

IV) Materials calculations with dynamical mean field theory (DMFT)
IV) Materials calculations with dynamical mean field theory (DMFT)
IV) Materials calculations with dynamical mean field theory (DMFT)

You also want an ePaper? Increase the reach of your titles
YUMPU automatically turns print PDFs into web optimized ePapers that Google loves.
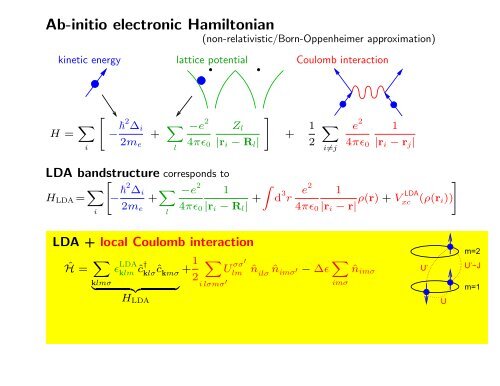
Ab-initio electronic Hamiltonian(non-relativistic/Born-Oppenheimer approximation)kinetic energy lattice potential Coulomb interactionH = X i"− 2 ∆ i2m e+ X l−e 24πɛ 0Z l|r i − R l |#+ 1 2Xi≠je 24πɛ 01|r i − r j |LDA bandstructure corresponds toH LDA = X i"− 2 ∆ i2m e+ X l−e 24πɛ 01|r i − R l | + Zd 3 r#e2 1LDAρ(r) + Vxc (ρ(r i ))4πɛ 0 |r i − r|LDA + local Coulomb interactionĤ = X klmσɛ LDAklm ĉ†klσĉkmσ+ 1 2| {z }H LDAXU σσ′lmi lσmσ ′ˆn ilσ ˆn imσ ′ − ∆ɛ X imσˆn imσU’Um=2U’−Jm=1






