3WCl 6 + 6AlMe 3 WMe 6 + 6AlClMe 2Problem: Based on core photoelectron spectroscopy, which complexis more electron-rich at the metal – W(CH 3 ) 6 or W(CO) 6 ?? Why?
The other common way of making M-alkyls is to react a moderatelyelectron-rich metal center with an alkyl halide (RCl, RBr or RI):ML n + RBrThis, once again, is called an oxidative addition.PPRhICO + MeIR–ML n BrPPCH 3IRhCOIThis will be discussed more fully in the oxidative addition reactionchapter.<strong>Aryl</strong> <strong>Ligands</strong><strong>Aryl</strong> ligands are relatively strong anionic twoelectron donors, essentially just like alkyls. MSince they cannot easily -hydride eliminate(formation of the benzyne intermediate istypically too unstable), metal aryl complexes are usually relativelystable compared to alkyls with -hydrogens. But “stable” is a relativeterm since transition metal aryl complexes are also quite air-sensitiveand reactive.<strong>Alkyl</strong>s 4<strong>Aryl</strong>s do have the potential for both -donation and -backbondingthrough the filled aryl -orbitals and empty * antibonding orbitals.This can provide additional stability to a metal complex, depending onwhether the metal needs additional electrons from the ligand or wants todump excess electron density onto the ligand.Problem: Cp 2 Re-CH 2 CH 3 is very stable under inert atmosphere,but Cp 2 Sc-CH 2 CH 3 readily decomposes. Why?
Fischer Carbenes<strong>Alkyl</strong>s 5In 1964 Fischer’s group prepared the first transitionmetal carbon double bond, which he called a carbene,after the very reactive neutral organic CR 2 fragment.(OC) 5 WOW(CO) 6 + CH 3 Li(OC) 5 WCH 3OCH 3(CH 3 ) 3 O +(OC) 5 WOCH 3CH 3Ernst O. FischerTechnical University of Munich,GermanyThe reaction of Cr(CO) 6 with Li[N(i-Pr) 2 ], followed by reaction withEt 3 O + generated the analogous Cr carbene complex with Et and N(i-Pr) 2groups on the carbene. A crystal structure of this complex revealed thefollowing unusual features of the Cr=C(Et)[N(i-Pr) 2 ] group:2.13 Å (Cr-R single bonddistances are 2.0-2.2 Å)CrOEtN(iPr) 21.35 Å (normal distance should be1.41 Å, a 0.06 Å shortening)1.33 Å (normal distance should be1.45 Å, a 0.12 Å shortening)Thus, the X-ray structure indicated that the actual electronic structure ofthis “carbene” was really more like one of these resonance hybrids:CrOEtCrOEtN(iPr) 2N(iPr) 2The presence of 5 electron-withdrawing CO ligands would certainlyhelp “suck up” the formal negative charge that these resonancestructures put on the metal.
<strong>Alkyl</strong>s 6The bonding description commonly used to describe Fischer Carbenes isto treat the carbene as a neutral 2e- donor ligand that really only makes asingle bond to the metal (BUT, we often draw it as a double bond!!). Inconsidering the carbene as a neutral ligand, it has one filled orbital (sp 2hybrid) that donates it’s lone pair to an empty orbital on the metal in atypical ligand fashion. But it also has one empty orbital (pure pcharacter) that wants to interact with a lone pair of electrons in order toform a stabilizing bonding interaction. This is a singlet state carbeneformalism and the possible orbital interactions are shown below:If the metal is electron deficient (perhaps due to all the good -acceptorCO ligands) then it can’t -donate very well to the carbene. Thus weend up with a M-C single bond (even though we draw a double bond!)and some multiple bond character between the carbene carbon and the-donor groups attached to it (like a NR 2 , OR, SR, Ph, etc).Most Fischer Carbenes have d 6 metal configurations (assuming that weelectron count the carbene ligand as a neutral 2 e- donor), but d 4 and d 8systems are known.
The bond strength in Fischer Carbenes depends on several factors:<strong>Alkyl</strong>s 7MetalCarbene groupsWeak M=CElectron-deficient(electron withdrawing ligandslike CO, NO, 1 st row metal,electronegative metal)Good donating functionalgroups that can -bond to thecarbene (like NR 2 , SR, OR,Ph); more than one donatinggroup really weakens the M-Cbond!!Strong M=CElectron-rich(electron donating ligands, 3 rdrow metal)Simple sigma donors like H orCH 3 that can’t -donate to theCarbene carbon atom.Note that most Fischer Carbenes favor the weak bonding situation,where the metal has a d 6 configuration (counting the carbene as neutralligand), CO ligands, and the carbene has -donating groups. The d 6configuration naturally favors the middle to late transition metals. Thestrong carbene bonding situation is actually considerably more reactive,much like the reactivity of a C=C double bond vs. a C-C single bond.The C=C double bond is stronger than the single bond, but it iskinetically considerably more reactive due to its unsaturation.Problem: Choose the complex that has the stronger M=C bond. Isthere a large or small difference in bond strengths? Explain.a) [Cp(CO) 2 (PPh 3 )Mo=CH 2 ]+ -or- [Cp(CO) 2 (PPh 3 )W=CH 2 ]+b) [Cp(CO) 2 (PPh 3 )W=CH 2 ]+ -or- [Cp(CO) 2 (PEt 3 )W=CH 2 ]+c) [Cp(dppe)Fe=CH 2 ]+ -or- [Cp(NO)(PPh 3 )Re=CH 2 ]+ (tricky!)
Problem: Order the following Fischer Carbenes from the weakestto the strongest M=C bond. Explain.<strong>Alkyl</strong>s 8a) Ph OMe b) Ph Me c)CCH 3 CCN P(OMe) 3Me 3 P PMe 3ReRe(MeO) 3 PCOMe 3 PClICO OOMeOCCCMnClOMeCCOOThe other reactivity characteristic of Fischer Carbenes is that becausethe carbene carbon atom formally has an empty p orbital, it is verysusceptible to nucleophillic attacks there. On the other hand,electrophiles tend to attack the metal center where there are a number ofmetal based lone pairs available.Schrock <strong>Alkyl</strong>idenesIn 1973 Richard Schrock, while working at DuPontcentral research, prepared the first early transitionmetal complex with a metal=carbon double bond:(t-butyl-CH 2 ) 3 TaCl 2+ 2Li(CH 2 -t-butyl)-eliminationTa(CH 2 -t-butyl) 5(t-butyl-CH 2 ) 3 Taunstable intermediateMeHMeMeRichard SchrockMIT+ neopentaneThis turned out to be a key development in early transition metalchemistry.
<strong>Alkyl</strong>s 9Unlike most Fischer Carbenes, these early transition metal alkylidenecomplexes did have clear-cut and strong metal=carbon double bonds.For example, the crystal structure of the Cp 2 Ta(=CH 2 )(CH 3 ) complexhas the following bond distances:138°TaCH 3CH 22.24 ÅThe Ta=CH 2 bond is distinctly shorterthan the Ta-CH 3 single bond!2.03 ÅThe reason that Schrock gave these “carbene” complexes a differentname (alkylidenes) was not just because of the structural differences.These early transition metal alkylidene complexes had very different(almost opposite) reactivities compared to Fischer Carbenes:Fischer CarbenesNucleophillic attacks at carbon atom ofcarbene (carbon is electron deficient)Electrophillic attacks on metal center(metal is more electron-rich, often d 6 18e- system)Carbene is stabilized by heteroatomgroups that can -bond to it. Likes NR 2 ,SR, OR, or Ph groups.Later transition metals favored,especially with d 6 counts (carbene asneutral 2e- donor ligand)Schrock <strong>Alkyl</strong>idenesElectrophillic attacks at carbon atom ofalkylidene (carbon is electron-rich)Nucleophillic attacks on metal center(metal is electron-deficient, usually d 2 ord 0 16 or 14 e- count)<strong>Alkyl</strong>idene is destabilized by heteroatomgroups that can -bond to it. Stronglyprefers H or simple alkyl groups.Early transition metals favored,especially with d 0 centers (alkylidene asdianionic 4e- donor)
The bonding description commonly used to describe Schrock<strong>Alkyl</strong>idenes is to treat the alkylidene as a dianionic 4e- donor ligand,which is what the electroncounting and valence rules fromthe first chapter would indicate.The filled p-orbital on thealkylidene carbon nicelyexplains the tendency forelectrophiles to attack at thissite, while in a Fischer carbenethis same orbital is formallyempty and thus susceptible to anucleophillic attack.<strong>Alkyl</strong>s 10Similarly, the d 0 metal center in the typical Schrock alkylidene usuallyonly has a 12 to 16 e- count (often 14 e-), this means that there areseveral empty low energy orbitals that are very attractive to anynucleophile that can sterically access the metal center. In Fischercarbenes, the metal is typically d 6 and 18e-, thus there are no emptyorbitals on the metal for a nucleophile to attack.One other way to view a Schrock alkylidene is as a neutral ligand, justas with a Fischer carbene, but that it is in the triplet carbene state andinteracting with a spin unpaired d 2 metalcenter:The view of an alkylidene as a neutraltriplet carbene forming a strong covalentdouble bond to a triplet metal center isvery analogous to the covalent C=Cdouble bond in organic chemistry.
<strong>Alkyl</strong>s 11Molecular orbital (MO) diagrams for generic Schrock alkylidene andFischer carbene ligands are shown below starting with both carbonfragments as neutral triplet (alkylidene) and singlet (carbene) groups:metal dorbitalsempty dorbitalsalkylidenep and sp2orbitalspmetal dorbitalscarbenethis extra dorbital used forbonding toother ligandssp 2Schrock alkylidene2Metal = dalkylidene triplet state(Ta 3+ , Nb 3+ , etc)Fischer carbeneMetal = d6carbene singlet state(Cr 0, Mo 0 , Re +1, etc)Note that the higher energy early transition metal orbitals match upmuch better with the higher energy triplet alkylidene orbitals – this leadsto considerably stronger covalent bonding (both MO diagrams are on thesame energy scale).
RR5: MATHEMATICS AND MIDDLE SCHOOL STUDENTS OF MEXICAN DESCENT6/4/09 5:04 PMreceive a great deal of attention in comparison classes, and that theme students would be exercisingcomputational skills in meaningful problem-solving contexts rather than practicing skills and algorithms asisolated ends in themselves. The fact that, overall, the heterogeneously grouped theme students didsomewhat better than those in more conventional classes served to alleviate the mathematics teachers'anxiety about teaching students in heterogeneous ability groupings, and they were extremely relieved thattheir students did not do less well in computation than they might have done in a more conventional class.The achievement data, then, provide considerable cause for optimism. With refinement and experience, webelieve that the thematic approach could become even more effective. In this regard, it is especiallyimportant to recall a major advantage of thematic and other approaches that include small group instruction:They provide the opportunity to engage students in instructional conversations (Goldenberg, 1991; Tharp &Gallimore, 1991). From a sociocultural perspective, learning takes place first on a social plane (intermentalfunctioning), with skills, concepts, and understandings acquired in social interaction later becominginternalized, enabling individual students then to function independently on the intramental plane (Wertsch,1991). Language is the vehicle of learning within this framework. Lacking opportunities for this kind oflinguistic engagement in their home environment, students from linguistic and cultural minoritybackgrounds will continue to be disproportionately at risk for failure in mathematics. In fact, as conventionalassessments of mathematics achievement give way to more varied approaches to assessment (Romberg,Zarinnia, & Collis, 1990), including more open-ended problems, it is reasonable to expect an increase in theachievement discrepancy between native speakers of English and minority students whose instruction failsto promote linguistic engagement in problem solving.The attitudinal data present an interesting characterization of this mainly poor, predominantly Hispanicsample of middle school students. On the basis of historical and demographic data, it seems safe to assumethat many of these students are at risk for educational failure and school drop out. If we expect, therefore, tofind among these students a high proportion who are alienated and consider themselves to be failures atmathematics, the data suggest we are mistaken. The data for both the first and second years of the studypresent a consistent picture. These students expressed a high degree of liking for mathematics. Moreover,they generally considered themselves to be good at it. The majority indicated that they wanted to take moremathematics, and, to a greater degree than the NAEP sample, they indicated that they expected to work inan occupation that would require mathematics. It was also encouraging to note that the majority did notexpress anxiety about the subject. Furthermore, relatively few considered mathematics to be boring.On the face of it then, these data seem to present an optimistic picture. The finding of positive schoolattitudes seems consistent with recent data from the California Identity Project showing that Hispanics inCalifornia, although disproportionately poor, do not demonstrate the characteristics usually associated withan underclass (Hayes-Bautista, Hurtado, Valdez, & Hernandez, 1990). However, it is disconcerting whenwe consider these data in relation to achievement outcomes. Although there was improvement over thecourse of both years, the absolute level of mathematics achievement of the students in our study wasactually quite low. The majority of students had some difficulty with simple arithmetic skills that shouldhave been mastered in elementary school. The juxtaposition of low achievement with the belief held bymost students that they were good at mathematics suggests that they may have gained misleadingexpectationsfrom theirelementary school experience. If teachers set lower goals for minority students thanfor non-minority students, as the literature suggests (Cocking & Chipman, 1988), students may be deprivedof bench marks from which to judge the level of their own performance. In this regard, it is interesting torecall Gandara's (1982) finding that many Hispanic females with exceptional educational accomplishmentsattributed part of their success to having attended integrated schools with high-achieving students whoestablished a standard of comparison. In the context of the theme approach, it is especially interesting tofile:///Users/morganenriquez/Desktop/untitled%20folder/BE021098.webarchivePage 12 of 17
NMR DataThere isn’t any clear cut way of distinguishing Fischer carbenes fromSchrock alkylidenes. Some complexes, of course, will fall in betweeneither category (shades of gray) and can’t be clearly identified.13 C NMR data is potentially one way of distinguishing betweencarbenes and alkylidenes because the chemical shift of the carbenecarbon is usually quite sensitive tothe chemical environment,electron density, and bondingfactors. Unfortunately, the NMRdata, although sometimes useful,generally won’t allow one toidentify when a system is acarbene or alkylidene, as shown inthe table to the right.Compound13 C (ppm)<strong>Alkyl</strong>s 13ClassCp 2 Ta(=CH 2 )(Me) 224 Schrock(t-BuCH 2 ) 3 Ta(=CH(t-Bu) 250 Schrock(OC) 5 Cr(=CH(NMe 2 )) 246 Fischer(OC) 5 Cr(=CPh(OMe)) 351 Fischer(OC) 5 Cr(=CPh 2 ) 399 Fischer1 H NMR data has provided useful information about carbene rotationalbarriers. As with most double bonds, there is a rotational barrier for theM=CR 2 bond. For Schrock alkylidenes this is usually quite high (G ‡> 100 kJ/mol), but for the more weakly bonded Fischer carbenes this canoften be readily determined from variable temperature 1 H NMR studies.The Cr carbene shown below actually has more double bond characterbetween the carbene carbon and the -OMe group relative to the Cr=Cbond. At 25ºC the methoxy CH 3 group shows a single 1 H NMRresonance indicating that there is relatively fast rotation about the C-OMe bond, while at 40ºCHthere are two resonances3 COOfor the methoxy CH 3 (OC) 5 Cr(OC) 5 Crgroup, one for the cis andCHtrans conformers,3CH 3consistent with partialtranscisdouble bond character.CH3
Carbynes/<strong>Alkyl</strong>idynes M C R<strong>Alkyl</strong>s 16E. O. Fischer accidentally prepared the first M≡C-R triple bondedcompound in 1973:MeORCOOC M COOCOCM = Cr, Mo, WX = Cl, Br, IR = Me, Et, Phplanned rxndidn't workCOOC M CO+ BX 3 OCCORCCOOC M COOCXHe called this a carbyne after alkyne, which refers to a C≡C triple bond.Early transition metal versions were prepared first by Schrock in 1978via -deprotonation of the alkylidene:XRClClTaRH1. PMe 3TaCl2. Ph 3 P=CH 2Me 3 PCR+ [Ph 3 PCH 3 ]ClThese were called alkylidynes by Schrock. Fortunately, while there aresome differences between early and later transition metal carbon triplebonds, we can treat them as basically being the same. Thus, one cansimply treat carbynes and alkylidynes as trianionic (-3) 6e- donatingligands. They are very strong donors as might be expected from therelatively low electronegativity of carbon and the -3 formal charge.CO = 1938 cm -1Ph HCOCl W PMe 3Me 3 PClPhCCOCl W PMe 3Me 3 PPyCO = 1870 cm -1