Liquefaction co-processing of coal shale oil at - Argonne National ...
Liquefaction co-processing of coal shale oil at - Argonne National ...
Liquefaction co-processing of coal shale oil at - Argonne National ...

You also want an ePaper? Increase the reach of your titles
YUMPU automatically turns print PDFs into web optimized ePapers that Google loves.
ABSTRACT<br />
LIQUEFACTION CO-PROCESSING OF COAL AND<br />
SHALE OIL AT LOW SEVERITY CONDITIONS<br />
R. L. Miller and R. M. Baldwin<br />
Chemical Engineering and Petroleum Refining Department<br />
Colorado School <strong>of</strong> Mines<br />
Golden. Colorado 80401<br />
Results are reported for a series <strong>of</strong> single stage b<strong>at</strong>ch reactor experiments<br />
in which Wyodak subbituminous <strong>co</strong>al and <strong>shale</strong> <strong>oil</strong> derived from<br />
medium grade Colorado <strong>oil</strong> <strong>shale</strong> were <strong>co</strong>-processed <strong>at</strong> low severity<br />
reaction <strong>co</strong>nditions using CO/H2O as reducing agent. Distill<strong>at</strong>e yields<br />
<strong>of</strong> over 85 wt% MAF <strong>co</strong>al with hydrogen equivalent <strong>co</strong>nsumptions <strong>of</strong> about<br />
1.0 wt% MAF <strong>co</strong>al were obtained <strong>at</strong> 600°F reaction temper<strong>at</strong>ure. Results<br />
from blank <strong>shale</strong> <strong>oil</strong> runs <strong>at</strong> the same mild reaction <strong>co</strong>nditions<br />
suggested th<strong>at</strong> <strong>shale</strong> <strong>oil</strong> residuum reactivity was enhanced in the<br />
presence <strong>of</strong> <strong>co</strong>al or primary <strong>co</strong>al-derived products.<br />
INTRODUCTION<br />
The possibility <strong>of</strong> liquefying <strong>co</strong>al <strong>at</strong> low severity reaction <strong>co</strong>nditions<br />
((700'F) has intriyiied researchers for many years. As early as 1921,<br />
Fischer and Schrader (1) reported production <strong>of</strong> an ether-soluble<br />
m<strong>at</strong>erial from <strong>co</strong>al <strong>at</strong> 660°F using carbon monoxide and w<strong>at</strong>er as reducing<br />
agent. More recently other groups including the Pittsburgh<br />
Energy Technology Center (PETC) (2) ~ the North Dakota Energy Research<br />
Center (3-4). SRI (5-7). and Carbon Resources, Inc. (8) have investig<strong>at</strong>ed<br />
various methods <strong>of</strong> utilizing H2, CO/H20 or CO/H2/H20 (synyas)<br />
in low severity liquefaction processes. Many incentives exist for<br />
<strong>co</strong>nverting <strong>co</strong>al <strong>at</strong> milder reaction <strong>co</strong>nditions. The most important <strong>of</strong><br />
these are listed below:<br />
1) Reduced H2 (or CO) <strong>co</strong>nsumption and hydrocarbon gas make<br />
2) Better distill<strong>at</strong>e and residuum product quality, since carboniz<strong>at</strong>ion<br />
and other retrogressive reactions are suppressed to<br />
a large extent<br />
3) Production <strong>of</strong> less refractory residuum which is more susceptible<br />
to hydrocracking in a <strong>co</strong>nventional se<strong>co</strong>nd stage<br />
hydrotre<strong>at</strong>er or hydrocracker<br />
4) Significant energy savings associ<strong>at</strong>ed with lower oper<strong>at</strong>ing<br />
temper<strong>at</strong>ures<br />
5) Less severe slurry handling and m<strong>at</strong>erials <strong>of</strong> <strong>co</strong>nstruction<br />
problems<br />
A number <strong>of</strong> studies have been reported in which <strong>co</strong>al and non<strong>co</strong>al-derived<br />
heavy <strong>oil</strong> have been <strong>co</strong>-processed <strong>at</strong> severe reaction<br />
<strong>co</strong>nditions to obtain valuable distillable liquid products (9-11). In<br />
some cases. larger distill<strong>at</strong>e yields were obtained by <strong>co</strong>-<strong>processing</strong><br />
<strong>co</strong>al and heavy <strong>oil</strong> than by <strong>processing</strong> each feed separ<strong>at</strong>ely. Shale <strong>oil</strong><br />
has been identified as a particularly promising feed, due in part to<br />
the high' heterocyclic basic nitrogen <strong>co</strong>ntent <strong>of</strong> the <strong>oil</strong> (12). In<br />
spite Of the advantages <strong>of</strong> oper<strong>at</strong>ing <strong>at</strong> milder <strong>co</strong>nditions, little work<br />
on low severity <strong>co</strong>-<strong>processing</strong> has been reported.<br />
152
The objective <strong>of</strong> this paper is to report yield and <strong>co</strong>nversion d<strong>at</strong>a<br />
from a series <strong>of</strong> single-stage low severity <strong>co</strong>-<strong>processing</strong> runs using<br />
Wyodak subbituminous <strong>co</strong>al and <strong>shale</strong> <strong>oil</strong> derived from medium grade<br />
Colorado <strong>shale</strong>. Blank <strong>shale</strong> <strong>oil</strong> runs (no <strong>co</strong>al added) were also <strong>co</strong>m-<br />
pleted <strong>at</strong> low severity <strong>co</strong>nditions to estim<strong>at</strong>e the individual distil-<br />
l<strong>at</strong>e yield <strong>co</strong>ntributions <strong>of</strong> <strong>co</strong>al and <strong>shale</strong> <strong>oil</strong>.<br />
EXPERIMENTAL PROCEDURE<br />
Wyodak subbituminous <strong>co</strong>al sample Wyo-3 was used as feed <strong>co</strong>al in the<br />
low severity liquefaction <strong>co</strong>-<strong>processing</strong> experiments. The ultim<strong>at</strong>e<br />
analysis for this sample is presented in Table I. Sampling and<br />
prepar<strong>at</strong>ion details <strong>of</strong> the <strong>co</strong>al have been reported elsewhere (13,14).<br />
Previous reactivity studies performed on four Wyodak subbituminous<br />
<strong>co</strong>als including WYO-3 indic<strong>at</strong>ed th<strong>at</strong> Wyo-3 was an extremely reactive<br />
<strong>co</strong>al <strong>at</strong> represent<strong>at</strong>ive direct liquefaction reaction <strong>co</strong>nditions<br />
(13,151. The high degree <strong>of</strong> reactivity was primarily <strong>at</strong>tributed to<br />
the high organic sulfur and reactive maceral (vitrinite and exinite)<br />
<strong>co</strong>ntent <strong>of</strong> Wyo-3 <strong>co</strong>al. Coal samples were dried to less than 1.0 wt%<br />
moisture <strong>co</strong>ntent before use.<br />
Two <strong>shale</strong> <strong>oil</strong> samples were used in low severity liquefaction <strong>co</strong>-<br />
<strong>processing</strong> runs. Solvent A-5 was a full b<strong>oil</strong>ing range sample <strong>of</strong> <strong>shale</strong><br />
<strong>oil</strong> obtained from the Western Research Institute (formerly the Laramie<br />
Energy Technology Center <strong>of</strong> the Department <strong>of</strong> Energy). This sample<br />
was produced from thermal retorting <strong>of</strong> medium grade (29 gal/ton)<br />
Colorado <strong>oil</strong> <strong>shale</strong>. Solvent A-6 was prepared by mildly hydrotre<strong>at</strong>ing<br />
a portion <strong>of</strong> sample A-5 in a two liter b<strong>at</strong>ch Autoclave Nagnedrive I1<br />
reactor <strong>at</strong> 650°F for one hour with an initial <strong>co</strong>ld hydrogen pressure<br />
<strong>of</strong> 2000 psig. , Nal<strong>co</strong>mo 477 <strong>co</strong>balt molybd<strong>at</strong>e c<strong>at</strong>alyst was used to<br />
hydrotre<strong>at</strong> the <strong>shale</strong> <strong>oil</strong>. C<strong>at</strong>alyst samples were thermactiv<strong>at</strong>ed <strong>at</strong><br />
100O0F for two hours in a muffle furnace prior to use. Approxim<strong>at</strong>ely<br />
0.6 wt% hydrogen was <strong>co</strong>nsumed by the <strong>shale</strong> <strong>oil</strong> during hydrotre<strong>at</strong>ing.<br />
Properties <strong>of</strong> <strong>shale</strong> <strong>oil</strong> samples A-5 and A-6 are presented in Table<br />
11. Approxim<strong>at</strong>ely 50 wt% <strong>of</strong> the nitrogen in these samples existed in<br />
quinoline-type or hydroquinoline-type molecular structures.<br />
Runs were carried out in a 60 cm3 stirred microautoclave reactor sys-<br />
tem designed and <strong>co</strong>nstructed <strong>at</strong> the University <strong>of</strong> Wyoming. This<br />
reactor was similar to larger Autoclave b<strong>at</strong>ch reactors except th<strong>at</strong><br />
he<strong>at</strong>ing was ac<strong>co</strong>mplished with an external high temper<strong>at</strong>ure furnace.<br />
AI. the end <strong>of</strong> each run, the reactor and its <strong>co</strong>ntents were quenched<br />
with an icew<strong>at</strong>er b<strong>at</strong>ch. This reactor system provided the benefits <strong>of</strong><br />
small tubing bomb reactors [quick he<strong>at</strong>up (-1 min. from room temper-<br />
<strong>at</strong>ure to 650°F) and <strong>co</strong>oldown (-30 sec. back to room temper<strong>at</strong>ure)],<br />
while <strong>at</strong> the same time insuring sufficient mechanical agit<strong>at</strong>ion <strong>of</strong> the<br />
reactants with an Autoclave Magnedrive I1 stirring assembly to min-<br />
imize hydrogen mass transfer effects. The system was also designed so<br />
th<strong>at</strong> the reactor pressure was very nearly <strong>co</strong>nstant throughout an<br />
experiment. Two iron-<strong>co</strong>nstantan thermo<strong>co</strong>uples <strong>at</strong>tached to a Fluke<br />
2175A digital thermometer were used for temper<strong>at</strong>ure measurements. One<br />
thermo<strong>co</strong>uple measured the temper<strong>at</strong>ure <strong>of</strong> the reactor <strong>co</strong>ntents, while<br />
the other measured the temper<strong>at</strong>ure <strong>of</strong> the reactor wall. Reactor<br />
pressure was monitored using a 0 - 5000 psi Marsh pressure gauge.<br />
In these runs, carbon monoxide and w<strong>at</strong>er were used as reducing agent.<br />
with hydrogen being produced via the aqueous phase w<strong>at</strong>er-gas shift<br />
reaction. Reaction <strong>co</strong>nditions were studied in the range: 600-650°F
eaction temper<strong>at</strong>ure, 1000-1500 psig initial <strong>co</strong>ld carbon monoxide<br />
pressure, and 15-60 minutes reaction time. Distilled w<strong>at</strong>er is an<br />
amount equal to 50 wt% <strong>of</strong> the dry feed <strong>co</strong>al was charged to each<br />
reactor run. Iron sulf<strong>at</strong>e (5 wt% dry feed <strong>co</strong>al) was used as a<br />
disposable c<strong>at</strong>alyst.<br />
Gaseous products were analyzed using gas chrom<strong>at</strong>ography. W<strong>at</strong>er and<br />
distill<strong>at</strong>e yields were measured by distilling portions <strong>of</strong> the <strong>co</strong>mbined<br />
liquid-solid product mixture to an 85OOF endpoint in a microdistill<strong>at</strong>ion<br />
appar<strong>at</strong>us. Additional portions <strong>of</strong> the liquid-solid<br />
product mixture were extracted in a Soxhlet extraction appar<strong>at</strong>us using<br />
cyclohexane, toluene, and pyridine. Details <strong>of</strong> the experimental<br />
procedures used in this work have been reported (9).<br />
RESULTS AND DISCUSSION<br />
Using d<strong>at</strong>a <strong>co</strong>llected with the analytical procedures described, detailed<br />
yield and <strong>co</strong>nversion results were <strong>co</strong>mputed for each liquefaction<br />
<strong>co</strong>-<strong>processing</strong> run. Details <strong>of</strong> the <strong>co</strong>mput<strong>at</strong>ional methods used<br />
in this study have been described previously (9). For purposes <strong>of</strong> the<br />
present discussion. process performance will be monitored using the<br />
following two parameters: C4-85O0F distill<strong>at</strong>e yield ,(wt% MAF <strong>co</strong>al<br />
basis), and pyi'idine <strong>co</strong>nversion (wt% MAF basis). Pyridine <strong>co</strong>nversion<br />
is defined as a measure <strong>of</strong> the extent <strong>of</strong> <strong>co</strong>nversion <strong>of</strong> all feeds (<strong>co</strong>al<br />
and non-<strong>co</strong>al-derived heavy <strong>oil</strong>) to pyridine soluble products. However,<br />
since both A-5 and A-6 <strong>shale</strong> <strong>oil</strong> samples were <strong>co</strong>mpletely soluble<br />
in pyridine and negligible <strong>co</strong>king <strong>of</strong> the <strong>shale</strong> <strong>oil</strong> occurred <strong>at</strong> low<br />
severity reaction <strong>co</strong>nditions, pyridine <strong>co</strong>nversion values reported in<br />
this paper.are direct measures <strong>of</strong> the extent <strong>of</strong> <strong>co</strong>al <strong>co</strong>nversion in the<br />
<strong>co</strong>-<strong>processing</strong> runs.<br />
Effect <strong>of</strong> Shale Oil Prehvdrotre<strong>at</strong>ment<br />
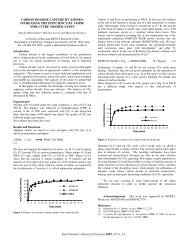
Figure 1 shows distill<strong>at</strong>e yield results from <strong>co</strong>-<strong>processing</strong> runs<br />
<strong>co</strong>mpleted using Wyo-3 <strong>co</strong>al and either A-5 or A-6 <strong>shale</strong> <strong>oil</strong> <strong>at</strong> 600°F<br />
and 1500 psig initial <strong>co</strong>ld CO pressure. This d<strong>at</strong>a clearly shows th<strong>at</strong><br />
mild hydrotre<strong>at</strong>ment <strong>of</strong> the <strong>shale</strong> <strong>oil</strong> gre<strong>at</strong>ly enhances <strong>co</strong>-<strong>processing</strong><br />
performance. Coal <strong>co</strong>nversion also increased significantly when A-6<br />
<strong>shale</strong> <strong>oil</strong> was used in place <strong>of</strong> A-5 <strong>shale</strong> <strong>oil</strong>. Distill<strong>at</strong>e yields <strong>of</strong><br />
over 85 wt% MAF <strong>co</strong>al (58 wt% MAF <strong>co</strong>al and 850°F+ <strong>shale</strong> <strong>oil</strong>) and pyridine<br />
soluble <strong>co</strong>al <strong>co</strong>nversions <strong>of</strong> nearly 60 wt% MAF basis were obtained<br />
with A-6. Previous high severity <strong>co</strong>-<strong>processing</strong> studies using Wyo-3<br />
and A-6 also demonstr<strong>at</strong>ed the beneficial effect <strong>of</strong> prehydrotre<strong>at</strong>ment.<br />
The enhancement <strong>at</strong> low severity <strong>co</strong>nditions can be <strong>at</strong>tributed to: 1)<br />
increased hydrogen donor ability <strong>of</strong> the hydrotre<strong>at</strong>ed <strong>shale</strong> <strong>oil</strong>s and 2)<br />
increased <strong>co</strong>ncentr<strong>at</strong>ion <strong>of</strong> partially hydrogen<strong>at</strong>ed basic nitrogen<br />
<strong>co</strong>mpounds such as tetrahydroquinoline and piperidinopyridine in the<br />
<strong>shale</strong> Oil. These <strong>co</strong>mpounds are known to promote <strong>co</strong>al dissolution and<br />
C<strong>at</strong>alyze the aqueous phase w<strong>at</strong>er-gas shift reaction.<br />
Effect <strong>of</strong> Reaction Temper<strong>at</strong>ure<br />
Figures 2 and 3 present yield and <strong>co</strong>nversion results for <strong>co</strong>-<strong>processing</strong><br />
runs <strong>co</strong>mpleted with Wyo-3 and A-6 <strong>at</strong> 600°F and 65O0F. These d<strong>at</strong>a show<br />
th<strong>at</strong> Process performance improved <strong>at</strong> lower reaction temper<strong>at</strong>ure. This<br />
effect can be <strong>at</strong> least partially <strong>at</strong>tributed to the favorable thermo-<br />
dynamic equilibrium <strong>of</strong> the w<strong>at</strong>er-gas shift reaction <strong>at</strong> lower temper-<br />
<strong>at</strong>ures.<br />
154<br />
I
1<br />
4<br />
t<br />
).<br />
+<br />
E<br />
Effect <strong>of</strong> Initial Carbon Monoxide Pressure<br />
The effect <strong>of</strong> varying the initial Co pressure is illustr<strong>at</strong>ed in Figure<br />
4. At 600°F reaction temper<strong>at</strong>ure, increasing the CO pressure from<br />
1000 to 1500 psig more than doubled the distill<strong>at</strong>e yield over the<br />
entire range <strong>of</strong> reaction times studied. These d<strong>at</strong>a indic<strong>at</strong>e th<strong>at</strong><br />
rel<strong>at</strong>ively high pressure is required to achieve sufficient CO sol-<br />
ubility in the aqueous phase for the w<strong>at</strong>er-gas shift reaction to<br />
proceed <strong>at</strong> a s<strong>at</strong>isfactory r<strong>at</strong>e.<br />
Results from Blank A-6 Shale Oil Runs<br />
In an <strong>at</strong>tempt to estim<strong>at</strong>e the amounts <strong>of</strong> distill<strong>at</strong>e derived from Coal<br />
and from <strong>shale</strong> <strong>oil</strong>, several blank <strong>shale</strong> <strong>oil</strong> runs (no <strong>co</strong>al added) were<br />
<strong>co</strong>mpleted. Results from both high severity and low severity blank<br />
runs are shown in Figure 5. These d<strong>at</strong>a were then used to estim<strong>at</strong>e the<br />
amount <strong>of</strong> distill<strong>at</strong>e <strong>at</strong>tributable to the <strong>shale</strong> <strong>oil</strong> feed in each c0-<br />
<strong>processing</strong> run. Estim<strong>at</strong>es <strong>of</strong> the <strong>co</strong>al-derived distill<strong>at</strong>e production<br />
were <strong>co</strong>mputed by assuming th<strong>at</strong> half <strong>of</strong> the total <strong>co</strong>al-derived cyclo-<br />
hexane soluble product was distill<strong>at</strong>e. Results <strong>of</strong> these calcul<strong>at</strong>ions<br />
are shown in Figure 6. Both low severity and high severity runs are<br />
included in this figure for <strong>co</strong>mparison purposes. In each <strong>co</strong>-pro-<br />
cessing run, additional distill<strong>at</strong>e in excess <strong>of</strong> th<strong>at</strong> predicted by the<br />
blank <strong>shale</strong> <strong>oil</strong> runs was obtained. Thus, it appears likely th<strong>at</strong> the<br />
reactivity <strong>of</strong> <strong>shale</strong> <strong>oil</strong> residuum towards distill<strong>at</strong>e production is<br />
enhanced in the presence <strong>of</strong> <strong>co</strong>al or primary <strong>co</strong>al-derived products.<br />
CONCLUSIONS<br />
A series <strong>of</strong> low severity liquefaction <strong>co</strong>-<strong>processing</strong> runs has been<br />
<strong>co</strong>mpleted using Wyodak subbituminous <strong>co</strong>al and two <strong>shale</strong> <strong>oil</strong> samples.<br />
Results indic<strong>at</strong>ed th<strong>at</strong> prehydrotre<strong>at</strong>ment <strong>of</strong> the <strong>shale</strong> <strong>oil</strong>, lower<br />
reaction temper<strong>at</strong>ure, and higher initial CO pressure all <strong>co</strong>ntributed<br />
to enhanced process performance. Distill<strong>at</strong>e yields in excess <strong>of</strong> 85<br />
wtO MAF <strong>co</strong>al were obtained <strong>at</strong> 60O0F, 1500 psig CO pressure, and 60<br />
minute reaction time. Results from blank <strong>shale</strong> <strong>oil</strong> experiments<br />
suggested th<strong>at</strong> overall distill<strong>at</strong>e yield <strong>co</strong>uld be maximized by <strong>co</strong>-<br />
<strong>processing</strong> <strong>co</strong>al and <strong>shale</strong> <strong>oil</strong> r<strong>at</strong>her than <strong>processing</strong> the two feeds<br />
separ<strong>at</strong>ely.<br />
ACKNOWLEDGEMENTS<br />
Financial support for this research was provided by the Electric Power<br />
Research Institute under Contract Number RP 2383-01. Mr. Conrad Kulik<br />
<strong>of</strong> EPRI provided helpful <strong>co</strong>mments.<br />
REFERENCES<br />
1. Fischer, F. and H. Schrader. Brennst<strong>of</strong>f-Chem., 2. 257. 1921.<br />
2. Appell. H. R., et al., ACS Div. Fuel Chem. Preprints, a, 1, 58,<br />
1975.<br />
3. Sondreal. E. A.. et al., Fuel, 61. 925. 1982.<br />
4. Farnam, S. A., et al., ACS Div. Fuel Chem. Preprints, s, 2, 354,<br />
1985.<br />
5. Ross, D. S., et al.. Fuel, 63. 1206, 1984.<br />
155
6.<br />
7.<br />
8.<br />
9.<br />
10.<br />
11.<br />
12.<br />
13.<br />
14.<br />
15 I<br />
ROSS, D. S., et al., ACS Div. Fuel Chem. Preprints, 30, 3.<br />
1985.<br />
94.<br />
ROSS, D. S.,<br />
1985<br />
et al.. ACS Div. Fuel Chem. Preprints. 30. 4. 339.<br />
Porter. C. R., and H. D. Kaesz. Thirteenth Biennial<br />
Symposium, Bismarck, North Dakota, May 21-23. 1985.<br />
Lignite<br />
Miller, R. L.. "Use <strong>of</strong> Non-Coal-Derived Heavy Solvents in Direct<br />
Coal <strong>Liquefaction</strong>," Interim Report for EPRI Project RP 2383-01,<br />
November 1985.<br />
Yan. T. Y., and Espenscheid. W. F., Fuel Proc. Tech., 7. 121,<br />
1983.<br />
Shinn, J. H., Dahlberg, A. J.. Kuehler, C. W., and Rosenthal. J.<br />
W.. "The Chevron Co-Refining Process." Proceedings <strong>of</strong> the Ninth<br />
Annual EPRI <strong>co</strong>ntractors' Conference on Coal <strong>Liquefaction</strong>. Palo<br />
Alto. California. May 1984.<br />
Miller, R. L., ACS Div. Fuel Chem. Preprints, 31. 1. 301, 1986.<br />
Miller, R. L., "Effect <strong>of</strong> Wyodak (Wyoming) Coal Properties on<br />
Direct <strong>Liquefaction</strong> Reactivity," Ph.D.<br />
School <strong>of</strong> Mines. Golden, Colorado, 1982.<br />
Dissert<strong>at</strong>ion, Colorado<br />
Silver, H. F.. Corry. R. G.. and Millnc. R. L.. "Coal LiqUefaction<br />
Studies," Final Report for EPRI Projects RP 779-23 and RP<br />
2210-1, December 1982.<br />
Miller, R. L., and Baldwin. R. M., Fuel, 64. 1235, 1985<br />
156
Sample<br />
Wt% Distilled<br />
Table I<br />
Ultim<strong>at</strong>e Analysis <strong>of</strong> Wyodak<br />
Subbituminous Coal Wyo-3<br />
Ultim<strong>at</strong>e Analysis, wt% dry basis<br />
Carbon<br />
Hyd r ogen<br />
Nitrogen<br />
Sulfur<br />
Sulf<strong>at</strong>e<br />
Pyrite<br />
Organic<br />
Oxygen<br />
Ash<br />
W<strong>at</strong>er<br />
350°F<br />
350°F-5000F<br />
500°F-6500F<br />
650°F-8500F<br />
BSOOF+<br />
(difference)<br />
Ultim<strong>at</strong>e Analysis. wt% dry basis<br />
Carbon<br />
Hydrogen<br />
Nitrogen<br />
Sulfur<br />
Oxygen (difference)<br />
Ash<br />
Table I1<br />
Properties <strong>of</strong> Shale Oil Samples<br />
157<br />
A- 5<br />
0.7<br />
4.2<br />
9.6<br />
18.8<br />
39.0<br />
27.7<br />
83.3<br />
12.1<br />
1.4<br />
0.5<br />
2.7<br />
0.0<br />
58.2<br />
4.3<br />
0.8<br />
2.9<br />
13.9<br />
19.9<br />
0.8<br />
0.9<br />
1.2<br />
A- 6<br />
0.1<br />
10.3<br />
18.3<br />
22.5<br />
29.8<br />
19.0<br />
84.7<br />
12.9<br />
1.2<br />
0.4<br />
0.8<br />
0.0
I<br />
0"<br />
100- I I I<br />
WyO-3<br />
P<br />
3:<br />
1500 psig CO<br />
I1<br />
0) FeSO,<br />
80- -<br />
2iiJ<br />
.- 2!z<br />
78k<br />
52 60-<br />
E8<br />
0-<br />
Ins<br />
(0<br />
I 40-<br />
0 600 OF<br />
0"<br />
20<br />
600 OF<br />
1500 psig CO<br />
20- I I<br />
0 15 30 - 45 60<br />
Reaction time, minutes<br />
Figure 1. Distill<strong>at</strong>e Yield as a Function <strong>of</strong> Reaction<br />
Time and Shale Oil Feed<br />
I I I<br />
A650 OF<br />
Figure 2. Distill<strong>at</strong>e Yield as a Function <strong>of</strong> Reaction<br />
Time and Temper<strong>at</strong>ure<br />
158
I<br />
.- m<br />
v)<br />
(D<br />
n<br />
L<br />
a<br />
z<br />
8<br />
U<br />
s<br />
r"<br />
.- 0<br />
E<br />
a2<br />
><br />
C<br />
0<br />
0<br />
P,<br />
.-<br />
c<br />
E<br />
L<br />
2,<br />
n.<br />
c WYO-3<br />
1500 psig CO<br />
FeSO,<br />
80<br />
I vu 1 I I I<br />
Reaction time, minutes<br />
600 OF<br />
A650 OF<br />
Figure 3. Pyridine Conversion as a Function <strong>of</strong> Reaction<br />
Time and Temper<strong>at</strong>ure<br />
Reaction time, minutes<br />
Figure 4. Distill<strong>at</strong>e Yield as a Function <strong>of</strong> Reaction<br />
Time and CO Pressure<br />
159
11.3<br />
A-6<br />
yJ 600 OF, 1500 psig CO,<br />
30 rnin<br />
825 OF, 2000 psig H,,<br />
60 min<br />
0.9<br />
C, - 850%F H, equivalent<br />
distill<strong>at</strong>e yield, <strong>co</strong>nsumption,<br />
wt % A-6<br />
wt % A-6<br />
Figure 5. Results from Blank Shale Oil Runs <strong>at</strong> Low<br />
and High Severity Reaction Conditions<br />
WyO-3, A-6<br />
600 OF, 1500 psig CO, 30 min<br />
825 OF, 2000 psig H,, 60 min<br />
Coal - derived Shale <strong>oil</strong> - derived<br />
Distill<strong>at</strong>e, %<br />
Coal or <strong>shale</strong> <strong>oil</strong><br />
- derived<br />
Figure 6. Estim<strong>at</strong>ed Distribution <strong>of</strong> Distill<strong>at</strong>e Production from wyo-3<br />
Coal and A-6 Shale O i l <strong>at</strong> Low and High Severity Reaction<br />
Conditions<br />
160
The Roles and Importance <strong>of</strong> Hydrogen<br />
Don<strong>at</strong>ion and C<strong>at</strong>alysis in Co<strong>processing</strong><br />
Christine W. Curtis, Frankie N. Cassell,<br />
and Donald R. Cahela<br />
Chemical Engineering Department<br />
Auburn University, Alabama 36849<br />
The roles and rel<strong>at</strong>ive importance <strong>of</strong> hydrogen don<strong>at</strong>ion and c<strong>at</strong>alysis in<br />
producing an upgraded product sl<strong>at</strong>e from <strong>co</strong><strong>processing</strong> are explored. These effects<br />
are examined in two types <strong>of</strong> systems: the upgrading <strong>of</strong> residuum and the<br />
<strong>co</strong><strong>processing</strong> <strong>of</strong> <strong>co</strong>al with residuum. In these experiments, the reactions were<br />
performed both thermally and c<strong>at</strong>alytically in the presence and absence <strong>of</strong> tetralin<br />
(TET) to elucid<strong>at</strong>e possible synergetic interactions between the c<strong>at</strong>alyst and TET<br />
for increased <strong>co</strong>al <strong>co</strong>nversion and an improved product sl<strong>at</strong>e.<br />
C<strong>at</strong>alytic <strong>co</strong><strong>processing</strong> which has been extensively reviewed by Monnier (1)<br />
generally produces an improved product sl<strong>at</strong>e and a higher degree <strong>of</strong> upgrading than<br />
does thermal <strong>co</strong><strong>processing</strong>. Previous work performed by Curtis et al. (2,3) has<br />
shown the necessity <strong>of</strong> having highly accessible c<strong>at</strong>alysts for <strong>co</strong><strong>processing</strong>.<br />
C<strong>at</strong>alyst accessibility affects both the amount <strong>of</strong> <strong>co</strong>al <strong>co</strong>nversion and the degree<br />
<strong>of</strong> upgrading <strong>of</strong> liquefied <strong>co</strong>al to soluble products and <strong>of</strong> petroleum asphaltenes to<br />
pentane solubles. In this work, two highly accessible c<strong>at</strong>alysts are investig<strong>at</strong>ed:<br />
an <strong>oil</strong> soluble c<strong>at</strong>alyst, Mo naphthen<strong>at</strong>e, and a presulfided <strong>co</strong>mmercial NiMo/AlzOg<br />
hydrotre<strong>at</strong>ing c<strong>at</strong>alyst which was ground to -200 mesh to increase its accessibility<br />
and, hence, its activity. Mo naphthen<strong>at</strong>e is a metal salt <strong>of</strong> an organic acid th<strong>at</strong><br />
is thought to be <strong>co</strong>nverted to an active c<strong>at</strong>alyst under typical <strong>co</strong><strong>processing</strong><br />
<strong>co</strong>nditions <strong>of</strong> high temper<strong>at</strong>ure and hydrogen <strong>at</strong>mosphere with a partial pressure <strong>of</strong><br />
hydrogen sulfide. The active species is believed to be a non<strong>co</strong>lloidal metal<br />
sulfide (4,5).<br />
The effect <strong>of</strong> hydrogen donor <strong>co</strong>mpounds on the products obtained from<br />
<strong>co</strong><strong>processing</strong> has been examined by Curtis et al. (6). They <strong>co</strong>ncluded th<strong>at</strong> hydrogen<br />
must be available for upgrading reactions to occur in <strong>co</strong><strong>processing</strong> and th<strong>at</strong> the<br />
hydrogen can be present either as molecular H2 or donable hydrogen. The type <strong>of</strong><br />
donor present affected the product sl<strong>at</strong>e; tetrahydroquinol ine promoted <strong>co</strong>al<br />
<strong>co</strong>nversion while tetra1 in and dihydrophenanthrene promoted the production <strong>of</strong><br />
lighter products. When reacted in either a H2 or N2 <strong>at</strong>mosphere, <strong>co</strong>ncentr<strong>at</strong>ions <strong>of</strong><br />
donable hydrogen <strong>at</strong> the 0.5 wt% level or higher were required to substantially<br />
affect <strong>co</strong>al <strong>co</strong>nversion.<br />
The objective <strong>of</strong> the current work is to investig<strong>at</strong>e the effect <strong>of</strong> hydrogen<br />
donor addition in <strong>co</strong>njunction with c<strong>at</strong>alytic hydrotre<strong>at</strong>ment on the products<br />
obtained from upgrading residuum and <strong>co</strong><strong>processing</strong> <strong>co</strong>al with petroleum residuum.<br />
The reaction products are evalu<strong>at</strong>ed in terms <strong>of</strong> solubility fractions, <strong>co</strong>al<br />
<strong>co</strong>nversion to solubles in the solvent extraction scheme used and <strong>oil</strong> production.<br />
Oil production is defined as the amount <strong>of</strong> pentane solubles after reaction minus<br />
the initial pentane solubles divided by the upgradeable m<strong>at</strong>erials which are the<br />
pentane insoluble m<strong>at</strong>erials from maf <strong>co</strong>al and the residuum. In addition, the<br />
amount <strong>of</strong> hydrogen transferred to the products via gas-phase molecular hydrogen<br />
and don<strong>at</strong>ion by hydrogen donors is examined.<br />
The efficacy <strong>of</strong> donable hydrogen<br />
in promoting <strong>co</strong>al <strong>co</strong>nversion and producing a high quality product sl<strong>at</strong>e is<br />
evalu<strong>at</strong>ed in thermal and c<strong>at</strong>alytic <strong>co</strong><strong>processing</strong>.<br />
161
Experimental<br />
UDqradinq and Coorocessinq Reactions. Upgrading and <strong>co</strong><strong>processing</strong> reactions were<br />
<strong>co</strong>nducted in 50 ml stainless steel reactors, charged with 1250 ps-ig H2 <strong>co</strong>ld<br />
(giving -3000 psi <strong>at</strong> reaction temper<strong>at</strong>ure), 39 mf <strong>co</strong>al and 69 solvent. In the<br />
c<strong>at</strong>alytic experiments, the c<strong>at</strong>alyst charge was lg <strong>of</strong> presulfided Shell 324<br />
NiMo/A1202 (0.0449 Mo/g mf <strong>co</strong>al) ground to -200 mesh from 1/16" inch extrud<strong>at</strong>es<br />
or, when using Mo naphthen<strong>at</strong>e, the charge was 0.0029 Mo/g mf <strong>co</strong>al. Reaction<br />
<strong>co</strong>nditions were 30 minutes <strong>at</strong> 4OO0C for the thermal reactions and <strong>at</strong> 400° and<br />
425OC for the NiMo/AlzO3 and Mo naphthen<strong>at</strong>e reactions. The reactors were agit<strong>at</strong>ed<br />
<strong>at</strong> 700 cpm with 2 steel balls as agit<strong>at</strong>ion aids. Maya topped long resid (TLR) was<br />
used in the upgrading reactions and as the solvent in the <strong>co</strong><strong>processing</strong> reactions.<br />
For <strong>co</strong><strong>processing</strong>, Western Kentucky 9/14 bituminous <strong>co</strong>al was used. In the<br />
reactions using TET, one weight percent donable hydrogen was introduced; the TET<br />
<strong>co</strong>mposed about one-third <strong>of</strong> the solvent. Each upgrading or <strong>co</strong><strong>processing</strong> reaction<br />
was, <strong>at</strong> least, duplic<strong>at</strong>ed and some were triplic<strong>at</strong>ed or quadruplic<strong>at</strong>ed. The<br />
activity <strong>of</strong> the NiMo/A1203 was tested by reacting naphthalene in H2 <strong>at</strong> 30OoC.<br />
Naphthalene (NAPH) was nearly totally hydrogen<strong>at</strong>ed forming TET and decalin (DEC),<br />
producing the expected amount (7). The activity <strong>of</strong> the Mo naphthen<strong>at</strong>e was also<br />
tested using naphthalene hydrogen<strong>at</strong>ion.<br />
Product Analysis.<br />
The liquid products obtained from the upgrading and<br />
<strong>co</strong><strong>processing</strong> reactions were analyzed using solvent extraction. The product<br />
fractions obtained were PS - pentane soluble; BS - benzene soluble, pentane<br />
insoluble; MCMS - methylene chloride/methanol soluble benzene soluble; THFS -<br />
tetrahydr<strong>of</strong>uran (THF) soluble, methylene chloride/methanol insoluble; IOM -<br />
insoluble organic m<strong>at</strong>ter, insoluble in THF. In the <strong>co</strong><strong>processing</strong> reactions, <strong>co</strong>al<br />
<strong>co</strong>nversion was calcul<strong>at</strong>ed on the basis <strong>of</strong> the amount <strong>of</strong> m<strong>at</strong>erial <strong>co</strong>nverted to<br />
soluble products and was <strong>co</strong>rrected for the amount <strong>of</strong> IOM formed in the upgrading<br />
reactions with equivalent reaction <strong>co</strong>nditions. The weight <strong>of</strong> the gas after<br />
reaction was measured and the hydrogen <strong>co</strong>nsumption was determined using PVT<br />
methods. The hydrogen <strong>co</strong>nsumption was calcul<strong>at</strong>ed for each reaction.<br />
The PS and BS fractions <strong>of</strong> the reaction products were analyzed by temper<strong>at</strong>ure<br />
programmed gas chrom<strong>at</strong>ography using a Varian Model 3700 equipped with a FID and a<br />
60 m DB-5 J and W fused silica capillary <strong>co</strong>lumn. The PS and BS fractions were<br />
analyzed for TET, DEC and NAPH to determine the amount <strong>of</strong> hydrogen transferred by<br />
TET to <strong>co</strong>al and the petroleum solvent. An internal standard, p-xylene, Waf used.<br />
Dihydronaphthalenes were not detected <strong>at</strong> detectability levels <strong>of</strong> 2.4 X 10- lg.<br />
Solvent extraction analysis <strong>of</strong> Maya TLR showed th<strong>at</strong> the residuum before<br />
reaction was <strong>co</strong>mposed <strong>of</strong> 77.6% PS and 22.4% BS. TET, NAPH, and DEC were not<br />
observed in fte chrom<strong>at</strong>ograms <strong>of</strong> the extracted residuum <strong>at</strong> the detectability levels<br />
<strong>of</strong> 2.4 X 10- g. Western Kentucky 9/14 <strong>co</strong>al was nearly insoluble <strong>at</strong> room<br />
temper<strong>at</strong>ure with less than 3% MCMS and 1% THFS being present.<br />
Discussion <strong>of</strong> Results<br />
Lbcrradina <strong>of</strong> Residuum. The results from the thermal ana c<strong>at</strong>alytic upgrading<br />
reactions performed in the presence and absence <strong>of</strong> tetralin are given in Table 1.<br />
After Maya TLR was thermally upgraded in H2, the product solubility fractions<br />
obtained devi<strong>at</strong>ed only slightly from the unreacted residuum. The major change<br />
observed was the decrease <strong>of</strong> the BS fraction and the form<strong>at</strong>ion <strong>of</strong> lighter and<br />
heavier fractions.<br />
Hydrogen <strong>co</strong>nsumption during the reaction was 17.7 mmole <strong>of</strong> H2<br />
Per 6 9 charge <strong>of</strong> residuum. No TET, NAPH, or DEC was observed in the upgraded<br />
residuum. When TET was added <strong>at</strong> the one percent donable hydrogen level, the<br />
product analysis was similar to th<strong>at</strong> without TET except th<strong>at</strong> the IOM was gre<strong>at</strong>er<br />
162<br />
I
and the <strong>oil</strong> fraction was slightly less (Table 1). Both hydrogen<strong>at</strong>ion and<br />
dehydrogen<strong>at</strong>ion reactions involving TET were observed in the reaction system:<br />
TET --> NAPH t 2H2<br />
TET t 3H2 --> DEC<br />
The dehydrogen<strong>at</strong>ion reaction was predominant and produced H2 while the<br />
hydrogen<strong>at</strong>ion reaction occurred to a lesser extent and <strong>co</strong>nsumed H . The net<br />
amount <strong>of</strong> hydrogen transferred was calcul<strong>at</strong>ed by subtracting the $2 <strong>co</strong>nsumed by<br />
reaction 2 from th<strong>at</strong> produced from reaction 1.<br />
Table 1<br />
Upgrading <strong>of</strong> Residuum<br />
Thermal<br />
400%<br />
Maya<br />
NtMo/Al?O?<br />
400 C 425 C<br />
Mo NaDhthen<strong>at</strong>e<br />
@Q°C 4 2 5 ° C<br />
Maya<br />
Product<br />
Distribution, %<br />
Gas<br />
PS<br />
BS<br />
MCMS<br />
THFS<br />
I OM<br />
Maya<br />
TLR<br />
1.9<br />
80.0<br />
17.1<br />
0.4<br />
0.5<br />
0.1<br />
TLR<br />
t TET<br />
2.0<br />
79.1<br />
17.4<br />
0.3<br />
0.4<br />
0.8<br />
Maya<br />
TLR<br />
2.1<br />
85.4<br />
7.7<br />
0.7<br />
0.7<br />
3.4<br />
Maya TLR<br />
t TET<br />
2.4<br />
88.1<br />
5.3<br />
1.2<br />
1.1<br />
1.9<br />
Maya<br />
TLR<br />
3.7<br />
86.0<br />
4.5<br />
0.4<br />
0.2<br />
5.2<br />
Maya<br />
TLR<br />
1.3<br />
80.5<br />
15.9<br />
0.6<br />
0.2<br />
1.5<br />
Maya<br />
TLR<br />
2.7<br />
86.7<br />
9.9<br />
0.2<br />
0.1<br />
0.4<br />
TLR<br />
t TET<br />
1.7<br />
85.2<br />
11.0<br />
0.9<br />
0.6<br />
0.6<br />
‘&AI es<br />
H Consumed, 19.5 14.2 31.3 28.5 44.3 23.1 19.5 12.82<br />
H2 Transferred,<br />
moles<br />
Total H2 Used,<br />
mmol es<br />
NA*<br />
19.5<br />
2.6<br />
16.8<br />
NA<br />
31.3<br />
0.5<br />
29.0<br />
NA<br />
44.3<br />
NA<br />
23.1<br />
NA<br />
19.5<br />
0.62<br />
13.44<br />
Oil Production, % 11.4 10.2 39.6 53.5 44.1 16.0 43.6 39.5<br />
NA: Not Applicable<br />
In the thermal upgrading reaction with TET, 2.6 mmoles <strong>of</strong> H were transferred<br />
from TET to the residuum and an average <strong>of</strong> 14.2 mmoles <strong>of</strong> molecufar hydrogen were<br />
<strong>co</strong>nsumed; therefore, the total hydrogen uti1 ized by the residuum was 16.8 mmoles.<br />
Although the total amount <strong>of</strong> H transferred to and <strong>co</strong>nsumed by the residuum was<br />
gre<strong>at</strong>er in the reaction without TET, the increased hydrogen utiliz<strong>at</strong>ion by the<br />
residuum did not result in higher <strong>co</strong>nversion <strong>of</strong> BS to PS; both reactions had nearly<br />
equivalent oi 1 production.<br />
C<strong>at</strong>alytic Umradina. In the c<strong>at</strong>alytic upgrading <strong>of</strong> Maya TLR with NiMo/A1203, two-<br />
thirds <strong>of</strong> the original BS were reacted, forming both lighter and heavier products<br />
(Table 1). A substantial amount <strong>of</strong> IOM, 3.4%, was produced <strong>co</strong>mpared to 0.1% in<br />
the thermal reaction. C<strong>at</strong>alytic hydrotre<strong>at</strong>ment increased <strong>oil</strong> production to - 40%<br />
<strong>co</strong>mpared to 11.4% i n the thermal reaction and hydrogen <strong>co</strong>nsumption was doubled.<br />
With NiMo/Al 0 trace but measurable quantities <strong>of</strong> DEC, TET, and NAPH were<br />
observed in 20% the PS and BS product fractions. These results indic<strong>at</strong>e th<strong>at</strong><br />
while substantial upgrading, i .e., high <strong>oil</strong> production and hydrogen <strong>co</strong>nsumption,<br />
occurred, <strong>co</strong>king also occurred, producing heavy products from the residuum.<br />
In Table 2, the hydrogen <strong>co</strong>ntents <strong>of</strong> the PS and BS fractions from the<br />
upgrading reactions are given. Comparing the hydrogen <strong>co</strong>ntent obtained from<br />
163
c<strong>at</strong>alytic to the thermal hydrogen<strong>at</strong>ion shows increases in the hydrogen <strong>co</strong>ntent in<br />
both PS and BS fractions with NiMo/Alz03. Thus, it appears th<strong>at</strong> the increased<br />
hydrogen <strong>co</strong>nsumption resulted in a direct increase <strong>of</strong> the hydrogen <strong>co</strong>ntent <strong>of</strong> the<br />
products. Comparison <strong>of</strong> the gas chrom<strong>at</strong>ograms obtained from both the PS and 8s<br />
fractions prior to the reaction to th<strong>at</strong> obtained after the thermal and c<strong>at</strong>alytic<br />
reactions, however, did not show any visible changes in the product fingerprint.<br />
Changes in the <strong>co</strong>mpounds present or the addition <strong>of</strong> new <strong>co</strong>mpounds to these product<br />
fractions was not discernible in the chrom<strong>at</strong>ograms.<br />
Table 2<br />
Elemental Composition <strong>of</strong> PS and 8s Fractions from Upgrading Fractions<br />
Temper<strong>at</strong>ure PS BS<br />
Reactants OC C<strong>at</strong>alyst % C % H % C XH<br />
Maya TLR No Reaction 84.7<br />
84.7<br />
11.5<br />
11.4<br />
NM*<br />
NM<br />
NM<br />
NM<br />
Maya TLR 400 None 85.7<br />
85.6<br />
11.2<br />
11.4<br />
82.8<br />
83.1<br />
7.0<br />
7.0<br />
Mava TLR 400 NiMo/A1902 ' &.I 87.1<br />
86.8<br />
12.0<br />
11.8 83.9 8.5<br />
Maya TLR 400 Mo Naphthen<strong>at</strong>e 84.9 11.3 82.7 6.5<br />
84.6 11.2 82.9 7.6<br />
-~ -.<br />
Maya TLR<br />
Maya TLR<br />
+ . TFT<br />
425<br />
No Reaction<br />
Mo Naphthen<strong>at</strong>e 86.1<br />
88.0<br />
86.9<br />
11.8<br />
12.1<br />
10.0<br />
85.3<br />
85.3<br />
NM<br />
7.4<br />
7.3<br />
NM<br />
Maya TLR<br />
+ TET<br />
Maya TLR<br />
+ TET<br />
Maya TLR<br />
+ TET<br />
400<br />
400<br />
425<br />
None<br />
NiMo/A1203<br />
Mo Naphthen<strong>at</strong>e<br />
86.6<br />
86.7<br />
87.9<br />
87.6<br />
86.8<br />
87.0<br />
10.2<br />
10.3<br />
10.9<br />
10.8<br />
10.3<br />
10.3<br />
83.6<br />
83.6<br />
79.3<br />
78.2<br />
84.9<br />
84.3<br />
7.3<br />
6.8<br />
8.7<br />
8.4<br />
7.3<br />
7.4<br />
*NM: Not measured<br />
When TET was added, a higher <strong>oil</strong> production (53.5%) and a decrease in BS were<br />
observed, although the total amount <strong>of</strong> heavy products (MCMS, THFS, and IOM) was<br />
similar to the reaction without TET. In these reactions, the c<strong>at</strong>alyst and TET<br />
were both positive factors in producing PS m<strong>at</strong>erials. The molecular hydrogen<br />
<strong>co</strong>nsumed by the residuum was nearly equivalent to the reaction without TET. Only<br />
0.39 mmoles <strong>of</strong> NAPH were produced which was approxim<strong>at</strong>ely one-sixth th<strong>at</strong> produced<br />
in the thermal reaction, indic<strong>at</strong>ing th<strong>at</strong> hydrogen<strong>at</strong>ion <strong>of</strong> NAPH formed during the<br />
reaction to TET occurred in the presence <strong>of</strong> NiMo/AlzO3.<br />
The c<strong>at</strong>alytic upgrading reactions with Mo naphthen<strong>at</strong>e were performed <strong>at</strong> 400<br />
and 425OC. Compared to the thermal reaction, only a small change in product<br />
sl<strong>at</strong>e was observed with Mo naphthen<strong>at</strong>e <strong>at</strong> 400 OC. At an increased reaction<br />
temper<strong>at</strong>ure <strong>of</strong> 425OC, the activity <strong>of</strong> the Mo naphthen<strong>at</strong>e c<strong>at</strong>alyst appeared to be<br />
enhanced since substantial increases in the upgrading <strong>of</strong> Maya TLR were observed;<br />
BS was <strong>co</strong>nverted to PS and only small amounts,
L<br />
h<br />
Coorocessinq <strong>of</strong> Coal with Residuum. In thermal <strong>co</strong><strong>processing</strong>, Maya TLR and Western<br />
Kentucky 9/14 <strong>co</strong>al were reacted in the presence and absence <strong>of</strong> TET <strong>at</strong> a one<br />
percent donable hydrogen level (Table 3). The reaction without TET achieved 47.9%<br />
<strong>co</strong>al <strong>co</strong>nversion which was <strong>co</strong>rrected to ac<strong>co</strong>unt for the IOM produced from the<br />
reaction using Maya TLR alone. The thermal <strong>co</strong><strong>processing</strong> reactions uti1 ized 12.3<br />
mmoles <strong>of</strong> H2 and achieved an <strong>oil</strong> production <strong>of</strong> 12.6%.<br />
<strong>co</strong>nversion increased to 69.7%; however, <strong>oil</strong> production was lowered to 4.1%.<br />
Although the <strong>co</strong>nsumption <strong>of</strong> molecular hydrogen in the reaction with TET was -2<br />
mmoles less than in the reaction without TET, an additional 6.5 moles <strong>of</strong> HZ was<br />
transferred from TET to the <strong>co</strong>al/petroleum system, yielding the total <strong>of</strong> 16.6<br />
mmoles <strong>of</strong> H utilized by the <strong>co</strong>al/residuum system. The increased H2 utiliz<strong>at</strong>ion<br />
by the <strong>co</strong>aljresiduum/TET system resulted in increased <strong>co</strong>al <strong>co</strong>nversion and in the<br />
production <strong>of</strong> the heavier product fractions but not in increased <strong>oil</strong> production.<br />
Table 3<br />
Co<strong>processing</strong> <strong>of</strong> Coal with Residuum <strong>at</strong> 4OO0C<br />
When TET was added, <strong>co</strong>al<br />
Mo<br />
Thermal Ni Mo/A120? Naohthen<strong>at</strong>e<br />
Product Maya TLR Maya TLR Maya TLR Maya TLR + Maya TLR<br />
Distribution, % + Coal Coal + TET + Coal Coal + TET + Coal<br />
Gas 1.7 1.9 -.. 1.8 -.- 1.7 1.9 _._<br />
PS<br />
53.2 48.3 63.6 60.1 54.7<br />
BS<br />
17.3 18.6 17.9 22.3 18.4<br />
MCMS<br />
5.8 9.9 3.1 5.5 8.6<br />
THFS<br />
5.9 9.0 1.6 1.9 8.6<br />
I OM<br />
16.1 12.3 12.0 8.5 7.8<br />
H Consumed,<br />
2ki11 es 12.3 10.1 30.2 45.7 26.7<br />
Hp Transferred,<br />
mol es<br />
Total Hp Used,<br />
mmoles<br />
Corrected Coal<br />
NA*<br />
12.3<br />
6.5<br />
16.6<br />
NA*<br />
30.2<br />
0.6<br />
46.3<br />
NA*<br />
26.7<br />
Conversion, % 4i.9 69.7 68.9 81 .a 78.5<br />
Oil Production, % 12.6 4.1 23.3 29.2 3.8<br />
BS Production, % 5.6 12.9 7.7 22.1 9.7<br />
NA: Not Applicable<br />
C<strong>at</strong>alvtic CoDrocessina. C<strong>at</strong>alytic <strong>co</strong><strong>processing</strong> <strong>of</strong> Western Kentucky <strong>co</strong>al with Maya<br />
TLR was performed in the presence <strong>of</strong> NiMo/A1203 and Mo naphthen<strong>at</strong>e c<strong>at</strong>alysts and<br />
also with and without TET. Analysis <strong>of</strong> the products achieved from these reactions<br />
are given in Tables 3 and 4. C<strong>at</strong>alytic tre<strong>at</strong>ment with NiMo/Al 0 achieved 68.9%<br />
<strong>co</strong>al <strong>co</strong>nversion which was gre<strong>at</strong>er than thermal <strong>co</strong><strong>processing</strong> (4f.d%) and nearly<br />
equivalent to thermal <strong>co</strong><strong>processing</strong> with TET (69.7%). The <strong>oil</strong> production from<br />
c<strong>at</strong>alytic <strong>co</strong><strong>processing</strong> was more than double th<strong>at</strong> <strong>of</strong> the thermal reactions with and<br />
without TET. In addition, higher hydrogen <strong>co</strong>nsumption and lower yields <strong>of</strong> the<br />
MCMS and THFS fractions were obtained, indic<strong>at</strong>ing a more highly upgraded product.<br />
The <strong>co</strong>mbined effect <strong>of</strong> hydrogen don<strong>at</strong>ion from TET and hydrotre<strong>at</strong>ment from<br />
NiMo/Al O3 synergetically promoted <strong>co</strong>al <strong>co</strong>nversion since the addition <strong>of</strong> TET<br />
produce8 a higher <strong>co</strong>al <strong>co</strong>nversion (81.8%) than did the c<strong>at</strong>alyst alone (68.9%) or<br />
the thermal reaction with TET (69.7%). High quality products were produced during<br />
165
the reaction with <strong>oil</strong> production reaching nearly 30%, higher BS and lower levels<br />
<strong>of</strong> MCMS and THFS fractions were also observed. A higher <strong>co</strong>nsumption <strong>of</strong> molecular<br />
hydrogen occurred with TET addition than without. During the reaction, three<br />
times more NAPH was produced than DEC; however, the NAPH production <strong>of</strong> 0.53 mmoles<br />
in the c<strong>at</strong>alytic reaction was low <strong>co</strong>mpared to 3.4 mmoles produced in the thermal<br />
reaction. As in the upgrading reactions, the presence <strong>of</strong> NiMo/A1203 caused NAPH to<br />
be rehydrogen<strong>at</strong>ed to TET and a <strong>co</strong>nsumer <strong>of</strong> H2. The total amount <strong>of</strong> H utilized by<br />
the <strong>co</strong>al/resid/TET system was 46.3 mmoles which was higher than the tierma1<br />
reaction or the NiMo/Al 0 reaction without TET. A small increase in the<br />
hydrogen <strong>co</strong>ntent <strong>of</strong> the%$ was observed <strong>co</strong>mpared to the thermal reaction as shown<br />
in Table 5.<br />
Table 4<br />
Co<strong>processing</strong> <strong>of</strong> Coal with Residuum <strong>at</strong> 425OC<br />
Thermal NiMo/AlTOi- Mo NaDhthen<strong>at</strong>e<br />
Maya TLR Maya L Maya TLR Maya TLR t<br />
Product Distribution, % + Coal t Coal + Coal Coal t TET<br />
Gas 4.1 3.5 4.0 3.6<br />
PS 55.1 61.8 66.8 61.9<br />
BS 15.0 19.8 19.3 21.8<br />
MCMS 3.4 2.8 4.8 6.0<br />
THFS 3.1 2.0 1.6 2.1<br />
IOM 19.4 9.6 3.5 4.6<br />
H Consumed,<br />
2Mbles<br />
H2 Transferred,<br />
mmol es<br />
Total H2 Used,<br />
mmol es<br />
Corrected Coal<br />
Conversion, X<br />
29.8<br />
NA*<br />
29.8<br />
52.4<br />
41.6<br />
NA<br />
41.6<br />
80.6<br />
57.5<br />
NA<br />
57.5<br />
89.5<br />
49.9<br />
2.6<br />
52.5<br />
89.3<br />
Oil Production, % 6.6 22.8 31.4 32.0<br />
BS Production, % -1.8 11.3 12.6 19.5<br />
*NA: Not applicable<br />
When Mo naphthen<strong>at</strong>e was used in <strong>co</strong><strong>processing</strong> <strong>at</strong> 4OO0C, <strong>co</strong>al <strong>co</strong>nversion<br />
increased <strong>co</strong>mpared to the thermal reaction but little other effect was observed<br />
(Table 3). When the temper<strong>at</strong>ure was increased to 425OC (Table 4), substantial<br />
increases in <strong>co</strong>al <strong>co</strong>nversion, hydrogen <strong>co</strong>nsumption, and <strong>oil</strong> production were<br />
observed in the reactions using Mo naphthen<strong>at</strong>e.<br />
c<strong>at</strong>alyst was added and the temper<strong>at</strong>ure was increased, the effect <strong>of</strong> the<br />
temper<strong>at</strong>ure increase on the reaction must be ascertained. This effect can be<br />
evalu<strong>at</strong>ed from the d<strong>at</strong>a given in Table 4, by <strong>co</strong>nparing the products produced<br />
during the thermal reaction <strong>at</strong> 425OC to those produced with Mo naphthen<strong>at</strong>e. Since<br />
both <strong>co</strong>al <strong>co</strong>nversion and <strong>oil</strong> production were low in the thermal reaction <strong>at</strong><br />
425OC, the high levels <strong>of</strong> <strong>co</strong>al <strong>co</strong>nversion and <strong>oil</strong> production can then be<br />
<strong>at</strong>tributed to the c<strong>at</strong>alytic activity <strong>of</strong> Mo naphthen<strong>at</strong>e not the temper<strong>at</strong>ure<br />
increase.<br />
Since in these reactions both a<br />
Comparing Mo naphthen<strong>at</strong>e to NiMo/Al O3 <strong>at</strong> 425OC, shows th<strong>at</strong> Mo<br />
naphthen<strong>at</strong>e is more active in terms <strong>of</strong> <strong>oil</strong> pro8uction and <strong>co</strong>al <strong>co</strong>nversion even<br />
though the <strong>co</strong>ncentr<strong>at</strong>ion level <strong>of</strong> Mo in the NiMo/A1203 reaction was 22 times th<strong>at</strong><br />
in Mo naphthen<strong>at</strong>e reaction.<br />
166
Table 5<br />
Elemental Composition <strong>of</strong> PS and BS Fractions from Co<strong>processing</strong><br />
Temper<strong>at</strong>ure ps BS<br />
Reactants QC C<strong>at</strong>al vst % C %H % C %H<br />
Maya TLR 400 None 84.5 11.4<br />
+ Coal 84.0 11.3 82.8<br />
Maya TLR 400 Ni Mo/A12O3 84.8 10.9 84.6<br />
+ Coal 84.8 10.9 86.8<br />
Maya TLR 400<br />
+ Coal<br />
Maya TLR 425<br />
+ Coal<br />
Maya TLR 400<br />
+ Coal + TET<br />
Maya-TLR 400<br />
+ Coal + TET<br />
Maya TLR 425<br />
+ Coal t TET<br />
Mo Naphthen<strong>at</strong>e 84.3 10.8 83.6<br />
84.3 10.8 84.1<br />
Mo Naphthen<strong>at</strong>e 85.7 11.0<br />
84.6 10.6 86.3<br />
84.9 10.7 85.1<br />
85.1 10.6<br />
None 86.3 10.1 83.8<br />
86.4 10.1 83.5<br />
NiMo/A1203 87.8 10.5<br />
87.1 10.5 86.0<br />
85.7 10.3 84.7<br />
87.4 10.5<br />
Mo Naphthen<strong>at</strong>e 86.9 10.5 85.6<br />
86.5 10.3 85.7<br />
The presence <strong>of</strong> TET in the <strong>co</strong>al/resid/Mo naphthen<strong>at</strong>e system did not<br />
substantially change the product sl<strong>at</strong>e. Coal <strong>co</strong>nversion, hydrogen <strong>co</strong>nsumption and<br />
<strong>oil</strong> production were the same as the reaction without TET. The total amount <strong>of</strong><br />
NAPH formed during the reaction was 1.5 mmoles which fell between th<strong>at</strong> for the<br />
thermal and NiMo/Al 03 reactions. Ten times as much NAPH was formed as DEC. The<br />
H2 utilized by the 60 naphthen<strong>at</strong>e systems, 57.5 mmoles without TET and 52.5 mmoles<br />
with TET, was high <strong>co</strong>mpared to the other reactions performed. The product sl<strong>at</strong>es<br />
from these Mo naphthen<strong>at</strong>e reactions show the effective utiliz<strong>at</strong>ion <strong>of</strong> H2 in terms<br />
<strong>of</strong> <strong>co</strong>al <strong>co</strong>nversion and <strong>oil</strong> production.<br />
1The addition <strong>of</strong><br />
c<strong>at</strong>alyst in the residuum upgrading reactions increased the amount <strong>of</strong> <strong>oil</strong><br />
production achieved and, in the case <strong>of</strong> NiMo/Al 03, substantially increased the<br />
IOM formed. The presence <strong>of</strong> TET had varying effect; but only in the reaction with<br />
NiMo/AlzOj did TET improve the <strong>oil</strong> production. In all upgrading reactions, the BS<br />
fraction was reduced during the reaction forming both 1 ighter and heavier<br />
products. The c<strong>at</strong>alytic reactions with NiMo/A1203 and Mo naphthen<strong>at</strong>e <strong>at</strong> 425OC<br />
reduced the BS fraction the most. The <strong>co</strong>mbin<strong>at</strong>ion <strong>of</strong> TET plus NiMo/A120 resulted<br />
in the gre<strong>at</strong>est reduction <strong>of</strong> the BS fraction and subsequent increase in $he PS<br />
fraction.<br />
In the <strong>co</strong><strong>processing</strong> experiments, both the addition <strong>of</strong> c<strong>at</strong>alyst and the<br />
addition <strong>of</strong> tetralin promoted <strong>co</strong>al <strong>co</strong>nversion. For the NiMo/Al2Og reactions, the<br />
<strong>co</strong>mbin<strong>at</strong>ion <strong>of</strong> c<strong>at</strong>alyst and tetra1 in synergetical ly promoted <strong>co</strong>al <strong>co</strong>nversion.<br />
With Mo naphthen<strong>at</strong>e, <strong>co</strong>al <strong>co</strong>nversion was high (89.5%) without TET addition and no<br />
change was observed with the addition <strong>of</strong> TET. Thus, with a highly accessible and<br />
active c<strong>at</strong>alyst, additional hydrogen don<strong>at</strong>ion from the solvent had 1 ittle<br />
influence on <strong>co</strong>al <strong>co</strong>nversion. Therefore, <strong>co</strong>al <strong>co</strong>nversion in <strong>co</strong><strong>processing</strong> appears<br />
to be dependent upon both c<strong>at</strong>alyst and hydrogen don<strong>at</strong>ion except in the case <strong>of</strong> a<br />
highly active c<strong>at</strong>alyst where c<strong>at</strong>alytic activity predomin<strong>at</strong>es.<br />
167<br />
7.1<br />
7.4<br />
7.3<br />
7.2<br />
7.2<br />
6.4<br />
6.8<br />
6.9<br />
7.0<br />
7.0<br />
7.1<br />
6.6<br />
6.6
TET did not promote the production <strong>of</strong> PS m<strong>at</strong>erials in either the thermal or<br />
c<strong>at</strong>alytic reactions. In fact, in the thermal reactions the presence <strong>of</strong> TET was<br />
detrimental to <strong>oil</strong> production.<br />
The effect <strong>of</strong> TET and c<strong>at</strong>alytic tre<strong>at</strong>ment on BS production is also<br />
instructive in examining the roles and rel<strong>at</strong>ive importance <strong>of</strong> these two factors in<br />
<strong>co</strong><strong>processing</strong>. BS production is defined as the difference between the final BS and<br />
the initial BS divided by the upgradeable m<strong>at</strong>erial which is maf <strong>co</strong>al. Compared to<br />
the thermal reaction <strong>at</strong> 4OO0C, the addition <strong>of</strong> TET increased the amount <strong>of</strong> BS<br />
production (Table 3). C<strong>at</strong>alytic tre<strong>at</strong>ment <strong>at</strong> 4OO0C did not increase the BS<br />
production; however, the addition <strong>of</strong> TET to reaction system with NiMo/A120j <strong>at</strong><br />
4OO0C did enhance BS production. At 425OC, the presence <strong>of</strong> NiMo/Al 03 and Mo<br />
naphthen<strong>at</strong>e increased the 8s production as shown in Table 4.<br />
The aidition <strong>of</strong> TET<br />
to the Mo naphthen<strong>at</strong>e reaction again increased the BS production. Since all <strong>of</strong><br />
these reactions showed positive <strong>oil</strong> production, the increases observed in the BS<br />
production were directly rel<strong>at</strong>ed to the upgrading <strong>of</strong> liquefied <strong>co</strong>al to BS<br />
products. Thus, the presence <strong>of</strong> TET assisted in the production <strong>of</strong> BS but not in<br />
the production <strong>of</strong> PS. The presence <strong>of</strong> a hydrotre<strong>at</strong>ing c<strong>at</strong>alyst was required for<br />
<strong>oil</strong> production in the <strong>co</strong><strong>processing</strong> reactions.<br />
References<br />
1. Monnier, J. CANMET Report 84-5E, "Review <strong>of</strong> the Co<strong>processing</strong> <strong>of</strong> Coals and<br />
Heavy Oils <strong>of</strong> Petroleum Origin", March 1984.<br />
2. Curtis, C.W., Tsai, K.J., Guin, J.A., Ind. Enq. Chem. Proc. Des. Dev., 1985,<br />
- 24, 1259.<br />
3. Curtis, C.W., Tsai, K.J., Guin, J.A., submitted to Ind. Ena. Prod. Res. and<br />
Dev., 1986.<br />
4. Kottensette, R.J., Sandia Report SANDB2-2495, March 1983.<br />
5. Bearden, R. and Aldridge, C.L., U.S. P<strong>at</strong>ent 4,134,824, 1979.<br />
6.<br />
7.<br />
Curtis, C.W., Tsai, K.J., and Guin, J.A., in press, Fuel Proc. Tech., 1986.<br />
Moody, T. Master's Thesis, Auburn University, 1985.<br />
Acknowledgments<br />
The authors gr<strong>at</strong>efully acknowledge the Department <strong>of</strong> Energy for support <strong>of</strong><br />
this work under Contract Nos. DEFG2282PC50793 and DEFG2285PC80502.<br />
168 j
i<br />
I<br />
REACTIVITY SCREENING OF FEEDSTOCKS FOR CATALYTIC COAL/OIL CO-PROCESSING<br />
J. B. McLean and J. E. Duddy<br />
Hydrocarbon Research, Inc.<br />
(A Subsidiary <strong>of</strong> Oynalectron Corpor<strong>at</strong>ion)<br />
P. 0. Box 6047<br />
Lawrencevil le, New Jersey 08648<br />
ABSTRACT<br />
HRI is currently <strong>co</strong>nducting a four-party funded program to develop and<br />
demonstr<strong>at</strong>e c<strong>at</strong>alytic <strong>co</strong>al/<strong>oil</strong> <strong>co</strong>-<strong>processing</strong> using HRI 's proven ebull<strong>at</strong>ed-bed<br />
reactor technology. The initial task in the research program was to determine<br />
reactivities <strong>of</strong> four <strong>co</strong>als (Illinois No. 6 and Ohio No. 5/6 bituminous. Alberta<br />
sub-bituminous and Texas lignite) and four petroleum residuums (Cold Lake, Maya,<br />
West Texas Sour, and Canadian IPL), both separ<strong>at</strong>ely and in <strong>co</strong>mbin<strong>at</strong>ion, using a<br />
20cc microautoclave reactor. Experimental <strong>co</strong>nditions and analytical procedures<br />
were developed to properly approxim<strong>at</strong>e ebull<strong>at</strong>ed bed <strong>co</strong>nditions <strong>at</strong> the small,<br />
b<strong>at</strong>ch scale and to allow estim<strong>at</strong>ion <strong>of</strong> both <strong>co</strong>al and petroleum residuum <strong>co</strong>nver-<br />
sions. Over 200 single-stage microautoclave tests were <strong>co</strong>nducted studying<br />
severity, feedstock r<strong>at</strong>io, and c<strong>at</strong>alyst effects. An interesting synergistic<br />
response was noted which indic<strong>at</strong>es optimum performance <strong>at</strong> 50/50 <strong>co</strong>al/<strong>oil</strong> r<strong>at</strong>io<br />
for one particular feedstock pair. Initial results from a single-stage run in a<br />
<strong>co</strong>ntinuous bench unit verified the trends noted in the microautoclave study.<br />
INTRODUCTION<br />
Hydrocarbon Research, Inc. (HRI) has developed and <strong>co</strong>mmercialized ebul l<strong>at</strong>ed-bed<br />
reactor technology for the c<strong>at</strong>alytic hydro<strong>co</strong>nversion <strong>of</strong> both <strong>co</strong>al and heavy <strong>oil</strong>.<br />
The H-Oil@ Process has been <strong>co</strong>mmercially demonstr<strong>at</strong>ed in both single- and two-<br />
stage process <strong>co</strong>nfigur<strong>at</strong>ions, and the H-Coal@ Process has been successfully<br />
scaled up through the 200 tonlday C<strong>at</strong>lettsburg pilot plant. While e<strong>co</strong>nomic <strong>co</strong>n-<br />
ditions have prevented the <strong>co</strong>mmerci a1 appliction <strong>of</strong> direct liquefaction tech-<br />
nology, <strong>co</strong>al/<strong>oil</strong> <strong>co</strong>-<strong>processing</strong> has gained increasing <strong>at</strong>tention as a mre<br />
<strong>co</strong>mmercially viable, nearer term way to introduce <strong>co</strong>al-derived liquid fuels into<br />
the market place. HRI's COILsm Process for <strong>co</strong>-<strong>processing</strong> was denonstr<strong>at</strong>ed on a<br />
bench unit Scale as early as 1974(1), and mre recently a two-stage process <strong>co</strong>n-<br />
figur<strong>at</strong>ion was denonstr<strong>at</strong>ed on a Canadian feedstock <strong>co</strong>mbin<strong>at</strong>ion <strong>of</strong> potential<br />
<strong>co</strong>mmercial interest(2). In 1985. a four-party funded program was started to<br />
further develop and demnstr<strong>at</strong>e c<strong>at</strong>alytic <strong>co</strong>al/<strong>oil</strong> <strong>co</strong>-<strong>processing</strong> using HRI's<br />
ebull<strong>at</strong>ed-bed reactor technology. The program sponsors, objectives and elements<br />
are listed in Table 1. This paper focuses on the results <strong>of</strong> the microautoclave<br />
reactivity screenfng program.<br />
169
MICROAUTOCLAVE DESCRIPTION AND PROCEDURES<br />
With the recent increase in interest in <strong>co</strong>-<strong>processing</strong>, numerws investi <strong>at</strong>on<br />
have reported results <strong>of</strong> b<strong>at</strong>ch reactor reactivity studies <strong>at</strong> various scales73-6).<br />
Many <strong>of</strong> these approaches tend to take a <strong>co</strong>nventional <strong>co</strong>al liquefaction approach,<br />
;y characterizing the effectiveness <strong>of</strong> petroleum Oils as <strong>co</strong>al liquefaction<br />
solvents". In <strong>co</strong>al/residuum <strong>co</strong>-<strong>processing</strong>, the <strong>oil</strong> is not a process solvent as<br />
such, but r<strong>at</strong>her a reactant, and it's reactions/<strong>co</strong>nversions are <strong>of</strong> equal - or<br />
even gre<strong>at</strong>er, depending on specific <strong>co</strong>nditions - importance than those <strong>of</strong> the<br />
<strong>co</strong>al. The opposite approach to this is to view <strong>co</strong>-<strong>processing</strong> as an extension <strong>of</strong><br />
refining technology, tre<strong>at</strong>ing the <strong>co</strong>al as an additive, usually in limited<br />
quantities(7.8). HRI's program was set up to <strong>co</strong>nsider a broad range <strong>of</strong> potential<br />
applic<strong>at</strong>ions using c<strong>at</strong>alytic ebul l<strong>at</strong>ed-bed <strong>co</strong>-<strong>processing</strong>. The microautoclave<br />
experimental and analytical procedures were speci fical ly developed to reflect<br />
this, and are in many respects <strong>co</strong>nsiderably different than those used by other<br />
workers. Some discussion is therefore necessary to explain the basis for these<br />
differences.<br />
The 20cc microautoclave reactor used in these studies is shown in Figure 1.<br />
Solvent, <strong>co</strong>al, residuum, and c<strong>at</strong>alyst are charged b<strong>at</strong>chwise in the appropri<strong>at</strong>e<br />
amounts prior to mounting the reactor. Following pressure-testing, the desired<br />
H2 (or Nz) pressure is established. Due to the volume <strong>of</strong> gas lines above the<br />
reactor itself, it is essentially an "infinite source" hydrogen system, and no<br />
adjustment <strong>of</strong> oper<strong>at</strong>ing pressure due to temper<strong>at</strong>ure is usually required. The<br />
entire assembly is shaken vertically with approxim<strong>at</strong>ely one-inch strokes <strong>at</strong><br />
460 rpm. with temper<strong>at</strong>ure <strong>co</strong>ntrol by imnersion in a fluidized sand b<strong>at</strong>h he<strong>at</strong>er.<br />
Dual sand b<strong>at</strong>hs are available for simul<strong>at</strong>ion <strong>of</strong> two-stage, close-<strong>co</strong>upled pro-<br />
cessing. Two identical microautoclave reactors are always oper<strong>at</strong>ed side-by-side.<br />
A <strong>co</strong>ld trap is provided to <strong>co</strong>llect any light liquids lost during oper<strong>at</strong>ion or<br />
depressuring.<br />
Following each run the reaction is quenched by inversion in a w<strong>at</strong>er b<strong>at</strong>h, and<br />
slowly depressured. The reactors and <strong>co</strong>ld traps are then removed, and the<br />
products are <strong>co</strong>mbined and subjected to the workup procedures described in Figure<br />
2. The use <strong>of</strong> the c<strong>at</strong>alyst basket allows separ<strong>at</strong>ion <strong>of</strong> product solids fran<br />
c<strong>at</strong>alyst extrud<strong>at</strong>es. Ash-balancing then a1 lows calcul<strong>at</strong>ion <strong>of</strong> <strong>co</strong>al <strong>co</strong>nversion.<br />
If necessary, product ashes can be checked for c<strong>at</strong>alyst metals to distinguish<br />
<strong>co</strong>al ash from <strong>at</strong>trited c<strong>at</strong>alyst. TGA simul<strong>at</strong>ed distill<strong>at</strong>ion is used to estim<strong>at</strong>e<br />
product residuum <strong>co</strong>ntents and calcul<strong>at</strong>e residuum <strong>co</strong>nversions. For selected runs,<br />
solvent precipit<strong>at</strong>ion was used to calcul<strong>at</strong>e asphaltene and preasphaltene<br />
<strong>co</strong>mponents in the product residuum. although this is <strong>of</strong> lesser utility in <strong>co</strong>-<br />
<strong>processing</strong> than in <strong>co</strong>al liquefaction since petroleum residua <strong>co</strong>ntains very low<br />
levels <strong>of</strong> insolubles.<br />
As noted above, HRI's microautoclave oper<strong>at</strong>ing procedures and <strong>co</strong>nditions are<br />
specifically designed to most properly approxim<strong>at</strong>e the <strong>co</strong>nditions <strong>of</strong> an<br />
ebull<strong>at</strong>ed-bed reactor, and are in many cases quite different than those<br />
"typically" used in the industry. Some specifics include:<br />
170
C<strong>at</strong>alyst Type and Loading - The ebull<strong>at</strong>ed-bed reactor oper<strong>at</strong>es with <strong>co</strong>nven-<br />
tional extrud<strong>at</strong>e c<strong>at</strong>alysts <strong>at</strong> very high loadings (up to 50% <strong>of</strong> the reactor<br />
volunre is occupied by c<strong>at</strong>alyst). Our microautoclave experiments typical ly<br />
charged a c<strong>at</strong>alyst/feedstock r<strong>at</strong>io <strong>of</strong> 1/1 t o reflect this. Techniques such<br />
as grinding <strong>of</strong> c<strong>at</strong>alyst are not represent<strong>at</strong>ive. The c<strong>at</strong>alyst used typically<br />
is process-presulfided in a pilot unit, and the use <strong>of</strong> the c<strong>at</strong>alyst basket<br />
allows separ<strong>at</strong>ion from reaction products. Some assumptions are necessary in<br />
calcul<strong>at</strong>ion procedures to ac<strong>co</strong>unt for items such as IOM deposition on c<strong>at</strong>a-<br />
lyst, c<strong>at</strong>alyst <strong>at</strong>trition, etc. during an experiment.<br />
Feedstock Dilution with Distill<strong>at</strong>e Products - The ebull<strong>at</strong>ed bed is a well-<br />
mixed reactor, due to the typically high r<strong>at</strong>io <strong>of</strong> internal recycle to fresh<br />
feed. As a result, reaction occurs in a <strong>co</strong>ncent<strong>at</strong>ion represented by the<br />
products. No b<strong>at</strong>ch reactor can properly model a CSTR from the standpoint <strong>of</strong><br />
fundamental kinetics, so a canpranise has to be made. Since the initial<br />
<strong>co</strong>nversion reactions in <strong>co</strong>al liquefaction are critical, an <strong>at</strong>tempt is made to<br />
simul<strong>at</strong>e the reactor environment in which they occur. Thus, microautoclave<br />
reactor charges are made up with a high level <strong>of</strong> distill<strong>at</strong>e diluent. An<br />
<strong>at</strong>tempt is made to approxim<strong>at</strong>e, to the extent possible, the properties <strong>of</strong> the<br />
distill<strong>at</strong>e m<strong>at</strong>erials which would be expected to be produced from the<br />
feedstocks and <strong>co</strong>nditions <strong>of</strong> interest. The distill<strong>at</strong>e solvents used are<br />
generally m<strong>at</strong>erials produced in substantial quantities from larger pilot<br />
plant oper<strong>at</strong>ions on the feedstocks <strong>of</strong> interest.<br />
Product Analyses - Coal <strong>co</strong>nversion to THF-solubles is calcul<strong>at</strong>ed in a fairly<br />
typical manner. As noted above, <strong>co</strong>nversions based on solubilities in other<br />
solvents are not <strong>co</strong>nsidered to be especially meaningful for <strong>co</strong>-<strong>processing</strong>. A<br />
simul<strong>at</strong>ed distill<strong>at</strong>ion procedure was developed using a Perkin-Elmer TGS-2<br />
Thermogravimetric Analyzer (TGA), which a1 lows estim<strong>at</strong>ion <strong>of</strong> 975OF+ <strong>co</strong>nversions.<br />
No <strong>at</strong>tempt is made to gener<strong>at</strong>e d<strong>at</strong>a such as gas yields or distill<strong>at</strong>e<br />
product distribution or quality. Such d<strong>at</strong>a are difficult to gener<strong>at</strong>e<br />
reliably on such a small scale. Even if this <strong>co</strong>uld be done, the results<br />
would not be meaningful for scaleup due to the large impacts <strong>of</strong> the<br />
distill<strong>at</strong>e diluents and the major differences between b<strong>at</strong>ch and <strong>co</strong>ntinuous<br />
units, on any scale.<br />
SCREENING STUDY CONDITIONS<br />
A five-point. low-to-mild severity <strong>co</strong>ndition m<strong>at</strong>rix was used to screen each<br />
feedstock and <strong>co</strong>mbin<strong>at</strong>ion <strong>of</strong> interest, as shown in Table 2. As noted, a 4/1/1<br />
charge r<strong>at</strong>io <strong>of</strong> distill<strong>at</strong>e solvent/reactant (<strong>co</strong>al and <strong>oil</strong> )/c<strong>at</strong>alyst was used.<br />
Severities ranged frm 2-20 STTU, based on HRI's <strong>co</strong>nversion model developed for<br />
<strong>co</strong>al <strong>co</strong>nversions. It is re<strong>co</strong>gnized th<strong>at</strong> the time/temper<strong>at</strong>ure rel<strong>at</strong>ionships for<br />
<strong>co</strong>-<strong>processing</strong> may not be truly represented by the STTU mdel, but it was used as<br />
a <strong>co</strong>nvenient way to express both severity parameters. The m<strong>at</strong>rix used provides a<br />
<strong>co</strong>mparison <strong>of</strong> three residence times <strong>at</strong> one temper<strong>at</strong>ure (8OO0F), and three tem-<br />
per<strong>at</strong>ures <strong>at</strong> one residence time (30 minutes). All severities are lower than<br />
those typically en<strong>co</strong>untered in larger scale oper<strong>at</strong>ions. This serves to keep<br />
<strong>co</strong>nversions low enough so th<strong>at</strong> kinetic reactivity differences can be properly<br />
observed.<br />
171
FEEDSTOCK PROPERTIES<br />
Some properties <strong>of</strong> the four <strong>co</strong>als and four Oils studied are listed in Table 3.<br />
The Cold Lake feedstock was available as a deep-cut A S from previous HRI H-Oil.<br />
studies, while the other three <strong>oil</strong>s were provided as crude <strong>oil</strong>s and were b<strong>at</strong>ch<br />
vacuum-distilled to approxim<strong>at</strong>ely the same residuum <strong>co</strong>ntent prior to the reac-<br />
tivity studies. All four <strong>co</strong>als were subjected to standard HRI bench unit pre-<br />
par<strong>at</strong>ion procedures (crushing, pulverizing to -70 mesh, drying to 2-10'7, moisture,<br />
and screening) and were further vacuum dried imdi<strong>at</strong>ely prior to microautoclave<br />
testing. Three diluent solvents were also used. as shown. The Illinois-derived<br />
solvent was used for Ohio and Illinois bituminous <strong>co</strong>als, the Wyodak solvent for<br />
Alberta sub-bituminous <strong>co</strong>al and Texas lignite, and the Cold Lake solvent for all<br />
petroleum <strong>oil</strong>s. Except for a few solvent-specific runs, solvents were blended in<br />
the same r<strong>at</strong>ios as the feedstocks for each run.<br />
PROGRAM OUTLINE<br />
Over two hundred tests were <strong>co</strong>nducted under the program, as noted in Table 4.<br />
The <strong>co</strong>-p,rocessing feedstock pairs chosen for evalu<strong>at</strong>ion were based on program<br />
sponsors <strong>co</strong>ncerns and represent meaningful canmerci a1 candid<strong>at</strong>es. No work was<br />
done on the Illinois No. 6 <strong>co</strong>al, since it was being extensively studied in HRI's<br />
parallel DOE funded <strong>co</strong>al liquefaction program. Most <strong>of</strong> the discussion to follow<br />
centers on the Ohio <strong>co</strong>al/Cold Lake ASB pair, which was the most extensively<br />
studied in 1985 (including both single- and two-stage process variable studies in<br />
the <strong>co</strong>ntinous bench unit). This canbin<strong>at</strong>ion has been selected by OOSFC as the<br />
basis for a prototype <strong>co</strong>mmercial facility to be loc<strong>at</strong>ed in Ohio.<br />
INDIVIDUAL FEEDSTOCK REACTIVITIES<br />
Figures 3 and 4 show STTU response curves for the Ohio <strong>co</strong>al and the Cold Lake<br />
ASB. Similar curves were gener<strong>at</strong>ed for each <strong>of</strong> the other feedstocks. In order<br />
to provide a quantit<strong>at</strong>ive reactivity ranking, kinetic r<strong>at</strong>e <strong>co</strong>nstants were back-<br />
calcul<strong>at</strong>ed from the d<strong>at</strong>a assuming various b<strong>at</strong>ch reactor models. For the <strong>oil</strong>s<br />
alone, a se<strong>co</strong>nd order fit was found to be the most s<strong>at</strong>isfactory, as shown in<br />
Figure 5. While it is unlikely th<strong>at</strong> the <strong>co</strong>nversion reactions are truly se<strong>co</strong>nd<br />
order, in the sense <strong>of</strong> being bimolecular, such a model fit is not unusual in<br />
systems <strong>of</strong> this type, where the "reactant" is not a single <strong>co</strong>mponent but r<strong>at</strong>her a<br />
range <strong>of</strong> <strong>co</strong>mponents with different reactivities. For the <strong>co</strong>als, a mre <strong>co</strong>mplex<br />
model would be required to separ<strong>at</strong>e the effects <strong>of</strong> <strong>co</strong>al <strong>co</strong>nversion to<br />
THF-solubles. the fraction <strong>of</strong> <strong>co</strong>nverted <strong>co</strong>al which fons 975OF+ residuum, and the<br />
kinetics <strong>of</strong> <strong>co</strong>nversion <strong>of</strong> the residuum. Realizing these deficiencies, the <strong>co</strong>al<br />
d<strong>at</strong>a were force-fit to the same simplified se<strong>co</strong>nd order 9750Ft <strong>co</strong>nversion model<br />
so th<strong>at</strong> a direct <strong>co</strong>mparison <strong>of</strong> <strong>oil</strong>s, <strong>co</strong>als, and <strong>co</strong>-<strong>processing</strong> pairs <strong>co</strong>uld be<br />
made. These results are shown in Table 5. As expected. the <strong>oil</strong>s are <strong>co</strong>n-<br />
siderably more reactive to total 975'Ft <strong>co</strong>nversion <strong>at</strong> low severities than the<br />
<strong>co</strong>als. It is notable th<strong>at</strong> the <strong>co</strong>-<strong>processing</strong> pairs do not necessarily fall in<br />
either the order or magnitude which would be expected fran the individual<br />
feedstocks, indic<strong>at</strong>ing th<strong>at</strong> synergistic interactions do occur. It is also<br />
notable th<strong>at</strong> the feedstock pair (Ohio/Cold Lake) studied most extensively in the<br />
172
program does not represent the "best" choice based on reactivities, but was cho-<br />
sen based on cannercia1 <strong>co</strong>nsider<strong>at</strong>tons.<br />
OHIO COAL/COLD LAKE ASB CO-PROCESSING<br />
Figure 6 shows the reactivity curves for a 50/50 blend <strong>of</strong> Ohio <strong>co</strong>al and Cold Lake<br />
ASB. The drop <strong>of</strong>f in 975'F+ <strong>co</strong>nversion <strong>at</strong> 20 STTU may be indic<strong>at</strong>ive <strong>of</strong> some<br />
regressive reaction due to poor solvent quality, as this is the highest<br />
temper<strong>at</strong>ure point (825OF) in the grid. The STTU axis has been extended to<br />
include a point <strong>at</strong> a typical bench unit oper<strong>at</strong>ing severity. It i s notable th<strong>at</strong><br />
there was no problem in achieving high (90% plus) <strong>co</strong>al <strong>co</strong>nversion to TW<br />
solubles. This was true <strong>of</strong> all the pairs studied, indic<strong>at</strong>ing th<strong>at</strong> the inherently<br />
poor hydrogen donor properties <strong>of</strong> the petroleum <strong>oil</strong>s can be oveccme by<br />
c<strong>at</strong>alytic, ebul l<strong>at</strong>ed-bed <strong>co</strong>-<strong>processing</strong>.<br />
Figure 7 shows the effect <strong>of</strong> <strong>co</strong>al to <strong>oil</strong> r<strong>at</strong>io on <strong>co</strong>nversions in a low severity<br />
test (10 STTU). As expected, the THF <strong>co</strong>nversion increases as the <strong>co</strong>al <strong>co</strong>n-<br />
centr<strong>at</strong>ion increases, since a higher percentage <strong>of</strong> the solvent is then <strong>co</strong>al-<br />
derived as well. The 975'F+ <strong>co</strong>nversion response is far less explainable. The<br />
individual feedstock points <strong>at</strong> 0 and loo$ are <strong>co</strong>nnected, to represent expected<br />
<strong>co</strong>nversions based on strict linear averaging. At <strong>co</strong>al <strong>co</strong>ncentr<strong>at</strong>ions up to 502,<br />
<strong>co</strong>nversions near or above this line occur, indic<strong>at</strong>ing a positive synergy.<br />
Surprisingly, <strong>at</strong> <strong>co</strong>al <strong>co</strong>ncentr<strong>at</strong>ions <strong>of</strong> 67-758, a large neg<strong>at</strong>ive interaction<br />
occurs, and 975OF+ <strong>co</strong>nversions are actually lower than those for <strong>co</strong>al alone.<br />
Each <strong>of</strong> these points was found to be reproducible. The most likely explan<strong>at</strong>ion<br />
for this phenomenon is th<strong>at</strong> the presence <strong>of</strong> the petroleum <strong>oil</strong>s sufficiently<br />
reduces the solvent quality in this range to cause a large drop in the <strong>co</strong>nversion<br />
<strong>of</strong> the <strong>co</strong>al residua. At the lower <strong>co</strong>al <strong>co</strong>ncentr<strong>at</strong>ions. this effect is <strong>of</strong>fset by<br />
the improved <strong>co</strong>nversions <strong>of</strong> the petroleum residua. Interestingly, this effect<br />
shows itself only in the 97S°F+ <strong>co</strong>nversions and not in the THF <strong>co</strong>nversions. It<br />
should be noted here th<strong>at</strong> <strong>co</strong>al/<strong>oil</strong> r<strong>at</strong>io studies with other feedstock pairs do<br />
not show this same neg<strong>at</strong>ive behavior (<strong>at</strong> least not to this extent), but in all<br />
cases the response is non-linear.<br />
Since this trend was interesting and unexpected, it was decided to repe<strong>at</strong> the<br />
<strong>co</strong>al/<strong>oil</strong> r<strong>at</strong>io studies <strong>at</strong> a higher severity, typical <strong>of</strong> bench unit process <strong>co</strong>n-<br />
ditions. This was done to <strong>co</strong>incide with the single-stage bench mn. which pro-<br />
vided <strong>co</strong>mpar<strong>at</strong>ive results in the single-stage, integr<strong>at</strong>ed bench unit <strong>at</strong> 33, 50<br />
and 67% <strong>co</strong>al. These results are shown in Figure 8. Note th<strong>at</strong> the cmplex r<strong>at</strong>io<br />
response curve for 975'F+ <strong>co</strong>nversion has been reproduced, although the extent <strong>of</strong><br />
the neg<strong>at</strong>ive devi<strong>at</strong>ions <strong>at</strong> 67-75% <strong>co</strong>al are reduced. The bench unit d<strong>at</strong>a, <strong>at</strong> 33<br />
and 50% <strong>co</strong>al, provide excellent agreement with the microautoclave d<strong>at</strong>a. The<br />
bench d<strong>at</strong>a <strong>at</strong> 67% <strong>co</strong>al do show some neg<strong>at</strong>ive effect, althought not as pronounced<br />
as in the microautoclave. One key difference is th<strong>at</strong> each bench d<strong>at</strong>a point<br />
represents several days <strong>of</strong> <strong>co</strong>ntinuous, integr<strong>at</strong>ed oper<strong>at</strong>ion with solvent quality<br />
equilibr<strong>at</strong>ion. while microautoclave solvents are artificially cmposited. It<br />
should also be noted th<strong>at</strong> the tie points <strong>at</strong> 0 and 1OG% <strong>co</strong>al were not detemined<br />
on the bench unit, so th<strong>at</strong> the extent <strong>of</strong> positive/neg<strong>at</strong>ive synergy may not be<br />
directly <strong>co</strong>mparable. The 50% <strong>co</strong>al case has been shown to be e<strong>co</strong>nomically pre-<br />
ferred t several severities <strong>at</strong> least in part due to synergistic reactivity<br />
effects 39)<br />
173
CONCLUSIONS<br />
HRI's microautoclave has been shown to be an effective tool for <strong>co</strong>mparing reac-<br />
tivjties <strong>of</strong> <strong>co</strong>als, <strong>oil</strong>s, and <strong>co</strong>mbin<strong>at</strong>ions for c<strong>at</strong>alytic <strong>co</strong>al/<strong>oil</strong> <strong>co</strong>-<strong>processing</strong>.<br />
Specific improvements in experimental and analytical procedures were implemented<br />
to expand the utility <strong>of</strong> the microautoclave from <strong>co</strong>al liquefaction into <strong>oil</strong> and<br />
<strong>co</strong>-<strong>processing</strong>. D<strong>at</strong>a gener<strong>at</strong>ed on the Ohio <strong>co</strong>al/Cold Lake ASB <strong>co</strong>mbin<strong>at</strong>ion led to<br />
some unexpected results, which were l<strong>at</strong>er <strong>co</strong>nfirmed by <strong>co</strong>ntinuous bench unit<br />
studies.<br />
ACKNOWLEDGEMENTS<br />
HRI wishes to acknowledge the program sponsors and their represent<strong>at</strong>ives who have<br />
<strong>co</strong>ntributed to the technical <strong>co</strong>ntent <strong>of</strong> this work. Some <strong>of</strong> the feedstock<br />
analytical d<strong>at</strong>a presented were gener<strong>at</strong>ed by the Alberta Research Council.<br />
REFERENCES<br />
1.<br />
2.<br />
3.<br />
4.<br />
5.<br />
6.<br />
7.<br />
8.<br />
9.<br />
U. S. P<strong>at</strong>ent 4,054,504.<br />
"Coal/Oil Co-Processing <strong>of</strong> Canadian Feedstocks" J. B. MacArthur, F. Boehm. A.<br />
Liron, R. H. Shannon. Proceedings: Tenth Annual EPRI Contractors'<br />
Conference on Clean Liquid and Solid Fuels, October, 1985.<br />
Evalu<strong>at</strong>ion <strong>of</strong> Process Parameters for Combined Processing <strong>of</strong> Coal with Heavy<br />
Crudes and Residua", C. U. Curtis, et al. Ind. Eng. Chem. Process Des.<br />
Oev., 1985, 24, 1259-1266.<br />
"Use <strong>of</strong> Non-Coal Derived Heavy Solvents in Direct Coal <strong>Liquefaction</strong>" R. L.<br />
Miller. Proceedings: Tenth Annual EPRI Contractors' Conference on Clean<br />
Liquid and Solid Fuels, October, 1985.<br />
"Lumus Co-Processing", M. Green. OOE's (PETC) Direct <strong>Liquefaction</strong><br />
Contractors' Conference. Pittsburgh, November 19, 1985<br />
"Coal <strong>Liquefaction</strong> Co-Processing". J. G<strong>at</strong>sis, et al. DOE'S (PETC) Direct<br />
<strong>Liquefaction</strong> Contractors' Conference, Pittsburgh, November 19, 1985<br />
"The Chevron Co-Refining Process", J. Shinn. et al. Proceedings <strong>of</strong> 9th<br />
Annual EPRI Contractors' Conference on Clean and Solid Fuels. March 1984.<br />
"Co-Processing <strong>of</strong> Canadian Lignites and Bitumen", S. Fouda, et al. AIChE<br />
N<strong>at</strong>ional Meeting, March 1985.<br />
"HRI's Coal/Oil Co-Processing Program - Phase I", J. E. Ouddy. Presented <strong>at</strong><br />
EPRI's 11th Annual Conference on Clean Liquid and Solid Fuels, May 1986.<br />
174
I<br />
SPONSORS: Electric Power Research Institute (EPRI)<br />
Ontario-Ohio Synthetic Fuels Corpor<strong>at</strong>ion, Ltd. (OOSFC)<br />
A1 berta Research Counci 1 (ARC)<br />
Dynalectron Corpor<strong>at</strong>ion/HRI<br />
OBJECTIVES:<br />
TABLE 1<br />
1. Produce incremental liquid fuels fran <strong>co</strong>al (including clean power plant<br />
2. Upgrade (desulfurize, demetallize) poor quality residuum fuels.<br />
3.<br />
fuel s ) .<br />
Utilize chemically <strong>co</strong>mbined hydrogen from residuum to produce incremental<br />
liquid fuels from <strong>co</strong>al.<br />
ELEMENTS (Labor<strong>at</strong>ory) : Feedstock Characteriz<strong>at</strong>ion (ARC/HRI)<br />
Microautoclave Reactivity Screening (HRI)<br />
B<strong>at</strong>ch Autoclave Screening (ARC)<br />
Continuous Bench Unit Oper<strong>at</strong>ions (HRI)<br />
MICROAUTOCLAVE STANDARD CONDITIO6<br />
REACTIVITY SCREENIN6 TESTS<br />
TABLE 2<br />
I 8 gms solvent Time, Temper<strong>at</strong>ure - Variable<br />
2 gms reactant (<strong>co</strong>al plus <strong>oil</strong>) 2 gms pretre<strong>at</strong>ed c<strong>at</strong>alyst (when used)<br />
2000 psig hydrogen<br />
I Standard Solvent - H-Coal@/H-Oil@ distill<strong>at</strong>es<br />
I Solvents blended in same r<strong>at</strong>io as feedstocks<br />
9<br />
Severity - Standard Time Temper<strong>at</strong>ure Units<br />
I 1 STTU = 1 minute <strong>at</strong> 84OOF<br />
Severity M<strong>at</strong>rix<br />
Temper<strong>at</strong>ure, OF Time Minutes<br />
7 50 +F<br />
800 15 5<br />
800 30 10<br />
800 45 15<br />
825 30 20<br />
175
176
MICROAUTOCLAVE REACTOR<br />
C<strong>at</strong>alyst<br />
Charge<br />
Basket<br />
FIGURE 1<br />
MECHANICAL DESIGN<br />
e 20cc Internal Volume<br />
e Maximum Inlet Hydrogen Pressure 3000 pst<br />
e Oper<strong>at</strong>ions to <strong>Liquefaction</strong> Temper<strong>at</strong>ure<br />
e Thermal and Gas Inlet Coupling<br />
e 347ss M<strong>at</strong>erial <strong>of</strong> Construction<br />
e External Cap Threads<br />
e Reactor Cap Redesign<br />
e Cold Traps<br />
0 C<strong>at</strong>alyst Basket<br />
0 Utilizes Whole Extrud<strong>at</strong>e C<strong>at</strong>alyst<br />
TESTING CAPABILITIES<br />
e Thermal Tests Varying Charge.<br />
feed R<strong>at</strong>tos. Temper<strong>at</strong>ure, Time<br />
e C<strong>at</strong>alytic Tests Varying Charge,<br />
Feed R<strong>at</strong>ios, Temper<strong>at</strong>ure, Time<br />
PRODUCT WORKUP PROCEDURES<br />
MASH REACTW<br />
C4lALYST BASKET<br />
"<br />
F ILTEU<br />
CALCUL4lE<br />
COAL CONYEUSION<br />
FILTRATE<br />
ROTO-EVAPOR<br />
THF<br />
- OIL - - - - - -<br />
1 (OPTIOML)<br />
CALC U T E I<br />
975'F' CONVERSION<br />
177<br />
ASPHALTENES. PREISPWLTENES<br />
FIGURE 2
100.<br />
2 8%.<br />
M<br />
= ?8.<br />
38.<br />
....<br />
. . . . . . . . . . . . . . . . . . . . . . . . . .<br />
+ + + + '.<br />
r.... :::: :::: ... .... .... .... .... .... ....<br />
-4. + -<br />
- -<br />
- -<br />
0 .<br />
- -<br />
-<br />
0<br />
I I I I I I I I I<br />
LL 8%. ..: : : . . . . . . . . . .. . . . . . . . . . . . . . . . . . . . . . . . . . . . .<br />
2<br />
h L.<br />
in<br />
4".<br />
1:; j<br />
CATALYTIC REACTIVITY OF OHIO NO. 5/6 COAL<br />
CATALYTIC REACTIVITY OF COLO LAKE AS6<br />
I: ;: : I . . i 1 . :. : I : j j j I: j j j I j j j 1 1 : ; j ~ 1<br />
-<br />
4 -<br />
-<br />
-<br />
-<br />
-1<br />
j j { j I j :; /._<br />
0. 5. 18. 15. 28. 25. 30. 35. 4%. 45. 50.<br />
178<br />
SEVERITY, STTU<br />
FIGURE 3<br />
FIGURE 4<br />
I
!<br />
.6<br />
.5<br />
-4<br />
y .?I<br />
ae<br />
.2<br />
.l<br />
0.<br />
CALCULATED SECOND DRDER RATE CONSTANTS FOR<br />
97!i°F+ CONVERSION OF OIL FEEDSTOCKS<br />
I I I b<br />
------->\ /'<br />
Lake<br />
1 I I I I<br />
0. 5. 10. 15. 20. 25.<br />
SEVERITY, STTU<br />
CATALYTIC CO-PROCESSIffi OF 50/50 BLEND OF<br />
OHIO n0. 5/6 COAL AND COLD LAKE ASB<br />
+ THF Conversion<br />
o Total Feed 975'F' Conversion<br />
100. I I I I I I I I I I<br />
M. + +<br />
* +<br />
88.<br />
40.<br />
0 ,<br />
I I , I I I I I 1<br />
0. 5. 18. 15. 20. 25. 30. 35. 40. 45. 50.<br />
SEVERITY, STTU<br />
179<br />
FIGURE 5<br />
FIGURE 6
CATALYTIC CO-PROCESSINS OF OHIO NO. 5/6 COAL AND COLD LAKE ASB<br />
EFFECT OF COAL CONCENTRATION<br />
!M.<br />
L3e.<br />
380. M<br />
20.<br />
10.<br />
(SEVERITY: 10 SrrU)<br />
+ THF Conversion<br />
o Total Feed 9750Ft Conversion<br />
1;: : : : I . . . . . . . . . . . .<br />
+ +<br />
+<br />
+ t<br />
t<br />
tj: I I I I I<br />
8. 20. 40 * 60. 80 * 100.<br />
PERCENT COAL<br />
CATALYTIC CO-PROCESSING OF OHIO NO. 5/6 COAL AND COLD LAKE ASB<br />
EFFECT OF COAL COWCENTRATION<br />
(BEWCH RUN SEVERITY)<br />
t THF Conversion<br />
o 975°F' Conversion<br />
x Sench Unit 975°F' Conversion D<strong>at</strong>a<br />
lea. I I I I I I<br />
1:<br />
0 7. <strong>co</strong>x<br />
-. .<br />
+ +<br />
50 * I I . . I I<br />
0. 20. 48. ce . 88. lee.<br />
PERCENT COAL<br />
t<br />
180<br />
FIGURE 7<br />
FIGURE 8<br />
i ]<br />
--I<br />
::!
I<br />
SINGLE-STAGE SLURRY CATALYZED CO-PROCESSING<br />
John G. G<strong>at</strong>sis<br />
Signal Research Center Inc<br />
Box 5016<br />
Des Plaines, Illinois 60017-5016<br />
INTRODUCTION<br />
UOP Inc. and the Signal Research Center are currently engaged in a Department<br />
<strong>of</strong> Energy (DOE) sponsored program to determine if a slurry c<strong>at</strong>alyzed, single-stage<br />
process involving the simultaneous <strong>co</strong>nversion <strong>of</strong> <strong>co</strong>al and petroleum resid <strong>of</strong>fers the<br />
potential for improved e<strong>co</strong>nomics.<br />
The program has been structured to ac<strong>co</strong>mplish the overall objectives <strong>of</strong><br />
evalu<strong>at</strong>ing the technical feasibility and establishing a process d<strong>at</strong>a base on the Co-<br />
<strong>processing</strong> <strong>co</strong>ncept. Specific objectives include the establishment <strong>of</strong> overall<br />
criteria for the selection <strong>of</strong> <strong>co</strong>al type and petroleum characteristics, evalu<strong>at</strong>ion <strong>of</strong><br />
process performance, and the <strong>co</strong>st estim<strong>at</strong>ion <strong>of</strong> a <strong>co</strong>nceptual <strong>co</strong>mmercial facility.<br />
This paper reviews results from the first phase <strong>of</strong> the program and early<br />
results from the <strong>co</strong>ntinuous bench-scale unit currently in oper<strong>at</strong>ion.<br />
PROPOSED PROCESS CONCEPT<br />
UOP Inc. and the Signal Research Center began development <strong>of</strong> the resid/<strong>co</strong>al Co<strong>processing</strong><br />
<strong>co</strong>ncept in 1970 and were issued a key p<strong>at</strong>ent in this area in 1972 (1).<br />
The inform<strong>at</strong>ion gained in this work plus the much longer and more extensive<br />
experience in petroleum resid upgrading and <strong>co</strong>al <strong>co</strong>nversion were used to formul<strong>at</strong>e a<br />
slurry c<strong>at</strong>alyzed, single-stage process for the simultaneous <strong>co</strong>nversion <strong>of</strong> <strong>co</strong>al and<br />
petroleum resid. This Co-<strong>processing</strong> process utilizes an active, well-dispersed<br />
c<strong>at</strong>alyst and oper<strong>at</strong>es <strong>at</strong> rel<strong>at</strong>ively low temper<strong>at</strong>ures.<br />
This allows high <strong>co</strong>al<br />
<strong>co</strong>nversion without cracking <strong>of</strong> resid and <strong>co</strong>al to light gases, and minimizes thermal<br />
degrad<strong>at</strong>ion reactions.<br />
FEEDSTOCK SELECTION<br />
Six vacuum resids, three bituminous <strong>co</strong>als and one subbituminous <strong>co</strong>al were<br />
selected for study.<br />
The vacuum resids were selected based on their <strong>co</strong>mmercial importance (availability)<br />
and to provide a wide range <strong>of</strong> chemical and physical properties. These<br />
resids were vacuum fraction<strong>at</strong>ed to 510°C <strong>at</strong> the 5 vol-% point so th<strong>at</strong> all would have<br />
similar b<strong>oil</strong>ing ranges, thus elimin<strong>at</strong>ing any process vari<strong>at</strong>ions due to different<br />
amounts <strong>of</strong> vacuum gas <strong>oil</strong> (VGO) in the feedstock.<br />
The chemical and physical properties are shown in Table 1. Figure 1 shows the<br />
rel<strong>at</strong>ionship <strong>of</strong> API gravity with respect to hydrogen, C7 insolubles and carbon<br />
residue <strong>co</strong>ntent. The <strong>co</strong>ntaminants (C7 insolubles and carbon residue) increase and<br />
hydrogen <strong>co</strong>ntent decreases with decreasing API gravity.<br />
The <strong>co</strong>al samples were selected primarily because <strong>of</strong> their use as references in<br />
other studies. The properties are shown in Table 2. The Wyodak Coal as received<br />
(C4.1) has a moisture <strong>co</strong>ntent <strong>of</strong> 14.7 wt-%. It was dried in the labor<strong>at</strong>ory to a<br />
moisture <strong>co</strong>ntent <strong>of</strong> 1.78 wt-% (C4.2).<br />
181
CATALYST COMPARISON STUDY<br />
The premise <strong>of</strong> this work involves the <strong>co</strong>ncept th<strong>at</strong> an active slurry c<strong>at</strong>alyst<br />
will efficiently promote and effect the necessary dissolution and upgrading<br />
reactions as <strong>co</strong>mpared with a less active c<strong>at</strong>alyst or a non-c<strong>at</strong>alytic process. and<br />
thus maximize <strong>co</strong>al <strong>co</strong>nversion and upgrading <strong>of</strong> the petroleum resid to produce a high<br />
quality syncrude.<br />
Disposable, iron-based slurry c<strong>at</strong>alysts, whose activities have been reported as<br />
being much lower than th<strong>at</strong> <strong>of</strong> other metal slurry c<strong>at</strong>alysts (2). have been shown to<br />
provide beneficial c<strong>at</strong>alytic effects in the upgrading <strong>of</strong> <strong>co</strong>al and <strong>co</strong>al/resid<br />
mixtures (3,4). An iron-based slurry c<strong>at</strong>alyst was tested to establish a <strong>co</strong>mparison<br />
with the active UOP slurry c<strong>at</strong>alyst. The iron-based disposable c<strong>at</strong>alyst selected<br />
was a porous iron oxide (Fe2O3) from Kerr-McGee (5). A run was also made without<br />
c<strong>at</strong>alyst.<br />
Lloydminster vacuum resid (R4) and Illinois No. 6 <strong>co</strong>al (CI) were used as feedstocks.<br />
The tests were <strong>co</strong>nducted in an 1800 cc rocker autoclave. The equipment and<br />
procedure have been described in previous work (6). The oper<strong>at</strong>ing <strong>co</strong>nditions are<br />
shown below:<br />
Resid/Coal R<strong>at</strong>io 2<br />
Pressure, psi! 3000<br />
Temper<strong>at</strong>ure, C Base<br />
Residence Time, hrs 2<br />
The iron-based c<strong>at</strong>alyst was tested <strong>at</strong> twice the c<strong>at</strong>alyst <strong>co</strong>ncentr<strong>at</strong>ion <strong>of</strong> the UOP<br />
slurry c<strong>at</strong>alyst to <strong>co</strong>mpens<strong>at</strong>e for its lower anticip<strong>at</strong>ed activity with respect to the<br />
active UOP slurry c<strong>at</strong>alyst.<br />
The results <strong>of</strong> this c<strong>at</strong>alyst <strong>co</strong>mparison study are summarized in Table 3. The<br />
addition <strong>of</strong> either c<strong>at</strong>alyst resulted in dram<strong>at</strong>ic increases in <strong>co</strong>al <strong>co</strong>nversion and<br />
heptane insoluble <strong>co</strong>nversion but had little effect on the non-distillable <strong>co</strong>nversion.<br />
The <strong>co</strong>al <strong>co</strong>nversion and heptane insoluble <strong>co</strong>nversion without the addition<br />
<strong>of</strong> c<strong>at</strong>alyst was 66.6 wt-% and 21.3 wt-%. respectively. The <strong>co</strong>al <strong>co</strong>nversion and<br />
heptane insoluble <strong>co</strong>nversion increased to 80.5 wt-% and 63.9 wt-% with the iron<br />
c<strong>at</strong>alyst and increased further with the UOP c<strong>at</strong>alyst to 92.2 wt-% and 81.3 wt-%,<br />
respectively. The non-distillable <strong>co</strong>nversion (510"C+) ranged from 69.3 to 73.6 wt.%<br />
for these three tests.<br />
Although the iron oxide c<strong>at</strong>alyst demonstr<strong>at</strong>ed some beneficial effects, its<br />
overall performance was inferior to the UOP slurry c<strong>at</strong>alyst. The differences<br />
between these two c<strong>at</strong>alysts be<strong>co</strong>mes even more apparent when hydrogen <strong>co</strong>nsumption and<br />
product quality are also included as part <strong>of</strong> the evalu<strong>at</strong>ion. The product properties<br />
<strong>of</strong> the total liquid product for each c<strong>at</strong>alyst system tested are surmnarized in<br />
Table 4.<br />
The UOP slurry c<strong>at</strong>alyst has the best hydrogen<strong>at</strong>ion capabilities <strong>of</strong> the three<br />
Systems tested. The hydrogen <strong>co</strong>nsumption with the UOP slurry c<strong>at</strong>alyst was 2.66<br />
&-%. <strong>co</strong>mpared to 1.84 wt-% and 1.68 wt-% using no c<strong>at</strong>alyst and the iron c<strong>at</strong>alyst,<br />
respectively. This higher hydrogen <strong>co</strong>nsumption yields a liquid product with the<br />
highest API gravity, highest hydrogen <strong>co</strong>ntent and the lowest heptane insoluble <strong>co</strong>ntent.<br />
The higher API gravity product is important because although the product has<br />
the same b<strong>oil</strong>ing range as products derived from no c<strong>at</strong>alyst and iron c<strong>at</strong>alyst, it is<br />
less arom<strong>at</strong>ic and more like petroleum fractions. Also, the lower heptane insoluble<br />
<strong>co</strong>ntent means th<strong>at</strong> the m<strong>at</strong>erial would have a lower tendency to poison or foul <strong>co</strong>nventional<br />
refinery upgrading c<strong>at</strong>alysts, thus making it more e<strong>co</strong>nomically <strong>at</strong>tractive<br />
to upgrade.<br />
182<br />
' I<br />
I
I<br />
COAL/RESID REACTIVITY EVALUATION<br />
The reactivities <strong>of</strong> different <strong>co</strong>al/resid <strong>co</strong>mbin<strong>at</strong>ions were evalu<strong>at</strong>ed. All the<br />
vacuum resids were tested with one <strong>co</strong>al (Illinois No. 6, C1) and all the <strong>co</strong>als were<br />
tested with one resid (Lloydminster. R4). The subbituminous <strong>co</strong>al (Wyodak) was<br />
tested as received (14.7 wt-% moisture <strong>co</strong>ntent, C4.1) and also dried (1.78 wt-%<br />
moisture <strong>co</strong>ntent, C4.2). The tests were made <strong>at</strong> the oper<strong>at</strong>ing <strong>co</strong>nditions st<strong>at</strong>ed<br />
above with the UOP slurry c<strong>at</strong>alyst.<br />
Resid reactivity screening results are summarized in Figure 2. Coal <strong>co</strong>nversions<br />
ranged from 87.9 to 92.5 wt-%. Hydrogen <strong>co</strong>nsumption generally decreased<br />
with increasing API gravity. The heptane insoluble and non-distillable <strong>co</strong>nversions<br />
followed a similar trend.<br />
Coal reactivity screening results are summarized in Figures 3 and 4. The three<br />
bituminous and the dried subbituminous (1.78 wt-% moisture <strong>co</strong>ntent) <strong>co</strong>als showed no<br />
particular trends. MAF <strong>co</strong>al <strong>co</strong>nversion and heptane insoluble <strong>co</strong>nversion for each<br />
<strong>co</strong>al were similar. The subbituminous <strong>co</strong>al as received (14.7 wt-% moisture <strong>co</strong>ntent),<br />
gave lower <strong>co</strong>al <strong>co</strong>nversion (78.3 vs 90.3 wt-% for dried Wyodak) and lower heptane<br />
insoluble <strong>co</strong>nversion (64.5 vs 78.8 wt-% for dried Wyodak).<br />
CONTINUOUS BENCH-SCALE OPERATIONS<br />
The objectives <strong>of</strong> the <strong>co</strong>ntinuous bench-scale oper<strong>at</strong>ions are to: 1) prove the<br />
process <strong>co</strong>ncept, 2) direct its development toward the goals <strong>of</strong> achieving maximum<br />
<strong>co</strong>al <strong>co</strong>ncentr<strong>at</strong>ion in the resid/<strong>co</strong>al feed and producing the gre<strong>at</strong>est distill<strong>at</strong>e<br />
yield, and 3) establish a firm experimental basis on which to evalu<strong>at</strong>e a <strong>co</strong>nceptual<br />
<strong>co</strong>mmercial facility. The early work reported here has been directed <strong>at</strong> the first<br />
and third objectives.<br />
A simplified block diagram <strong>of</strong> the pilot plant is shown in Figure 5. The slurry<br />
feed (finely ground <strong>co</strong>al, petroleum resid and c<strong>at</strong>alyst) is <strong>co</strong>mbined with hydrogenrich<br />
recycle gas and is then prehe<strong>at</strong>ed before it enters the bottom <strong>of</strong> the upflow<br />
reactor. The products from the reactor are then separ<strong>at</strong>ed into a gas and <strong>oil</strong> stream<br />
<strong>at</strong> the high pressure separ<strong>at</strong>or. The gas stream from the high pressure separ<strong>at</strong>or is<br />
<strong>co</strong>mbined with make-up hydrogen before being recycled back to the in<strong>co</strong>ming fresh<br />
feed. The <strong>oil</strong> stream from the high pressure separ<strong>at</strong>or is sent to a stripper where<br />
the lighter hydrocarbons are separ<strong>at</strong>ed from the heavier fraction. The lighter<br />
hydrocarbon stream is separ<strong>at</strong>ed further in the debutanizer into C4 minus and C4 plus<br />
products. The heavier hydrocarbon stream from the stripper is sent to a vacuum<br />
fraction<strong>at</strong>or to obtain appropri<strong>at</strong>e fractions.<br />
A temper<strong>at</strong>ure and space velocity survey was <strong>co</strong>nducted <strong>processing</strong> Illinois No. 6<br />
<strong>co</strong>al (C1.2) and a <strong>co</strong>mmercially fraction<strong>at</strong>ed Lloydminster resid (RE) with the UOP<br />
slurry c<strong>at</strong>alyst. The <strong>co</strong>mmercially fraction<strong>at</strong>ed Lloydminster resid is lighter than<br />
the Lloydminster (R4) used in the autoclave studies, <strong>co</strong>ntaining 15 vol-% more 510°C<br />
minus m<strong>at</strong>erial. The tests were made <strong>at</strong> the oper<strong>at</strong>ing <strong>co</strong>nditions st<strong>at</strong>ed below.<br />
Three temper<strong>at</strong>ures and three space velocities were run.<br />
1<br />
,<br />
><br />
I<br />
Resid<br />
Coal<br />
Resid/Coal R<strong>at</strong>io<br />
Pressure, psi!<br />
Temper<strong>at</strong>ure, C<br />
Oper<strong>at</strong>inq Conditions<br />
R8, Lloydminster Vacuum Bottoms<br />
C1.2. Illinois No. 6<br />
2<br />
3000<br />
Varied<br />
WHSV, G/Hr/cc Varied<br />
i 183<br />
The effects <strong>of</strong> temper<strong>at</strong>ure on product distribution and <strong>co</strong>nversions are shown in<br />
Table 5. The product distributions give the expected trends, an increase <strong>of</strong> lighter
fractions and a decrease <strong>of</strong> heavier fractions with increasing temper<strong>at</strong>ure. Coal<br />
<strong>co</strong>nversion and heptane insoluble <strong>co</strong>nversion exhibited an interesting trend in the<br />
higher temper<strong>at</strong>ure range. At the lowest temper<strong>at</strong>ure, 83.0 wt-% <strong>of</strong> the MAF <strong>co</strong>al was<br />
<strong>co</strong>nverted. Coal <strong>co</strong>nversion increased to 91.8 wt-% <strong>at</strong> the mid-temper<strong>at</strong>ure, and then<br />
decreased slightly to 90.7 wt-% <strong>at</strong> the highest temper<strong>at</strong>ure. Heptane insoluble <strong>co</strong>nversion<br />
behaved similarly, increasing from 72.8 wt-% <strong>at</strong> the lowest temper<strong>at</strong>ure to<br />
82.2 wt-% <strong>at</strong> the mid-temper<strong>at</strong>ure, then decreasing to 72.5 wt-% <strong>at</strong> the highest<br />
temper<strong>at</strong>ure. The fact th<strong>at</strong> both <strong>co</strong>al <strong>co</strong>nversion and heptane insoluble <strong>co</strong>nversion<br />
decreased <strong>at</strong> the highest temper<strong>at</strong>ure suggests th<strong>at</strong> the highest temper<strong>at</strong>ure is too<br />
severe. resulting in thermal degrad<strong>at</strong>ion reactions.<br />
c<strong>at</strong>alytic effects predomin<strong>at</strong>e over thermal effects.<br />
At lower temper<strong>at</strong>ures,<br />
The effects <strong>of</strong> residence time on product distribution and <strong>co</strong>nversion are shown<br />
in Table 6. The product distributions show an increase <strong>of</strong> lighter fractions and a<br />
decrease <strong>of</strong> heavier fractions with longer residence time. However, <strong>co</strong>al <strong>co</strong>nversion<br />
and heptane insoluble <strong>co</strong>nversion show adverse responses to the longest residence<br />
time. At 1.01 WHSV (g/hr/cc reactor volume), 86.8 wt-% <strong>of</strong> the MAF <strong>co</strong>al was <strong>co</strong>nverted.<br />
Coal <strong>co</strong>nversion increased to 91.8 wt-% <strong>at</strong> 0.78 WHSV, and then decreased<br />
slightly to 90.5 wt-% <strong>at</strong> the 0.62 WHSV. Heptane insoluble <strong>co</strong>nversion behaved<br />
similarly , increasing from 75.7 wt-% <strong>at</strong> 1.01 WHSV to 82.2 wt-% <strong>at</strong> 0.78 WHSV, then<br />
decreasing significantly to 69.9 wt-% <strong>at</strong> 0.62 WHSV. Analogous to the high temper<strong>at</strong>ure<br />
experiment, both decreased <strong>co</strong>al <strong>co</strong>nversion and decreased heptane insoluble<br />
<strong>co</strong>nversion <strong>at</strong> the lowest space velocity suggest th<strong>at</strong> too severe an oper<strong>at</strong>ing <strong>co</strong>ndition,<br />
in this case residence time, is resulting in thermal degrad<strong>at</strong>ion reactions.<br />
CONCLUSIONS<br />
The single-stage, slurry-c<strong>at</strong>alyzed Co-<strong>processing</strong> <strong>co</strong>ncept was successfully<br />
demonstr<strong>at</strong>ed in labor<strong>at</strong>ory b<strong>at</strong>ch experiments. The active UOP c<strong>at</strong>alyst gave high<br />
<strong>co</strong>al <strong>co</strong>nversion and high <strong>co</strong>nversion to liquid product <strong>at</strong> rel<strong>at</strong>ively low temper<strong>at</strong>ure<br />
and, as a result, thermal degrad<strong>at</strong>ion reactions and cracking <strong>of</strong> resid- and <strong>co</strong>al-<br />
derived liquid to light gases were minimized. The liquid hydrocarbon product is <strong>of</strong><br />
high quality and can be efficiently utilized as a feedstock in existing refineries.<br />
The <strong>co</strong>ntinuous bench-scale oper<strong>at</strong>ion gave similar performance to the labor<strong>at</strong>ory<br />
b<strong>at</strong>ch experiments, s<strong>at</strong>isfying the pro<strong>of</strong>-<strong>of</strong>-<strong>co</strong>ncept objective. In addition, d<strong>at</strong>a<br />
gener<strong>at</strong>ed to d<strong>at</strong>e initi<strong>at</strong>e a firm experimental basis on which to evalu<strong>at</strong>e a <strong>co</strong>nceptual<br />
<strong>co</strong>mmercial facility. These d<strong>at</strong>a show th<strong>at</strong> the Co-<strong>processing</strong> process is<br />
sensitive to high severity <strong>co</strong>nditions (temper<strong>at</strong>ure, residence time).<br />
High <strong>co</strong>al <strong>co</strong>n-<br />
version and high <strong>co</strong>nversion to high quality liquid product can be achieved by<br />
oper<strong>at</strong>ing <strong>at</strong> rel<strong>at</strong>ively mild <strong>co</strong>nditions where thermal degrad<strong>at</strong>ion reactions are<br />
minimized.<br />
ACKNOWLEDGMENl<br />
The author expresses his thanks to Beckay J. Nelson, John G. Sikonia and Carl<br />
Lea <strong>of</strong> the Signal Research Center and Michael J. Humbach and Charles P. Luebke <strong>of</strong><br />
UOP Inc. for their <strong>co</strong>ntributions to this study; and to Burtron H. Davis <strong>of</strong> the<br />
Kentucky Center for Energy Research Labor<strong>at</strong>ory for the acquisition and prepar<strong>at</strong>ion<br />
<strong>of</strong> the <strong>co</strong>al samples. This work,is supported by DOE Contract DE-AC22-84PC70002,<br />
"Coal <strong>Liquefaction</strong> Co-Processing".<br />
1.<br />
2.<br />
REFERENCES<br />
J. G. G<strong>at</strong>sis. U.S. P<strong>at</strong>ent 3.705.092. "Solvent Extraction <strong>of</strong> Coal by a Heavy<br />
Of 1 " (1972).<br />
s. w. Weller, "C<strong>at</strong>alysis in the Liquid Phase Hydrogen<strong>at</strong>ion <strong>of</strong> Coal and Tars,"<br />
Ch 7 in "C<strong>at</strong>alysis," Vol 4, P. H. Emmett (ed) Reinhold. New York (1956).<br />
184
F. Freidrich and B. Strobel, "<strong>Liquefaction</strong> <strong>of</strong> Coal in the Federal Republic <strong>of</strong><br />
Germany," Intern<strong>at</strong>ional Seminar on the Replacement <strong>of</strong> Fuels, University <strong>of</strong><br />
Liege, May 25-27, 1981.<br />
"St<strong>at</strong>us <strong>of</strong> Synfuels Development in Japan." NEDO, Synfuels, 3rd Worldwide<br />
Symposium. Washington, DC, November 1-3, 1983.<br />
A.'S. Paranjape and D. E. Rhodes, "Use <strong>of</strong> Iron Oxide and Hydrogen Sulfide to<br />
Improve Integr<strong>at</strong>ed Two-Stage <strong>Liquefaction</strong>." Proceedinqs <strong>of</strong> DOE Direct Coal<br />
<strong>Liquefaction</strong> Contractors' Review Meeting, October 17-18, 1984.<br />
J. G. G<strong>at</strong>sis et al., "Coal <strong>Liquefaction</strong> Co-Processing," Proceedings <strong>of</strong> DOE<br />
Direct Coal <strong>Liquefaction</strong> Contractors' Review Meeting, November 19-21, 1985.<br />
TABLE 1<br />
U.S. Alaskan<br />
Mid- North<br />
Continent Kuwait Slope Lloydminster Hondo Maya Lloydminster<br />
Resid Name (Rl) (R2) (R3) (R4) (R5) (R6) (R8)<br />
Total Sample<br />
API Gravity<br />
Specific Gravity<br />
0- 1160, "C<br />
IBP, VO~-%<br />
5<br />
10<br />
20<br />
30<br />
EP<br />
Overhead, vol-%<br />
Analysis, wt-%<br />
Carbon<br />
Hydrogen<br />
Oxygen<br />
Sulfur<br />
Nitrogen<br />
Carbon Residue<br />
Petroleum Ash<br />
C7 Insolubles<br />
Nickel, ppm<br />
Vanadium, ppm<br />
Iron, ppm<br />
Molecular Weight<br />
Furol Visc.,<br />
sec (121°C)<br />
pour Point. "C<br />
Salt, lb/1000 bbls<br />
12.70<br />
0.9813<br />
473.0<br />
510.0<br />
525.0<br />
546.0<br />
,568.0<br />
568.0<br />
30.0<br />
87.30<br />
10.25<br />
0.30<br />
1.0<br />
0.45<br />
16.50<br />
0.030<br />
8.29<br />
35.0<br />
113.0<br />
62.0<br />
839.0<br />
755.0<br />
38.00<br />
2.90<br />
7.90 8.90<br />
1.0151 1.0078<br />
472.0 422.0<br />
505.0 494.0<br />
517.0 515.0<br />
542.0 541.0<br />
556.0 550.0<br />
26.0 24.0<br />
84.15 84.10<br />
10.55 10.85<br />
0.35 0.27<br />
4.9 2.3<br />
0.35 0.55<br />
18.00 17.30<br />
0.020 0.020<br />
5.95 4.80<br />
28.0 38.0<br />
100.0 79.0<br />
4.5 2.0<br />
1054.0 810.0<br />
1016.0 ~~ 1295.0<br />
~~<br />
38.00 32.00<br />
3.50 1.20<br />
185<br />
3.60<br />
1.0474<br />
406.0<br />
509.0<br />
509.0<br />
6.0<br />
82.70<br />
10.15<br />
0.29<br />
5.6<br />
0.62<br />
22.20<br />
0.090<br />
18.10<br />
122.0<br />
278.0<br />
82.0<br />
1444.0<br />
1921.0<br />
91.00<br />
3.30<br />
3.70 2.80<br />
1.0466 1.0536<br />
478.0 452.0<br />
512.0 515.0<br />
524.0 532.0<br />
-<br />
-<br />
524.0 532.0<br />
10.0 10.0<br />
81.20 83.90<br />
10.10 9.15<br />
0.36 0.48<br />
6.6 4.9<br />
1.10 0.71<br />
19.90 26.10<br />
0.110 0.126<br />
17.80 22.40<br />
157.0 116.0<br />
435.0 595.0<br />
42.0 29.0<br />
1125.0 1015.0<br />
1126.0 2217.0<br />
79.00 91.00<br />
4.00 20.70<br />
6.50<br />
1.0254<br />
369 2 0<br />
432.0<br />
463.0<br />
505.0<br />
-<br />
523.0<br />
26.5<br />
83.70<br />
10.00<br />
-<br />
5.1<br />
0.48<br />
17.30<br />
0.051<br />
13.91<br />
83.0<br />
165.0<br />
3.6<br />
255.0<br />
266.1<br />
120.0<br />
5.2
TABLE 2<br />
Coal Analyses<br />
I1 1 i noi s Kentucky Indiana Wyodak Wyodak Illinois<br />
No. 6 No. 9 No. V (As-Received) (Dried) No. 6<br />
Coal Name 0 (C2) (C3) (C4.1) IC4.2) (C1.2)<br />
Ultim<strong>at</strong>e Analysis, wt-%<br />
Ash 9.65<br />
Carbon 68.60<br />
Hydrogen 4.51<br />
Nitrogen 1.39<br />
Sulfur 3.04<br />
Oxygen* 9.66<br />
Proxim<strong>at</strong>e Analysis, wt-%<br />
Moisture 3.15<br />
Ash 9.65<br />
Vol<strong>at</strong>ile M<strong>at</strong>ter 39.95<br />
Fixed Carbon 47.25<br />
*Difference<br />
Oper<strong>at</strong>ing Conditions<br />
C<strong>at</strong>alyst Type<br />
Concentr<strong>at</strong>ion<br />
8.68 8.12 10.30 12.00 10.56<br />
71.95 69.70 54.70 63.01 68.77<br />
4.78 5.40 3.83 4.50 4.84<br />
1.54 1.42 0.69 0.90 1.37<br />
2.97 4.28 0.99 1.08 3.34<br />
8.53 9.37 14.79 16.73 7.03<br />
1.55 1.71 14.70 1.78 4.09<br />
8.68 8.12 10.30 12.00 10.56<br />
42.35 48.25 37.00 42.60 39.90<br />
47.42 41.92 38.00 43.62 45.45<br />
TABLE 3<br />
C<strong>at</strong>alyst Comparison Study<br />
None Fe 0 UOP C<strong>at</strong>alyst<br />
0 2 dase Base<br />
Performance<br />
Conversions, wt-%<br />
Coal 66.6 80.5 92.2<br />
Heptane insolubles 21.3 63.9 81.3<br />
Non-di st i 1 1 ables (510"C+) 69.3 73.6 72.1<br />
Hydrogen Consumption, wt-% 1.84 1.68 2.66<br />
186<br />
,<br />
' I
C<strong>at</strong>alyst Type<br />
API Gravity <strong>at</strong> 15.6"C<br />
Specific Gravity<br />
Carbon, wt-%<br />
Hydrogen, wt-%<br />
Oxygen, wt-%<br />
Sulfur, wt-%<br />
Nitrogen, wt-%<br />
Ash, wt-%<br />
Heptane Insolubles, wt-%<br />
Carbon Residue, wt-%<br />
Vanadium and Nickel, wt-ppm<br />
*Estim<strong>at</strong>ed<br />
TABLE 4<br />
C<strong>at</strong>alyst Comparison Study<br />
Total Liquid Product Properties<br />
None<br />
9.3<br />
1.0050<br />
85.15<br />
10.05<br />
1.00<br />
2.75<br />
0.60<br />
0.005<br />
37.03<br />
14.6<br />
19<br />
TABLE 5<br />
Fe203<br />
8.5<br />
1.0107<br />
84.40<br />
9.6*<br />
-<br />
2.30<br />
0.90<br />
Temper<strong>at</strong>ure<br />
WHSV, G/hr/cc<br />
TABLE 6<br />
Continuous Bench-Scale Oper<strong>at</strong>ions<br />
Effect <strong>of</strong> Residence Time<br />
Base + 4<br />
0.62<br />
Product Distribution<br />
Hetero Gases + HvO, wt-% 6.3<br />
Hcbn. Gas C4-, wt-% 3.1<br />
C - 371"C, wt-% 42.0<br />
391 - 510°C, wt-% 43.4<br />
510°C + 4.1<br />
MAF Coal<br />
Total, wt-%<br />
3.3<br />
102.2<br />
Conversion<br />
Coal, wt-% MAF Coal 90.5<br />
C Insolubles, wt-% 69.9<br />
51OoC+, wt-% 67.7<br />
371"C+, wt-% 38.7<br />
H2 Consumption, wt-% 2.19<br />
Base + 6<br />
0.78<br />
8.9<br />
2.5<br />
38.0<br />
40.1<br />
10.1<br />
3.0<br />
102.6<br />
91.8<br />
82.2<br />
64.2<br />
40.1<br />
2.58<br />
Base +5<br />
1.01<br />
8.4<br />
2.3<br />
31.1<br />
41.9<br />
13.9<br />
4.7<br />
102.3<br />
86.8<br />
75.7<br />
56.8<br />
33.2<br />
2.29
FIGURE 1<br />
VACUUM RESID FEEDSTOCKS<br />
COMWsmoN<br />
FIGURE 2<br />
RESID REACTIVITY SCREENING<br />
(ILUNOIS COAL NO. 6)<br />
189
z<br />
FIGURE 3<br />
COAL REACTIVITY SCREENING<br />
(UOVDMINSTER RESID)<br />
90 -<br />
$80i<br />
170-<br />
Y<br />
E 60-<br />
0<br />
50 -<br />
40-<br />
MAF COAL c7 INSOL<br />
FIGURE 4<br />
COAL REACTlVlTV SCREENING<br />
(LLOVDMINSTER RESID)<br />
- 1<br />
JIWC+ 371"C+<br />
CONW CONK<br />
c2 m c 3 0 c4.1 r-JC4.2<br />
190<br />
"os 12a%3?<br />
YO, ,=>a
c<br />
RECICLE<br />
COMPRESSOR<br />
MAKE-UP H2<br />
LEGEND<br />
R = RwmR<br />
S = HIGH PRESSURE SEPARATOR<br />
Sl = KIRIPPER<br />
D = DEBUTANIZER<br />
VF = VACUUM FRACTIONATOR<br />
FIGURE 5<br />
PILOT PLANT FLOW SCHEME<br />
I . I<br />
191<br />
vF I<br />
Cq MINUS<br />
cq PLUS<br />
PRODUCT<br />
DISTILLATE<br />
10M'F EP<br />
HEAVY<br />
PROWCT
COPROCESSING USING HzS AS A PROMOTER<br />
P.M. Rahimi, S.A. Fouda and J.F. Kelly<br />
CANMET, Energy, Mines and Resources Canada, 555 Booth Street,<br />
Ottawa, Ontario K1A OG1<br />
INTRODUCTION<br />
Co<strong>processing</strong> heavy <strong>oil</strong>s, bitumens or petroleum residues with <strong>co</strong>al<br />
can be <strong>co</strong>nsidered as a bridge between <strong>co</strong>al liquefaction and<br />
hydrocracking. The existing technologies <strong>of</strong> liquefaction and<br />
hydrocracking can be applied with modific<strong>at</strong>ion to <strong>co</strong><strong>processing</strong>. In<br />
terms <strong>of</strong> oper<strong>at</strong>ion, <strong>co</strong><strong>processing</strong> is less <strong>co</strong>mplic<strong>at</strong>ed than liquefaction<br />
because recylce solvent is elimin<strong>at</strong>ed. Since the <strong>co</strong><strong>processing</strong> solvent<br />
is upgraded simultaneously with <strong>co</strong>al the reactor volume is utilized<br />
more effectively. If residuum <strong>co</strong>nversion levels during <strong>co</strong><strong>processing</strong><br />
are as high as those in hydrocracking then <strong>co</strong><strong>processing</strong> also <strong>of</strong>fers a<br />
significant saving in feedstock <strong>co</strong>sts by substituting a significant<br />
portion <strong>of</strong> the heavy <strong>oil</strong> with less expensive <strong>co</strong>al.<br />
CANMEl <strong>co</strong><strong>processing</strong> involves the simultaneous upgrading <strong>of</strong> <strong>co</strong>al<br />
and heavy <strong>oil</strong> or bitumen in a once-through mode <strong>of</strong> oper<strong>at</strong>ion using a<br />
disposable iron c<strong>at</strong>alyst. The CANMET additive (pulverized <strong>co</strong>al<br />
impregn<strong>at</strong>ed with iron sulph<strong>at</strong>e), hereinafter referred to as FeS04, ha8<br />
been identified as both an hydrogen<strong>at</strong>ion and <strong>co</strong>ke-reducing c<strong>at</strong>alyst.<br />
Process feasibility has been investig<strong>at</strong>ed using a variety <strong>of</strong> <strong>co</strong>als and<br />
heavy <strong>oil</strong>s/bitumens(l). Also, it has been demonstr<strong>at</strong>ed th<strong>at</strong> in terms<br />
<strong>of</strong> product yields for subbituminous <strong>co</strong>als, CANMET <strong>co</strong><strong>processing</strong> is<br />
superior to liquefaction and is <strong>co</strong>mparable to hydrocracking (2-3).<br />
The effect <strong>of</strong> H2S in hydrocracking <strong>of</strong> model <strong>co</strong>mpounds and in<br />
liquefaction is well documented (4-11). The ability <strong>of</strong> H2S to reduce<br />
<strong>co</strong>ke form<strong>at</strong>ion and increase liquid yield in <strong>co</strong>al liquefaction has been<br />
p<strong>at</strong>ented by Exxon Research and Engineering Company (12). It has also<br />
been shown th<strong>at</strong> HpS has benefical effects in non-c<strong>at</strong>alytic crude <strong>oil</strong><br />
hydrorefining processes (13).<br />
In a previous b<strong>at</strong>ch autoclave study the use <strong>of</strong> H2S in<br />
<strong>co</strong><strong>processing</strong> subbituminous <strong>co</strong>al and bitumen resulted in high <strong>co</strong>al<br />
<strong>co</strong>nversion and distill<strong>at</strong>e yield (14). The increase in product yields<br />
in the presence <strong>of</strong> H2S was <strong>at</strong>tributed to its ability to don<strong>at</strong>e its<br />
hydrogen to radicals derived from <strong>co</strong>al and bitumen (15).<br />
The objective <strong>of</strong> the present study was to verify the positive<br />
effects <strong>of</strong> H2S under <strong>co</strong><strong>processing</strong> <strong>co</strong>nditions using a <strong>co</strong>ntinuous-flow<br />
bench scale pilot plant and to <strong>co</strong>mpare the activity <strong>of</strong> H2S with FeSO4<br />
under similar oper<strong>at</strong>ing <strong>co</strong>nditions.<br />
192
I<br />
Process Unit<br />
- 2 -<br />
EXPERIMENTAL<br />
Co<strong>processing</strong> experiments were carried out in a I-L<br />
<strong>co</strong>ntinuous-flow stirred tank reactor with a nominal capacity <strong>of</strong> 1 kg/h<br />
<strong>of</strong> slurry feed while product samples were <strong>co</strong>llected over I-h periods<br />
<strong>at</strong> steady st<strong>at</strong>e. For all the experimental runs reported in thls<br />
paper, the m<strong>at</strong>erial balances were within i5 wt X.<br />
purposes, all the d<strong>at</strong>a were normalized to 100% m<strong>at</strong>erial balance by<br />
proportioning the losses over each <strong>of</strong> the product fractions. Other<br />
details <strong>of</strong> the experimental unit are available elsewhere (2).<br />
Feedstocks<br />
For <strong>co</strong>mparison<br />
The analysis <strong>of</strong> Forestburq subbituminous C <strong>co</strong>al and Cold Lake<br />
vacuum bottoms (CLVB) is shown in Table 1. Additives or promoters<br />
were FeS04 or H2S or both. The HzS was obtained from M<strong>at</strong>heson and<br />
used as received. In experiments where H2S was used it was pumped as<br />
a liquid using a W<strong>at</strong>ers LC pump model 6000A.<br />
Product Yields<br />
RESULrS AND DISCUSSION<br />
Previous b<strong>at</strong>ch autoclave experiments indic<strong>at</strong>ed th<strong>at</strong> H2S is most<br />
effective <strong>at</strong> low tp moder<strong>at</strong>e severities in terms <strong>of</strong> improving product<br />
yields when <strong>co</strong>mpared to <strong>co</strong><strong>processing</strong> without any additive. AT<br />
moder<strong>at</strong>e-high severity, using the feedstocks reported in this<br />
paper,rel<strong>at</strong>ively high <strong>co</strong>ke form<strong>at</strong>ion was observed even in the presence<br />
<strong>of</strong> H2S. For this reason, the CSTR experiments which involved H2S only<br />
were performed <strong>at</strong> low to moder<strong>at</strong>e severities. For moder<strong>at</strong>e-high<br />
severity experiments, iron sulph<strong>at</strong>e was used to assure smooth process<br />
oper<strong>at</strong>ion and to prevent <strong>co</strong>ke form<strong>at</strong>ion. Attempts to perform<br />
<strong>co</strong><strong>processing</strong> experiments in the CSTR unit using CLVB and Forestburg<br />
<strong>co</strong>al without any c<strong>at</strong>alyst even <strong>at</strong> low severity resulted in <strong>co</strong>ke<br />
form<strong>at</strong>ion and plant shutdown. Thus, it is not possible to <strong>co</strong>mpare the<br />
results <strong>of</strong> experimental runs using H2S only with those using no<br />
additive or promoter as was done in the b<strong>at</strong>ch autoclave studies (15).<br />
The fact th<strong>at</strong> <strong>co</strong><strong>processing</strong> experiments <strong>co</strong>uld be performed in the CSTR<br />
with H2S and no other c<strong>at</strong>alyst <strong>at</strong> low and moder<strong>at</strong>e severities is<br />
significant and verifies earlier b<strong>at</strong>ch results which indic<strong>at</strong>ed th<strong>at</strong><br />
HpS prevents <strong>co</strong>ke form<strong>at</strong>ion under the <strong>co</strong>nditions employed (14-15).<br />
Table 2 <strong>co</strong>mpares <strong>co</strong><strong>processing</strong> results obtained in the presence <strong>of</strong><br />
HpS and iron sulph<strong>at</strong>e <strong>at</strong> two levels <strong>of</strong> severities. At both levels<br />
replacement <strong>of</strong> FeSO4 with H2S resulted in higher distill<strong>at</strong>e yield,<br />
pitch and <strong>co</strong>al <strong>co</strong>nversions. The results <strong>of</strong> b<strong>at</strong>ch studies indic<strong>at</strong>ed<br />
193
- 3 -<br />
th<strong>at</strong> product yields depend on HpS <strong>co</strong>ncentr<strong>at</strong>ion. At moder<strong>at</strong>e<br />
temper<strong>at</strong>ure maximum <strong>co</strong>al <strong>co</strong>nversion and distill<strong>at</strong>e yield were obtained<br />
<strong>at</strong> about 3.5 w t X HzS based on maf slurry feed (15). However, the<br />
results reported in this work are based on only one experimental run<br />
and are <strong>at</strong> approxim<strong>at</strong>ely 8 w t X HpS based on maf slurry feed. No<br />
optimiz<strong>at</strong>ion <strong>of</strong> HpS <strong>co</strong>ncentr<strong>at</strong>ion on product yields was carried out in<br />
this CSTR study.<br />
The increase in <strong>co</strong>nversions and distill<strong>at</strong>e yield in the presence<br />
<strong>of</strong> HzS can be r<strong>at</strong>ionalized by its hydrogen-donor ability. Hydrogen<br />
sulphide can don<strong>at</strong>e its hydrogen directly to <strong>co</strong>al and bitumen-derived<br />
radicals or the available hydrogens in H2S can be transferred to<br />
radicals via <strong>co</strong>al-derived liquids. The evidence for direct hydrogen<br />
don<strong>at</strong>ion by HzS <strong>co</strong>mes from b<strong>at</strong>ch autoclave hydrocracking studies using<br />
CLVB (14). At low severity, the presence <strong>of</strong> HpS resulted in a<br />
substantial improvement and <strong>at</strong> moder<strong>at</strong>e severity a slight increase in<br />
distill<strong>at</strong>e yield. The <strong>co</strong>nsiderable increase in distill<strong>at</strong>e yield in<br />
the presence <strong>of</strong> HpS suggests th<strong>at</strong> <strong>at</strong> least in part, hydrogen from H S<br />
is transferred to bitumen-derived radicals. The previous study (14%<br />
also showed th<strong>at</strong> while the <strong>co</strong>nversion <strong>of</strong> Forestburg <strong>co</strong>al in anthracene<br />
<strong>oil</strong> increased with HpS, distill<strong>at</strong>e yield did not. However, in<br />
<strong>co</strong>p<strong>co</strong>cessing (b<strong>at</strong>ch and CSIR) both <strong>co</strong>al <strong>co</strong>nversion and distill<strong>at</strong>e<br />
yield improved substantially when HpS was used. These results may<br />
indic<strong>at</strong>e th<strong>at</strong> H2S promotes upgrading <strong>of</strong> bitumen during <strong>co</strong><strong>processing</strong>.<br />
An apparent synergism between <strong>co</strong>al and HpS is also suggested by less<br />
<strong>co</strong>ke form<strong>at</strong>ion during <strong>co</strong><strong>processing</strong> in the presence <strong>of</strong> HpS rel<strong>at</strong>ive to<br />
the hydrocracking <strong>of</strong> bitumen only using H2S as a promoter (15). Table<br />
2 indic<strong>at</strong>es th<strong>at</strong> <strong>at</strong> least <strong>at</strong>.low and moder<strong>at</strong>e severities the<br />
performance <strong>of</strong> HpS under <strong>co</strong><strong>processing</strong> <strong>co</strong>nditions in a CSTR is as good<br />
as or better than FeS04.<br />
Table 3 <strong>co</strong>mpares the activities <strong>of</strong> iron sulph<strong>at</strong>e with and without<br />
H2S. At very low and low severities addition <strong>of</strong> HpS to FeS04 resulted<br />
in an increase in <strong>co</strong>al <strong>co</strong>nversions whereas distill<strong>at</strong>e yields and pitch<br />
<strong>co</strong>nversions did not change. However, <strong>at</strong> moder<strong>at</strong>e severity, HpS had a<br />
significant effect on distill<strong>at</strong>e yield, <strong>co</strong>al and pitch <strong>co</strong>nversions.<br />
The presence <strong>of</strong> HzS <strong>at</strong> moder<strong>at</strong>e-high severity had small effect on<br />
distill<strong>at</strong>e yield and pitch <strong>co</strong>nversion but no effect on <strong>co</strong>al<br />
<strong>co</strong>nversion. It appears th<strong>at</strong> <strong>at</strong> higher severities the positive effect<br />
<strong>of</strong> H2S is masked by the presence <strong>of</strong> FeSOq. A <strong>co</strong>mparison <strong>of</strong> lables 2<br />
and 3 reveals th<strong>at</strong> <strong>at</strong> low severity a higher distill<strong>at</strong>e yield was<br />
obtained with the HpS only run. However, <strong>at</strong> moder<strong>at</strong>e severity, no<br />
improvement was observed using HpS+FeS04 <strong>co</strong>mpared to HpS only.<br />
Also,<br />
the CGmpeiiSGii clearly shows th<strong>at</strong> <strong>at</strong> moder<strong>at</strong>e severity <strong>co</strong>al <strong>co</strong>nversion<br />
in the presence <strong>of</strong> HpS only (80.4 wt X) approaches th<strong>at</strong> <strong>at</strong><br />
moder<strong>at</strong>e-high severity using FeS04 only (86.3 wt %).<br />
194
i<br />
Product Characteristics<br />
a) Distill<strong>at</strong>es<br />
-* -<br />
Table 4 shows the characteristics <strong>of</strong> distill<strong>at</strong>e products <strong>at</strong> four<br />
different severities and <strong>co</strong>mpares product qualities obtained in the<br />
presence <strong>of</strong> H2S with those obtained using FeS04 or H2S + FeSOq.<br />
At very low severity it appears th<strong>at</strong> the distill<strong>at</strong>e products obtained<br />
using H2S + FeSOq are rel<strong>at</strong>ively heavier than those obtained in the<br />
presence <strong>of</strong> FeSOq only. The sulphur <strong>co</strong>ntent <strong>of</strong> the distill<strong>at</strong>e did not<br />
change when H2S was added to FeS04, however, the arom<strong>at</strong>icity increased<br />
from 26 to 31.<br />
This increase parallels th<strong>at</strong> <strong>of</strong> increased <strong>co</strong>al<br />
<strong>co</strong>nversion upon H2S addition (see Table 3) and may imply th<strong>at</strong> some<br />
<strong>co</strong>al-derived liquid <strong>co</strong>ntributed to the distill<strong>at</strong>e. Again <strong>at</strong> low<br />
severity, higher <strong>co</strong>al <strong>co</strong>nversion in the presence <strong>of</strong> H2S only <strong>co</strong>mpared<br />
to FeS04 or H2S + FeSOq resulted in a rel<strong>at</strong>ively heavier distill<strong>at</strong>e.<br />
The sulphur <strong>co</strong>ntent <strong>of</strong> the distill<strong>at</strong>e decreased slightly in the H2S<br />
only run. At moder<strong>at</strong>e severity, the use <strong>of</strong> H2S alone resulted in a<br />
heavier liquid product, lower H/C r<strong>at</strong>io and higher molecular weight<br />
than the distill<strong>at</strong>e obtained using either FeS04 or H2S + FeSO4.<br />
From the results shown in Table 4 it appears th<strong>at</strong> the effect <strong>of</strong><br />
FeS04 as a hydrogen<strong>at</strong>ion c<strong>at</strong>alyst is more pronounced <strong>at</strong> rel<strong>at</strong>ively<br />
higher severities. At moder<strong>at</strong>e severity, although similar <strong>co</strong>al<br />
<strong>co</strong>nversions and distill<strong>at</strong>e yields were obtained with both H2S and H2S<br />
+ FeSOq, a better distill<strong>at</strong>e quality was obtained with H2S + FeS04.<br />
Again the higher molecular weight in the H2S only run may suggest th<strong>at</strong><br />
more <strong>co</strong>al-derived liquid <strong>co</strong>ntributes to the distill<strong>at</strong>e but the product<br />
is not upgraded to the same degree as when FeS04 is used. The oxygen<br />
<strong>co</strong>ntent <strong>of</strong> the distill<strong>at</strong>e decreased when HzS was used instead <strong>of</strong><br />
FeS04. Addition <strong>of</strong> HzS to FeS04 further reduced the oxygen<br />
<strong>co</strong>ntent which indic<strong>at</strong>es th<strong>at</strong> H2S reacts with the oxygen<br />
functionalities in <strong>co</strong>al. However, sulphur <strong>co</strong>ntent <strong>of</strong> the distill<strong>at</strong>e<br />
increased slightly in the presence <strong>of</strong> H2S only.<br />
severity, the product quality improved slightly in the presence <strong>of</strong> H2S<br />
in terms <strong>of</strong> higher H/C r<strong>at</strong>io, lower oxygen <strong>co</strong>ntent, arom<strong>at</strong>icity and<br />
molecular weight.<br />
b) Residues<br />
At moder<strong>at</strong>e-high<br />
The <strong>co</strong>mpositions <strong>of</strong> residues obtained under different process<br />
severities are shown in Fig. 1. At very low severity, the addition <strong>of</strong><br />
H2S to FeS04 resulted in slightly higher yields <strong>of</strong> asphaltenes,<br />
preasphaltenes and lower THF insolubles. At low severity, the lowest<br />
yield <strong>of</strong> THF insolubles was obtained with H2S which reflects a higher<br />
<strong>co</strong>al <strong>co</strong>nversion than the FeSOq and HzS+FeS04 runs. Under these<br />
<strong>co</strong>nditions, the rel<strong>at</strong>ive yields <strong>of</strong> <strong>oil</strong>s, asphaltenes, and<br />
preasphaltenes remained unchanged. At moder<strong>at</strong>e severity, the residue<br />
in the FeS04 run <strong>co</strong>ntained more residual <strong>oil</strong> than the HzS run.<br />
However, as shown in Table 2 the total distill<strong>at</strong>e yield as well as the<br />
pitch <strong>co</strong>nversion in the H2S run are higher. This suggests th<strong>at</strong> the<br />
upgrading <strong>of</strong> heavy m<strong>at</strong>erial in <strong>co</strong><strong>processing</strong> is more efficient using<br />
H2S than FeS04 <strong>at</strong> least <strong>at</strong> moder<strong>at</strong>e severities. Little or no change<br />
195
- 5 -<br />
occurs in the yields <strong>of</strong> asphaltenes and preasphaltenes <strong>at</strong> moder<strong>at</strong>e<br />
severity using the different additives. At moder<strong>at</strong>e-high severity,<br />
adding H2S to FeSOq resulted in slightly higher pitch <strong>co</strong>nversion and<br />
<strong>co</strong>nsequently lower residue yield (Table 3). The drop in residue yield<br />
is reflected mainly in lower preasphaltenes and asphaltenes yields.<br />
The toluene insolubles <strong>of</strong> some <strong>of</strong> the <strong>co</strong><strong>processing</strong> residues were<br />
also examined using optical micros<strong>co</strong>py. This technique, supplemented<br />
by semi-quantit<strong>at</strong>ive elemental analysis by scanning electron<br />
micros<strong>co</strong>py has shown th<strong>at</strong> it is possible to distinguish the<br />
originality <strong>of</strong> <strong>co</strong>al-derived and bitumen-derived solids in <strong>co</strong><strong>processing</strong><br />
residues (16). At moder<strong>at</strong>e severity, the toluene insolubles <strong>of</strong> the<br />
<strong>co</strong><strong>processing</strong> residue obtained using H2S <strong>co</strong>ntains 22.5 vol %<br />
<strong>co</strong>al-derived solids (altered <strong>co</strong>al or unreacted <strong>co</strong>al) whereas the<br />
residue from the FeS04 run <strong>co</strong>ntains 52.1 vol t <strong>co</strong>al-derived<br />
m<strong>at</strong>erials. These results are <strong>co</strong>nsistent with the higher <strong>co</strong>al<br />
<strong>co</strong>nversion in the H2S run rel<strong>at</strong>ive to the FeSOq run. Also a small<br />
amount, (0.9 vol %) <strong>of</strong> anisotropic solids in both the H2S and FeSQ<br />
runs was detected whereas none was detected in the H2S + FeSUq run.<br />
CONCLUSIONS<br />
Hydrogen sulphide has been shown to be an effective promoter in<br />
achieving high <strong>co</strong>al <strong>co</strong>nversions and distill<strong>at</strong>e yields when<br />
<strong>co</strong><strong>processing</strong> subbituminous <strong>co</strong>al with bitumen vacuum bottoms in a<br />
<strong>co</strong>ntinuous-flow bench scale oper<strong>at</strong>ion. Results indic<strong>at</strong>e th<strong>at</strong>, <strong>at</strong><br />
least, <strong>at</strong> low and moder<strong>at</strong>e severities <strong>of</strong> oper<strong>at</strong>ion H2S performs as<br />
good as or better than FeS04 in terms <strong>of</strong> product yields as well as<br />
qualities. However, <strong>at</strong> higher severities, FeS04 is superior to H2S.<br />
REFERENCES<br />
1. Fouda, S.A. and Kelly, J.F. "CANMET <strong>co</strong><strong>processing</strong> <strong>of</strong> low-rank<br />
Canadian <strong>co</strong>als'*; Division Report ERP/ERL 85-63(0PJ), CANMET, Energy<br />
Mines and Resources Canada, 1985; presented <strong>at</strong> the U.S. Dept. <strong>of</strong><br />
Energy Direct <strong>Liquefaction</strong> Contractors' Review Meeting, Pittsburg, PA,<br />
November 19-21, 1985.<br />
2. Kelly, J.F., Fouda, S.A., Rahimi, P.M. and Ikura, M. "CANMET<br />
<strong>co</strong><strong>processing</strong> - A st<strong>at</strong>us report"; Proceedings <strong>of</strong> the Coal Conversion<br />
Contractors' Review Meeting, Calgary, Alberta, 1984; Kelly, J.F.<br />
(editor), pp. 397-423, CANMET public<strong>at</strong>ion SP85-4,Supply and Services<br />
Canada, 19R5.<br />
3. Kelly, J.F. "Development <strong>of</strong> Co<strong>processing</strong> Technology - A Canadian<br />
Synthetic Fuels Opportunity" presented as the 1985 ERCO Award lecture<br />
<strong>at</strong> the 35th Canadian Chemical Engineerinq Conference, October 6-9,<br />
Calgary, Alberta, 1985.<br />
196
4.<br />
5.<br />
6.<br />
7.<br />
8.<br />
9.<br />
-6-<br />
Lambert, J.M. Jr., Fuel 61,777 (1982)<br />
Stenberg, V.I., Baltisberger, R.J., Ogaura, T., Raman, K. and<br />
Woolsey, N.F., Am Chem Soc Div Fuel Chem Preprints 27,22 (1982)<br />
Stenberg, V.I., Tanabe, K. Ogaura, T., Sweeny, P. and Hei, R.,<br />
Am Chem SOC Div Fuel Chem Preprints 28,183 (1983)<br />
Baldwin, R.M. and Vinciguerra, S., 62,498 (1983)<br />
Sondreal, E.A., Wilson, W.G., Stenberg, V.I., 61,925 (1982)<br />
Stenberg, V.I., Srinivas, V.R., Sweeny, P. Baltisberger, R.J. and<br />
Woolsey, N.F., 62,913 (1983)<br />
IO. S<strong>at</strong>terfield, C.N., Modell, M. and Mayer, J.F., AICHE J 21,1100<br />
(1975)<br />
11. Rollman, L.O.J., C<strong>at</strong>al 46,243 (1977)<br />
12. Exxon Research and Engineering Company, U.S. P<strong>at</strong>ent 4,149,959,<br />
April 17, 1979.<br />
13. Gleim, W.K.T. U.S. P<strong>at</strong>ent 3,303,126, June 17, 1964.<br />
14. Rahimi, P.M. and Kelly, J.F. "Coprocessinq behaviour <strong>of</strong> Cold Lake<br />
vacuum bottoms and Forestburg subbituminous C <strong>co</strong>al using H2S as a<br />
c<strong>at</strong>alyst"; Division Report ERP/ERL 84-21 (Confidential)<br />
CANMEI,Energy, Mines and Resources Canada, 1984.<br />
15. Rahimi, P.M. and Kelly, J.F. " The use <strong>of</strong> H2S as a promoter in<br />
<strong>co</strong><strong>processing</strong> low rank Canadian <strong>co</strong>als and hitumen";<br />
proceedingsIntern<strong>at</strong>iona1 Conference on Coal Science, Sydney,<br />
Australia, October 1985, pp. 43-46.<br />
16. Potter, J., Kybett, B.D., McDougall, W.M. Nambudiri, E.M.V.<br />
Rahimi, P.M. and Price, J.T. "Petrographic characteriz<strong>at</strong>ion <strong>of</strong> the<br />
solid products <strong>of</strong> <strong>co</strong>al-pitch <strong>co</strong><strong>processing</strong>"; submitted for<br />
public<strong>at</strong>ion in Canadian Mineralogist.<br />
197
Table 1<br />
Analysis <strong>of</strong> Feedstocks<br />
Forestburg Coal Cold-Lake<br />
vacum bottolns<br />
Proxim<strong>at</strong>e analysis Specific gravity, 15/15'C 1.038<br />
(wt %, as received) Pentane insolubles, wt % 23.8<br />
Pitch <strong>co</strong>ntent, wt % 83.2<br />
Moisture 19.7 Conradson Carbon, wt I 17.1<br />
Vol<strong>at</strong>ile 37.2 Elemental ccunposition,wt I<br />
Fixed carbon 36.1 C 83.34<br />
Ash 7.0 H<br />
N<br />
9.69<br />
0.45<br />
Ultim<strong>at</strong>e analysis S 5.84<br />
(wt I DAF) 0 0.68<br />
C 74.34 Metals, ppm<br />
H<br />
N<br />
4.81<br />
1.78<br />
V<br />
Ni<br />
235<br />
93<br />
0 18.58 Fe 18<br />
S 0.49<br />
Table 2<br />
Comparison <strong>of</strong> the Effects <strong>of</strong> H2S w ith FeSO(,<br />
Severity Low Moder<strong>at</strong>e<br />
H2'5 (1) no yes<br />
FeSOq yes no<br />
Distill<strong>at</strong>e yield (2) 22.9 27.3<br />
Coal <strong>co</strong>nversion (3) 53.7 67.9<br />
Pitch <strong>co</strong>nversion (4) 15.8 20.3<br />
(1)<br />
(3)<br />
no Yes<br />
Yes no<br />
36.3 43.2<br />
70.4 80.4<br />
34.1 42.5<br />
8 wt I, based on maf slurry feed (2) w t I, based on maf slurry feed<br />
w t I, based on maf <strong>co</strong>al, (4) maf (+525 "C) in - maf (+525'C) out<br />
defined as THF solubility<br />
maf (+525"C) in<br />
Table 3<br />
Comparison <strong>of</strong> The Effects <strong>of</strong> FeS04 and HpS + FeSO4<br />
-<br />
Severity Very low Low Moder<strong>at</strong>e Moder<strong>at</strong>e-hiqh<br />
HgS (1) no yes no yes no yes no yes<br />
FeSO4 yes yes yes yes yes yes yes yes<br />
Distill<strong>at</strong>e yield (2) 15.8 16.5 22.9 22.9 36.3 42.1 60.9 63.1<br />
Coal <strong>co</strong>nversion (3) 27.4 41.1 53.7 61.3 70.4 83.7 86.3 85.9<br />
Pitch <strong>co</strong>nversion (4) 8.1 7.5 15.8 18.6 34.1 42.4 64.8 69.7<br />
(1) 8 wt %, based on maf slurry feed (2) wt I, based on maf slurry feed<br />
(3) wt %, based on maf <strong>co</strong>al, (4) maf (+525'C) in - maf (+525"C) out<br />
defined as THF solubility maf (+525"C) in<br />
198
Table 4<br />
Distill<strong>at</strong>e Characteristics<br />
Severity Very low - Low Moder<strong>at</strong>e Moder<strong>at</strong>e-hiqh<br />
H2S<br />
FeSOg<br />
API<br />
H/C<br />
N,wt A<br />
S,wt A<br />
0,wt A<br />
- fa<br />
Mn,g/mole<br />
no yes<br />
yes yes<br />
15.2 13.3<br />
1.59 1.53<br />
0.26 0.37<br />
3.15 3.11<br />
0.89 1.30<br />
26<br />
-<br />
31<br />
-<br />
no<br />
Yes<br />
17.0<br />
1.56<br />
0.39<br />
2.98<br />
1.28<br />
29<br />
107<br />
yes yes<br />
no yes<br />
15.9 15.8<br />
1.57 1.54<br />
0.41 0.41<br />
2.A3 2.98<br />
1.23 1.31<br />
28 30<br />
320 322<br />
no<br />
yes<br />
22.4<br />
1.62<br />
0.44<br />
2.30<br />
1.40<br />
25<br />
272<br />
yes yes<br />
no yes<br />
19.8 22.9<br />
1.58 1.63<br />
0.47 0.43<br />
2.44 2.27<br />
1.28 1.02<br />
29 24<br />
305 279<br />
FIGURE 1 RESIDUE COMPOSITION<br />
no<br />
yes<br />
25.4<br />
1.58<br />
0.50<br />
1.64<br />
1.46<br />
30<br />
293<br />
-<br />
-very low low<br />
SEVERITY<br />
mderste mder<strong>at</strong>c-high<br />
199<br />
H2S H2S+FeS04<br />
insoluble!<br />
FeS04<br />
U<br />
0 v)<br />
LL<br />
L<br />
Yes<br />
Yes<br />
25.9<br />
1.62<br />
0.50<br />
1.69<br />
0.85<br />
25<br />
278
THO-STAGE COPROCESSING OF SUBBITUMINOUS COALS AND BITLMEN OR HEAVY OIL*<br />
ABSTRACT<br />
B. Ignasiak, T. Ohuchi, P. Clark, D. Aitchison and T. Lee<br />
Coal Research Department<br />
Alberta Research Council<br />
1 O i l P<strong>at</strong>ch Drive<br />
P.O. Bag #1310<br />
Devon, A1 berta, Canada<br />
TOC 1EO<br />
Pretre<strong>at</strong>ment <strong>of</strong> subbituminous <strong>co</strong>al with an appropri<strong>at</strong>ely formul<strong>at</strong>ed mix <strong>of</strong><br />
carbon monoxide and w<strong>at</strong>er, in presence <strong>of</strong> bitumen or heavy <strong>oil</strong>, results in very<br />
fast reactions characterized by a high degree <strong>of</strong> <strong>co</strong>al solubiliz<strong>at</strong>ion and deoxy-<br />
gen<strong>at</strong>ion. The reaction is c<strong>at</strong>alysed by a mixture <strong>of</strong> alkali metal carbon<strong>at</strong>es<br />
and proceeds readily <strong>at</strong> 380-400OC. The first-stage reaction product appears to<br />
be susceptible to further c<strong>at</strong>alytic hydrogen<strong>at</strong>ion <strong>at</strong> 420-460T with gaseous<br />
hydrogen yielding 65-70% (on daf feed) <strong>of</strong> hydrogen-rich distillable <strong>oil</strong>, <strong>co</strong>m-<br />
posed mainly <strong>of</strong> naphtha and middle <strong>oil</strong>.<br />
The process flowsheet is presented and the <strong>co</strong>mpar<strong>at</strong>ive e<strong>co</strong>nomics <strong>of</strong> two-stage<br />
carbon nonoxide/steam-hydrogen and hydrogen-hydrogen <strong>co</strong><strong>processing</strong> schemes are<br />
discussed.<br />
INTRODUCTION<br />
Alberta is endowed with immense reserves <strong>of</strong> subbituminous <strong>co</strong>als (11, bitumen<br />
and heavy <strong>oil</strong> (2). The <strong>co</strong>ncept <strong>of</strong> <strong>co</strong><strong>processing</strong> <strong>co</strong>al and petroleum derived<br />
solvents is not a new one (3.4) and there is a <strong>co</strong>nsensus th<strong>at</strong> this approach is<br />
more <strong>at</strong>tractive e<strong>co</strong>nomically than <strong>co</strong>nventional <strong>co</strong>al 1 iquefaction (5). The most<br />
<strong>at</strong>tractive fe<strong>at</strong>ure <strong>of</strong> the <strong>co</strong><strong>processing</strong> <strong>co</strong>ncept is its potential for elimin<strong>at</strong>ion<br />
<strong>of</strong> <strong>oil</strong> recycle which may increase the output <strong>of</strong> the install<strong>at</strong>ion by up to three<br />
times.<br />
It has to be emphasized th<strong>at</strong> under Alberta <strong>co</strong>nditions the e<strong>co</strong>nomics <strong>of</strong> a<br />
<strong>co</strong><strong>processing</strong> plant have to be <strong>co</strong>mpared to a heavy <strong>oil</strong> and/or bitumen hydro-<br />
cracking plant. The major advantage <strong>of</strong> <strong>co</strong><strong>processing</strong> as opposed to bitumen or<br />
heavy <strong>oil</strong> hydrocracking is the low <strong>co</strong>st <strong>of</strong> <strong>co</strong>al. This has to be weighed<br />
against the increased hydrogen <strong>co</strong>nsumption, increased plant <strong>co</strong>mplexity<br />
(<strong>co</strong>nversion <strong>of</strong> <strong>co</strong>al to distill<strong>at</strong>e <strong>oil</strong> requires more severe <strong>co</strong>nditions <strong>co</strong>mpared<br />
to bitumen) and the element <strong>of</strong> risk associ<strong>at</strong>ed with implement<strong>at</strong>ion <strong>of</strong> the new<br />
<strong>co</strong><strong>processing</strong> technology.<br />
A factor which may have a substantial effect on the e<strong>co</strong>nomics <strong>of</strong> <strong>co</strong><strong>processing</strong><br />
as <strong>co</strong>mpared to bitumen or heavy <strong>oil</strong> hydrocracking is th<strong>at</strong> <strong>of</strong> purely chemical<br />
n<strong>at</strong>ure. It has not been firmly established whether the interaction among <strong>co</strong>al-<br />
and bitumen-derived radical intermedi<strong>at</strong>es leads to an increase or a reduction<br />
in <strong>oil</strong> yield or its quality.<br />
On the other hand, it has been demonstr<strong>at</strong>ed th<strong>at</strong> hydrocracking <strong>of</strong> bitumen in a<br />
one-stage process in the presence <strong>of</strong> small (1-3% by weight) quantities <strong>of</strong> sub-<br />
bituminous <strong>co</strong>al results in significant improvement in <strong>oil</strong> yield (6). Similar<br />
results can be obtained by employing chars gener<strong>at</strong>ed from brown <strong>co</strong>als (4,7) and<br />
this furnishes a strong evidence th<strong>at</strong> c<strong>at</strong>alytic effects and not the chemistry<br />
<strong>of</strong> the <strong>co</strong>mponents o f the substr<strong>at</strong>e play a dominant role in a one-stage bituen<br />
* To be presented <strong>at</strong> the Fuel Division "Reactions <strong>of</strong> Coal in Novel Systems",<br />
Anaheim ACS Meeting, September 7-12. 1986<br />
200
hydrocracking process with low <strong>co</strong>al <strong>co</strong>ncentr<strong>at</strong>ions. However, the ohjective <strong>of</strong><br />
<strong>co</strong><strong>processing</strong> is to naximize the <strong>co</strong>al <strong>co</strong>ncentr<strong>at</strong>ion in the feedstock without<br />
sacrificing the distillable product yield and quality.<br />
The two-stage process developed <strong>at</strong> Alberta Research Council is based on solubi-<br />
liz<strong>at</strong>ion <strong>of</strong> high oxygen subbituminous <strong>co</strong>al in bitumen (heavy <strong>oil</strong>) using a mix-<br />
ture <strong>of</strong> carbon monoxide/steam <strong>at</strong> 380-409OC in presence <strong>of</strong> alkali metal c<strong>at</strong>a-<br />
lyst. followed by c<strong>at</strong>alytic hydrocracking <strong>at</strong> temper<strong>at</strong>ures <strong>of</strong> 420-460'C and<br />
pressures up to 18.0 MPa.<br />
EXPERIMENTAL<br />
The experiments were carried out in standard b<strong>at</strong>ch autoclave system and in a<br />
hot charge/discharge unit (a system developed for studying two-stage lique-<br />
faction processes).<br />
B<strong>at</strong>ch Autoclave Simul<strong>at</strong>ed Two-Stage Studies<br />
The b<strong>at</strong>ch autoclave experiments were carried out in 1 l itre magnedrive auto-<br />
claves (manufactured by Autoclave Engineers Ltd.) with internal <strong>co</strong>oling <strong>co</strong>ils.<br />
The <strong>co</strong>al/bitumen slurry was charged into an autoclave <strong>at</strong> room temper<strong>at</strong>ure<br />
followed by pressurizing the system with carbon monoxide (5.2 MPa) or hydrogen<br />
(8.3 MPa). The autoclave was he<strong>at</strong>ed up to 390°C. maintained <strong>at</strong> this tempera-<br />
ture for 30 min. and depressurized <strong>at</strong> elev<strong>at</strong>ed temper<strong>at</strong>ures. Gas samples were<br />
analysed uslng a CARLE gas chrom<strong>at</strong>ograph. The se<strong>co</strong>nd stage (hydrogen<strong>at</strong>ion)<br />
c<strong>at</strong>alyst and sulfur additive were then introduced to the <strong>co</strong>ld reactor which was<br />
subsequently repressurized to 8.3 MPa with hydrogen. The reactor was he<strong>at</strong>ed to<br />
440°C and held <strong>at</strong> this temper<strong>at</strong>ure for 60 min. Subsequently, the reactor was<br />
depressurized as before, <strong>co</strong>oled to room temper<strong>at</strong>ure and discharged. The pro-<br />
duct work-up procedure was the same as described before (8).<br />
Hot Charge/Discharge Unit (HCOU)<br />
The HCDU <strong>co</strong>nsists <strong>of</strong> two magnetically stirred reactors <strong>of</strong> one and two litre<br />
capacity and a high pressure vessel to <strong>co</strong>llect the product slurry. The first<br />
reactor oper<strong>at</strong>es in b<strong>at</strong>ch mode and the se<strong>co</strong>nd one in a semi-<strong>co</strong>ntinuous mode.<br />
Details regarding <strong>co</strong>nstruction and oper<strong>at</strong>ion <strong>of</strong> the system were given elsewhere<br />
(8). The product work-up procedure and product analyses were the same as for<br />
b<strong>at</strong>ch autoclave tests.<br />
D I KUSSION<br />
Sufficient evidence has been accumul<strong>at</strong>ed to show th<strong>at</strong> two-stage <strong>co</strong>al liquefac-<br />
tion process yields better results <strong>co</strong>npared to <strong>co</strong>nventional single-stage pro-<br />
cesses (9).<br />
The importance <strong>of</strong> the first (solubiliz<strong>at</strong>ion) stage in the overall liquefaction<br />
process had been ignored until it became evident th<strong>at</strong> depending on the results<br />
<strong>of</strong> the solubiliz<strong>at</strong>ion. the se<strong>co</strong>nd (hydrogen<strong>at</strong>ion) stage proceeds more or less<br />
efficiently. Though no results <strong>of</strong> system<strong>at</strong>ic research on the solubiliz<strong>at</strong>ion-<br />
hydrogen<strong>at</strong>ion rel<strong>at</strong>ionship are available, one can specul<strong>at</strong>e th<strong>at</strong> the mechanism<br />
<strong>of</strong> the initial disintegr<strong>at</strong>ion <strong>of</strong> <strong>co</strong>al and the character and properties <strong>of</strong> the<br />
intermedi<strong>at</strong>e soluble product may have a major influence on the effectiveness <strong>of</strong><br />
the hydrogen<strong>at</strong>ion step.<br />
The influence <strong>of</strong> solubiliz<strong>at</strong>ion <strong>of</strong> low rank <strong>co</strong>als on their hydrogen<strong>at</strong>ion may be<br />
particularly important due to their high oxygen <strong>co</strong>ntent and high reactivity <strong>of</strong><br />
a major fraction <strong>of</strong> this oxygen <strong>at</strong> temper<strong>at</strong>ures significantly below the<br />
hydrogen<strong>at</strong>ion tenper<strong>at</strong>ure. Presence <strong>of</strong> highly reactive oxygen in the <strong>co</strong>al may<br />
201
esult in retrogressive reactions taking place during soluhil iz<strong>at</strong>ion <strong>at</strong> Cig$er<br />
temper<strong>at</strong>ures. Therefore, for Alberta subbituminous <strong>co</strong>als a mixture <strong>of</strong> carbon<br />
monoxide/steam or hydrogen was tested in low temper<strong>at</strong>ure solubiliz<strong>at</strong>ion<br />
studies. The work carried out <strong>at</strong> Alberta Research Council on solubiliz<strong>at</strong>ion <strong>of</strong><br />
indigenous subbituminous <strong>co</strong>als in CO/steam in bitumen and/or heavy <strong>oil</strong> showed<br />
th<strong>at</strong> these <strong>co</strong>als are readily solubilized <strong>at</strong> a low temper<strong>at</strong>ure <strong>of</strong> 38O-40O0C with<br />
<strong>co</strong>nversion 85-96s (10,ll). Although the <strong>co</strong>nversion was ac<strong>co</strong>mpanied by low<br />
hydrocarbon gas gener<strong>at</strong>ion and advanced deoxygen<strong>at</strong>ion for both gases tested<br />
(see Table 1). CO/steam appeared to be superior <strong>co</strong>mpared with hydrogen in terms<br />
<strong>of</strong> reaction kinetics measured as <strong>co</strong>al <strong>co</strong>nversions <strong>at</strong> 390°C (see Table 2).<br />
The susceptibility <strong>of</strong> the <strong>co</strong>al solubilized under mild <strong>co</strong>nditions with either<br />
hydrogen or carbon monoxide/steam to further hydrogen<strong>at</strong>ion in presence <strong>of</strong><br />
potassium molybd<strong>at</strong>e is presented in Table 3.<br />
Analysis <strong>of</strong> the results obtained in simul<strong>at</strong>ed two-stage autoclave experiments<br />
and presented in Table 3 indic<strong>at</strong>es, th<strong>at</strong> in terms <strong>of</strong> distillable <strong>oil</strong> yield the<br />
solubiliz<strong>at</strong>ion <strong>of</strong> <strong>co</strong>al in bitumen in presence <strong>of</strong> CO/H 0-K C03 followed by<br />
c<strong>at</strong>alytic hydrogen<strong>at</strong>ion yields slightly better results c&pa?ed to solubil iz<strong>at</strong>ion<br />
in hydrogen and followed by c<strong>at</strong>alytic hydrogen<strong>at</strong>ion. Furthermore, twostage<br />
<strong>co</strong>-<strong>processing</strong> where solubiliz<strong>at</strong>ion was ac<strong>co</strong>mplished by action <strong>of</strong> either<br />
CO/H 0 or CO/H 0-K CO seems to result in sovewh<strong>at</strong> lower gener<strong>at</strong>ion <strong>of</strong> gaseous<br />
hydr%carbons ibmpaqed to solubiliz<strong>at</strong>ion with hydrogen (Table 3). The <strong>co</strong>al<br />
<strong>co</strong>nversion values are by far the highest (98%) for the sample solubilized using<br />
CO/H20-K,C03.<br />
In <strong>co</strong>nclusion, on the basis <strong>of</strong> autoclave studies the two-stage CO/H 0-K CO<br />
H route appears to be marginally more appealing than the H2-H2 rout6 in’tehn;<br />
03 product yields and <strong>co</strong>nversion.<br />
The Alberta Research Council route requires th<strong>at</strong> CO be used as reducing gas in<br />
the first stage <strong>of</strong> the liquefaction process. It is noteworthy th<strong>at</strong> the<br />
refonning technology for <strong>co</strong>nversion <strong>of</strong> n<strong>at</strong>ural gas (CH to either H or CO is<br />
well known and in both cases is equally efficient in tehs <strong>of</strong> the quahities <strong>of</strong><br />
the reducing gas produced.<br />
CH4 + 2H20 C02 + 4H2 (1)<br />
CH4 + 3C02 ~<br />
4CO<br />
+ 2H20 (2)<br />
The <strong>co</strong>nversion <strong>of</strong> methane to CO instead <strong>of</strong> H is more <strong>at</strong>tractive in view <strong>of</strong> the<br />
elimin<strong>at</strong>ion <strong>of</strong> the demand for w<strong>at</strong>er and tt?e potential for recycling the CO<br />
produced in the first stage <strong>of</strong> the <strong>co</strong><strong>processing</strong>. The disadvantage <strong>of</strong> reformins<br />
with C02 lies in endothermic n<strong>at</strong>ure <strong>of</strong> this reaction and in a need for separa-<br />
tion <strong>of</strong> gases (namely CO, C02 and H2).<br />
The block diagram <strong>of</strong> the <strong>co</strong><strong>processing</strong> plant based on the <strong>co</strong>ncept <strong>of</strong> C0/H20-<br />
K2C03 - H2 reaction is presented in Figure 1.<br />
The Process is <strong>co</strong>mposed <strong>of</strong> three trains: 1) distill<strong>at</strong>ion <strong>of</strong> bitumen and<br />
agglomer<strong>at</strong>ion <strong>of</strong> <strong>co</strong>al; 2) gener<strong>at</strong>ion and separ<strong>at</strong>ion <strong>of</strong> reaction gases; and 3)<br />
sol ubi1 iz<strong>at</strong>i on, hydrogen<strong>at</strong>ion, distil 1 <strong>at</strong>ion and refining <strong>of</strong> vol <strong>at</strong>i 1 e products .<br />
Earlier work showed th<strong>at</strong> bitumen based bridging liquid was very effective in<br />
removal <strong>of</strong> a major portion <strong>of</strong> mineral m<strong>at</strong>ter (particularly silica and clays)<br />
from subbituminous <strong>co</strong>als during their agglomer<strong>at</strong>ion (12). It i s expected th<strong>at</strong><br />
deashing <strong>of</strong> <strong>co</strong>al may have a beneficial influence on liquefaction c<strong>at</strong>alyst per-<br />
formance and resolve the problems associ<strong>at</strong>ed with erosion <strong>of</strong> pressure let-down<br />
valves (13).<br />
202<br />
I ‘~<br />
i<br />
I
The overall mass balance <strong>of</strong> the optimized two-stage CO/H 0-K CO - H <strong>co</strong>al/<br />
bitumen process is presented in Table 4. The soluble extrast (3523°C) &<strong>co</strong>unts<br />
for 12.3% <strong>of</strong> the feedstock (<strong>co</strong>al + bitumen) as <strong>co</strong>mpared with about 19% (see<br />
Table 3) obtained from simul<strong>at</strong>ed two-stage autoclave tests. The reduction in<br />
gener<strong>at</strong>ion <strong>of</strong> extractable m<strong>at</strong>ter was achieved through more advanced hydrogen<strong>at</strong>ion<br />
<strong>co</strong>mpared to autoclave tests. As a result the distillable <strong>oil</strong> yield after<br />
optimiz<strong>at</strong>ion was increased to 70.1% (see Table 5) <strong>co</strong>mpared to 66.2% obtained in<br />
an autoclave (Table 3). Equally important, the process gener<strong>at</strong>es mainly light<br />
(-375OC) <strong>oil</strong>. which ac<strong>co</strong>unts for about 90% <strong>of</strong> total <strong>oil</strong> produced. It is expected<br />
th<strong>at</strong> in a <strong>co</strong>ntinuous oper<strong>at</strong>ion higher yields <strong>of</strong> distillable <strong>oil</strong>s can be ob-<br />
tained.<br />
c<strong>at</strong>alyst should have a major impact on further improvement <strong>of</strong> the ARC process<br />
<strong>co</strong>ncept.<br />
Recently <strong>co</strong>mpleted e<strong>co</strong>nomic feasibility studies on two-stage <strong>co</strong>al/bitumen<br />
<strong>co</strong><strong>processing</strong> (14) indic<strong>at</strong>e th<strong>at</strong> the Alberta Research Council <strong>co</strong>ncept to carry<br />
out the solubiliz<strong>at</strong>ign stage in CO/steam <strong>at</strong>mosphere adds about $100 million to<br />
the <strong>co</strong>st <strong>of</strong> the <strong>co</strong><strong>processing</strong> plant and this ac<strong>co</strong>unts for approxim<strong>at</strong>ely 8% <strong>of</strong><br />
total plant <strong>co</strong>st. However, when the feasibility studies were <strong>co</strong>mpleted (early<br />
1985) the d<strong>at</strong>a indic<strong>at</strong>ing th<strong>at</strong> the CO/steam-K CO solubiliz<strong>at</strong>ion results in<br />
higher yield <strong>of</strong> distillable <strong>oil</strong>s <strong>co</strong>mpared to hy&og%n solubiliz<strong>at</strong>ion (see Table<br />
3) were not available. It is noteworthy th<strong>at</strong> 4% higher <strong>oil</strong> yield in plant<br />
production <strong>co</strong>uld readily <strong>of</strong>fset the additional <strong>co</strong>st associ<strong>at</strong>ed with CO/steam<br />
solubiliz<strong>at</strong>ion. Furthermore, there are other factors (like reaction kinetics)<br />
which seem to favor CO/steam solubiliz<strong>at</strong>ion and which do not seem to be fully<br />
ac<strong>co</strong>unted for in the feasibility studies.<br />
Under the circumstances it is <strong>co</strong>ncluded th<strong>at</strong> there is a need for further<br />
verific<strong>at</strong>ion <strong>of</strong> the effectiveness <strong>of</strong> the CO/steam-K CO versus H solubiliz-<br />
<strong>at</strong>ion. It is essential to carry out <strong>co</strong>ntinuous two-s?ag$ <strong>co</strong>al/bit&en tests in<br />
both (CO/steam-K CO - H versus H -H modes in order to obtain more reliable<br />
yield d<strong>at</strong>a and cbnv&sior? values f& Aonomic analysis.<br />
ACKNOULEDGEUENT<br />
The authors would like to thank Mr. C. Gietz and Dr. S. Chakrabartty for<br />
reviewing the manuscript.<br />
REFERENCES<br />
Progress in development <strong>of</strong> an active, inexpensive and disposable<br />
1. Reserves <strong>of</strong> Coal, Province <strong>of</strong> Alberta, December 31, 1980, Energy Resources<br />
Conserv<strong>at</strong>ion Board.<br />
2. Alberta's Reserves <strong>of</strong> Crude Oil, O i l Sands, Gas, N<strong>at</strong>ural Gas Liquids and<br />
Sulphur, December 31, 1983. Energy Resources Conserv<strong>at</strong>ion Board Report No.<br />
84-18.<br />
3. E. Boomer and A. Saddington, Canadian Journal <strong>of</strong> Research, 1935, 2, p.<br />
825.<br />
4. J. Varga, J. Karolyi, Gy. Rabo, P. Steingaszner, A. Szekely and A. Zalay,<br />
Petroleum Refiner, 1957, Sept., p. 198.<br />
5. United St<strong>at</strong>es P<strong>at</strong>ent. 4,330,393, May 18. 1982.<br />
6. Canadian P<strong>at</strong>ent, 269,020, December 31. 1976.<br />
7. J. Varga, J. Karolyi, P. Steingasmer. A. Zalay, R. Birthler and Gy. Rabo,<br />
Petroleum Refiner, 1960, April, p. 182.<br />
203
8. 6. Ignasiak, L. Lewkowicz, G. Kovacik, T. Ohuchi and 14.P. du Plessis,<br />
5. Kaliaguine and A. Makay (Editors), C<strong>at</strong>alysis on the Energy Scene, 1984<br />
Elsevier Science Publishers B.V., Amsterdam.<br />
9. J.H. Shinn, A.J. Dahlberg, C.W. Kuehler. J.W. Rosenthal Proceedings; Ninth<br />
Annual EPRI Contractor's Conference on Coal <strong>Liquefaction</strong>, EPRI AP-3825-SR,<br />
March 1985, p. 33-1.<br />
10. M. Gawlak, D. Carson, 14. Holuszko and D. Vernon, Report No. YCLQ-7, April<br />
1982, Alberta Research Council, Edmonton, Canada.<br />
11. G. Kovacik. P.D. Clark, and T. Ohuchi; Studies on Co<strong>processing</strong> Highvale<br />
Coal and Bitumen, ENR-ARC Coal Conversion Research Program Final Reports<br />
for 1984/85, Volume I, June 27, 1985, Alberta Research Council, Edmonton,<br />
Canada.<br />
12. W. Pawlak, J. Janiak. K. Szymocha, A. Turak and B. Ignasiak; Pipeline<br />
Agglomer<strong>at</strong>ion <strong>of</strong> Coal, Final Report for the Alberta/Canada Energy Resources<br />
Research Fund, July, 1984, Alberta Research Council, Edmonton, Canada.<br />
13. G.J. Perry, Coal Corpor<strong>at</strong>ion <strong>of</strong> Victoria - personal <strong>co</strong>mmunic<strong>at</strong>ion.<br />
14. CoallHeavy Oil Hydrogen<strong>at</strong>ion Plant Feasibility Study, Volume 11, Cost<br />
Estim<strong>at</strong>e and Financial Evalu<strong>at</strong>ion, April 1985, Prepared for Contar Systems<br />
Engineering Ltd. by Kilborn Kellogg Rust Ltd.<br />
Table 1<br />
DEOXYGENATION OF THE FEEDSTOCK (COAL AND BITUMEN) AND<br />
HYDROCARBON GAS YIELDS ON SOLUBILIZATION OF HIGHVALE COAL<br />
I N BITUMEN USING CO/STEAM OR He AT 390°C<br />
Hydrocagbon Gas Yield<br />
(C1-C4) (XI<br />
CO/Steam H,<br />
1 .o 0.9<br />
Deoxygen<strong>at</strong>ionb (%) 86 94<br />
a) on daf feedstock<br />
b) defined as: _.<br />
- 0 in (distillable + extractable) products<br />
0 in (<strong>co</strong>al + bitumen) feedstock<br />
204
2nd Stage C<strong>at</strong>alyst<br />
Table 2<br />
THE EFFECT OF RESIDENCE TIME ON<br />
CONVERSION FOR THE SOLUBILIZATION OF<br />
HIGHVALE COAL IN BITUMEN WITH<br />
CO/STEAM OR H2 AT 390°C<br />
Time (min') Coal Conversion<br />
(on daf <strong>co</strong>al )<br />
0<br />
15<br />
30<br />
60<br />
CO/Steam<br />
Table 3<br />
H7<br />
58 10<br />
65 34<br />
85 65<br />
86 68<br />
PRODUCT YIELDS AND COAL CONVERSIONS FROM THE<br />
TWO-STAGE CO-PROCESSING OF BITUMEN AND HIGHVALE COAL<br />
K2Mo04 - CH3SSCH3<br />
1st Stage Reducing Gas H2 CO/H20 CO/H20-K2C03<br />
- Yielda<br />
Hydrocarbon Gas (C,-C5) 7.2 (20.8) 5.2(+0.2) 5.3c<br />
Distillable Oil (IBP-525"C) 62.3(+0.9) 57.7 (+0.8 ) 66.2 (20.7 )<br />
Soluble Extract 18.0 (20. 2 ) 19.3 (20. 9) 18.7 (+O .3 )<br />
M<strong>at</strong>erial Balance 94.1 (23.1 ) 90.4 (20.2) 96.1'<br />
Coal Conversion (X daf <strong>co</strong>al) 90 (21 ) 91 (21) 98 (20 1<br />
a) Yields are presented as % daf organic feed (bitumen + <strong>co</strong>al).<br />
b) All d<strong>at</strong>a are quoted as the average values <strong>of</strong> two duplic<strong>at</strong>e experiments.<br />
Figures in brackets show the spreads for the two experiments.<br />
c) Single d<strong>at</strong>a point.<br />
205<br />
~
Table 4<br />
OVERALL MASS BALANCE FOR THE OPTIMIZED ARC TWO-STAGE<br />
CO/H2O-K2CO3 - H2 COALDITUMEN PROCESS<br />
Bitumen Coal - 2.5/1<br />
Basis 100 kg - feed (daf)<br />
Component Input output<br />
c5-2000c 3.1 11.0<br />
200-375OC 15.0 40.1<br />
375-525OC 16.1 19.0(2)<br />
+ 525OC 37.2 12.3<br />
Coal 28.6<br />
Ash 1.5(1)<br />
Un<strong>co</strong>nverted Coal<br />
Residue<br />
W<strong>at</strong>er<br />
<strong>co</strong><br />
c02<br />
10.26<br />
0.6<br />
26.1<br />
H2S<br />
NH4<br />
'1-'4<br />
C<strong>at</strong>alyst 0.34<br />
2.7<br />
1.84<br />
44.7<br />
1.5<br />
0.46<br />
5.2<br />
138.8 138.8<br />
(1) Ash reduced to 5% by deashing.<br />
(2) Estim<strong>at</strong>e <strong>of</strong> yield after optimiz<strong>at</strong>ion.<br />
206
GAS GENERATION<br />
AND SEPARATION<br />
COAL<br />
SOLUBILIZATION<br />
HYDROGENATION<br />
DISTILLATION<br />
AND<br />
AGGLOMERATION<br />
SteaE (Optional) N<strong>at</strong>ural Gas<br />
<strong>co</strong> Rich<br />
Bi twn Bi tmen<br />
(andlor<br />
(andlor<br />
heavy <strong>of</strong>1 1<br />
Coal<br />
heavy oi 1 )<br />
Recycle M<strong>at</strong>er<br />
Figure 1: Block Diagram <strong>of</strong> the ARC Two-Stage CO/Steam-K2C03 - H2<br />
Coal/Bitumen Process Concept<br />
207
BACKGROUND<br />
COAL LIQUEFACTION/RESID HYDROCRACKING<br />
VI A THO - STAGE INTEGRATED CO- PROCESS I NG<br />
Marvin Greene<br />
Avinash Gupta<br />
William Moon<br />
LUMMUS CREST INC.<br />
1515 BROAD STREET<br />
BLOOMFIELD, NEW JERSEY 07003<br />
Lummus Crest Inc. (LCI), a subsidiary <strong>of</strong> Combustion Engineering Inc., has been<br />
developing technology for the simultaneous <strong>processing</strong> <strong>of</strong> <strong>co</strong>al and heavy petroleum<br />
liquids under a joint development <strong>co</strong>ntract with the U. S. Department <strong>of</strong> Energy.<br />
The LCI <strong>co</strong>-<strong>processing</strong> route is an outgrowth <strong>of</strong> its Integr<strong>at</strong>ed Two-Stage<br />
<strong>Liquefaction</strong> (ITSL) technology developed over the past decade by LCI for <strong>co</strong>al<br />
liquefaction. A 33-month R&D <strong>co</strong>ntract was initi<strong>at</strong>ed in October 1984 with the<br />
objective <strong>of</strong> determining the technical and e<strong>co</strong>nomic feasibility <strong>of</strong> <strong>co</strong>al<br />
liquefaction via the LCI <strong>co</strong>-<strong>processing</strong> route.<br />
The project was formul<strong>at</strong>ed into five major program tasks as follows:<br />
Task 1: Project Management Plan<br />
Task 2: Feedstock Analysis<br />
Task 3: Co-Processing Reactivity Screening<br />
Task 4: Continuous Bench-Scale Oper<strong>at</strong>ions<br />
Task 5: Cost Estim<strong>at</strong>e <strong>of</strong> Conceptual Commercial Facility<br />
The first three tasks have been <strong>co</strong>mpleted and the <strong>co</strong>ntinuous Bench-Scale<br />
Oper<strong>at</strong>ions task has recently been initi<strong>at</strong>ed. The balance <strong>of</strong> this paper will<br />
describe experimental methods, the LCI <strong>co</strong>-<strong>processing</strong> approach and the results <strong>of</strong><br />
recent bench-scale unit oper<strong>at</strong>ions.<br />
SOME JUSTIFICATIONS FOR CO-PROCESSING<br />
Since <strong>co</strong>-<strong>processing</strong> inherently requires two separ<strong>at</strong>e feedstocks, namely <strong>co</strong>al and<br />
petroleum resid, it is possible to assess any potential process advantages from<br />
two viewpoints. From the refiner's viewpoint, the arom<strong>at</strong>ic-rich, <strong>co</strong>al-derived<br />
extracts, being well known hydrogen donor solvents, can improve the<br />
hydro<strong>processing</strong> <strong>co</strong>nversion <strong>of</strong> heavy, low grade petroleum feedstocks. On a<br />
<strong>co</strong>nstant energy <strong>co</strong>st basis, the syncrude <strong>co</strong>st <strong>co</strong>ntribution from a <strong>co</strong>al feedstock<br />
may be less than th<strong>at</strong> from a petroleum feedstock.<br />
For example, if one assumes a typical net syncrude yield <strong>of</strong> 0.0005 M3/Kg (3.0<br />
bbl/ton) for a run-<strong>of</strong>-mine bituminous <strong>co</strong>al priced <strong>at</strong> $0.033[5g ($30/ton), then the<br />
<strong>co</strong>al feedstock <strong>co</strong>st <strong>of</strong> the <strong>co</strong>al syncrude is about f62/M (SlO.OO/bbl). This<br />
<strong>co</strong>mpares to petroleum cfude prices, even under the current suppressed spot market,<br />
in excess <strong>of</strong> $74-100/M ($12-16/bbl). The situ<strong>at</strong>ion is even more pronounced in<br />
the case <strong>of</strong> a typical subbituminous, <strong>co</strong>al. Although the net yield <strong>of</strong> liq9ds from<br />
subbitumjnous <strong>co</strong>al is lower than 'th<strong>at</strong> from bituminous <strong>co</strong>al (0.0003 M /Kg vs.<br />
0.0005 M /Kg for bituminous), the <strong>co</strong>rresponding subbituminous R.O.M. <strong>co</strong>al <strong>co</strong>st is<br />
not proportion<strong>at</strong>ely lower but r<strong>at</strong>her about 73 percent lower than th<strong>at</strong> <strong>of</strong> the<br />
bituminous <strong>co</strong>al ($O.OOSS/Kg vs. $0.033/Kg). This translyes to a subbituminous<br />
Coal feedstock <strong>co</strong>st <strong>of</strong> the <strong>co</strong>al liquids <strong>of</strong> about $32.6/M ($5.20/bbl). In both<br />
cases, it is envisioned th<strong>at</strong> a rel<strong>at</strong>ively low level <strong>of</strong> <strong>co</strong>al-derived liquids would<br />
be blended with petroleum resid feedstock so as not to gre<strong>at</strong>ly alter the<br />
downstream refinery processability <strong>of</strong> the petroleum-<strong>co</strong>al liquid mixtures.<br />
208
I<br />
From a <strong>co</strong>al liquefaction plant owner’s viewpoint, the introduction <strong>of</strong> petroleum<br />
resid allows for a reduction in the large and <strong>co</strong>stly solvent recycle systems.<br />
Additional advantages include:<br />
o Reduces net hydrogen <strong>co</strong>nsumption and <strong>co</strong>rrespondingly reduced hydrogen<br />
production <strong>co</strong>sts;<br />
o Avoids the need for a <strong>co</strong>stly deashing step;<br />
o Provides for a more rapid introduction <strong>of</strong> <strong>co</strong>al into the domestic energy<br />
networks; and<br />
o Allows the <strong>co</strong>nsider<strong>at</strong>ion <strong>of</strong> smaller-scale, less capital intensive plant<br />
sizes in an over-the-fence <strong>co</strong>ncept r<strong>at</strong>her than in a grass-roots,<br />
mega-project <strong>co</strong>ncept.<br />
LCI CO-PROCESSING APPROACH<br />
Of the three key <strong>co</strong>-<strong>processing</strong> routes - thermal, thermoc<strong>at</strong>alytic, biochemical -<br />
the LCI approach represents a hybrid <strong>of</strong> the first two in th<strong>at</strong> it <strong>co</strong>nsists <strong>of</strong> a<br />
two-stage method. The first-stage is a thermal reaction system paralleling the<br />
Short Contact Time (SCT) reaction system developed for LCI’s ITSL Process. The<br />
se<strong>co</strong>nd-stage <strong>co</strong>nsists <strong>of</strong> a c<strong>at</strong>alytic reac&on system based on LCI’s proprietary<br />
expanded-bed &,chnology known as LC-Fining . The SCT reactor is close-<strong>co</strong>upled to<br />
the LC-Fipang reactor to allow for rapid stabiliz<strong>at</strong>ion <strong>of</strong> <strong>co</strong>al extracts by the<br />
LC-Fining c<strong>at</strong>alyst thereby minimizing undesirable free radical <strong>co</strong>ndens<strong>at</strong>ion<br />
reactions.<br />
Figures 1 and 2 show two altern<strong>at</strong>ive flowschemes depending upon the source and<br />
type <strong>of</strong> the petroleum resid. The scheme shown in Figure 1 is predic<strong>at</strong>ed on the<br />
use <strong>of</strong> a heavy refinery stream such as the un<strong>co</strong>nverted resid from a c<strong>at</strong>alytic<br />
hydrocracker. In this scheme, the petroleum feedstock is blended with the <strong>co</strong>al I<br />
and a recycle gas <strong>oil</strong> stream prior to the first-stage, SCT thermal reactor.<br />
The scheme shown in Figure 2 is predic<strong>at</strong>fd on the use <strong>of</strong> a virgin vacuum residua<br />
which is fed directly into the LC-F’ er along with the SCT <strong>co</strong>al extract. The<br />
first-stage reactor <strong>of</strong> the LC-Finer” can be oper<strong>at</strong>ed to simultaneously optimize<br />
the production <strong>of</strong> a) a donor solvent-rich gas <strong>oil</strong> recycle stream; b) an<br />
un<strong>co</strong>nverted but hydrotre<strong>at</strong>ed rehycle reskd stream having improved solvency for<br />
<strong>co</strong>al; and c) hydrocracked C5-524 C (C5-975 F) distill<strong>at</strong>es.<br />
EXPERIMENTAL APPROACH<br />
The experimental approach to obtaining key process d<strong>at</strong>a required for the<br />
preliminary design and estim<strong>at</strong>e <strong>of</strong> a <strong>co</strong>nceptual <strong>co</strong>mmercial facility has been<br />
ac<strong>co</strong>mplished in a variety <strong>of</strong> test units. Initial work for screening candid<strong>at</strong>e<br />
<strong>co</strong>al and petroleum feedstocks was carried out in microautoclave reaction systems<br />
shown schem<strong>at</strong>ically in Figure 3. This was followed by testing in a <strong>co</strong>ntinuous,<br />
close-<strong>co</strong>upled test unit under once-through <strong>co</strong>nditions utilizing solvents<br />
characteristic in <strong>co</strong>mposition to wh<strong>at</strong> is expected <strong>at</strong> steady-st<strong>at</strong>e, but<br />
synthetically gener<strong>at</strong>ed. The test unit, shown schem<strong>at</strong>ically in Figure 4, <strong>co</strong>nsists<br />
<strong>of</strong> an SCT reaction system <strong>co</strong>mprised <strong>of</strong> a 6.2 mm i.d. by 343 cm long horizontal<br />
<strong>co</strong>il he<strong>at</strong>er followed by a vertical SCT reactor having a volume <strong>of</strong> 118 cc. The SCT<br />
(sm) LC-Fining is a service mark <strong>of</strong> Lumus Crest Inc. for engineering, marketing<br />
and technical services rel<strong>at</strong>ed to hydrocracking and hydrodesulfuriz<strong>at</strong>ion processes<br />
for reduced crude and residual <strong>oil</strong>s.<br />
209
system, which <strong>co</strong>uld be oper<strong>at</strong>ed with either SCT reactors described above, was<br />
close-<strong>co</strong>upled to a stirred autoclave c<strong>at</strong>alytic hydrocracker. The <strong>co</strong>ntinuous<br />
bench-scale unit being oper<strong>at</strong>ed in Task 4 <strong>co</strong>nsists <strong>of</strong> the above SEA reactor<br />
systems close-<strong>co</strong>up1 ed to a small diameter, expanded-bed LC-Fining reactor<br />
system. This unit can oper<strong>at</strong>e in a recycle mo& which <strong>co</strong>nsists <strong>of</strong> b<strong>at</strong>ch<br />
<strong>co</strong>llection <strong>of</strong> hydrotre<strong>at</strong>ed 1 iquids from the LC-Finer followed by b<strong>at</strong>ch slurryiQg<br />
<strong>of</strong> the <strong>co</strong>al and the thus <strong>co</strong>llected recycle liquids. Both SCT and LC-Finer<br />
reactors are oper<strong>at</strong>ed <strong>co</strong>ntinuously.<br />
RESULTS OF BENCH-SCALE TESTING<br />
Process variables studies, carried out in the <strong>co</strong>ntinuous close-<strong>co</strong>upled test units,<br />
have recently been <strong>co</strong>mpletfd. The bulk <strong>of</strong> the test scans <strong>co</strong>ncentr<strong>at</strong>ed on s<strong>co</strong>uting<br />
<strong>of</strong> both SCT and LC-Fining reaction <strong>co</strong>nditions close to th<strong>at</strong> <strong>of</strong> the anticip<strong>at</strong>ed<br />
region <strong>of</strong> preferred <strong>co</strong>mmercial oper<strong>at</strong>ions. Tests were made with two<br />
<strong>co</strong>al -petroleum <strong>co</strong>mbin<strong>at</strong>ions th<strong>at</strong> were selected from the analysis <strong>of</strong> the<br />
microautoclave test d<strong>at</strong>a. These <strong>co</strong>mbin<strong>at</strong>ions <strong>co</strong>nsisted <strong>of</strong> a bituminuous <strong>co</strong>al -<br />
Pittsburgh seam with Athabasca vacuum resid - and a subbituminous <strong>co</strong>al - Wyodak<br />
with Arab Heavy vacuum resid. Dughng the scans, two sets <strong>of</strong> d<strong>at</strong>a were obtained<br />
indic<strong>at</strong>ing the effect <strong>of</strong> LC-Fining reactor temper<strong>at</strong>ure and solvent/<strong>co</strong>al r<strong>at</strong>io on<br />
<strong>co</strong>-<strong>processing</strong> performance.<br />
EFFECT OF TEMPERATURE<br />
The impact <strong>of</strong> LC-FiningSm temper<strong>at</strong>ure <strong>at</strong> <strong>co</strong>nstant SCT reaction <strong>co</strong>nditions is shown<br />
in Table 1. From these d<strong>at</strong>a, we have calcul<strong>at</strong>ed pseudo-kinetic r<strong>at</strong>e <strong>co</strong>nstants for<br />
each <strong>of</strong> the performance criteria based on simplified kinetic models. These values<br />
are indic<strong>at</strong>ed below:<br />
Co-Processinq (SCT & LC-F) Performance Parameter Pseudo-R<strong>at</strong>e Constant, hr-I<br />
Desulfuriz<strong>at</strong>ion<br />
Demetalliz<strong>at</strong>ion<br />
524Ct Conversion<br />
Denitrogen<strong>at</strong>ion<br />
- 404C - 416C __ 432C<br />
0.75 1.09 1.67<br />
0.55 0.90 1.12<br />
0.54 0.72 1.11<br />
0.29 0.43 0.90<br />
The feedstock in all tests <strong>co</strong>nsisted <strong>of</strong> 25 percent <strong>co</strong>a1/37.5 percent hydrotre<strong>at</strong>ed<br />
petroleum resid/37.5 percent <strong>co</strong>al -derived gas <strong>oil</strong>. The order <strong>of</strong> reactivity as<br />
measured by the specific <strong>co</strong>-<strong>processing</strong> performance parameters in descending order<br />
is:<br />
Desulfuriz<strong>at</strong>ion > Demetall iz<strong>at</strong>ion > Conversion > Deni trogen<strong>at</strong>ion<br />
It is also interesting to note th<strong>at</strong> in all thrfie tests, the preasphaltene <strong>co</strong>ntent<br />
in the product <strong>of</strong> the single-stage LC-Finer has been reduced to less than 2<br />
percent.<br />
EFFECT OF SOLVENT QUALITY AND SOLVENT/COAL RATIO<br />
The impa$m <strong>of</strong> solvent/<strong>co</strong>al r<strong>at</strong>io and solvent quality <strong>at</strong> <strong>co</strong>nstant SCT and<br />
LC-Fining reaction <strong>co</strong>nditions is shown in Table 2. The following interesting<br />
observ<strong>at</strong>ions have been made based on the d<strong>at</strong>a shown:<br />
0 At cgnstant <strong>co</strong>al slurry <strong>co</strong>ncentr<strong>at</strong>ion, reducing the r<strong>at</strong>io <strong>of</strong> the solvent<br />
(524 C- b<strong>oil</strong>ing range) to <strong>co</strong>al from 1.5/1 to 1.0/1 had a minimal effect<br />
on observed <strong>co</strong>al <strong>co</strong>nversions.<br />
210
i<br />
0 However, the reduction in the said solvent/<strong>co</strong>al r<strong>at</strong>io did adversely<br />
affect syncrude characteristics and net distill<strong>at</strong>e yields.<br />
It should be kept in mind th<strong>at</strong> the high LC-FiningSm severity used in this<br />
particular test campaign was aimed <strong>at</strong> assessing per pass performance (e.g.,<br />
<strong>co</strong>nversion, desulfuriz<strong>at</strong>ion, etc.) 1 imits during <strong>co</strong>-<strong>processing</strong>. In the <strong>co</strong>nceptual<br />
<strong>co</strong>mmercial <strong>co</strong>ncept Qgpicted in Figures 1 and 2, it is anticip<strong>at</strong>ed th<strong>at</strong> the<br />
first-stage LC-Finer loc<strong>at</strong>ed immedi<strong>at</strong>ely downstream <strong>of</strong> the SCT reactor will be<br />
oper<strong>at</strong>ed under optimal <strong>co</strong>nditions for gener<strong>at</strong>ing recycle gas <strong>oil</strong> solvents having<br />
higb hydrogen donor capacity. At these severities, it is anticip<strong>at</strong>ed th<strong>at</strong> the<br />
524 Ct <strong>co</strong>nversions would be lower than the values shown in Table 4. The<br />
additional required feed <strong>co</strong>nversions, i .e., overall swversions in exce~ss <strong>of</strong> 80<br />
percent, <strong>co</strong>uld be achieved in a se<strong>co</strong>nd-stage LC-Finer oper<strong>at</strong>ed <strong>at</strong> higher, more<br />
<strong>co</strong>nventional petroleum hydrocracking severities.<br />
In test BSCL-20, a 524OC- recycle solvent during <strong>co</strong>-<strong>processing</strong> was simul<strong>at</strong>ed by<br />
blending a ne<strong>at</strong> <strong>co</strong>al-derived hydrogen<strong>at</strong>ed solvent with a ne<strong>at</strong> petroleum-derived<br />
hydrogen<strong>at</strong>ed solvent in the same r<strong>at</strong>io as th<strong>at</strong> <strong>of</strong> the <strong>co</strong>al and petroleum resid fed<br />
in th<strong>at</strong> run. By <strong>co</strong>mparing these results to those <strong>of</strong> test BSCL-9 made only with<br />
<strong>co</strong>al-derived 524OC- solvent, it is possible to estim<strong>at</strong>e the rel<strong>at</strong>ive solvent<br />
quality index (SQI) <strong>of</strong> the petroleum-derived gas <strong>oil</strong> solvent in <strong>co</strong>mparison to th<strong>at</strong><br />
<strong>of</strong> the <strong>co</strong>al-derived gas <strong>oil</strong> solvent. The SQI <strong>of</strong> the particular petroleum gas <strong>oil</strong><br />
utilized in this test has been estim<strong>at</strong>ed to be about 60 percent <strong>of</strong> th<strong>at</strong> <strong>of</strong> the<br />
<strong>co</strong>al -derived solvent based on rel<strong>at</strong>ive solvent performance as measured by observed<br />
net distill<strong>at</strong>e yields. Optimiz<strong>at</strong>ion <strong>of</strong> the petroleum resid hydrocracking step in<br />
which the simul<strong>at</strong>ed gas <strong>oil</strong> solvent was gener<strong>at</strong>ed has the potential to increase<br />
the l<strong>at</strong>ter's SQI value closer to th<strong>at</strong> <strong>of</strong> the <strong>co</strong>al-based solvent.<br />
FUTURE WORK<br />
The <strong>co</strong>ntinuous bench-scale unit will be oper<strong>at</strong>ed in the recycle mode to<br />
demonstr<strong>at</strong>e the effect <strong>of</strong> solvent maintenance on <strong>co</strong>-<strong>processing</strong> performance as a<br />
function <strong>of</strong> feedstock types (Pittsburgh seam and Wyodak <strong>co</strong>als; Arab Heavy and<br />
Athabasca residua); <strong>co</strong>al/resid r<strong>at</strong>ios; solvent/<strong>co</strong>al r<strong>at</strong>io and c<strong>at</strong>alyst age. These<br />
d<strong>at</strong>a will serve as the basis for formul<strong>at</strong>ing a <strong>co</strong>nceptual <strong>co</strong>mmercial plant design.<br />
Towards the end <strong>of</strong> the Task 4 experimental program, a c<strong>at</strong>alyst life test <strong>at</strong><br />
preferred <strong>co</strong>-<strong>processing</strong> <strong>co</strong>nditions will be made to demonstr<strong>at</strong>e the technical<br />
feasibility <strong>of</strong> the base case design.<br />
ACKNOWLEDGEMENTS<br />
The LCI <strong>co</strong>-<strong>processing</strong> program is being jointly funded by the Department <strong>of</strong> Energy<br />
under Contract No. DE-AC22-84PC70042. We would like to acknowledge the technical<br />
support <strong>of</strong> Ness Mazzoc<strong>co</strong>, DOE Project Manager; Dr. Eneo Moroni, DOE Program<br />
Manager; and Dr. Harvey Schindler, Science Applic<strong>at</strong>ions Intern<strong>at</strong>ional Corpor<strong>at</strong>ion.<br />
211
TABLE 1<br />
EFFECT OF LC-FINING~"' TEMPERATURE ON CLOSE-COUPLED CO-PROCESSING PERFORMANCE<br />
RunNo.<br />
(8SCL-)<br />
1. Test Conditions<br />
SCT Tempeg,ture, OC (OF)<br />
LC-Fining Temper<strong>at</strong>ure, OC (OF)<br />
Coal Space Velocity to SCT, Kg/Hr/M3<br />
Solvent/Coal Ut. R<strong>at</strong>io<br />
Resid/Coal Wt. R<strong>at</strong>io<br />
11. Test Results<br />
SUMMARY OF TEST CONDITIONS AND RESULTS<br />
Coal Conversion. Wt.% MAF<br />
524OCt (975Ft) Conversion, W8.X<br />
Net Distill<strong>at</strong>e Yield, Kg 524 C-/lo0 Kg 524OC+<br />
Desulfuriz<strong>at</strong>ion, %<br />
Denitrogen<strong>at</strong>ion, %<br />
Resid Demetalliz<strong>at</strong>ion, %<br />
Preasphaltenes Concentr<strong>at</strong>ion, %<br />
- 9<br />
449 (840)<br />
432(810)<br />
3400<br />
1.5<br />
1.5<br />
96.0<br />
75.8<br />
58.1<br />
82.5<br />
71.7<br />
76.0<br />
1.7<br />
- 10<br />
449(840)<br />
416 (780)<br />
3400<br />
1.5<br />
1.5<br />
93.8<br />
67.3<br />
55.9<br />
75.7<br />
55.0<br />
72.0<br />
1.5<br />
- 11<br />
449(840)<br />
404(760)<br />
3400<br />
1.5<br />
1.5<br />
93.7<br />
58.4<br />
48.3<br />
66.0<br />
43.0<br />
59.0<br />
1.9<br />
(1) Feedstocks: Pittsburgh seam <strong>co</strong>al ; Prehydrotre<strong>at</strong>ed 524OC+ Athabasca resid<br />
(2) Solvent: 524OC- gas <strong>oil</strong> characteristic <strong>of</strong> a <strong>co</strong>al-derived recycle<br />
solvent produced in LCI's ITSL PDU during Wyodak <strong>co</strong>al<br />
oper<strong>at</strong>ions<br />
(3) SCT Condition:<br />
137 <strong>at</strong>m; 360 M3 H2/M3 feed<br />
(4) LC-FininqSm<br />
Conditions: Shell 324M c<strong>at</strong>alyst; 137 <strong>at</strong>m; 0.4R hr-'; 530 M3 H,/M3 feed<br />
212
Run No.<br />
(BSCL-)<br />
TABLE 2<br />
EFFECT OF SOLVENT OUALITY AN0 SOLVENT/COAL RATIO ON CLOSE-COUPLE0<br />
CO-PROCESSING PERFORMANCE<br />
I. Test Conditions<br />
Solvent/Coal Ut. R<strong>at</strong>io<br />
Coal/Resid Ut. R<strong>at</strong>io<br />
Coal Slurry Concentr<strong>at</strong>ion, Ut.%<br />
524OC- Solvent Comoosition. Ut.%<br />
ITSL*<br />
Petroleum Gas Oil**<br />
SCT Tempejdture, OC (OF)<br />
LC-Fining Temper<strong>at</strong>ure, OC (OF)<br />
Coal Space Velocity to SCT, Kg/Hr/M3<br />
11. Test Results<br />
SUMMARY OF TEST CONDITIONS AND RESULTS<br />
Coah Conversion, Wt.% MAF<br />
524 Ct (975Ft) Conversion, Wh.%<br />
Net Distill<strong>at</strong>e Yield, Kg 524 C-/lo0 Kg 524OCt<br />
Desul furiz<strong>at</strong>ion, %<br />
Denitrogen<strong>at</strong>ion, %<br />
Resid Demetalliz<strong>at</strong>ion, %<br />
Preasphaltenes Concentr<strong>at</strong>ion, %<br />
*<br />
**<br />
1.5<br />
0.67<br />
25.0<br />
- 9 - 21 - 20<br />
100<br />
0<br />
449 (840)<br />
432(810)<br />
3400<br />
96.0<br />
75.8<br />
58.1<br />
82.5<br />
71.7<br />
76.0<br />
1.7<br />
1.0<br />
0.50<br />
25.0<br />
100<br />
0<br />
449(840)<br />
432(810)<br />
3400<br />
93.4<br />
66.2<br />
51.0<br />
75.0<br />
65.0<br />
69.0<br />
2.2<br />
1.5<br />
0.67<br />
25.0<br />
40<br />
60<br />
449(840)<br />
432(810)<br />
3400<br />
94.2<br />
64.2<br />
46.0<br />
77.2<br />
67.1<br />
71.0<br />
2.0<br />
524OC- gas <strong>oil</strong> characteristic <strong>of</strong> a <strong>co</strong>al-derived recycle solvent produced in<br />
LCI's ITSL PDU during Wyodak <strong>co</strong>al oper<strong>at</strong>ions.<br />
524OC- gas <strong>oil</strong> cQgracteristic <strong>of</strong> a petroleum-derived recycle solvent produced<br />
during LC-Fining <strong>of</strong> virgin Athabasca bitumen.<br />
1. Feedstocks: Pittsburgh seam <strong>co</strong>al ; prehydrotre<strong>at</strong>ed 524OCt<br />
Athabasca resjd<br />
2. SCT Conditims: 137 <strong>at</strong>m; 360 M H2/M3 feed<br />
3. LC-Fininq"<br />
Conditions: Shell 324M c<strong>at</strong>alyst; 137 <strong>at</strong>m; 0.4R Hr-'; 530 M3 H2/M3 feed<br />
213
Figure 1. SCHEMATIC OF LCI CO-PROCESSING CONCEPT WITH HYDROCRACKED PETROLEUM RESIDUA<br />
0-:+<br />
COAL<br />
-<br />
SCT<br />
c 1st-Stage<br />
REACTION I<br />
SYSTEM LC- F I NER<br />
* V/L<br />
SEPARATOR -<br />
0<br />
Hydrotre<strong>at</strong>ed Vac Resid<br />
- \<br />
2nd-Stage<br />
Hydrocracked<br />
LC-FINER OISTILLA- DDistil 1 <strong>at</strong>es<br />
1-D Drag Stream To Gasifier<br />
- -<br />
Figure 2. SCHEMATIC OF LCI CO-PROCESSING CONCEPT WITH VIRGIN PETROLEUM RESIDUA<br />
COAL<br />
SYSTEM<br />
I<br />
'Virgin Vac Resid<br />
-<br />
-<br />
Recycl e Hydrocracked Resid<br />
2nd-Stage<br />
LC-FINER p DISTILLA-<br />
TION<br />
214<br />
r #<br />
SEPARATOR<br />
- Hydrocracked I<br />
Distill<strong>at</strong>e<br />
Drag Stream To<br />
Gasifier<br />
1
I<br />
Fl- 3<br />
9C)IEMTIC OF MICROAUTOCLAM TEST WIT<br />
THERMOCOUPLE<br />
215<br />
H2<br />
N2
SIMULATION OF A COAL/PETROLEuII RESID COPROCESSING PILOT PLANT SCHEME<br />
George W. Pukanic, Dennis N. Smith, and John A. Ruether<br />
U.S. Department <strong>of</strong> Energy<br />
Pittsburgh Energy Technology Center<br />
Pittsburgh, Pennsylvania 15236<br />
ABSTRACT<br />
Co<strong>processing</strong> involves the <strong>co</strong>nversion <strong>of</strong> <strong>co</strong>al and heavy <strong>oil</strong> in the presence <strong>of</strong><br />
hydrogen to products th<strong>at</strong> can be further upgraded into <strong>co</strong>mmercial fuels; the up-<br />
grading can be carried out in an existing refining oper<strong>at</strong>ion. This rel<strong>at</strong>ively new<br />
<strong>co</strong>ncept elimin<strong>at</strong>es or significantly reduces the need for expensive solvent<br />
recycle, and thus has the potential for improved e<strong>co</strong>nomic performance over current<br />
direct <strong>co</strong>al liquefaction processes.<br />
A simul<strong>at</strong>ion <strong>of</strong> a single-stage <strong>co</strong><strong>processing</strong> pilot plant involving the simultaneous<br />
<strong>co</strong>nversion <strong>of</strong> resid and <strong>co</strong>al has been carried out using the ASPEN PLUS simul<strong>at</strong>or.<br />
Limited experimental d<strong>at</strong>a were available for only one run, and results should be<br />
<strong>co</strong>nsidered preliminary. The Assay D<strong>at</strong>a Analysis and Pseudo<strong>co</strong>mponent Correl<strong>at</strong>ion<br />
System <strong>of</strong> ASPEN PLUS has been used to develop a set <strong>of</strong> pseudo<strong>co</strong>mponents for <strong>co</strong>al/<br />
resid liquids and to estim<strong>at</strong>e <strong>co</strong>rresponding physical and thermodynamic properties.<br />
Correl<strong>at</strong>ions based on <strong>co</strong>al liquids and petroleum liquids have been utilized. For<br />
some process equipment, petroleum liquids <strong>co</strong>rrel<strong>at</strong>ions are better than <strong>co</strong>al<br />
liquids <strong>co</strong>rrel<strong>at</strong>ions. Testing for the presence <strong>of</strong> a free w<strong>at</strong>er phase and the<br />
tre<strong>at</strong>ment <strong>of</strong> heavy resid as a single high-b<strong>oil</strong>ing pseudo<strong>co</strong>mponent has improved<br />
simul<strong>at</strong>or performance.<br />
INTRODUCTION<br />
Serious research efforts are under way to develop altern<strong>at</strong>ive energy sources in<br />
order to prevent petroleum supply disruptions from having adverse impact upon the<br />
e<strong>co</strong>nomy <strong>of</strong> those <strong>co</strong>untries dependent on external supplies <strong>of</strong> petroleum. During<br />
the last twenty years, <strong>co</strong>nsiderable work has been done in <strong>at</strong>tempts to understand<br />
the scientific and technological applic<strong>at</strong>ions <strong>of</strong> <strong>co</strong>al <strong>co</strong>nversion schemes for pro-<br />
duction <strong>of</strong> liquid fuels to supplement dwindling petroleum reserves. A rel<strong>at</strong>ively<br />
new <strong>co</strong>ncept addressing this issue involves <strong>co</strong><strong>processing</strong> heavy <strong>oil</strong> with rel<strong>at</strong>ively<br />
low-<strong>co</strong>st <strong>co</strong>als to produce liquid distill<strong>at</strong>es. A review <strong>of</strong> <strong>co</strong>al-<strong>oil</strong> <strong>co</strong><strong>processing</strong><br />
technology has been given by Cugini [l]. This review addresses the st<strong>at</strong>e <strong>of</strong> the<br />
technology, and several important research efforts required to advance the tech-<br />
nology beyond the current level <strong>of</strong> knowledge. One important area <strong>of</strong> research<br />
required to improve understanding <strong>of</strong> <strong>co</strong><strong>processing</strong> technology is the characteriza-<br />
tion <strong>of</strong> the heavy nondistillable feedstock and product <strong>oil</strong>s. Estim<strong>at</strong>ions <strong>of</strong><br />
physical and thermodynamic properties <strong>of</strong> feedstock and product <strong>oil</strong>s are required<br />
to design the <strong>co</strong>mmercial reactor and the product separ<strong>at</strong>ion train and to estim<strong>at</strong>e<br />
m<strong>at</strong>erial flows and <strong>co</strong>mpositions for internal and external streams <strong>of</strong> the <strong>co</strong>proces-<br />
sing plant.<br />
There are several sources <strong>of</strong> inform<strong>at</strong>ion rel<strong>at</strong>ed to <strong>co</strong><strong>processing</strong> <strong>of</strong> <strong>co</strong>al and heavy<br />
<strong>oil</strong> in bench-scale and pilot-plant oper<strong>at</strong>ions [2-41. However, insufficient infor-<br />
m<strong>at</strong>ion is given in these reports to properly characterize the thermodynamic and<br />
physical properties <strong>of</strong> the liquid products as a function <strong>of</strong> oper<strong>at</strong>ing <strong>co</strong>nditions.<br />
Coal and heavy <strong>oil</strong> <strong>co</strong><strong>processing</strong> in a <strong>co</strong>ntinuous bench-scale plant has recently<br />
been initi<strong>at</strong>ed <strong>at</strong> UOP, Inc. and the Signal Research Center, Inc. [51. The work<br />
rel<strong>at</strong>ed to this study has been described in sufficient detail to allow characteri-<br />
z<strong>at</strong>ion <strong>of</strong> the products. The present effort describes the results <strong>of</strong> a simul<strong>at</strong>ion<br />
<strong>of</strong> the single-stage <strong>co</strong><strong>processing</strong> bench-scale unit <strong>at</strong> Signal Research Center, Inc.,<br />
using the ASPEN PLUS simul<strong>at</strong>or to determine physical and thermodynamic properties<br />
216
<strong>of</strong> the liquid products. This bench-scale work forms a preliminary basis to<br />
<strong>co</strong>nduct process modeling studies for a range <strong>of</strong> oper<strong>at</strong>ing <strong>co</strong>nditions used with<br />
this <strong>co</strong>ncept.<br />
EXPERIMENTAL.<br />
The simul<strong>at</strong>ion studies were based on one c<strong>at</strong>alytic <strong>co</strong><strong>processing</strong> run <strong>at</strong> the UOP<br />
pilot plant (Figure 1). Two parts <strong>of</strong> Lloydminster resid to one part <strong>of</strong> Illinois<br />
No. 6 <strong>co</strong>al <strong>co</strong>nstituted the feed. The Lloydminster resid was fraction<strong>at</strong>ed so th<strong>at</strong><br />
95% <strong>of</strong> the resid <strong>co</strong>nsisted <strong>of</strong> 950°F+ m<strong>at</strong>erial. Total feed r<strong>at</strong>e to the reactor was<br />
3.75 lb in 12 hours. Coal <strong>co</strong>nversion defined as toluene insolubles was 86%.<br />
Oper<strong>at</strong>ing <strong>co</strong>nditions for the separ<strong>at</strong>ors include the following: high-pressure<br />
separ<strong>at</strong>or--temper<strong>at</strong>ure = 3020F, pressure = 3114.7 psia; three-phase separ<strong>at</strong>or--<br />
temper<strong>at</strong>ure = 860F, pressure = 3014.7 psia; low-pressure separ<strong>at</strong>or--temper<strong>at</strong>ure =<br />
2840F, pressure = 19.7 psia; debutanizer--temper<strong>at</strong>ure = 43OF, pressure = 17.7<br />
psia; vacuum fraction<strong>at</strong>or--temper<strong>at</strong>ure = 6080F, pressure = 0.94 psia. The<br />
effluent from the low-pressure separ<strong>at</strong>or and gases from the vacuum pump are sent<br />
to the debutanizer, where c6+ m<strong>at</strong>erial is <strong>co</strong>ndensed and <strong>of</strong>f-gases are sampled.<br />
The vacuum fraction<strong>at</strong>or is a packed <strong>co</strong>lumn th<strong>at</strong> oper<strong>at</strong>es with an overhead reflux.<br />
Dimensions <strong>of</strong> the debutanizer and vacuum fraction<strong>at</strong>or towers and <strong>of</strong> their packing<br />
were not available. For lack <strong>of</strong> better inform<strong>at</strong>ion on the number <strong>of</strong> equivalent<br />
theoretical pl<strong>at</strong>es in the debutanizer and vacuum fraction<strong>at</strong>or, simul<strong>at</strong>ions for<br />
both units were performed as simple flash calcul<strong>at</strong>ions.<br />
Experimental values were available for the three-phase separ<strong>at</strong>or vapor stream, the<br />
debutanizer vapor stream, the vacuum fraction<strong>at</strong>or bottoms, and a <strong>co</strong>mbined <strong>co</strong>mposi-<br />
tion for the vacuum fraction<strong>at</strong>or overhead and debutanizer bottoms. Gas phase <strong>co</strong>m-<br />
ponents were analyzed by gas chrom<strong>at</strong>ography. B<strong>oil</strong>ing point distributions were<br />
obtained by gas chrom<strong>at</strong>ographic simul<strong>at</strong>ed distill<strong>at</strong>ion.<br />
FLOWSHEET SIMULATION IUXHODOLOGY<br />
The ASPEN PLUS simul<strong>at</strong>or [61 has been used for the flowsheet analysis <strong>of</strong> the sepa-<br />
r<strong>at</strong>or system downstream from the reactor in the UOP <strong>co</strong><strong>processing</strong> pilot plant<br />
(Figure 1). ASPEN PLUS was originally designed for the analysis <strong>of</strong> fossil fuel<br />
<strong>co</strong>nversion processes, although it has proven useful for many process industries.<br />
It has been used to develop a reference d<strong>at</strong>a base system <strong>of</strong> thermophysical<br />
properties <strong>of</strong> <strong>co</strong>al liquids needed for vapor-liquid equilibrium and for he<strong>at</strong> and<br />
m<strong>at</strong>erial balance calcul<strong>at</strong>ions [2], and to simul<strong>at</strong>e the preliminary separ<strong>at</strong>or<br />
system downstream from the reactor in the SRC-I1 <strong>co</strong>al liquefaction process [TI.<br />
A preliminary step in the simul<strong>at</strong>ion process is the development <strong>of</strong> an ASPEN flow-<br />
sheet. Figure 2 represents the ASPEN flowsheet for the separ<strong>at</strong>or system following<br />
the reactor in the <strong>co</strong><strong>processing</strong> pilot-plant flow diagram in Figure 1. The outlet<br />
stream from the reactor is the process feed stream for the simul<strong>at</strong>ion. The five<br />
separ<strong>at</strong>ors include a high-pressure separ<strong>at</strong>or, a three-phase separ<strong>at</strong>or, a low-<br />
pressure separ<strong>at</strong>or, a debutanizer, and a vacuum fraction<strong>at</strong>or. All are modeled as<br />
flash units where vapor-liquid equilibrium calcul<strong>at</strong>ions are performed to produce<br />
vapor and liquid outlet streams. Mixer units are used to <strong>co</strong>mbine m<strong>at</strong>erial streams<br />
into one stream. Names associ<strong>at</strong>ed with the streams, and unit oper<strong>at</strong>ion models for<br />
ASPEN identific<strong>at</strong>ion purposes are design<strong>at</strong>ed in Figure 2.<br />
Thermophysical properties used for the simul<strong>at</strong>ions were based on three ASPEN PLUS<br />
option sets. The option set used for lighter <strong>co</strong>mponents up to c6 is based pri-<br />
marily on the Redlich-Kwong-Soave thermophysical model. Heavier <strong>co</strong>mponents were<br />
analyzed using the ASPEN PLUS Assay D<strong>at</strong>a Analysis and Pseudo<strong>co</strong>mponent Correl<strong>at</strong>ion<br />
System for petroleum liquids and <strong>co</strong>al liquids. Standard API procedures were used<br />
for the petroleum liquids thermophysical models, and <strong>co</strong>rrel<strong>at</strong>ions developed pri-<br />
marily <strong>at</strong> Aspen Technology, Inc., were used for the <strong>co</strong>al liquids thermophysical<br />
models. From assay analyses <strong>of</strong> the vacuum fraction<strong>at</strong>or process streams, a set <strong>of</strong><br />
I 217
20 pseudo<strong>co</strong>mponents was developed to represent the heavier <strong>co</strong>mponents; each<br />
pseudo<strong>co</strong>mponent represents about a 50°F cut <strong>of</strong> liquid distill<strong>at</strong>e. Since the<br />
process feed stream included both <strong>co</strong>al and petroleum liquids, separ<strong>at</strong>e simul<strong>at</strong>ion<br />
runs were made where thermophysical properties for each pseudo<strong>co</strong>mponent were<br />
analyzed using the <strong>co</strong>rrel<strong>at</strong>ion option set for <strong>co</strong>al liquids and the set ,for<br />
petroleum liquids. Comparisons have been made for the performance <strong>of</strong> each option<br />
set.<br />
RESULTS AND DISCUSSION<br />
The <strong>co</strong>mposition for the process feed stream to the high-pressure separ<strong>at</strong>or is<br />
estim<strong>at</strong>ed from experimental product sl<strong>at</strong>es for the vacuum fraction<strong>at</strong>or, the<br />
debutanizer, and the three-phase separ<strong>at</strong>or (see Table 1). Gases and identified<br />
<strong>co</strong>mpounds range from HZ to pentane. Liquid distill<strong>at</strong>es are represented as pseudo-<br />
<strong>co</strong>mponents and range from an average b<strong>oil</strong>ing point <strong>of</strong> lll°F to 1091OF (pseudo-<br />
<strong>co</strong>mponents are prefaced by PC and followed by the average b<strong>oil</strong>ing point <strong>of</strong> the<br />
approxim<strong>at</strong>ely 50°F cut). The initial b<strong>oil</strong>ing point (IBP) <strong>of</strong> the liquid distill<strong>at</strong>e<br />
is 69.80F. For <strong>co</strong>nciseness in the present<strong>at</strong>ion <strong>of</strong> results, the pseudo<strong>co</strong>mponents<br />
have been expressed as four distill<strong>at</strong>e fractions (see Table 2).<br />
TABLE 1. Process Feed Stream Composition for High Pressure Separ<strong>at</strong>or<br />
Components' Flows (lb/hr) Components* Flows (lb/hr)<br />
Ht<br />
<strong>co</strong><br />
HzS<br />
CH +<br />
0.0593<br />
0.0051<br />
0.0552<br />
0.0637<br />
PC428<br />
PC477<br />
PC525<br />
PC575<br />
0.0075<br />
0.0168<br />
0.0163<br />
0.0163<br />
C2H6 0.0364 PC625 0.0167<br />
CsHe 0.0255 PC675 0.0174<br />
CIHio 0.0105 PC725 0.0158<br />
I-C~HI o 0.0028 PC774 0.0139<br />
CsHi z<br />
I-CSHIZ<br />
HzO<br />
PClll<br />
PC176<br />
PC226<br />
PC276<br />
PC327<br />
PC376<br />
0.0034<br />
0.0034<br />
0.0108<br />
0.0052<br />
0.0017<br />
0.0020<br />
0.0025<br />
0.0041<br />
0.0053<br />
PC825<br />
PC876<br />
PC924<br />
pc973<br />
PC1022<br />
PC1091<br />
Ash<br />
Un<strong>co</strong>nverted Coal<br />
Nondistill<strong>at</strong>e Solids<br />
0.0128<br />
0.0113<br />
0.0113<br />
0.0048<br />
0.0034<br />
0.0503<br />
0.0108<br />
0.0135<br />
0.0456<br />
*Pseudo<strong>co</strong>mponents are 50°F cuts and are represented by PC followed by the average<br />
b<strong>oil</strong>ing point.<br />
TABLE 2. Process Feed Stream Distill<strong>at</strong>e Fractions Expressed as Pseudo<strong>co</strong>mponents.<br />
Distill<strong>at</strong>e Fraction Pseudo<strong>co</strong>mponent Range Flows (lb/hr)<br />
IBP-3500F PC111-PC327 0.0155<br />
350°-4500F PC376-PC428 0.0128<br />
45O0-95OOF PC477-PC924 0.149<br />
9500F+ PC973-PC1091 0.0585<br />
218<br />
~<br />
\
I<br />
i<br />
Three-phase SeDar<strong>at</strong>or<br />
For the three-phase separ<strong>at</strong>or overheads, the effect <strong>of</strong> the <strong>co</strong>rrel<strong>at</strong>ion option is<br />
given in Table 3. There is some improvement using the petroleum-liquids option<br />
set. Use <strong>of</strong> the <strong>co</strong>al-liquids option set results in an overall error for total<br />
mass flow <strong>of</strong> overhead <strong>of</strong> 1.862, and the petroleum-liquids option set results in an<br />
error <strong>of</strong> 0.73%.<br />
TABU 3. Comparison <strong>of</strong> Calcul<strong>at</strong>ed and Experimental Overhead Flows (lb/hr) for the<br />
Three-phase Separ<strong>at</strong>or as a Function <strong>of</strong> Physical Properties Calcul<strong>at</strong>ions Method<br />
Correl<strong>at</strong>ion Option Set<br />
Components Coal Liquids Petroleum Liquids Experimental<br />
Hz 0.0591 0.0589 0.0587<br />
<strong>co</strong> 0.0051 0.0051 0.00513<br />
HIS 0.0516 0.0511 0.0498<br />
CH z 0.0629 0.0626 0.0621<br />
CZH6 0.0349 0.0346 0.0340<br />
CsHs 0.0233 0.0229 0.0225<br />
C*HIO 0.0086 0.0084 0.00849<br />
I-C~HI o 0.0024 0.0023 0.00212<br />
CSHI z 0.0023 0.0022 0.00263<br />
I-CSHI z 0.0025 0.0023 0.00263<br />
Debutanizer<br />
For the debutanizer overheads, the effect <strong>of</strong> the <strong>co</strong>rrel<strong>at</strong>ion option is given in<br />
Table 4. There is improvement using the petroleum-liquids option set. Use <strong>of</strong> the<br />
<strong>co</strong>al-liquids option set results in an overall error for total mass flow <strong>of</strong> over-<br />
head <strong>of</strong> 16.42, and the petroleum-liquids option set results in an error <strong>of</strong> only<br />
1.37%.<br />
TABLE 4. Comparison <strong>of</strong> Calcul<strong>at</strong>ed and Experimental Overhead Flows (lb/hr) for<br />
the Debutanizer as a E’unction <strong>of</strong> Physical Properties Calcul<strong>at</strong>ions Method<br />
Correl<strong>at</strong>ions Options Set<br />
Components Coal Liquids Petroleum Liquids Experimental<br />
2.31 x lo-’<br />
2.79 10-~<br />
3.5 io-’<br />
7.84 x lo-’<br />
1.4 10-~<br />
2.1 io-’<br />
1.5 x lo-:<br />
3.34 x 10-<br />
6.15 x lo-:<br />
6.10 x 10-<br />
1.05 x lo-’<br />
3.91 x lo-*<br />
4.59 10-~<br />
3.9 x lo-’<br />
1.1 x lo-’<br />
1.8 x lo-’<br />
2.5 x io-’<br />
1.8 x lo-;<br />
4.02 x 10-<br />
7.41 x lo-*<br />
7.31 x lo-*<br />
1.35 x lo-’<br />
6.09 x io-*<br />
--<br />
--<br />
1.60 x lo-’<br />
2.35 x 10-~<br />
2.96 x lo-:<br />
1.95 x 10-<br />
6.51 x lo-*<br />
8.08 x lo-*<br />
8.08 x lo-’<br />
2.88 x lo-’<br />
Vacuum Fraction<strong>at</strong>or<br />
As a measure <strong>of</strong> simul<strong>at</strong>ion adequacy, total vapor and, liquid flows <strong>co</strong>mputed by<br />
simul<strong>at</strong>ion were <strong>co</strong>mpared with experimental d<strong>at</strong>a. Total mass flow <strong>of</strong> overhead and<br />
219
ottoms is less sensitive to the number <strong>of</strong> theoretical stages than are the<br />
individual distill<strong>at</strong>e fraction flows. The effect Of the <strong>co</strong>rrel<strong>at</strong>ion option set<br />
(<strong>co</strong>al-liquids vs. petroleum-liquids) on the vacuum fraction<strong>at</strong>or simul<strong>at</strong>ion perfor-<br />
mance was determined. Since simul<strong>at</strong>ed distill<strong>at</strong>e flows were in <strong>co</strong>nsiderable dis-<br />
agreement with experimental values for the reported oper<strong>at</strong>ing pressure <strong>of</strong> 0.94<br />
psia, additional simul<strong>at</strong>ion runs were made to observe the effect <strong>of</strong> assumed <strong>co</strong>lumn<br />
pressures. For the petroleum-liquids option set, a value <strong>of</strong> 4.5 psia gave the<br />
best m<strong>at</strong>ch <strong>of</strong> calcul<strong>at</strong>ed total overhead and bottom flows to experimental flows.<br />
Correspondingly, a value <strong>of</strong> 6.0 psia was found for the <strong>co</strong>al-liquids option set.<br />
Given th<strong>at</strong> the reported <strong>co</strong>lumn pressure, 0.94 psia, is closer to 4.5 psia than to<br />
6.0 psia, this result gives ail indirect indic<strong>at</strong>ion th<strong>at</strong> the petroleum-based option<br />
set better describes the experimental system. The actual oper<strong>at</strong>ing pressure for<br />
the vacuum fraction<strong>at</strong>or was known to increase above 0.94 psia during the experi-<br />
mental run, but no inform<strong>at</strong>ion is available as to the extent <strong>of</strong> increase. The<br />
results for the flows <strong>of</strong> the distill<strong>at</strong>e fractions are presented in Tables 5-7.<br />
Table 5 represents results for vacuum bottoms flows, and Table 6, for vacuum over-<br />
head flows (calcul<strong>at</strong>ed vacuum overhead flows also include values for debutanizer<br />
bottoms flows in order to agree with experimental measurements). Table 7 gives a<br />
<strong>co</strong>mparison <strong>of</strong> the pressure and <strong>co</strong>rrel<strong>at</strong>ion option set in terms <strong>of</strong> an overall per-<br />
centage error for both bottoms and overhead <strong>at</strong> the oper<strong>at</strong>ing pressure <strong>of</strong><br />
0.94 psia. Use <strong>of</strong> the petroleum-liquids option set gives better agreement,<br />
although the percentage <strong>of</strong> error rel<strong>at</strong>ive to experimental error is still<br />
<strong>co</strong>nsiderable.<br />
TABLE 5. Comparison <strong>of</strong> Calcul<strong>at</strong>ed and Experimental Vacuum Bottoms Flows (lbhr)<br />
as a Function <strong>of</strong> Physical Properties Calcul<strong>at</strong>ion kthod and System Pressure<br />
P = 6.0 psia<br />
Petroleum Liquids<br />
P = 0.94 psia<br />
P = 4.5 psia<br />
~ ~<br />
B<strong>oil</strong>ing Point<br />
Correl<strong>at</strong>ion Option Set Range, OF Calcul<strong>at</strong>ed Exper imen tal<br />
Coal Liquids<br />
P = 0.94 psia IBP-350 1.52 x lo-' --<br />
350-450 8.46 x 10-~ --<br />
450-950<br />
950+<br />
5.60 x lo-'<br />
3.24 x lo-'<br />
5.16 x lo-'<br />
5.92 x lo-'<br />
Total 3.80 x lo-' 1.11 x lo-'<br />
IBP-350 5.38 x 10-5 --<br />
350-450 2.94 x lo-* --<br />
450-950<br />
950+<br />
5.41 x lo-'<br />
5.67 x lo-'<br />
5.16 x lo-'<br />
5.92 x lo-'<br />
Total 1.12 x 10-I 1.11 x 10-I<br />
IBP-350 4.92 x lo-' --<br />
350-450 2.47 x 10-~ --<br />
450-950<br />
950+<br />
1.40 x lo-'<br />
5.38 x 10-2<br />
5.16 x lo-'<br />
5.92 x lo-'<br />
Total 6.78 x lo-' 1.11 x lo-'<br />
IBP-350 5.59 x 10-~ --<br />
350-450 2.75 x io-' --<br />
450-950 5.21 x lo-' 5.16 x lo-'<br />
950+ 5.79 x lo-' 5.92 x lo-'<br />
Total 1.10 x 10-1 1.11 x lo-'<br />
220
TABLE 6. Comparison <strong>of</strong> Calcul<strong>at</strong>ed and Experimental Vacuum Overhead Flows (lb/hr)<br />
as a Function <strong>of</strong> physical Properties Calcul<strong>at</strong>ion Method and System Pressure<br />
B<strong>oil</strong>ing Point<br />
Correl<strong>at</strong>ion Option Set Range, OF Calcul<strong>at</strong>ed Experimental<br />
Coal Liquids<br />
P = 0.94 psia<br />
P = 6.0 psia<br />
Petroleum Liquids<br />
P = 0.94 psia<br />
P = 4.5 psia<br />
IBP-350 1.07 x lo-' 1.34 x lo-'<br />
3 5 0 - 4 5 0 1.28 x lo-' 1.90 x 10-2<br />
450-950 1.43 x lo-' 8.97 x lo-'<br />
950+ 2.60 x lo-' 8.17 x 10-~<br />
Total 1.93 x lo-' 1.22 x 10-1<br />
IBP-350 1.07 x lo-' 1.34 x lo-'<br />
350-450 1.25 x lo-' 1.90 x<br />
450-950 9.38 x lo-' 8.97 x lo-'<br />
950+ 1.70 x lo-' 8.17 x 10-~<br />
Total 1.19 x 10-1 1.22 x lo-'<br />
IBP-350<br />
350-450<br />
4 5 0 - 9 5 0<br />
950+<br />
Total<br />
1.05 x lo-'<br />
1.28 x lo-'<br />
1.34 x lo-'<br />
4.60 x lo-'<br />
1.62 x lo-'<br />
1.34 x lo-'<br />
1.90 x lo-'<br />
8.97 x lo-'<br />
8.17 x 10-~<br />
1.22 x lo-'<br />
IBP-350<br />
350-450<br />
4 5 0 - 9 5 0<br />
950+<br />
Total<br />
1.05 x lo-'<br />
1.25 x lo-'<br />
9.64 x lo-'<br />
5.08 x lo-'<br />
1.24 x lo-'<br />
1.34 x 10-i<br />
1.90 x 10-<br />
8.97 x lo-'<br />
8.i~ x 10-~<br />
1.22 x 10-f<br />
TABLE 7. Comparison <strong>of</strong> Physical Properties Calcul<strong>at</strong>ion Method on Vacuum<br />
Fraction<strong>at</strong>or Effluent Stream Hass Flow R<strong>at</strong>es<br />
Correl<strong>at</strong>ion Option Set<br />
~ ~~<br />
Coal Liauids<br />
$ Error"<br />
Vacuum Bottoms Vacuum Overhead<br />
P = 0.94 psia 65.8 58.2<br />
Petroleum Liquids<br />
P = 0.94 38.9 32.8<br />
*Absolute value <strong>of</strong> (Calcul<strong>at</strong>ed - Experimental)/Experimental.<br />
Effects <strong>of</strong> pressure and <strong>co</strong>rrel<strong>at</strong>ion option set on pseudo<strong>co</strong>mponent <strong>co</strong>mposition for<br />
the vacuum fraction<strong>at</strong>or are reflected in Figures 3-6. Figures 3 and 4 represent<br />
the effect on the vacuum bottoms stream, and Figures 5 and 6, the effect on the<br />
overhead stream. These figures indic<strong>at</strong>e th<strong>at</strong> the petroleum-liquids option set<br />
gives better values than the <strong>co</strong>al-liquids option set and th<strong>at</strong> the effect <strong>of</strong> pres-<br />
sure is <strong>co</strong>nsiderable.<br />
221<br />
~~~
Low Pressure Separ<strong>at</strong>or<br />
A temper<strong>at</strong>ure <strong>of</strong> 284OF was used for the oper<strong>at</strong>ing <strong>co</strong>ndition <strong>of</strong> the low-pressure<br />
separ<strong>at</strong>or in the simul<strong>at</strong>or. Since, experimentally, a temper<strong>at</strong>ure range <strong>of</strong> 2480F-<br />
2840F was given, it was decided to make a simul<strong>at</strong>ion run <strong>at</strong> the lower temper<strong>at</strong>ure<br />
to determine any effects <strong>of</strong> the assumed temper<strong>at</strong>ure on the effluent flow r<strong>at</strong>es.<br />
Results for the lower temper<strong>at</strong>ure indic<strong>at</strong>e only a small increase in bottoms flows<br />
<strong>of</strong> 1.3% and an decrease in overhead flows <strong>of</strong> 11.8%.<br />
Effect <strong>of</strong> Free W<strong>at</strong>er Phase on the Debutanizer<br />
Simul<strong>at</strong>ions were carried out on the debutanizer to determine the impact <strong>of</strong> the<br />
presence <strong>of</strong> a free w<strong>at</strong>er phase (Table 8). Without the invoc<strong>at</strong>ion <strong>of</strong> the option to<br />
test for the presence <strong>of</strong> free w<strong>at</strong>er, no distill<strong>at</strong>e and only a small amount <strong>of</strong><br />
lighter gases are predicted by the simul<strong>at</strong>or to be present in the debutanizer<br />
bottoms. With the test for the presence <strong>of</strong> w<strong>at</strong>er invoked, the presence <strong>of</strong> a free<br />
w<strong>at</strong>er phase is <strong>co</strong>nfirmed, and results indic<strong>at</strong>e a substantial increase in bottoms<br />
flow and a decrease in overhead. Both <strong>of</strong> these predictions agree with experiment,<br />
as shown in Table 8. All simul<strong>at</strong>ions have tested for the presence <strong>of</strong> a free w<strong>at</strong>er<br />
phase.<br />
TABLE 8. Effect <strong>of</strong> Tre<strong>at</strong>ment <strong>of</strong> W<strong>at</strong>er on the Debutanizer Effluent Product<br />
Flows (lb/hr)<br />
Assumed Absence <strong>of</strong> Free<br />
W<strong>at</strong>er Phase<br />
Assumed Presence <strong>of</strong> Free<br />
W<strong>at</strong>er Phase<br />
Experimental<br />
Products Overhead Bottoms Overhead Bottoms Overhead<br />
Gases-Cs 6.93 x lo-’ 3.90 x 1.11 x lo-* 1.16 x lo-’ 1.18 x lo-*<br />
IBP-1500F 3.79 x lo-’ 0 1.05 10-3 4.86 IO-’)<br />
35003500~ i.53 x 10-3 0 1.08 10-7 iIi6 X 10-3 }<br />
450°-9500F 1.31 x 0 4.69 x lo-’ 8.46 x lo-* 2‘88 lo-’<br />
950°F+ 1.02 10-7 0 0 6.52 x IO-~J<br />
TABLE 9. Effect <strong>of</strong> Tre<strong>at</strong>ment <strong>of</strong> Heavy Resid on Vacuum Bottoms<br />
Heaw Residue Tre<strong>at</strong>ed as<br />
Solid M<strong>at</strong>erial Pseudo<strong>co</strong>mponent<br />
Product (Flows, lb/hr) (Flows, lb/hr)<br />
Gases-Cs<br />
I BP-3500F<br />
350°-4500F<br />
450°-9500F<br />
950°F+<br />
Nondistill<strong>at</strong>e Solid<br />
0<br />
0<br />
0<br />
0<br />
0<br />
0.0456<br />
1.44 x 10-a<br />
1.52 x<br />
8.47 x lo-‘<br />
5.59 10-~<br />
3.25 x lo-‘<br />
0<br />
Effect <strong>of</strong> Tre<strong>at</strong>ment <strong>of</strong> Heavy Resid on Vacuum Bottoms<br />
The method <strong>of</strong> tre<strong>at</strong>ment <strong>of</strong> heavy resid has an impact on predicted <strong>co</strong>mposition and<br />
flow r<strong>at</strong>e <strong>of</strong> vacuum bottoms as reflected in Table 9. The d<strong>at</strong>a are for the simula-<br />
tion <strong>of</strong> the vacuum fraction<strong>at</strong>or <strong>at</strong> the pressure <strong>of</strong> 0.94 psia and using <strong>co</strong>al-<br />
liquids <strong>co</strong>rrel<strong>at</strong>ions. When the heavy resid is tre<strong>at</strong>ed as a nondistill<strong>at</strong>e solid,<br />
i.e., m<strong>at</strong>erial with negligible vapor pressure, no distill<strong>at</strong>es are predicted to<br />
appear in the vacuum bottoms. When the heavy resid is tre<strong>at</strong>ed as a 1091°F pseudo-<br />
222<br />
I
<strong>co</strong>mponent, distill<strong>at</strong>e products are predicted in the bottoms. This tre<strong>at</strong>ment <strong>of</strong><br />
the resid fraction <strong>co</strong>rresponds to th<strong>at</strong> <strong>of</strong> McKeegan and Klunder [El, who also<br />
assigned a single normal b<strong>oil</strong>ing point to the nondistill<strong>at</strong>e m<strong>at</strong>erial in their<br />
simul<strong>at</strong>ion <strong>of</strong> the separ<strong>at</strong>or system in the SRC-I1 <strong>co</strong>al liquefaction process<br />
(although they used a much higher temper<strong>at</strong>ure). The plot in Figure 4 for P 0.94<br />
psia, as well as for P = 6.0 psia, reflects the presence <strong>of</strong> heavy resid tre<strong>at</strong>ed as<br />
the 10910F pseudo<strong>co</strong>mponent. These plots are in line with the experimental obser-<br />
v<strong>at</strong>ion <strong>of</strong> the presence <strong>of</strong> significant amounts <strong>of</strong> liquid distill<strong>at</strong>e product in the<br />
vacuum bottoms stream. All simul<strong>at</strong>ion results for the vacuum fraction<strong>at</strong>or pre-<br />
sented in Tables 5-7 and Figures 3-6 have tre<strong>at</strong>ed the -heavy resid as a 1091OF<br />
pseudo<strong>co</strong>mponent.<br />
CONCLUSIONS<br />
The very preliminary results reported here indic<strong>at</strong>e th<strong>at</strong> the use <strong>of</strong> petroleum-<br />
liquids <strong>co</strong>rrel<strong>at</strong>ions may result in an improvement over <strong>co</strong>al-liquids <strong>co</strong>rrel<strong>at</strong>ions<br />
in the simul<strong>at</strong>ion <strong>of</strong> the <strong>co</strong><strong>processing</strong> <strong>of</strong> Lloydminster with Illinois No. 6 <strong>co</strong>al.<br />
Agreement between simul<strong>at</strong>ion and experiment is improved by using a higher assumed<br />
pressure than the experimental pressure for the vacuum fraction<strong>at</strong>or, by tre<strong>at</strong>ing<br />
the presence <strong>of</strong> w<strong>at</strong>er as a free w<strong>at</strong>er phase, and by tre<strong>at</strong>ing the heavy resid as<br />
109l0F distill<strong>at</strong>e r<strong>at</strong>her than an inert solid m<strong>at</strong>erial. It is necessary to obtain<br />
better definition <strong>of</strong> the separ<strong>at</strong>ion equipment used and the oper<strong>at</strong>ing <strong>co</strong>nditions<br />
employed, and to acquire a larger d<strong>at</strong>a set in order to evalu<strong>at</strong>e the present<br />
capability for simul<strong>at</strong>ing the separ<strong>at</strong>ion steps in <strong>co</strong><strong>processing</strong>.<br />
ACKNOWLEDCUENT<br />
The authors would like to thank UOP, Inc., for supplying the experimental d<strong>at</strong>a<br />
th<strong>at</strong> served as the basis for the simul<strong>at</strong>ion studies, and Charles Luebke <strong>of</strong> UOP,<br />
Inc., and Carl Lea <strong>of</strong> Signal Research Center, Inc., for many discussions rel<strong>at</strong>ive<br />
to process oper<strong>at</strong>ion and d<strong>at</strong>a analysis.<br />
DISCLAIMER<br />
Reference in this report to any specific product, process, or service is to<br />
facilit<strong>at</strong>e understanding and does not necessarily imply its endorsement or favor-<br />
ing by the United St<strong>at</strong>es Department <strong>of</strong> Energy.<br />
1.<br />
2.<br />
3.<br />
4.<br />
5.<br />
6.<br />
REFERENCES<br />
Cugini, A.V., "A Review <strong>of</strong> Coal-Oil Co<strong>processing</strong> Technology," M.S. Thesis,<br />
Department <strong>of</strong> Chemical and Petroleum Engineering, University <strong>of</strong> Pittsburgh,<br />
Pittsburgh, Pennsylvania, 1985.<br />
Rhodes, D., "Comparison <strong>of</strong> Coal and Bitumen-Coal Process Configur<strong>at</strong>ion," pre-<br />
sented <strong>at</strong> the Tenth Annual EPRI' Contractors' Conference, Palo Alto,<br />
California, April 23-25, 1985.<br />
Duddy, J.E., and MacArthur, J.B., "Coal/Oil Co-Processing,'' presented <strong>at</strong> the<br />
AIChE Summer N<strong>at</strong>ional Meeting, paper no. 16b, Philadelphia, Pennsylvania,<br />
August 21, 1984.<br />
Kelly, J., Fouda, S., Rahimi, P., and Ikura, M., "CANMET Co-Processing: A<br />
St<strong>at</strong>us Report," Synthetic Fuels Research Labor<strong>at</strong>ory, September, 1984.<br />
C<strong>at</strong>sis, J.C., Sikonia, J.C., Nelson, B.J., Luebke, C.P., and Humbach, M.J.,<br />
"Coal <strong>Liquefaction</strong> Co-Processing," presented <strong>at</strong> the Direct <strong>Liquefaction</strong> Con-<br />
tractors' Review Meeting, Pittsburgh, Pennsylvania, November 19-21, 1985.<br />
Aspen Technology, Inc., ASPEN PLUS Introductory Manual, 1985.<br />
223
7. Gallier, P.W., Boston, J.F., Wu, P.C., and Yoon, E.S., Development <strong>of</strong> a<br />
Reference D<strong>at</strong>a System for the <strong>Liquefaction</strong> Technology D<strong>at</strong>a Base, DOE/PC/<br />
5005 1-T4 (DE84004620), 1983.<br />
8. McKeegan, D.P., and Klunder, E.B., "An ASPEN Simul<strong>at</strong>ion <strong>of</strong> the SRC-I1 Process<br />
as Conducted in Gulf's P-99 Process Development Unit," presented <strong>at</strong> the AIChE<br />
N<strong>at</strong>ional Meeting, paper no. 33f, San Francis<strong>co</strong>, California, November 25-30,<br />
1984.<br />
Figwe I - UOP Co<strong>processing</strong> Pilot-plont Flow Diogrom.<br />
FlQURE 2. ASPEN FLOWSHEET OF SEPARATOR SYSTEM FOR<br />
UOP COPROCESSINQ PILOT-PLANT FLOW SCHEME.<br />
224<br />
VFOlL
100<br />
80<br />
$<br />
c<br />
3<br />
9 60-<br />
-<br />
t<br />
J<br />
240-<br />
5<br />
0<br />
- Calcul<strong>at</strong>ed<br />
AVERAGE PSEUDOCOMPONENT BOILING POINT, O F<br />
Figure 3 -Composition <strong>of</strong> Vacuum Bottoms Pseudo<strong>co</strong>mponents Using Petroleum<br />
Liquids Correl<strong>at</strong>ions.<br />
20 -<br />
P= Pressure, psia<br />
0<br />
- --L<br />
-<br />
I I I I I I I I I I<br />
Experimental<br />
- Calcul<strong>at</strong>ed<br />
I _ - 8 - - I - -<br />
400 600 800 1000<br />
AVERAGE PSEUDOCOMPONENT BOILING POINT, O F<br />
Figure 4 - Composition <strong>of</strong> Vacuum Bottoms Pseudo<strong>co</strong>mponents Using Coal<br />
Liquids Correl<strong>at</strong>ions.<br />
225
I I I I I I I I I<br />
100- --- Experimental - Calcula tad<br />
$80-<br />
c<br />
1<br />
G<br />
c60<br />
$40-<br />
3<br />
V<br />
-<br />
20 -<br />
P= Pressure, psi0<br />
0*-- 200 400 600 800 IO00<br />
AVERAGE PSEUDOCOMPONENT BOILING POINT, O F<br />
Figure 5 - Composition <strong>of</strong> Vacuum Overhead Using Petroleum Liquids<br />
Correl<strong>at</strong>ions.<br />
-<br />
I I I I I I<br />
100- --- Experimental<br />
Calcul<strong>at</strong>ed<br />
$ 80-<br />
c<br />
3<br />
20 -<br />
P= Pressure, psia<br />
200 400 600 800 1000<br />
AVERAGE PSEUDOCOMPONENT BOILING POINT, OF Figure 6-Composltlon <strong>of</strong> Vacuum Overhead Using Coal Liquids Correl<strong>at</strong>ions.<br />
226<br />
-<br />
-<br />
d<br />
I<br />
!<br />
1<br />
E<br />
f
AN ASSESSMENT OF THE POTENTIAL FOR COAL/RESIDUAL OIL COPROCESSING<br />
D.A. Huber. Q. Lee and R.L. Thomas K. Frye and G. Rudins<br />
Burns and Roe<br />
Oradell, NJ<br />
07649<br />
Abstract<br />
U.S. DOE<br />
Germantown, MD<br />
20585<br />
Among the promising new techniques to produce liquid hydrocarbon fuels from <strong>co</strong>al is<br />
<strong>co</strong>al/petroleum <strong>co</strong><strong>processing</strong> based upon the use <strong>of</strong> heavy <strong>oil</strong>, tar sand bitumen and<br />
petroleum residua as "solvents" for the <strong>co</strong>nversion <strong>of</strong> <strong>co</strong>al. Co<strong>processing</strong> is the<br />
simultaneous hydrogen<strong>at</strong>ion <strong>of</strong> <strong>co</strong>al and heavy <strong>oil</strong> fractions in specially designed<br />
reactors with <strong>co</strong>al <strong>co</strong>ntents by weight ranging from as low as 1% to potentially as<br />
high as 50-60% depending upon the technology employed. The results <strong>of</strong> a study on<br />
the potential for <strong>co</strong>al/residual <strong>oil</strong> <strong>co</strong><strong>processing</strong> in the United St<strong>at</strong>es are addressed<br />
in this paper.<br />
Introduction<br />
E<strong>co</strong>nomics, the desire for less dependence upon the import<strong>at</strong>ion <strong>of</strong> foreign <strong>oil</strong>, and<br />
the depletion <strong>of</strong> lighter crudes in the United St<strong>at</strong>es has led the refining industry<br />
to process heavier crudes and bitumens. Upgrading and <strong>co</strong>nverting these heavy <strong>oil</strong>s<br />
to distill<strong>at</strong>e liquids using <strong>co</strong>nventional petroleum thermal cracking, c<strong>at</strong>alytic<br />
cracking and/or hydrocracking technologies has required the install<strong>at</strong>ion <strong>of</strong> <strong>co</strong>stly<br />
equipment to handle the heavier <strong>oil</strong>s.<br />
There exists in the liter<strong>at</strong>ure sufficient<br />
evidence to suggest th<strong>at</strong> heavy <strong>oil</strong> <strong>co</strong>nverts more readily in the presence <strong>of</strong> <strong>co</strong>al and<br />
th<strong>at</strong> significant demetalliz<strong>at</strong>ion, desulfuriz<strong>at</strong>ion, denitrific<strong>at</strong>ion and <strong>co</strong>nversion <strong>of</strong><br />
asphaltenes to <strong>oil</strong>s also occurs. Thus the simultaneous <strong>co</strong>nversion <strong>of</strong> <strong>co</strong>al and<br />
petroleum heavy <strong>oil</strong> fractions to produce distill<strong>at</strong>e liquid products while upgrading<br />
the remaining heavy <strong>oil</strong> merits further investig<strong>at</strong>ion. This type <strong>of</strong> process, termed,<br />
<strong>co</strong>al/<strong>oil</strong> <strong>co</strong><strong>processing</strong> has the potential for being an effective method for <strong>co</strong>nverting<br />
<strong>co</strong>al to liquids and for introducing <strong>co</strong>al liquids into the market place in a <strong>co</strong>st<br />
effective evolutionary manner while gre<strong>at</strong>ly reducing the capital investment<br />
associ<strong>at</strong>ed with the historical approach for establishing a liquefaction industry.<br />
Among the additional potential benefits for the implement<strong>at</strong>ion and utiliz<strong>at</strong>ion <strong>of</strong><br />
the <strong>co</strong><strong>processing</strong> <strong>co</strong>ncept are:<br />
Provision <strong>of</strong> a link or bridge between present day refining technology and a<br />
total <strong>co</strong>al based synfuels industry.<br />
Improved e<strong>co</strong>nomics <strong>co</strong>mpared to direct <strong>co</strong>al liquefaction due to smaller plant<br />
sizes, due to lower hydrogen requirements and the elimin<strong>at</strong>ion <strong>of</strong> the use <strong>of</strong><br />
process derived solvent recycle.<br />
Residuum demetalliz<strong>at</strong>ion, improved product yields and mix.<br />
Minimiz<strong>at</strong>ion <strong>of</strong> the production <strong>of</strong> gases and undesirable by-products; such as<br />
high sulfur <strong>co</strong>ke.<br />
Continued use <strong>of</strong> the U.S. hydrocarbon fuel infrastructure.<br />
A means <strong>of</strong> extending petroleum reserves by reducing crude utiliz<strong>at</strong>ion<br />
requirements.<br />
227
Co<strong>processing</strong> Schemes<br />
The <strong>co</strong><strong>processing</strong> schemes under <strong>co</strong>nsider<strong>at</strong>ion are generally an extension <strong>of</strong> two-stage<br />
<strong>co</strong>al liquefaction and applic<strong>at</strong>ion <strong>of</strong> residuum hydrocracking technology. It has been<br />
re<strong>co</strong>gnized th<strong>at</strong> a possible synergism exists between <strong>co</strong>al derived 1 iquids and<br />
petroleum derived residua. Co<strong>processing</strong> improves the quality <strong>of</strong> synthetic liquid<br />
fuel products from <strong>co</strong>al by diluting them directly with petroleum-derived liquids.<br />
Coal liquids <strong>co</strong>ntain a much higher proportion <strong>of</strong> arom<strong>at</strong>ics <strong>co</strong>mpared to <strong>co</strong>nventional<br />
petroleum-derived liquids, and the non-arom<strong>at</strong>ic portion tends to be naphthenic<br />
r<strong>at</strong>her than paraffinic. Coal liquids <strong>co</strong>ntain significant amounts <strong>of</strong> highly-polar<br />
<strong>co</strong>mpounds, and asphaltenes, but a rel<strong>at</strong>ively low amount <strong>of</strong> sulfur <strong>co</strong>ntaining<br />
<strong>co</strong>mpounds.<br />
Further, petroleum-derived naphtha, is low in nitrogen and oxygen. Coal-derived<br />
naphtha, on the other hand, has higher nitrogen and oxygen <strong>co</strong>ntents, is easier to<br />
reform, and has a higher octane number. Thus, <strong>co</strong>mbining <strong>co</strong>al-derived liquids with<br />
petroleum-derived liquid can provide some positive impacts on the overall product<br />
quality.<br />
Broadly speaking, the <strong>co</strong><strong>processing</strong> processes can be divided into four c<strong>at</strong>egories:<br />
o Hydro-c<strong>at</strong>alytic processes<br />
o Extractive processes<br />
o Thermal processes (non-c<strong>at</strong>alytic)<br />
o Hydro-thermal processes<br />
The first c<strong>at</strong>egory includes HRI, Lumnus, CANMET, UOP, Chevron and Kerr-McKee<br />
processes. The se<strong>co</strong>nd c<strong>at</strong>egory includes <strong>processing</strong> vari<strong>at</strong>ions in<strong>co</strong>rpor<strong>at</strong>ed for<br />
solids removal and deasphalting by Kerr-McGee, UOP and Lumnus. The Cherry-P-process<br />
falls into the thermal process c<strong>at</strong>egory. The process <strong>co</strong>nditions are somewh<strong>at</strong><br />
between those visbreaking and delayed <strong>co</strong>king. The Pyrosol process falls into the<br />
last c<strong>at</strong>egory above and utilizes a mild hydrogen<strong>at</strong>ion <strong>of</strong> <strong>co</strong>al and heavy <strong>oil</strong> in the<br />
first stage. The se<strong>co</strong>nd stage processes residuum under hydrogen pressure to<br />
produce more oi 1.<br />
Refinery Integr<strong>at</strong>ion Consider<strong>at</strong>ions<br />
Since the l<strong>at</strong>e 1970's intensive capital investments in'residuum upgrading and<br />
hydrotre<strong>at</strong>ing capacity have been made by the refinery industry for the <strong>co</strong>nversion <strong>of</strong><br />
heavier crude <strong>oil</strong> fractions to gasoline and distill<strong>at</strong>e fuels. At the same time, the<br />
number <strong>of</strong> oper<strong>at</strong>ing refineries in the United St<strong>at</strong>es has decreased from 319 to 191.<br />
As shown in Figure 1, this decrease has been ac<strong>co</strong>mplished primarily by the deactiv<strong>at</strong>ion<br />
<strong>of</strong> a number <strong>of</strong> low capacity refineries oper<strong>at</strong>ing in the hydroskimning or<br />
topping mode. The major driving force for this realignment in refining capacity has<br />
been largely due to a growing imbalance between the residuum <strong>co</strong>ntent <strong>of</strong> available<br />
crude <strong>oil</strong> and a decrease in demand for residual fuel <strong>oil</strong>. Residual fuels such as<br />
No. 6 Fuel Oil, Bunker C, etc., are by-products <strong>of</strong> refining. As such, their<br />
production and availability are based on the demand for transport<strong>at</strong>ion and distill<strong>at</strong>e<br />
fuels. Based upon d<strong>at</strong>a in the O i l and Gas Journal, residuum <strong>processing</strong><br />
(thermal and hydrocracking) capacity as a percent <strong>of</strong> overall refining capacity has<br />
essentially increased 20% since 1980 to provide supply elasticity for the changing<br />
residual fuel demand, representing about 19% <strong>of</strong> the today's U.S. crude <strong>processing</strong><br />
capacity. The future outlook is for this trend to <strong>co</strong>ntinue as fuel <strong>oil</strong> is replaced<br />
by other energy forms such as <strong>co</strong>al, nuclear and n<strong>at</strong>ural gas.<br />
It is important to<br />
note th<strong>at</strong> this <strong>processing</strong> <strong>of</strong> the heavy ends to yield prime products represents a<br />
reduction in the amount <strong>of</strong> crude <strong>oil</strong> required to meet gasoline and distill<strong>at</strong>e fuel<br />
demand. Table 1 presents a pr<strong>of</strong>ile <strong>of</strong> the Refining Industry in the U.S.<br />
While <strong>co</strong>al liquefaction research and development has demonstr<strong>at</strong>ed significant<br />
Progress in recent years, it has not addressed the fundamental causes for the high<br />
228
<strong>co</strong>st <strong>of</strong> <strong>co</strong>al liquefaction, the high recycle <strong>oil</strong> requirements. In all direct<br />
liquefaction processes <strong>co</strong>al is slurried with a process derived recycle <strong>oil</strong> <strong>at</strong> a<br />
mica1 r<strong>at</strong>io <strong>of</strong> 2:l recycle <strong>oil</strong> to <strong>co</strong>al feed. Co<strong>processing</strong> <strong>of</strong> residuum and <strong>co</strong>al<br />
reduces the high <strong>co</strong>st associ<strong>at</strong>ed with recycle <strong>oil</strong> by elimin<strong>at</strong>ing or reducing the<br />
requirements for recycle <strong>oil</strong>.<br />
Co<strong>processing</strong> hydrocracking technology was originally developed for <strong>processing</strong> heavy<br />
crude with <strong>co</strong>al additives as a means <strong>of</strong> inhibiting the form<strong>at</strong>ion <strong>of</strong> <strong>co</strong>ke. The<br />
CANMET hydrocracking process is based upon this <strong>co</strong>mcept. This emerging technology<br />
shows promise <strong>of</strong> I]iqh demetalliz<strong>at</strong>ion, residuum hydrocracking, and high <strong>co</strong>nversion<br />
<strong>of</strong> the pitch (975 F ) fraction. Coal additives include 1.0 - 2.0 wt.% <strong>of</strong> fine <strong>co</strong>al<br />
and ferrous sulf<strong>at</strong>e.<br />
Integr<strong>at</strong>ion <strong>of</strong> the CANMET type process initially to an existing refinery and/or idle<br />
units is a first step toward utiliz<strong>at</strong>ion <strong>of</strong> <strong>co</strong>al and heavy <strong>oil</strong>s (pitch and<br />
asphaltenes). A once through process arrangement without the use <strong>of</strong> a recycle<br />
stream may also be possible <strong>at</strong> lower <strong>co</strong>al <strong>co</strong>ncentr<strong>at</strong>ions.<br />
Co<strong>processing</strong> technologies to be included in a staged approach are HRI, Lumnus, and<br />
the Cherry-P processes which can process up to 50 wt.% <strong>of</strong> <strong>co</strong>al in heavy <strong>oil</strong><br />
fraction.<br />
Implement<strong>at</strong>ion <strong>of</strong> <strong>co</strong><strong>processing</strong> will likely require additional refinery hydrogen<br />
gener<strong>at</strong>ion. This will probably be based upon steam reforming <strong>of</strong> hydrocarbon gases<br />
and light naphtha. Steam reforming is a well established and adopted method <strong>of</strong><br />
gener<strong>at</strong>ing hydrogen. The expansion <strong>of</strong> reforming units can be ac<strong>co</strong>mplished more<br />
easily than integr<strong>at</strong>ing gasific<strong>at</strong>ion units into refineries.<br />
Hydrostabiliz<strong>at</strong>ion <strong>of</strong> product distill<strong>at</strong>es are in<strong>co</strong>rpor<strong>at</strong>ed into a refinery to<br />
provide hydrotre<strong>at</strong>ment and product stabiliz<strong>at</strong>ion prior to distribution outside the<br />
refinery <strong>co</strong>mplex. Further pretre<strong>at</strong>ments for hetero<strong>at</strong>om removal may be required in a<br />
refinery utilizing <strong>co</strong><strong>processing</strong> derived liquids.<br />
The introduction <strong>of</strong> <strong>co</strong>al/residuum <strong>co</strong><strong>processing</strong> will tend to reduce crude re-<br />
quirements. The extent <strong>of</strong> reduction will be dict<strong>at</strong>ed by market demands as well as<br />
product yields and qualities <strong>of</strong> the <strong>co</strong><strong>processing</strong> distill<strong>at</strong>e liquids. Other<br />
positive factors are 1) the use <strong>of</strong> existing refinery and infrastructure, 2) better<br />
e<strong>co</strong>nomics than direct liquefaction, 3) <strong>co</strong>mp<strong>at</strong>ability with the use <strong>of</strong> heavier crudes,<br />
and 4) the capability <strong>of</strong> installing a <strong>co</strong><strong>processing</strong> unit independently from existing<br />
refinery oper<strong>at</strong>ions.<br />
Potential Coal Requirements<br />
An estim<strong>at</strong>e <strong>of</strong> the potential <strong>co</strong>al requirements for <strong>co</strong>al/<strong>oil</strong> <strong>co</strong><strong>processing</strong> for the<br />
general refinery types in the United St<strong>at</strong>es is presented in Table 2. These<br />
capacities represent an upper limit for the applic<strong>at</strong>ion <strong>of</strong> <strong>co</strong>al/residua <strong>co</strong><strong>processing</strong><br />
as fuel <strong>oil</strong> production was assumed to be zero. It was also assumed th<strong>at</strong> CO<strong>processing</strong><br />
is more e<strong>co</strong>nomic than vacuum distill<strong>at</strong>ion (both cases are highly<br />
unlikely). Based upon a 1985 production capacity <strong>of</strong> 890 million tons <strong>of</strong> <strong>co</strong>al in the<br />
U.S.; <strong>co</strong>al producing capacity would have to increase by one-third if the upper<br />
limits <strong>of</strong> <strong>co</strong>al/residua <strong>co</strong><strong>processing</strong> were achieved.<br />
The most likely near term applic<strong>at</strong>ion for <strong>co</strong>al/<strong>oil</strong> <strong>co</strong><strong>processing</strong> appears to be for<br />
residuum <strong>co</strong>nversion capacity additions to low <strong>co</strong>nversion refineries to improve<br />
pr<strong>of</strong>itability and to high <strong>co</strong>nversion refineries to provide the capability for<br />
handling future feedstocks with increasingly higher residuum <strong>co</strong>ntent. This premise<br />
is based on the assumption th<strong>at</strong> present trends toward a heavier crude feedstocks and<br />
lighter reduced fuel <strong>oil</strong> requirements will <strong>co</strong>ntinue. In terms <strong>of</strong> refinery capacity,<br />
average size Hydroskimming or Topping and High Conversion Refineries<br />
229
<strong>processing</strong> 25,000 and 150,000 bbl/day <strong>of</strong> heavy crude (25' API), respectively, w i l l<br />
require approxim<strong>at</strong>ely 850 and 4,000 tons/day <strong>of</strong> <strong>co</strong>al, respectively, when<br />
<strong>co</strong><strong>processing</strong> <strong>at</strong> slurry <strong>co</strong>ncentr<strong>at</strong>ions <strong>of</strong> 50%. These <strong>co</strong>al capacities are well within<br />
existing transport<strong>at</strong>ion and handling experience for <strong>co</strong>al fired industriallutility<br />
b<strong>oil</strong>er applic<strong>at</strong>ions.<br />
Process E<strong>co</strong>nomics<br />
While the detailed engineering required to develop definitive <strong>co</strong><strong>processing</strong> e<strong>co</strong>nomics<br />
was beyond the s<strong>co</strong>pe <strong>of</strong> the effort, this paper would not be <strong>co</strong>mplete without<br />
presenting some guidelines. For this purpose, the install<strong>at</strong>ion <strong>of</strong> a <strong>co</strong><strong>processing</strong><br />
plant with a residuum throughput <strong>of</strong> 10,000 bbl/day (600-700 tons/day <strong>of</strong> <strong>co</strong>al) to a<br />
refinery (Figure 2) is estim<strong>at</strong>ed to <strong>co</strong>st <strong>of</strong> the order <strong>of</strong> fl70MM. This <strong>co</strong>st includes<br />
<strong>co</strong>al hand1 ing and prepar<strong>at</strong>ion, <strong>co</strong>al/residuum <strong>co</strong>nversion and allowances for<br />
hydrocarbon steam reforming for hydrogen gener<strong>at</strong>ion (- 40% <strong>of</strong> the <strong>co</strong>st). Land and<br />
owner <strong>co</strong>sts are not included in the estim<strong>at</strong>e. In addition, it must be stressed th<strong>at</strong><br />
actual <strong>co</strong>sts are refinery specific and w i l l vary gre<strong>at</strong>ly, depending upon the<br />
adequacy and availability <strong>of</strong> refinery utility systems and the degree <strong>of</strong> integr<strong>at</strong>ion<br />
capabi 1 ity.<br />
Oevel opment Program Requirements<br />
A potentially broad variety <strong>of</strong> <strong>co</strong>als and petroleum residua are candid<strong>at</strong>es for<br />
<strong>co</strong><strong>processing</strong>. The properties <strong>of</strong> these feedstocks will have to be investig<strong>at</strong>ed in<br />
bench scale experiments to define product quality. In addition, better<br />
characteriz<strong>at</strong>ion <strong>of</strong> hydrogen requirements are required to improve e<strong>co</strong>nomies. These<br />
d<strong>at</strong>a are required to facilit<strong>at</strong>e the design and integr<strong>at</strong>ion <strong>of</strong> <strong>co</strong><strong>processing</strong> units<br />
into existing refineries.<br />
Conclusions<br />
Although <strong>co</strong>ntinued Research and Development are required to define product quality<br />
and yields, <strong>co</strong><strong>processing</strong> <strong>of</strong> <strong>co</strong>al and residual <strong>oil</strong> shows promise. It is anticip<strong>at</strong>ed<br />
th<strong>at</strong> initial applic<strong>at</strong>ion <strong>of</strong> <strong>co</strong><strong>processing</strong> will involve the utiliz<strong>at</strong>ion <strong>of</strong> small<br />
amounts <strong>of</strong> <strong>co</strong>al (1-2 wt.%) in existing refineries. This will be followed by<br />
demonstr<strong>at</strong>ion units (10,000-15,000 bbl/day) utilizing a staged approach, <strong>processing</strong><br />
30-40 wt.% <strong>co</strong>al. Commercial units should be able to process up to 50-60 wt.% <strong>co</strong>al<br />
and will be integr<strong>at</strong>ed into high and low <strong>co</strong>nversion refineries using vacuum residua<br />
as feedstocks and th<strong>at</strong> there is a potential for the install<strong>at</strong>ion <strong>of</strong> upwards <strong>of</strong> 100<br />
units <strong>of</strong> 10-15,000 bbl/day capacity.<br />
230
TABLE 1<br />
OIL REFINING PROFILE (CONTINENTIAL USA)<br />
Low High Specialty<br />
Hydroskimming Conversion Conversion Plants<br />
Number <strong>of</strong><br />
Refineries 24 59 61 41<br />
Capacity,<br />
K BBLlday 560 4,685 9,215 475<br />
% Capacity 4 31 62 3<br />
Major's<br />
Oper<strong>at</strong>e, % 30 25 90 Low<br />
Source - Oil and Gas Journal<br />
TABLE 2<br />
IMPACT OF CONVERSION OF EXISTING REFINERY<br />
CAPACITY TO ADVANCE0 COP ROCESSING OF FUTURE HEAVY CRUDES<br />
Coal Consumption, MMTY<br />
Feedstock<br />
Existing Refinery Type Atmospheric Residuum Residuum<br />
Hydroskimning 9 - 11 3 - 6<br />
Low Conversion<br />
High Conversion<br />
74 - 94*<br />
146 - 185*<br />
229 - 290<br />
* Requires Shutdown <strong>of</strong> Existing Units<br />
--Prime Applic<strong>at</strong>ion for Co<strong>processing</strong><br />
231<br />
58 - 99*<br />
91 - 155
FIGURE 1<br />
I DISTRIBUTION OF REFINERIES BY SIZE j<br />
I 1980 to 1985<br />
I THOUSAND BARRELS PER DAY<br />
FIGURE 2<br />
FLOW DIAGRAM<br />
HIGH CONVERSION REFINERIES WITH COPROCESSING<br />
-<br />
232
CHEMICAL AND TOXICOLOGIC CHARACTERIZATION OF CO-PROCESSlh% AND<br />
TWO-STAGE DIRECT COAL LIQUEFAcIlON MATERIALS<br />
Cherylyn W. Wright, Dorothy L. Stewart, D. Dennis Mahlum,<br />
Edward K. Chess, and Bary W. Wilson<br />
Pacific Northwest Labor<strong>at</strong>ory<br />
P. 0. Box 999<br />
Richland, WA 99352<br />
Research and development <strong>of</strong> advanced <strong>co</strong>al liquefaction technology is being supported by the U.S.<br />
Department <strong>of</strong> Energy (DOE) as a means <strong>of</strong> utilizing domestic supplies <strong>of</strong> <strong>co</strong>al to produce petroleum-<br />
substitute fuels. As a <strong>co</strong>mponent <strong>of</strong> this effort, the U.S. DOE has supported the chemical analyses v d<br />
toxi<strong>co</strong>logical evalu<strong>at</strong>ions <strong>of</strong> <strong>co</strong>al <strong>co</strong>nversion products and internal process m<strong>at</strong>erials to assess the potenhal<br />
health effects and industrial hygiene <strong>co</strong>ncerns associ<strong>at</strong>ed with <strong>co</strong>al liquefaction technology.<br />
Recent advances in <strong>co</strong>al liquefaction have included two-stage direct <strong>co</strong>al liquefaction processes and<br />
petroleum residkoa1 <strong>co</strong>-<strong>processing</strong> technology. Two-stage <strong>co</strong>al liquefaction processes are generally<br />
<strong>co</strong>mprised <strong>of</strong> a first-stage thermal or liquefaction reactor followed by a se<strong>co</strong>nd-stage hydrogen<strong>at</strong>ion step.<br />
Petroleum resids and <strong>co</strong>al are simultaneously <strong>co</strong>nverted to liquefaction products in <strong>co</strong>-<strong>processing</strong><br />
technology. The purpose <strong>of</strong> this paper is to report the preliminary results <strong>of</strong> the chemical analysis and<br />
toxi<strong>co</strong>logical testing <strong>of</strong> a <strong>co</strong>al liquefaction <strong>co</strong>-<strong>processing</strong> sample set, and to <strong>co</strong>mpare. these results to those<br />
obtained from two-stage <strong>co</strong>al liquefaction m<strong>at</strong>erials.<br />
SAMPLES<br />
Samples for <strong>co</strong>mpar<strong>at</strong>ive chemical analysis and toxi<strong>co</strong>logical evalu<strong>at</strong>ion were provided from the<br />
proprietary UOP, Inc. <strong>co</strong>-<strong>processing</strong> technology (Des Plaines, Illinois) and the integr<strong>at</strong>ed, non-integr<strong>at</strong>ed,<br />
re<strong>co</strong>nfigured integr<strong>at</strong>ed, and close-<strong>co</strong>upled re<strong>co</strong>nfigured integr<strong>at</strong>ed two-stage <strong>co</strong>al liquefaction processes<br />
(ITSL, NTSL, RITSL, and CCRITSL respectively) from the Wilsonville Advanced Coal <strong>Liquefaction</strong><br />
Research and Development Facility (Wilsonville, Alabama) oper<strong>at</strong>ed by C<strong>at</strong>alytic, Inc. A summary is<br />
<strong>co</strong>ntained in Table 1 <strong>of</strong> the <strong>co</strong>-<strong>processing</strong> samples received from UOP, Inc. and the two-stage <strong>co</strong>al<br />
liquefaction m<strong>at</strong>erials analyzed for <strong>co</strong>mpar<strong>at</strong>ive purposes. Since the samples provided were from pilot<br />
plant or bench-scale advanced <strong>co</strong>al liquefaction origins, they may not necessarily be represent<strong>at</strong>ive <strong>of</strong><br />
m<strong>at</strong>erials produced on a <strong>co</strong>mmercial basis.<br />
A description <strong>of</strong> the proprietary UOP, Inc. c<strong>at</strong>alyzed, slurry, single-stage <strong>co</strong>al liquefaction <strong>co</strong>-<br />
<strong>processing</strong> technology has been given by G<strong>at</strong>sis, et al. (1). ITSL and NTSL processes have been<br />
described by L<strong>at</strong>er (2). For a brief overview <strong>of</strong> the RlTSL and CCRlTSL, see Gough et a1 . (3).<br />
EXPERIMENTAL<br />
Samples were chemically characterized by chemical class fraction<strong>at</strong>ion, gas chrom<strong>at</strong>ography, gas<br />
chrom<strong>at</strong>ography-mass spectrometry, and low-voltage probe-inlet mass spectrometry. Toxi<strong>co</strong>logical<br />
activity was measured using the standard histidine reversion microbial mutagenicity test and an<br />
initi<strong>at</strong>iodpromotion assay for mouse skin tumorigenesis. A brief description <strong>of</strong> these methods follow.<br />
. .<br />
-Class F~aXxmtm<br />
Samples were fraction<strong>at</strong>ed ac<strong>co</strong>rding to the method described by L<strong>at</strong>er er a1 . (4) and L<strong>at</strong>er and Lee (5)<br />
by sequential elution <strong>of</strong> standardized alumina (1.5% w<strong>at</strong>er, L<strong>at</strong>er er al., 6) with 20 ml hexane, 50 ml<br />
233
Table 1. Samples Analyzed<br />
PNLNumber Process Description<br />
5 1396-005<br />
51396-004<br />
51396-001<br />
5 1396-003<br />
51396-002<br />
5226-059<br />
5226-022<br />
50378-100<br />
50378-101<br />
50378-139<br />
UOP<br />
UOP<br />
UOP<br />
UOP<br />
UOP<br />
lTSL<br />
NTSL<br />
RITSL<br />
RlTSL<br />
CCRITSL<br />
Lloydminster Peholeum Resid. Nominal b<strong>oil</strong>ing point (bp) >840"F,<br />
including non-distillables, no solids.<br />
Illinois No. 6 Coal and Lloydminster Pemleum Resid Slurry Feed.<br />
Nominal bp >840"F, including solids and non-distillables.<br />
Liquid Process Solvent (LPS). Nominal bp >212'F, including solids and<br />
non-distillables.<br />
Vacuum Fraction<strong>at</strong>or Overhead Product. Nominal bp -212-910'F.<br />
Vacuum Fraction<strong>at</strong>or Bottoms Product. Nominal bp >910'F, including<br />
solids and non-distillables.<br />
Hydrotre<strong>at</strong>er (HTR) Distill<strong>at</strong>ion Column Bottoms. Nominal bp -450-<br />
850'F. Run #242.<br />
HTRDistill<strong>at</strong>ion Column Bottoms. Nominal bp -450450°F. Run #241.<br />
HTR Heavy Distill<strong>at</strong>e Product. Nominal bp >500'F, including resids and<br />
ash. Run #247.<br />
HTR Heavy Distill<strong>at</strong>e Product. Nominal bp >500'F, less resids and ash.<br />
Run #247.<br />
HTR Heavy Distill<strong>at</strong>e Product. Nominal bp >500T. Run #249.<br />
benzene, 70 ml ch1or<strong>of</strong>orm:ethanol (99:1), and 50 ml methanol to produce aliph<strong>at</strong>ic hydrocarbon (AH),<br />
polycyclic arom<strong>at</strong>ic hydrccarbon (PAH), nitrogen-<strong>co</strong>ntaining polycyclic arom<strong>at</strong>ic <strong>co</strong>mpound WAC), and<br />
hydroxy-substituted PAH (hydroxy-PAH) fractions, respectively. The weight percent <strong>co</strong>ntribution <strong>of</strong><br />
each fraction was determined gravimemcally after solvent removal by rotoevapor<strong>at</strong>ion <strong>at</strong> 40°C and drying<br />
under a stream <strong>of</strong> nitrogen.<br />
Bas Chroma-<br />
Selected fractions were analyzed by gas chrom<strong>at</strong>ography using a Hewlett-Packard (HP) 5880A gas<br />
chrom<strong>at</strong>ograph equipped with a 30-m x 0.25-mm-ID fused silica capillary <strong>co</strong>lumn <strong>co</strong><strong>at</strong>ed with 0.25-pm<br />
film thickness DB-5 (J & W Scientific). The oven was temper<strong>at</strong>ure-programmed to 280'C <strong>at</strong> 4'Clmin after<br />
2 minutes isothermal <strong>at</strong> 50°C with a 5 minute isothermal period <strong>at</strong> the upper temper<strong>at</strong>ure limit. Splitless<br />
injection was used with hydrogen as carrier gas <strong>at</strong> 100 cm/sec linear velocity. The injection port and flame<br />
ioniz<strong>at</strong>ion detector were oper<strong>at</strong>ed <strong>at</strong> 275 and 300'C, respectively. Fractions were analyzed <strong>at</strong> 5.0 mglml<br />
dilutions with 2-chlomanthracene added as an internal standard <strong>at</strong> a fmal <strong>co</strong>ncentr<strong>at</strong>ion <strong>of</strong> 25 ng/@<br />
Gas Chrom<strong>at</strong>ograDhvlMasstmmehv CGCMs1<br />
GCMS analyses were performed on an HP-5982A quadrupole mass spectrometer interfaced to an<br />
HP-5710 gas chrom<strong>at</strong>ograph equipped with a 15-m x 0.25-mm-ID DB-5 fused silica capillary <strong>co</strong>lumn (J<br />
& W Scientific). Gas chrom<strong>at</strong>ographic <strong>co</strong>nditions were similar to those described above, except the oven<br />
was temper<strong>at</strong>ure-programmed <strong>at</strong> 8 Clmin. The MS was oper<strong>at</strong>ed in the electron impact mode <strong>at</strong> 70 eV,<br />
and scan r<strong>at</strong>es were typically 100 <strong>at</strong>omic mass units (amu)/sec.<br />
Low-Volm Probe-- Smhometrv<br />
A VG ZAB 2-F double-focusing MS oper<strong>at</strong>ed in the electrom impact mode using ionizing electron<br />
energies <strong>of</strong> 10-12 eV was used for the LVMS analyses. Each sample (10 to 20 pg) was loaded into a<br />
glass capillary tube, which was then inserted into the source affixed to the end <strong>of</strong> a direct insertion probe.<br />
The probe was he<strong>at</strong>ed in a linear fashion from ambient to 250-280°C while the MS scanned repe<strong>at</strong>edly<br />
throughout the desorption period. The MS was oper<strong>at</strong>ed with an acceler<strong>at</strong>ing voltage <strong>of</strong> 6OOO or 7000 V, a<br />
magnet scan r<strong>at</strong>e <strong>of</strong> 2 to 3 seclmass decade, a source temper<strong>at</strong>ure <strong>of</strong> 250'C, and a dynamic resolving<br />
power (as determined by the VG 2035 d<strong>at</strong>a system) <strong>of</strong> 1:2OOO. The intensities <strong>of</strong> each mass across the<br />
entire pr<strong>of</strong>ile were summed, gener<strong>at</strong>ing an average spectrum th<strong>at</strong> was represent<strong>at</strong>ive <strong>of</strong> the entire sample.<br />
234
Microbial Mu-<br />
..<br />
Standard agar-pl<strong>at</strong>e mutagenicity assays were perfomed as described by Ames er d. (7) using<br />
Sa~moneh typhimurium, TA98 microbial tester strain with S9 metabolic activ<strong>at</strong>ion. Revertant <strong>co</strong>lonies<br />
per petri pl<strong>at</strong>e were <strong>co</strong>unted using a Biotrm II autom<strong>at</strong>ed <strong>co</strong>lony <strong>co</strong>unter. The specific mutagenic activities<br />
<strong>of</strong> samples are expressed as revertant <strong>co</strong>lonies <strong>of</strong> S. typhimurium, TA98 per pg <strong>of</strong> test m<strong>at</strong>erial as<br />
estim<strong>at</strong>ed by linear regression analysis <strong>of</strong> dose-response d<strong>at</strong>a. The following criteria were used for<br />
selecting the best dose range for estim<strong>at</strong>ing a linear dose response: <strong>at</strong> least a four-point dose range;<br />
approxim<strong>at</strong>e doubling <strong>of</strong> response for doubled dose <strong>co</strong>ncentr<strong>at</strong>ion; a <strong>co</strong>rrel<strong>at</strong>ion <strong>co</strong>efficient <strong>of</strong> 0.8 or<br />
gre<strong>at</strong>er; and an intercept on the response (ordin<strong>at</strong>e) axis within 20% <strong>of</strong> the neg<strong>at</strong>ive <strong>co</strong>ntrol for the day.<br />
for Mouse Skin Tu-<br />
. ..<br />
The VP mouse skin tumorigenicity assays were performed on selected samples as described by<br />
Mahlum (8) using female CD-1 mice (Charles River Labor<strong>at</strong>ories, Portage, MI), approxim<strong>at</strong>ely 6 to 8<br />
weeks <strong>of</strong> age with 30 animals per test group. Each test m<strong>at</strong>erial was diluted 1: 1 with acetone or methylene<br />
chloride, and 50 fl <strong>of</strong> the diluted m<strong>at</strong>erial was applied to the shaved backs <strong>of</strong> the mice (approxim<strong>at</strong>ely 25<br />
mg dose per mouse). Two weeks after initi<strong>at</strong>ion, 5-pg doses <strong>of</strong> phorbol myrist<strong>at</strong>e acet<strong>at</strong>e (0.1 mglml<br />
acetone) were applied to the initi<strong>at</strong>ed area, twice weekly for 24 weeks. The mice were shaved as necessary<br />
throughout the study, usually weekly. Animals were observed regularly for tumor growth, and the<br />
number <strong>of</strong> tumors per animal was <strong>co</strong>unted biweekly. The d<strong>at</strong>a are expressed as the total number <strong>of</strong> tumors<br />
per mouse normalized to groups <strong>of</strong> 30 mice.<br />
The results <strong>of</strong> the chemical class fraction<strong>at</strong>ion by alumina <strong>co</strong>lumn chrom<strong>at</strong>ography are given in Table<br />
2 for the advanced <strong>co</strong>al liquefaction products and internal process m<strong>at</strong>erials studied. The Lloydminster<br />
petroleum resid and the slurry feed from the UOP <strong>co</strong>-<strong>processing</strong> technology had low levels <strong>of</strong> AH<br />
<strong>co</strong>mpared to the other m<strong>at</strong>erials fraction<strong>at</strong>ed using these methods. The UOP slurry feed also gave a lower<br />
total re<strong>co</strong>very <strong>of</strong> m<strong>at</strong>erial from the neutral alumina than did the other m<strong>at</strong>erials, indic<strong>at</strong>ing there was a high<br />
<strong>co</strong>ncentr<strong>at</strong>ion <strong>of</strong> insoluble or intractable <strong>co</strong>mponents in the slmy feed presumably due to the presence <strong>of</strong><br />
Table 2. Chemical Class Fraction<strong>at</strong>ion D<strong>at</strong>a<br />
Fraction Weight Percenta<br />
Sampleb Process AH PAH WAC Hydroxy-PAH Total<br />
51396-005 UOP 12 50 36 3 101<br />
51396-004 UOP 8 26 22 2 58<br />
5 1396-001 UOP 26 26 23 10 85<br />
5 1396-003 UOP 53 27 8 6 94<br />
5 1396-002 UOP 19 27 30 11 87<br />
5226-059 ITSL 63 26 5 9 103<br />
5226-022 NTSL 45 34 7 15 101<br />
50378-100 RITSL 58 36 4 2 100<br />
50378-101 RITSL 57 39 4 2 102<br />
50378-139 CCRITSL 60 43 5 4 112<br />
aAverage <strong>of</strong> two determin<strong>at</strong>ions<br />
bFor description, see Table 1<br />
235
the <strong>co</strong>al itself. The chemical <strong>co</strong>mposition <strong>of</strong> the UOP vacuum fraction<strong>at</strong>or overhead product (51396-003)<br />
was <strong>co</strong>mparable to an average <strong>co</strong>mposition <strong>of</strong> the two-stage <strong>co</strong>al liquefaction products, as determined by<br />
this chemical class fraction<strong>at</strong>ion. The UOP bottoms product had a decreased AH <strong>co</strong>mposition and an<br />
increased MAC and hydroxy-PAH <strong>co</strong>ntent <strong>co</strong>mpared to the lower b<strong>oil</strong>ing UOP overhead product.<br />
Similar results have been noted for both single- and two-stage <strong>co</strong>al liquefaction m<strong>at</strong>erials, namely, th<strong>at</strong><br />
higher b<strong>oil</strong>ing fractions have had decreased AH <strong>co</strong>ntent and increased hetero<strong>at</strong>om <strong>co</strong>ntent <strong>co</strong>mpared to<br />
their lower b<strong>oil</strong>ing <strong>co</strong>unterparts (9).<br />
The PAH fractions isol<strong>at</strong>ed from the samples were analyzed in gre<strong>at</strong>er detail since this chemical<br />
fraction has historically been the most tumorigenic fraction isol<strong>at</strong>ed from <strong>co</strong>al liquefaction products and<br />
internal process m<strong>at</strong>erials when analyzed using these methods. High-resolution gas chrom<strong>at</strong>ograms <strong>of</strong> the<br />
PAH fractions isol<strong>at</strong>ed from the UOP, ITSL, and NTSL distilled products are. shown in Figure 1. Many<br />
<strong>of</strong> the major <strong>co</strong>mponents in each <strong>of</strong> these fractions are labelled with their identific<strong>at</strong>ions from retention time<br />
and GCNS d<strong>at</strong>a. Thmajor <strong>co</strong>mponents identified in the UOP vacuum fraction<strong>at</strong>or overheads were<br />
similar to the major <strong>co</strong>mponents identified in both the ITSL and NTSL hydrotre<strong>at</strong>er distill<strong>at</strong>ion <strong>co</strong>lumn<br />
bottoms; PAH <strong>co</strong>mpounds were present ranging from two to four arom<strong>at</strong>ic rings in size. Alkyl-substituted<br />
PAH and some hydroarom<strong>at</strong>ics (particularly <strong>of</strong> m/z 168 and 182, the parent and methyl-substituted<br />
dihydr<strong>of</strong>luorenes or dihydrophenalenes) were also detected in all three products. The <strong>co</strong>mponents<br />
identified in the RITSL and CCRITSLPAH fractions were similar to those detected in the UOP, ITSL,<br />
and NTSL PAH fractions <strong>of</strong> Figure 1, except they were <strong>of</strong> a higher molecular weight range; the<br />
methylchrysene isomer was the <strong>co</strong>mponent <strong>of</strong> highest <strong>co</strong>ncentr<strong>at</strong>ion in both the RITSL and CCRITSL<br />
distilled products.<br />
The LVMS spectra from the analyses <strong>of</strong> the PAH fractions isol<strong>at</strong>ed from the UOP, ITSL, and NTSL<br />
distilled products are shown in Figure 2. The UOP product PAH fraction was more <strong>co</strong>mplex than either<br />
<strong>of</strong> the two-stage <strong>co</strong>al liquefaction PAH fractions shown. For example, there were signals for a gre<strong>at</strong>er<br />
number <strong>of</strong> masses representing 40% or more <strong>of</strong> the total ion current (nC) in the UOP product PAH<br />
fractions than there were for the ITSL and NTSL PAH fractions. There was also rel<strong>at</strong>ively more m<strong>at</strong>erials<br />
th<strong>at</strong> gave rise to the series including masses 232, 246, 260, and 274 amu in the UOP distilled product<br />
PAH fractions as <strong>co</strong>mpared to the ITSL and NTSL distilled product PAH fractions, showing some<br />
differences in the <strong>co</strong>mposition <strong>of</strong> the <strong>co</strong>-<strong>processing</strong> and two-stage <strong>co</strong>al liquefaction samples.<br />
Table 3 <strong>co</strong>ntains the results <strong>of</strong> microbial mutagenicity testing <strong>of</strong> the crudes and chemical class<br />
fractions isol<strong>at</strong>ed from some <strong>of</strong> the advanced <strong>co</strong>al liquefaction samples studied. No mutagenic activity was<br />
detected in any <strong>of</strong> the AH or PAH fractions isol<strong>at</strong>ed from the UOP petroleum residkoa1 <strong>co</strong>-<strong>processing</strong><br />
m<strong>at</strong>erials, as was also the case for the distilled two-stage <strong>co</strong>al liquefaction products. Regardless <strong>of</strong><br />
process, the majority <strong>of</strong> the microbial mutagenicity was expressed by the isol<strong>at</strong>ed WAC fractions, with<br />
Table 3. Microbial Mutagenicity D<strong>at</strong>a<br />
Response (rev&); Chemical Class Fraction<br />
Samplea Process Crude AH PAH NPAC Hydr~xy-PAH<br />
51396-004 UOP 0 0 0 0
Some mutagenic response also expressed by the hydroxy-PAH fractions (particularly in the UOP vacuum<br />
fraction<strong>at</strong>or bottoms product, 5 1396-002). The microbial mutagenic response <strong>of</strong> the UOP vacuum<br />
fraction<strong>at</strong>or overhead product more closely resembled the response <strong>of</strong> the NTSL and RITSL distilled<br />
products, showing increased mutagenic activity as <strong>co</strong>mpared to the lTSL distilled product.<br />
hiti<strong>at</strong>idpromotion results, given as total number <strong>of</strong> tumors per mouse f standard error, for the lTSL<br />
and NTSL hydrotre<strong>at</strong>er distill<strong>at</strong>ion <strong>co</strong>lumn bottoms were 1.3 f 1.2 and 1.1 f 1.4, respectively.<br />
ACKNOWLEDGMENTS<br />
This work was supported by the U. S. Department <strong>of</strong> Energy, Office <strong>of</strong> Fossil Energy under Contract<br />
NO. DE-AC06-76RLO-1830 to the Pacific Northwest Labor<strong>at</strong>ory. The authors thank UOP, Inc. and<br />
C<strong>at</strong>alytic, Inc. for supplying the advanced <strong>co</strong>al liquefaction products and internal process m<strong>at</strong>erials.<br />
REFERENCES<br />
1. G<strong>at</strong>sis, J. G., J. G. Sikonia, B. J. Nelson, C. P. Luebke, and M. J. Humbach. 1986. Coal<br />
<strong>Liquefaction</strong> Co-Processing. In: Proceedings Direct <strong>Liquefaction</strong> Contractors' Review Meeting,<br />
November 19-21, 1985, Pittsburgh, Pennsylvania, U. S. Department <strong>of</strong> Energy Pittsburgh Energy<br />
Technology Center.<br />
2. L<strong>at</strong>er, D. W. 1985. Two-Stage Coal Liquefnction Process M<strong>at</strong>erials from the Wilsonville Facility<br />
Oper<strong>at</strong>ed in the Nonintegr<strong>at</strong>ed and Integr<strong>at</strong>ed Modes: Chemical Analyses and Biological Testing . PNL-<br />
5215, Pacific Northwest Labor<strong>at</strong>ory, Richland, Washington, NTIS, Springfield, Virginia.<br />
3. Gough, J. R., C. W. Lamb, R. V. Nalitham, H. M. Risbud, and T. W. Johnson. 1986. Recent<br />
Developments in Two-Stage Coal <strong>Liquefaction</strong> <strong>at</strong> Wilsonville. In: Proceedings Direct <strong>Liquefaction</strong><br />
Contractors' Review Meeting, November 19-21, 1985, Pittsburgh, Pennsylvania, U. S. Department <strong>of</strong><br />
Energy Pittsburgh Energy Technology Center.<br />
4. L<strong>at</strong>er, D. W., M. L. Lee, K. D. Bartle, R. C. Kong, and D. L. Vassilaros. 1981. Chemical class<br />
separ<strong>at</strong>ion and chracteriz<strong>at</strong>ion <strong>of</strong> organic <strong>co</strong>mpounds in synthetic fuels. Anal. Chem. 53: 1612-1620.<br />
5. L<strong>at</strong>er, D. W. and M. L. Lee. 1983. Chrom<strong>at</strong>ographic methods for the chemical and biological<br />
characteriz<strong>at</strong>ion <strong>of</strong> polycyclic arom<strong>at</strong>ic <strong>co</strong>mpounds in synfuel m<strong>at</strong>erials, pp. 44-73. In: Advanced<br />
Techniques in Synthetic Fuels Analysis , C. W. Wright, W. C. Weimer, and W. D. Felix, eds., CONF-<br />
81 160, NTIS, Springfield, Virginia.<br />
6. L<strong>at</strong>er, D. W., B. W. Wilson, and M. L. Lee. 1985. Standardiz<strong>at</strong>ion <strong>of</strong> Alumina and Silica<br />
Adsorbents Used for Chemical Class Separ<strong>at</strong>ions <strong>of</strong> Polycyclic Arom<strong>at</strong>ic Compounds. Anal. Chem. 57:<br />
2979-2984.<br />
7. Ames, B. N., J. McCann, and E. Yamasaki. 1975. Methods for detecting carcinogens and mutagens<br />
with the Salmone[lu/mammalian-microsome mutagenicity test Mur<strong>at</strong>. Res. 35: 247-364.<br />
8. Mahlum, D. D. 1983. Initi<strong>at</strong>ion/promotion studies with <strong>co</strong>al-derived liquids. J. Appl. Toxi<strong>co</strong>l. 3: 31-<br />
34.<br />
9. Wright, C. W., D. W. L<strong>at</strong>er, D. D. Dauble, and B. W. Wilson. 1985. Fractionally DistilledSRC-I,<br />
SRC-II, EDS, H-Coal andlTSL Direct Coal <strong>Liquefaction</strong> Process M<strong>at</strong>erials: A Compar<strong>at</strong>ive Summary <strong>of</strong><br />
Chemical Analysis and Biological Testing. PNL-5528, Pacific Northwest Labor<strong>at</strong>ory, Richland, WA,<br />
NTIS, Springfield, VA.<br />
237
- -<br />
ITSL<br />
PAH Fraction<br />
PAH Fraction<br />
I I<br />
0 20 Time, minutes 40 60<br />
I I I I I<br />
50 100 150 200 250<br />
Temper<strong>at</strong>ure, OC<br />
Figure 1. High Resolution Gas Chrom<strong>at</strong>ograms <strong>of</strong> the PAH Fractions Isol<strong>at</strong>ed from the UOP Vacuum<br />
Fraction<strong>at</strong>or Overhead Product (top), ITSL (middle) and NTSL (bottom) Hydrotre<strong>at</strong>er Distill<strong>at</strong>ion Column<br />
Bottoms. See Text for Conditions.
%I<br />
,100<br />
60<br />
40<br />
20<br />
0<br />
100 222<br />
%I<br />
EO -<br />
60<br />
40<br />
20<br />
0<br />
100<br />
20<br />
I .^^ I II I II 11111 11111111111l lllll 111, I I<br />
UOP<br />
PAH Fraction<br />
ITSL<br />
PAH Fraction<br />
NTSL<br />
PAH Fraction<br />
0<br />
100 200 300 400 5 0<br />
amu<br />
Figure 2. LVMS Spectra. See Figure 1 for Sample Descriptions. See Text for Conditions.<br />
239
PROCESS DEVELOPMENT STUDIES OF<br />
TWO-STAGE LIQUEFACTION AT WILSONVILLE<br />
C. W. Lamb, R. V. Nalitham<br />
C<strong>at</strong>alytic, Inc., Wilsonville, AL 35186<br />
and T. W. Johnson<br />
Southern Company Services, Inc., Wilsonville, AL 35186<br />
INTRODUCTION<br />
The Advanced Coal <strong>Liquefaction</strong> RAD Facility <strong>at</strong> Wilsonville. Alabama, has been in oper<strong>at</strong>ion<br />
for over 12 years. It is the largest direct <strong>co</strong>al liquefaction pilot plant still in<br />
oper<strong>at</strong>ion in the United St<strong>at</strong>es. Process investig<strong>at</strong>ions have evolved from the original<br />
study <strong>of</strong> the Solvent Refined Coal Process for making a clean solid fuel to the recent<br />
investig<strong>at</strong>ion <strong>of</strong> two-stage liquefaction processes for making clean distill<strong>at</strong>e fuels. This<br />
paper presents results from the current study <strong>of</strong> various <strong>processing</strong> schemes designed to<br />
reduce the <strong>co</strong>st <strong>of</strong> fuels produced by two-stage liquefaction plants.<br />
Most important among these <strong>co</strong>nfigur<strong>at</strong>ions is direct <strong>co</strong>upling <strong>of</strong> the thermal and c<strong>at</strong>alytic<br />
reactors. Such close-<strong>co</strong>upled oper<strong>at</strong>ion should lower the <strong>co</strong>st <strong>of</strong> two-stage fuels by<br />
increasing overall thermal efficiency and improving product quality. Results from<br />
W i l sonville runs are characterized by discussion <strong>of</strong> represent<strong>at</strong>ive product yield and<br />
product quality d<strong>at</strong>a. Also, a <strong>co</strong>mparison <strong>of</strong> performance and stability <strong>of</strong> Shell 324 and<br />
Amoc<strong>at</strong> 1C c<strong>at</strong>alysts is presented.<br />
Other pertinent tests in the close-<strong>co</strong>upled mode are discussed with particular emphasis on<br />
the effect <strong>of</strong> higher system space velocity and on the impact <strong>of</strong> solids recycle.<br />
PROCESS CONFIGURATION COMPARISON<br />
The integr<strong>at</strong>ed TSL (ITSL) mode and the close-<strong>co</strong>upled (CC-ITSL) mode are shown in Figures 1<br />
and 2, respectively. In both modes, the reaction stages are integr<strong>at</strong>ed by the recycle <strong>of</strong><br />
hydrotre<strong>at</strong>ed resid with the goal <strong>of</strong> producing "all-distill<strong>at</strong>e'' yield sl<strong>at</strong>es. The CC-ITSL<br />
<strong>co</strong>nfigur<strong>at</strong>ion differs from ITSL in th<strong>at</strong> for the CC-ITSL mode, products from the thermal<br />
reactor are fed directly to the c<strong>at</strong>alytic reactor without pressure letdown or significant<br />
<strong>co</strong>oling. Ac<strong>co</strong>rdingly, vacuum-flashed product from CC-ITSL is deashed downstream <strong>of</strong> the<br />
c<strong>at</strong>alytic reactor r<strong>at</strong>her than upstream as in the ITSL mode. Close-<strong>co</strong>upled oper<strong>at</strong>ion thus<br />
elimin<strong>at</strong>es thermal inefficiency <strong>of</strong> the <strong>co</strong>ol down/rehe<strong>at</strong> cycle associ<strong>at</strong>ed with deashing<br />
between reaction stages.<br />
PROCESS PERFORMANCE<br />
A wide range <strong>of</strong> thermal and c<strong>at</strong>alytic stage oper<strong>at</strong>ing <strong>co</strong>nditions were investig<strong>at</strong>ed in the<br />
CC-ITSL mode to determine their effects on product yield structure and quaiity. For<br />
<strong>co</strong>mparison purposes, Table 1 l ists five sets <strong>of</strong> CC-ITSL <strong>co</strong>nditions and yields rel<strong>at</strong>ive to<br />
one typical set <strong>of</strong> ITSL <strong>co</strong>nditions and yields. Illinois No. 6 <strong>co</strong>al from Burning Star Mine<br />
was used in all the runs.<br />
An initial baseline run (250B) was <strong>co</strong>nducted using aged Shell 324 c<strong>at</strong>alyst to quantify the<br />
impact <strong>of</strong> <strong>co</strong>upling the reactors. No interstage-vapor separ<strong>at</strong>ion was utilized in Run 250B,<br />
although a high pressure separ<strong>at</strong>or was installed and oper<strong>at</strong>ed during the remainder <strong>of</strong> the<br />
run. The major effect <strong>of</strong> close-<strong>co</strong>upled oper<strong>at</strong>ion was an increase in hydrogen <strong>co</strong>nsumption<br />
with a <strong>co</strong>rresponding improvement in distill<strong>at</strong>e product quality (Table 2). Hydrogen and<br />
sulfur values were significantly better for CC-ITSL (250B) when <strong>co</strong>mpared to ITSL (244).<br />
This improvement resulted from the fact th<strong>at</strong> all the TSL product was derived from the<br />
240
c<strong>at</strong>alytic stage in Run 2506, whereas in Run 244 approxim<strong>at</strong>ely 45 wt % was produced by the<br />
c<strong>at</strong>alytic hydrotre<strong>at</strong>er (Table 3). Higher quality products are made by the c<strong>at</strong>alytic<br />
stage.<br />
Subsequent oper<strong>at</strong>ion in CC-ITSL was devoted to study <strong>of</strong> performance <strong>of</strong> a new c<strong>at</strong>alyst,<br />
Amoc<strong>at</strong> 1C. As noted previously, interstage vapor separ<strong>at</strong>ion was employed throughout this<br />
period. A <strong>co</strong>mparison <strong>of</strong> "all-distill<strong>at</strong>e'' oper<strong>at</strong>ion <strong>at</strong> the same <strong>co</strong>al feed r<strong>at</strong>e for Shell<br />
324 and Amoc<strong>at</strong> 1C revealed th<strong>at</strong> distill<strong>at</strong>e and hydrocarbon gas yields were similar (Run<br />
2508 versus Run 250C). An altern<strong>at</strong>e deashing solvent was used in Run 250C in order to<br />
maintain stable perfonnance while <strong>processing</strong> the highly soluble feed gener<strong>at</strong>ed by the<br />
fresh Amoc<strong>at</strong> c<strong>at</strong>alyst. This resulted in gre<strong>at</strong>er rejection <strong>of</strong> resid to ash <strong>co</strong>ncentr<strong>at</strong>e<br />
with <strong>co</strong>n<strong>co</strong>mitantly less TSL resid produced. A lower hydrogen <strong>co</strong>nsumption was observed<br />
for Run 250C. This phenomenon <strong>co</strong>uld be <strong>at</strong>tributed to the effect <strong>of</strong> interstage separ<strong>at</strong>ion<br />
(i.e., not hydrotre<strong>at</strong>ing all distill<strong>at</strong>e products) and/or to rel<strong>at</strong>ively lower hydrogen<strong>at</strong>ion<br />
activity <strong>of</strong> the Amoc<strong>at</strong> c<strong>at</strong>alyst.<br />
Higher system space velocities were explored by increasing <strong>co</strong>al feed r<strong>at</strong>es. The goal was<br />
to demonstr<strong>at</strong>e the production <strong>of</strong> high quality (CC-ITSL) distill<strong>at</strong>es <strong>at</strong> reduced <strong>co</strong>st. Yield<br />
d<strong>at</strong>a are reported for Run 250 Periods C, 0, and E in Table 1. Product quality d<strong>at</strong>a and<br />
unit <strong>co</strong>ntributions to distill<strong>at</strong>e production for Period D are given in Tables 2 and 3,<br />
respectively. The d<strong>at</strong>a clearly indic<strong>at</strong>e th<strong>at</strong> "all-distill<strong>at</strong>e'' yield sl<strong>at</strong>es were produced<br />
<strong>at</strong> increased throughputs by <strong>co</strong>mpens<strong>at</strong>ory increases in reactor temper<strong>at</strong>ures. Product<br />
quality did not change significantly <strong>at</strong> the higher r<strong>at</strong>es. It should also be noted th<strong>at</strong><br />
products from higher space velocity CC-ITSL were substantially better than those from<br />
lower space velocity ITSL.<br />
Near the <strong>co</strong>nclusion <strong>of</strong> Run 250, a test <strong>of</strong> solids (un<strong>co</strong>nverted <strong>co</strong>al and ash-cresol insolu-<br />
bles) recycle was performed (Figure 3). The objective was to decrease size <strong>of</strong> the<br />
critical solvent deashing unit by deashing a higher solids <strong>co</strong>ntent vacuum bottoms stream.<br />
Deashed resid was recycled as a <strong>co</strong>mponent <strong>of</strong> the liquefaction solvent. Lower organic<br />
rejection to the ash <strong>co</strong>ncentr<strong>at</strong>e was demonstr<strong>at</strong>ed with the <strong>co</strong>ncentr<strong>at</strong>ed feed. Based on<br />
this result, the <strong>co</strong>ncept <strong>of</strong> solids recycle may be investig<strong>at</strong>ed in a future close-<strong>co</strong>upled<br />
run using c<strong>at</strong>alyst in both reactors.<br />
B<strong>at</strong>ch deactiv<strong>at</strong>ion trends for resid <strong>co</strong>nversion in the c<strong>at</strong>alytic reactor were developed for<br />
the ITSL (deashed bituminous)- Shell 324 and CC-ITSL (close-<strong>co</strong>upled bituminous)-Amoc<strong>at</strong> 1C<br />
modes, using a first order resid <strong>co</strong>nversion model (1). The trends are plotted in Figure 4<br />
together with b<strong>at</strong>ch deactiv<strong>at</strong>ion d<strong>at</strong>a from Wilsonville Run 247 ("simul<strong>at</strong>ed" close-<br />
<strong>co</strong>upled). The trends showed initial periods <strong>of</strong> rapid deactiv<strong>at</strong>ion, followed by slower<br />
deactiv<strong>at</strong>ion r<strong>at</strong>es. Higher resid <strong>co</strong>nversion r<strong>at</strong>e <strong>co</strong>nstants were observed for close-<br />
<strong>co</strong>upled oper<strong>at</strong>ion using the Amoc<strong>at</strong> 1C c<strong>at</strong>alyst. The close-<strong>co</strong>upled residlhoc<strong>at</strong> <strong>co</strong>mbina-<br />
tion was more reactive than the other feedlc<strong>at</strong>alyst (Shell 324) <strong>co</strong>mbin<strong>at</strong>ions. Figure 5<br />
further illustr<strong>at</strong>es this point in an Arrhenius plot.<br />
RELATIVE ECONOMICS<br />
Results from an e<strong>co</strong>nomic screening study performed by Lummus Crest Inc. indic<strong>at</strong>ed a<br />
reduction in the required product selling price for CC-ITSL products <strong>co</strong>mpared to ITSL<br />
products (Table 41. The study was based on a <strong>co</strong>nceptual <strong>co</strong>mnercial 10,000 TPD plant using<br />
Illinois No. 6 <strong>co</strong>al. The rel<strong>at</strong>ively lower price was due to higher distill<strong>at</strong>e production<br />
and improved distill<strong>at</strong>e quality for the close-<strong>co</strong>upled case (2).<br />
241
SUMMARY<br />
e The major effect <strong>of</strong> close-<strong>co</strong>upled oper<strong>at</strong>lon was an increase in hydrogen <strong>co</strong>n-<br />
sumption with a <strong>co</strong>rresponding improvement in distill<strong>at</strong>e product quality.<br />
a At the same <strong>co</strong>al feed r<strong>at</strong>e, C4+ distill<strong>at</strong>e and hydrocarbon gas yields were<br />
similar for close-<strong>co</strong>upled oper<strong>at</strong>ion with Shell 324 and Amoc<strong>at</strong> 1C c<strong>at</strong>alysts. A<br />
lower hydrogen <strong>co</strong>nsumption was observed in the Amoc<strong>at</strong> oper<strong>at</strong>ion. This phenome-<br />
non <strong>co</strong>uld be <strong>at</strong>tributed to the effect <strong>of</strong> interstage separ<strong>at</strong>ion and/or to the<br />
rel<strong>at</strong>ively lower hydrogen<strong>at</strong>ion activity <strong>of</strong> the Amoc<strong>at</strong> c<strong>at</strong>alyst.<br />
a<br />
"All-distill<strong>at</strong>e'' yield sl<strong>at</strong>es were produced <strong>at</strong> higher system space velocities by<br />
<strong>co</strong>mpens<strong>at</strong>ory increases in reactor temper<strong>at</strong>ures.<br />
e Product quality was not significantly affected <strong>at</strong> higher system space veloci-<br />
ties.<br />
a Oper<strong>at</strong>ing with solids recycle in the close-<strong>co</strong>upled mode, lower organic rejection<br />
to ash <strong>co</strong>ncentr<strong>at</strong>e was demonstr<strong>at</strong>ed <strong>at</strong> a rel<strong>at</strong>ive reduction in feed r<strong>at</strong>e to the<br />
critical solvent deashing unft.<br />
a<br />
A <strong>co</strong>mparison <strong>of</strong> resid <strong>co</strong>nversion r<strong>at</strong>e <strong>co</strong>nstants indic<strong>at</strong>ed th<strong>at</strong> the close-<strong>co</strong>upled<br />
resid/Amoc<strong>at</strong> 1C <strong>co</strong>mbin<strong>at</strong>ion was more reactive than other feed/c<strong>at</strong>alyst (Shell<br />
324) <strong>co</strong>mbin<strong>at</strong>ions.<br />
a Results from an e<strong>co</strong>nomic screening study for a <strong>co</strong>nceptual <strong>co</strong>mmercial 10,000 TPD<br />
plant indic<strong>at</strong>ed a reduction in the required product selling price for CC-ITSL<br />
plant products <strong>co</strong>mpared to ITSL plant products.<br />
ACKNOWLEDGMENTS<br />
This work was supported by the U. S. Department <strong>of</strong> Energy under Contract<br />
DE-AC22-82PC50041, and by the Electric Power Research Institute under Contract RP1234-1-2.<br />
The author also gr<strong>at</strong>efully appreci<strong>at</strong>es the assistance <strong>of</strong> Ms. Joyce Spearman and Messrs.<br />
Bill Hollenack, Johnny Gill, and Dave Edwards.<br />
REFERENCES<br />
1. Rao, A. K., Gadiyar, H. J., P<strong>at</strong>e, F. L., "C<strong>at</strong>alytic Hydrogen<strong>at</strong>ion <strong>of</strong> SRC-I Product <strong>at</strong><br />
the Wilsonville Pilot Plant", Proceedings <strong>of</strong> the Seventh Annual EPRI Contractors<br />
Conference on Coal <strong>Liquefaction</strong>, May 1982.<br />
2.<br />
Corrypondence from M. Peluso to W. Weber, "RP832-11, Final Technical and E<strong>co</strong>nomic<br />
D<strong>at</strong>a , January 23, 1986.<br />
242
I<br />
OlnO?<br />
mum<br />
m m-<br />
N<br />
mho?<br />
N m W<br />
w m-<br />
N<br />
zsga?<br />
m m-<br />
N<br />
zgz?<br />
m 0-<br />
N<br />
243<br />
-<br />
m<br />
0-0<br />
d -V)<br />
hNh<br />
4<br />
hUNN W I N W *<br />
V)+NN?<br />
W NV)<br />
I
omw<br />
'??h<br />
4NO<br />
WNID<br />
?N?<br />
000<br />
wmm<br />
c9'1'9<br />
Nom<br />
4-4<br />
.+e-<br />
???<br />
mwm<br />
mwm<br />
244<br />
-4W<br />
,??<br />
00<br />
000<br />
NNm<br />
N??<br />
000<br />
ulrnl.<br />
NhN
Configur<strong>at</strong>ion<br />
Run No.<br />
C<strong>at</strong>alyst Type<br />
Unit Contributions(3)<br />
Thermal Stage<br />
C<strong>at</strong>alytic Stage<br />
Table 3<br />
UNIT CONTRIBUTIONS TO DISTILLATE PRODUCTION<br />
ITSL<br />
244<br />
Shell 324<br />
55<br />
45<br />
(1) Without interstage vapor separ<strong>at</strong>ion.<br />
(2) With interstage vapor separ<strong>at</strong>ion.<br />
(3) W t % <strong>of</strong> total distill<strong>at</strong>e production.<br />
Rel<strong>at</strong>ive Capital Cost<br />
Rel<strong>at</strong>ive Oper<strong>at</strong>ing Cost<br />
Re1 <strong>at</strong>ive Production R<strong>at</strong>e(2)<br />
Rel<strong>at</strong>ive Required Product Selling Price(3)<br />
Thermal Efficiency ($1<br />
Table 4<br />
CC-ITSL(~)<br />
2508<br />
Shell 324<br />
0<br />
100<br />
ECONOMIC SCREENING STUDY(^)<br />
(10,000 TPD PLANT)<br />
CC-ITSL (2)<br />
250D<br />
Amoc<strong>at</strong> 1C<br />
25<br />
75<br />
ITSL (e SE \ cc- TSL<br />
1 .oo<br />
1 .oo<br />
1 .oo<br />
1.00<br />
(1) Performed by Lumnus Crest, Inc. under EPRI RP832-11.<br />
(2) Based on barrels <strong>of</strong> crude <strong>oil</strong> equivalent with <strong>co</strong>nsider<strong>at</strong>ion for quality <strong>of</strong><br />
di sti 11 <strong>at</strong>e products.<br />
(3) First year.<br />
245<br />
68.1<br />
1.04<br />
1.01<br />
1.10<br />
0.93<br />
69.6
tl f<br />
246
I<br />
i<br />
T<br />
247
248
I-YH'Y 'lNVlSN03 31W NOISa3AN03 alS3U<br />
249
qkCqQ r! o!<br />
0 0 0 0 0 0 0<br />
I-UH ')I 'INUSNO3 31VU NOISU3hN03 OIS3M<br />
250<br />
-<br />
0 W<br />
s a<br />
c3<br />
i
?HE IMPACT OF THE CHEMICAL CONSTITUENTS OF HYDROTREATER<br />
FEED ON CATALYST ACTIVITY*<br />
Frances V. Stohl and Howard P. Stephens<br />
Sandia N<strong>at</strong>ional Labor<strong>at</strong>ories, Albuquerque, NM 87185<br />
INTRODUCTION<br />
The deposition <strong>of</strong> carbonaceous m<strong>at</strong>erial on direct <strong>co</strong>al<br />
liquefaction c<strong>at</strong>alysts is known to cause rapid and significant<br />
c<strong>at</strong>alyst deactiv<strong>at</strong>ion (1,2). Studies <strong>of</strong> hydrotre<strong>at</strong>er c<strong>at</strong>alyst<br />
samples from several different runs <strong>at</strong> the Wilsonville Advanced<br />
Coal <strong>Liquefaction</strong> R & D Facility have shown th<strong>at</strong> gre<strong>at</strong>er than 75%<br />
<strong>of</strong> their hydrogen<strong>at</strong>ion activity and 50% <strong>of</strong> their hydrodesulfuriz<strong>at</strong>ion<br />
activity were lost within the first few days <strong>of</strong> <strong>co</strong>al<br />
<strong>processing</strong> (3). Hydrotre<strong>at</strong>ing light thermal resid from the third<br />
stage <strong>of</strong> the Kerr McGee critical solvent deasher yielded the least<br />
deactiv<strong>at</strong>ion whereas hydrotre<strong>at</strong>ing the heavier nondeashed resid<br />
yielded the largest buildup <strong>of</strong> carbonaceous deposits and the<br />
gre<strong>at</strong>est deactiv<strong>at</strong>ion. These trends were due to differences in<br />
the <strong>co</strong>mpositions <strong>of</strong> the hydrotre<strong>at</strong>er feeds. Previously reported<br />
work (4) has shown th<strong>at</strong> carbonaceous deposits cause homogeneous<br />
poisoning <strong>of</strong> active sites and about a 50% decrease in the c<strong>at</strong>alyst<br />
effective diffusivity, which is the diffusion <strong>co</strong>efficient within<br />
the extrud<strong>at</strong>es.<br />
As a result <strong>of</strong> the work on the Wilsonville c<strong>at</strong>alysts, we have<br />
initi<strong>at</strong>ed a program to identify the hydrotre<strong>at</strong>er feed <strong>co</strong>mponents<br />
th<strong>at</strong> are most detrimental to c<strong>at</strong>alyst activity. Studies <strong>of</strong> the<br />
effect <strong>of</strong> hydrotre<strong>at</strong>er feed b<strong>oil</strong>ing point cut on c<strong>at</strong>alyst activity<br />
(5) have shown th<strong>at</strong> <strong>processing</strong> a -550F <strong>co</strong>mponent yields a 23%<br />
decrease in extrud<strong>at</strong>e hydrogen<strong>at</strong>ion activity whereas hydrotre<strong>at</strong>ing<br />
an 850F+ <strong>co</strong>mponent results in an 82% loss. Although hydro-<br />
desulfuriz<strong>at</strong>ion activity was not affected by the low b<strong>oil</strong>ing<br />
fraction, a 70% loss resulted from hydrotre<strong>at</strong>ing the highest<br />
b<strong>oil</strong>ing fraction.<br />
In this paper we report the impacts on c<strong>at</strong>alyst activity <strong>of</strong><br />
four different chemical classes <strong>of</strong> <strong>co</strong>mpounds found in hydrotre<strong>at</strong>er<br />
feeds. These chemical classes included the aliph<strong>at</strong>ic hydro-<br />
carbons, neutral polycyclic arom<strong>at</strong>ic <strong>co</strong>mpounds (PAC), nitrogen<br />
polycyclic arom<strong>at</strong>ic <strong>co</strong>mpounds (N-PAC) and hydroxy polycyclic<br />
arom<strong>at</strong>ic hydrocarbons (HPAH).<br />
EXPERIMENTAL PROCEDURES<br />
A hydrotre<strong>at</strong>er process stream obtained from the Wilsonville<br />
facility and four classes <strong>of</strong> chemical <strong>co</strong>mpounds separ<strong>at</strong>ed from<br />
this stream were each c<strong>at</strong>alytically hydrogen<strong>at</strong>ed in microreactors.<br />
The starting feeds and used c<strong>at</strong>alysts from these experiments were<br />
then characterized and the c<strong>at</strong>alysts were tested for hydrogen<strong>at</strong>ion<br />
activity.<br />
* This work supported by the U. S. Dept. <strong>of</strong> Energy <strong>at</strong> Sandia<br />
N<strong>at</strong>ional Labor<strong>at</strong>ories under Contract DE-AC04-76DP00789.<br />
251
M<strong>at</strong>erials<br />
The c<strong>at</strong>alyst was shell 324M with 12.4 wt% Mo and 2.8 wt% N i on<br />
an alumina support in the form <strong>of</strong> extrud<strong>at</strong>es measuring about 0.8<br />
mm in diameter and 4 mm in length. Prior to use, the c<strong>at</strong>alyst was<br />
presulfided with a 10 mol% H2S in H2 mixture <strong>at</strong> 400C and<br />
<strong>at</strong>mospheric pressure for two hours. The V-178 hydrotre<strong>at</strong>er<br />
process stream used in this study was obtained from the<br />
Wilsonville facility's run 247, which processed Illinois #6<br />
bituminous <strong>co</strong>al in the Re<strong>co</strong>nfigured Integr<strong>at</strong>ed Two-Stage<br />
<strong>Liquefaction</strong> process <strong>co</strong>nfigur<strong>at</strong>ion(6). The V-178 stream,<br />
identified by the number <strong>of</strong> the storage tank from which it was<br />
derived prior to entering the hydrotre<strong>at</strong>er, is the light portion<br />
<strong>of</strong> the hydrotre<strong>at</strong>er feed and <strong>co</strong>mprises about 35 wt% <strong>of</strong> the total<br />
feed. Distill<strong>at</strong>ion <strong>of</strong> the V-178 showed th<strong>at</strong> the initial b<strong>oil</strong>ing<br />
point was 400F and 96.1 wt% b<strong>oil</strong>ed below 850F (5).<br />
The V-178 process stream was separ<strong>at</strong>ed into four chemical<br />
classes by adsorption <strong>co</strong>lumn chrom<strong>at</strong>ography using neutral aluminum<br />
oxide (7). A 10 g sample was dissolved in chlor<strong>of</strong>orm and adsorbed<br />
onto 50 g alumina, which was then dried and placed on top <strong>of</strong> 100 g<br />
alumina in a 22 mm id <strong>co</strong>lumn. The aliph<strong>at</strong>ic hydrocarbon fraction<br />
was eluted first using hexane, then the PAC using benzene,<br />
followed by the N-PAC using chlor<strong>of</strong>orm and the HPAH using 10%<br />
ethanol in tetrahydr<strong>of</strong>uran. Solvent was removed from each<br />
fraction by evapor<strong>at</strong>ion under vacuum.<br />
Hydrotr e<strong>at</strong> ing Fxper iments<br />
Each chemical class and the V-178 process stream were<br />
hydrotre<strong>at</strong>ed with presulfided c<strong>at</strong>alyst in 26 cc b<strong>at</strong>ch<br />
microreactors <strong>at</strong> 300C for 2 hours with a 1100 psig H2 <strong>co</strong>ld<br />
charge pressure. The microreactors were charged with 0.5 g feed,<br />
0.17 g presulfided c<strong>at</strong>alyst and 1.5 g hexadecane, which was added<br />
to provide adequ<strong>at</strong>e mixing in the reactors because <strong>of</strong> the small<br />
amounts <strong>of</strong> feed available. The aged c<strong>at</strong>alysts were Soxhlet<br />
extracted with tetrahydr<strong>of</strong>uran prior to analysis or activity<br />
testing. Elemental analyses <strong>of</strong> the V-178 stream, the four<br />
fractions and the aged c<strong>at</strong>alysts were performed using standard<br />
methods.<br />
Activity Testing<br />
Hydrogen<strong>at</strong>ion activities <strong>of</strong> fresh and aged c<strong>at</strong>alysts were<br />
determined by measuring the r<strong>at</strong>e <strong>of</strong> hydrogen<strong>at</strong>ion <strong>of</strong> pyrene to<br />
dihydropyrene (4) in 26 cc microreactors <strong>at</strong> 300C with 450 psig H<br />
<strong>co</strong>ld charge pressure. Experiments with c<strong>at</strong>alyst ground to -200<br />
mesh and whole extrud<strong>at</strong>es enabled determin<strong>at</strong>ion <strong>of</strong> the losses <strong>of</strong><br />
both intrinsic and extrud<strong>at</strong>e activities respectively.<br />
Feed and C<strong>at</strong>alyst Compositions<br />
RESULTS AND DISCUSSION<br />
The <strong>co</strong>mpositions <strong>of</strong> the V-178 stream and the amounts and<br />
<strong>co</strong>mpositions <strong>of</strong> the four separ<strong>at</strong>ed chemical classes, given in<br />
Table 1, show th<strong>at</strong> the V-178 <strong>co</strong>ntains significant amounts <strong>of</strong><br />
aliph<strong>at</strong>ic hydrocarbons and the PAC fraction, and only low<br />
<strong>co</strong>ncentr<strong>at</strong>ions <strong>of</strong> nitrogen and hydroxy <strong>co</strong>mpounds. The 95% total<br />
re<strong>co</strong>very for the four chemical classes is good for this type <strong>of</strong><br />
252
separ<strong>at</strong>ion. The high quality <strong>of</strong> the separ<strong>at</strong>ions <strong>of</strong> the aliph<strong>at</strong>ic<br />
hydrocarbons and PAC fraction is indic<strong>at</strong>ed by the much higher H/C<br />
r<strong>at</strong>io <strong>of</strong> the aliph<strong>at</strong>ic fraction (1.70) <strong>co</strong>mpared to the PAC<br />
fraction (1.16) and the low <strong>co</strong>ncentr<strong>at</strong>ions <strong>of</strong> nitrogen and oxygen<br />
in these two fractions. In <strong>co</strong>ntrast, the N-PAC and HPAH fractions<br />
both have significant amounts <strong>of</strong>' oxygen and nitrogen indic<strong>at</strong>ing<br />
the presence <strong>of</strong> <strong>co</strong>mpounds th<strong>at</strong> <strong>co</strong>ntain both hetero<strong>at</strong>oms or<br />
possibly some overlap <strong>of</strong> the fractions.<br />
Results <strong>of</strong> analyses <strong>of</strong> the aged c<strong>at</strong>alysts, given in Table 2,<br />
show th<strong>at</strong> c<strong>at</strong>alytic hydrotre<strong>at</strong>ing <strong>of</strong> the aliph<strong>at</strong>ic and PAC<br />
fractions yielded lower carbon accumul<strong>at</strong>ions on the c<strong>at</strong>alysts than<br />
hydrotre<strong>at</strong>ing the V-178 or the N-PAC and HPAH fractions.<br />
Likewise, the c<strong>at</strong>alysts used to hydrotre<strong>at</strong> the N-PAC and HPAH<br />
fractions have significantly higher accumul<strong>at</strong>ions <strong>of</strong> nitrogen than<br />
the c<strong>at</strong>alysts used to hydrotre<strong>at</strong> the aliph<strong>at</strong>ics and the PAC<br />
fractions. The 0.6 and 0.5 wt% accumul<strong>at</strong>ions resulting from our<br />
two hour experiments are <strong>co</strong>mparable to the levels (0.5 to 0.6 wt%)<br />
observed on the first c<strong>at</strong>alysts, with c<strong>at</strong>alyst ages <strong>of</strong> about 20 lb<br />
resid/lb c<strong>at</strong>alyst, withdrawn from Wilsonville runs. These results<br />
show th<strong>at</strong> the nitrogen buildup on the c<strong>at</strong>alyst under process<br />
<strong>co</strong>nditions must be very rapid.<br />
Hydrogen<strong>at</strong>ion Activity<br />
The measured intrinsic activity losses (a) and the measured<br />
remaining extrud<strong>at</strong>e activities (F) are given in Table 3. A<br />
quantit<strong>at</strong>ive m<strong>at</strong>hem<strong>at</strong>ical expression, reported previously (E),<br />
rel<strong>at</strong>es F to a and effective diffusivity. Use <strong>of</strong> this equ<strong>at</strong>ion<br />
enabled us to determine the c<strong>at</strong>alyst effective diffusivities. The<br />
c<strong>at</strong>alysts used to hydrotre<strong>at</strong> the V-178 and the aliph<strong>at</strong>ic<br />
hydrocarbon and PAC fractions showed a 20% decrease in effective<br />
diffusivit from the fresh c<strong>at</strong>alyst value <strong>of</strong> 5x10-6<br />
cm2/sec/cm3, whereas those used to hydrotre<strong>at</strong> the HPAH and<br />
N-PAC fractions had gre<strong>at</strong>er than a 50% decrease. Recalcul<strong>at</strong>ing<br />
the F values without these changes in effective diffusivity (i.e.<br />
with fresh c<strong>at</strong>alyst effective diffusivity) (Table 3 ) shows th<strong>at</strong><br />
less than 10% <strong>of</strong> the loss <strong>of</strong> fresh extrud<strong>at</strong>e activity is due to<br />
the changes in effective diffusivity.<br />
The rel<strong>at</strong>ionship between F (<strong>co</strong>rrected for changes in effective<br />
diffusivity) and a also enabled us to differenti<strong>at</strong>e the two<br />
limiting modes <strong>of</strong> deactiv<strong>at</strong>ion -- homogeneous and shell-<br />
progressive poisoning. A plot <strong>of</strong> F vs a for the results from the<br />
hydrogen<strong>at</strong>ion activity testing <strong>of</strong> the V-178 and the four chemical<br />
classes is shown in Figure 1. Since the a values increase more<br />
rapidly than the F values, the dominant mode <strong>of</strong> deactiv<strong>at</strong>ion for<br />
these c<strong>at</strong>alysts is homogeneous poisoning <strong>of</strong> active sites (9). As<br />
can be seen in Figure 1, the aliph<strong>at</strong>ic hydrocarbons and the PAC<br />
fraction caused less deactiv<strong>at</strong>ion than the V-178, whereas the<br />
N-PAC and HPAH caused more deactiv<strong>at</strong>ion. This trend in<br />
deactiv<strong>at</strong>ion is inversely <strong>co</strong>rrel<strong>at</strong>ed with the carbon <strong>co</strong>ntents <strong>of</strong><br />
the aged c<strong>at</strong>alysts given in Table 2. The c<strong>at</strong>alysts used to process<br />
the aliph<strong>at</strong>ics and the PAC fraction have lower carbon <strong>co</strong>ntents<br />
than the c<strong>at</strong>alyst used to hydrotre<strong>at</strong> the V-178, whereas the<br />
c<strong>at</strong>alysts used to hydrotre<strong>at</strong> the N-PAC and HPAH fractions have<br />
higher carbon <strong>co</strong>ntents. Hydrotre<strong>at</strong>ing the N-PAC fraction yields<br />
the gre<strong>at</strong>est deactiv<strong>at</strong>ion with about an 87% loss <strong>of</strong> extrud<strong>at</strong>e<br />
hydrogen<strong>at</strong>ion activity and 98% <strong>of</strong> the active sites poisoned.<br />
253
CONCLUSIONS<br />
Separ<strong>at</strong>ion <strong>of</strong> a light hydrotre<strong>at</strong>er process stream into four<br />
chemical classes has shown th<strong>at</strong> about half <strong>of</strong> the stream is<br />
<strong>co</strong>mposed <strong>of</strong> aliph<strong>at</strong>ic hydrocarbons. There are also small amounts<br />
<strong>of</strong> N-PAC and HPAH fractions. Hydrotre<strong>at</strong>ing each <strong>of</strong> these four<br />
fractions and the whole process stream with c<strong>at</strong>alyst has shown<br />
th<strong>at</strong> all four fractions cause deactiv<strong>at</strong>ion. The gre<strong>at</strong>est<br />
deactiv<strong>at</strong>ion is due to the N-PAC fraction, although the HPAH<br />
fraction also yields gre<strong>at</strong>er deactiv<strong>at</strong>ion than the whole process<br />
stream. The deactiv<strong>at</strong>ion is caused primarily by active site<br />
poisoning, with a lesser amount due to a decrease in effective<br />
dif fusivity.<br />
REFERENCES<br />
1.<br />
2.<br />
3.<br />
4.<br />
5.<br />
6.<br />
7.<br />
8.<br />
9.<br />
Ocampo, A., Schrodt, J. T. and Kovach, S. M.,<br />
Prod. Res. Dev. 17(3), 266, 1978.<br />
Ind. Eng. Chem.<br />
Furimsky, E., Erzl Kohle, Petrochem %(E), 383, 1979.<br />
Stohl, F. V. and Stephens, H. P., Proc. Tenth Annual EPRI<br />
Contractors' Conference on Clean Liquid and Solid Fuels, April<br />
23-25, 1985, Palo Alto, CA.<br />
Stephens, H. P. and Stohl, F. V., ACS Division <strong>of</strong> Fuel<br />
Chemistry Preprints 29(6), 79, 1984.<br />
Stohl F. V. and Stephens, H. P., ACS Division <strong>of</strong> Fuel<br />
Chemistry Preprints 30(4), 148, 1985.<br />
Lamb, C. W., Lee, J. M., Moniz, M. J., Risbud, H. M.,<br />
Johnson, T. W., Proc. Tenth Annual EPRI Contractors'<br />
and<br />
Conference on Clean Liquid and Solid Fuels, April 23-25, 1985,<br />
Palo Alto, CA.<br />
L<strong>at</strong>er, D. w., Lee, M. L., Bartle, K. D., Kong, R. C. and<br />
Vassilaros, D. L., Anal. Chem. 53, 1612, 1981.<br />
Stephens, H. P. and Stohl, F. V y ACS Division <strong>of</strong> Petroleum<br />
Chemistry Preprints 30(3), 465, 1985.<br />
Wheeler, A., C<strong>at</strong>alysis 2, 105, P.H. Emmett ed., Reinhold,<br />
N.Y., 1955.<br />
254
i<br />
Table 1. Compositions <strong>of</strong> the V-178 stream and the four chemical classes in<br />
weight percents.<br />
---<br />
C H N o H / C<br />
V-178 87.69 10.05 0.23 1.08 1.38<br />
wt a<br />
<strong>of</strong> V-178<br />
Aliph<strong>at</strong>ic hydrocarbons 46 87.27 12.36 cO.10 0.10 1.70<br />
Neutral polycyclic 35 90.15 8.70 0.10 0.17 1.16<br />
arom<strong>at</strong>ic <strong>co</strong>mpounds (PAC)<br />
Nitrogen polycyclic<br />
arom<strong>at</strong>ic <strong>co</strong>mpounds<br />
(N-PAC)<br />
5 83.60 7.93 3.61 2.81 1.14<br />
Hydroxy polycyclic 9 78.18 8.25 1.13 9.44 1.27<br />
arom<strong>at</strong>ic <strong>co</strong>mpounds<br />
(HPEH)<br />
Table 2. Analyses <strong>of</strong> aged c<strong>at</strong>alysts from<br />
microreactor runs (reported as<br />
weight percents).<br />
C<strong>at</strong>alyst<br />
--<br />
C N<br />
V-178 run 3.87 0.3<br />
Aliph<strong>at</strong>ic hydrocarbon run 2.36 0.1<br />
PAC run 2.71 0.1<br />
N-PAC run 4.53 0.6<br />
HPAH run 5.02 0.5<br />
255
Table 3. Results <strong>of</strong> activity testing experiments<br />
(for fresh c<strong>at</strong>alyst F = 1.00, a = 0.0)<br />
F a F*<br />
Reactor Feed measured measured -<br />
V-178 0.44 0.72 0.52<br />
Aliph<strong>at</strong>ic 0.72 0.39 0.78<br />
hydrocarbons<br />
PAC<br />
m-PAC<br />
0.59 0.58 0.64<br />
0.05 0.90 0.13<br />
HPAH 0.19 0.92 0.26<br />
* Corrected for changes in effective diffusivity.<br />
F<br />
0.2<br />
O - t<br />
01<br />
0<br />
hydrocarbons<br />
a<br />
V-l78\ a,<br />
N-PAC<br />
0.2 0.4 0.8 0.8 1 .o<br />
Figure 1. F vs a plot for V-178 and the four chemical<br />
classes.<br />
256<br />
I
i ~<br />
IMPROVEMENT IN COAL LIQUEFACTION SOLVENT QUALITY BY DEWAXING<br />
ABSTRACT<br />
R. A. Winschel, G. A. Robbins and F. P. Burke<br />
CONOCO COAL RESEARCH DIVISION<br />
4000 Brownsville Road<br />
Library, PA 15129<br />
Recycle <strong>oil</strong>s from the Integr<strong>at</strong>ed Two-Stage <strong>Liquefaction</strong> (ITSL), H-Coal and Solvent<br />
Refined Coal (SRC) processes were dewaxed by variants <strong>of</strong> <strong>co</strong>mmercial dewaxing<br />
processes yielding up to 47 wt % "wax". Dewaxing methods used include the ketone<br />
and the urea adduction techniques. The clean waxes are reasonably pure<br />
paraffins. The dewaxed <strong>oil</strong>s were substantially better <strong>co</strong>al liquefaction solvents<br />
than the original (non-dewaxed) <strong>oil</strong>s in b<strong>at</strong>ch liquefaction tests. For example, in<br />
one case, dewaxing improved the <strong>co</strong>nversion <strong>of</strong> a standard <strong>co</strong>al to tetrahydr<strong>of</strong>uran<br />
solubles <strong>at</strong> standard reaction <strong>co</strong>nditions from 71% with the original <strong>oil</strong> to 87% with<br />
the dewaxed <strong>oil</strong>. These d<strong>at</strong>a provide a direct indic<strong>at</strong>ion <strong>of</strong> the inimical effect <strong>of</strong><br />
paraffinic <strong>co</strong>mponents on solvent quality. The impact <strong>of</strong> solvent quality is particu-<br />
larly relevant to two-stage liquefaction, in which thermal first-stage reactions<br />
proceed in a recycle solvent. In addition, these results indic<strong>at</strong>e the technical<br />
feasibility <strong>of</strong> dewaxing <strong>co</strong>al liquefaction recycle <strong>oil</strong>s by <strong>co</strong>mmercially available<br />
technology to improve solvent quality and to produce a useful by-product.<br />
Dewaxing <strong>co</strong>uld be applied to any liquefaction process th<strong>at</strong> uses a deashed (prefer-<br />
ably distill<strong>at</strong>e) recycle stream.<br />
INTRODUCTION<br />
Paraffinic and other s<strong>at</strong>ur<strong>at</strong>ed hydrocarbons are well known <strong>co</strong>mponents <strong>of</strong> <strong>co</strong>al and<br />
<strong>co</strong>al liquefaction products (1). The presence <strong>of</strong> substantial quantities <strong>of</strong> s<strong>at</strong>ur<strong>at</strong>ed<br />
hydrocarbons in <strong>co</strong>al liquefaction recycle solvents has been reported (2).<br />
Increasing <strong>co</strong>ncentr<strong>at</strong>ions <strong>of</strong> these <strong>co</strong>mpounds (as well as <strong>of</strong> highly alkyl<strong>at</strong>ed<br />
<strong>co</strong>mpounds) have been linked to a decreasing quality <strong>of</strong> the recycle <strong>oil</strong> as a donor<br />
solvent for <strong>co</strong>al liquefaction (2). Other work has demonstr<strong>at</strong>ed th<strong>at</strong> the effective-<br />
ness <strong>of</strong> <strong>co</strong>al liquids as <strong>co</strong>al liquefaction donor solvents shows a neg<strong>at</strong>ive <strong>co</strong>rrel<strong>at</strong>ion<br />
with the paraffinic n<strong>at</strong>ure <strong>of</strong> the <strong>co</strong>al liquid (3). In the development <strong>of</strong> the Consol<br />
Synthetic Fuels (CSF) (4) and Exxon DonorSolvent (EDS) (5) processes, it was<br />
re<strong>co</strong>gnized th<strong>at</strong> the bund-up <strong>of</strong> s<strong>at</strong>ur<strong>at</strong>ed hydrocarbons in -the recycle solvent<br />
resulted in deterior<strong>at</strong>ing solvent quality. Paraffinic and other s<strong>at</strong>ur<strong>at</strong>ed hydro-<br />
carbons are known to be non-donors (6) or <strong>at</strong> least very poor donors (7.8) <strong>at</strong> <strong>co</strong>al<br />
liquefaction <strong>co</strong>nditions. Their presence in recycle <strong>oil</strong>s reduces solventquality, <strong>at</strong><br />
least by diluting the active solvent molecules and <strong>at</strong> worst by acting as a detri-<br />
mental antisolvent th<strong>at</strong> leads to reduced solvent effectiveness.<br />
S<strong>at</strong>ur<strong>at</strong>ed hydrocarbons in <strong>co</strong>al liquefaction recycle <strong>oil</strong>s are formed in part by<br />
<strong>co</strong>mplete hydrogen<strong>at</strong>ion <strong>of</strong> arom<strong>at</strong>ics to form naphthenes. However, the majority <strong>of</strong><br />
the paraffins (and particularly the n-paraffins) must ultim<strong>at</strong>ely arise either<br />
unchanged directly from the <strong>co</strong>al (9) or as products <strong>of</strong> the cracking <strong>of</strong> alkyl<br />
side-chains or <strong>of</strong> larger paraffins. lfthe recycle solvent is higher b<strong>oil</strong>ing than the<br />
major liquefaction products, s<strong>at</strong>ur<strong>at</strong>es must exit the recycle loop by cracking to<br />
lighter products. Paraffins, however, tend to crack selectively to gases (2) thus<br />
<strong>co</strong>nsuming expensive hydrogen while producing undesirable gas.<br />
The quality and paraffin <strong>co</strong>ntent <strong>of</strong> the recycle solvent <strong>at</strong> equilibrium is fixed for<br />
each liquefaction process by the plant <strong>co</strong>nfigur<strong>at</strong>ion, feed <strong>co</strong>al and oper<strong>at</strong>ing<br />
<strong>co</strong>nditions in use <strong>at</strong> any time. To reduce the paraffin <strong>co</strong>ntent <strong>of</strong> the recycle<br />
solvent in order to improve its quality, oper<strong>at</strong>ing <strong>co</strong>nditions must be changed if the<br />
257
feed <strong>co</strong>al and plant <strong>co</strong>nfigur<strong>at</strong>ion are heid <strong>co</strong>nstant. However, by changing<br />
oper<strong>at</strong>ing <strong>co</strong>nditions, product yield sl<strong>at</strong>e and/or product quality may be undesirably<br />
affected.<br />
This paper presents a novel applic<strong>at</strong>ion <strong>of</strong> <strong>co</strong>mmonly used <strong>co</strong>mmercialized technology<br />
to improve the quality <strong>of</strong> <strong>co</strong>al liquefaction recycle solvents. Th<strong>at</strong> technology, in<br />
<strong>co</strong>mmon use in the petroleum refining industry, is dewaxing. When applied to <strong>co</strong>al<br />
liquefaction recycle solvents, dewaxing improves donor solvent quality by removing<br />
predominantly the paraffins and other s<strong>at</strong>ur<strong>at</strong>ed hydrocarbons th<strong>at</strong> are undesirable<br />
<strong>co</strong>mponents. If applied <strong>co</strong>mmercially, a high-value by-product wax <strong>co</strong>uld be sold.<br />
For example, recent price ranges (101 <strong>of</strong> rel<strong>at</strong>ed products follow: paraffin wax,<br />
$0.16-0.46/lb; petrol<strong>at</strong>um, $0.30-07iO/lb; montan wax, $0.58-0.65/lb; micro-<br />
crystalline wax, $0.36-0.48 Ib; and mineral <strong>oil</strong>, $2.68-3.101gal. These prices are<br />
<strong>co</strong>nsiderably gre<strong>at</strong>er than <strong>co</strong>mmanded by fuels. Removal <strong>of</strong> paraffins and other<br />
s<strong>at</strong>ur<strong>at</strong>es from the liquefaction process this way may also reduce gas production and<br />
hydrogen <strong>co</strong>nsumption and would reduce the occurrance <strong>of</strong> wax precipit<strong>at</strong>ion <strong>at</strong> low<br />
temper<strong>at</strong>ure from products with b<strong>oil</strong>ing points similar to the dewaxed stream.<br />
Various dewaxing methods are now, or have been, in <strong>co</strong>mmercial use in the<br />
petroleum industry (11 ) including pressing and swe<strong>at</strong>ing, centrifug<strong>at</strong>ion, solvent<br />
dewaxing (e.g., the propane and ketone processes) and urea adduction methods.<br />
The bulk <strong>of</strong> the experiments reported here used a labor<strong>at</strong>ory version <strong>of</strong> the ketone<br />
process, though the urea adduction process was also tested, both with promising<br />
results. Commercially, methyl ethyl ketone is typically employed in the ketone<br />
process (11); acetone was used in our experiments for <strong>co</strong>nvenience. These experi-<br />
ments demonstr<strong>at</strong>e on a small scale the technical feasibility <strong>of</strong> improving <strong>co</strong>al<br />
liquefaction solvent quality by dewaxing using <strong>co</strong>mmercially available technology.<br />
The impact <strong>of</strong> solvent quality is particularly relevant to processes using a thermal<br />
reactor in which <strong>co</strong>al liquefaction proceeds in and depends upon a recycle solvent<br />
such as the ITSL, SRC-I, SRC-II and EDS processes. Dewaxing should be directly<br />
applicable to processes th<strong>at</strong> recycle <strong>at</strong> least one distill<strong>at</strong>e-only stream such as ITSL<br />
(the re<strong>co</strong>nfigured mode in use <strong>at</strong> Wilsonvilie), SRC-I, EDS (without bottoms recycle)<br />
and H-Coal. ,it should also be possible to dewax any intermedi<strong>at</strong>e distill<strong>at</strong>e stream<br />
if the recycle does not <strong>co</strong>ntain a separ<strong>at</strong>e distill<strong>at</strong>e <strong>co</strong>mponent. One experiment<br />
demonstr<strong>at</strong>ed th<strong>at</strong> a deasphalted residual <strong>oil</strong> <strong>co</strong>uld also be dewaxed.<br />
EXPERIMENTAL<br />
Ketone Dewaxing<br />
A weighed amount (about 85 g) <strong>of</strong> the <strong>oil</strong> to be dewaxed was mixed with acetone<br />
(Fisher HPLC grade) in the desired r<strong>at</strong>io (112 to 1/3.3 by volume) in a beaker<br />
equipped with a magnetic stirrer. All <strong>oil</strong>s tested, except the single 85OoF+ resid,<br />
dissolved readily <strong>at</strong> room temper<strong>at</strong>ure. The <strong>co</strong>ntents were <strong>co</strong>oled while stirring in a<br />
dry icelacetone b<strong>at</strong>h to the desired temper<strong>at</strong>ure to precipit<strong>at</strong>e the waxes. When the<br />
desired temper<strong>at</strong>ure (-20 or -5OOC) was reached, the mixture was immedi<strong>at</strong>ely<br />
filtered. while still <strong>co</strong>ld, in a Buchner funnel equipped with a glass-fiber filter<br />
(Reeve Ange! 8934AH). The filter cake (wax) was washed with additional <strong>co</strong>ld<br />
acetone approxim<strong>at</strong>ely equaling the volume <strong>of</strong> the original <strong>oil</strong>lacetone mixture. This<br />
filtr<strong>at</strong>e was set aside. The wax cake was washed through the filter with freshly<br />
distilled tetrahydr<strong>of</strong>uran (THF). Each filtr<strong>at</strong>e (acetone and THF) was rotary<br />
evapor<strong>at</strong>ed <strong>at</strong> about 6OoC to <strong>co</strong>nstant weight to remove all traces <strong>of</strong> solvents,<br />
leaving the dewaxed <strong>oil</strong> and the waxes, respectively, which were then weighed and<br />
analyzed. The product waxes were usually a white solid though some <strong>of</strong> the less<br />
pure waxes were dis<strong>co</strong>lored.<br />
One sample, a solid deasphalted 85OoF+ resid, was ketone dewaxed using a somewh<strong>at</strong><br />
different method. The solid (329) was dissolved in lZOg <strong>of</strong> an 80/20 v/v solution <strong>of</strong><br />
acetone and freshly distilled toluene, then dewaxed as above <strong>at</strong> -5OOC. These<br />
258
I<br />
waxes (12% <strong>of</strong> <strong>oil</strong>) were very Impure and so were subjected to a se<strong>co</strong>nd dewaxing<br />
procedure similar to the first except th<strong>at</strong> the solvent used was 100 mL <strong>of</strong> a 70/30<br />
vlv mixture <strong>of</strong> acetone and toluene. This product was a very hard brown waxy<br />
sol id.<br />
Urea Dewaxinq<br />
I<br />
One <strong>oil</strong> sample was dewaxed by the urea adduction method. About 509 <strong>of</strong> the <strong>oil</strong><br />
was weighed and diluted with an equal weight <strong>of</strong> CH,CI, (MCB reagent). To this<br />
mixture was slowly added about 50 mls <strong>of</strong> an aqueous solution <strong>of</strong> urea (Fisher<br />
certified) s<strong>at</strong>ur<strong>at</strong>ed <strong>at</strong> 80°C which crystallized upon <strong>co</strong>oling on <strong>co</strong>ntact with the<br />
I<br />
<strong>oil</strong>/CH,CI, solution. The mixture was stirred for one hour, filtered in a Buchner<br />
funnel equipped with a glass-fiber filter, then washed with several aliquots <strong>of</strong><br />
CH,CI,. This filtr<strong>at</strong>e was set aside. The filter-cake was washed with warm w<strong>at</strong>er<br />
to dissolve the urea leaving the waxes on the filter. The w<strong>at</strong>er wash was<br />
I discarded. The waxes were washed through the filter with freshly distilled THF.<br />
The CH,CI, and THF filtr<strong>at</strong>es were stripped <strong>of</strong> solvent by rotary evapor<strong>at</strong>ion to<br />
produce the dewaxed <strong>oil</strong> and the waxes, respectively, which were weighed and<br />
analyzed. The product wax was a white solid.<br />
Solvent Quality (Microautoclave) Tests<br />
Samples were tested for their effectiveness as <strong>co</strong>al liquefaction donor solvents using<br />
a standard microautoclave test. This test, called the modified equilibrium test, has<br />
been described in detail previously (3). Briefly, 69 <strong>of</strong> a standard <strong>co</strong>al and 9g <strong>of</strong><br />
the sample to be tested are he<strong>at</strong>ed t5 75OOF for 30 min without added gas in a 30<br />
mL microautoclave. The <strong>co</strong>ntents are <strong>co</strong>oled and extracted with THF to determine<br />
the <strong>co</strong>nversion <strong>of</strong> <strong>co</strong>al to solubles. This test is an authentic <strong>co</strong>al liquefaction<br />
experiment. Solvent effects can be tested easily because there is no interfering<br />
c<strong>at</strong>alyst or extraneous gas present. The results <strong>of</strong> this test serve as an empirical<br />
measure <strong>of</strong> donor solvent quality. Coal <strong>co</strong>nversions obtained with several pure<br />
model <strong>co</strong>mpounds follow: n-tetradecane, 25.4%, 1-methylnaphthalene, 48.2%.<br />
tetralin, 85.4%. This test is reproducible to 1.2% absolute [standard devi<strong>at</strong>ion).<br />
Other Analyses<br />
Samples were analyzed by IH-NMR using a procedure described in detail elsewhere<br />
(2) to determine the effectiveness <strong>of</strong> dewaxing and to determine the purity <strong>of</strong> the<br />
waxes. Briefly, the H-NMR spectrum is divided in regions roughly <strong>co</strong>rresponding<br />
to different proton types. For example, the region between 10.0 and 4.7 ppm is<br />
assigned to arom<strong>at</strong>ic protons and the region between 1.4 and 0.5 ppm is assigned to<br />
to paraffinic protons. Paraffinic protons are protons on internal -CH,- groups or<br />
-CH, groups <strong>of</strong> paraffins and long alkyl chains. Reproducibility is 0.2% absolute<br />
(standard devi<strong>at</strong>ion).<br />
Gas chrom<strong>at</strong>ography [CC) was performed with a Perkin-Elmer Sigma 2000 instrument<br />
equipped with dual flame ioniz<strong>at</strong>ion detectors. The <strong>co</strong>lumn, a 30m x 0.25mm DB-5<br />
<strong>co</strong>lumn from JLW Scientific, was initially <strong>at</strong> 5OoC for 4 min, then programmed to<br />
280OC <strong>at</strong> 4OClmin and held for 20 min. Carrier gas was 20 psig H,. injector and<br />
detector temper<strong>at</strong>ures were 300OC. A split injection <strong>of</strong> 0.2pL <strong>of</strong> sample (O.lg/mL in<br />
THF) was used.<br />
internal standard.<br />
Quantit<strong>at</strong>ion was based on peak areas referred to an n-decane<br />
I Elemental analyses (C, H, N and S) were performed with Le<strong>co</strong> CHN-600 and SC32<br />
Instruments. Though these instruments were designed for the analysis <strong>of</strong> <strong>co</strong>als,<br />
not <strong>oil</strong>s, reliable N and S determin<strong>at</strong>ions can be made. C and H values are less<br />
reliable, but are probably accur<strong>at</strong>e to ?l% absolute.<br />
Feed Oils<br />
All samples were authentic <strong>co</strong>al liquefaction recycle <strong>oil</strong>s.<br />
259
P1<br />
#2<br />
#3<br />
114<br />
115<br />
#6<br />
ITSL subbituminous distill<strong>at</strong>e - A <strong>co</strong>mposite <strong>of</strong> the 850OF- distill<strong>at</strong>e portions <strong>of</strong><br />
twenty daily samples taken between 5/6 and 8/6/84 <strong>of</strong> the recycle solvent<br />
(V-13lB) from Wilsonville Run 246 made with Wyoming (Clovis Point mine) sub-<br />
bituminous <strong>co</strong>al.<br />
ITSL subbituminous distill<strong>at</strong>e - The 850OF- distill<strong>at</strong>e portion <strong>of</strong> a sample <strong>of</strong><br />
hydrotre<strong>at</strong>er flashed bottoms (V-1067) taken 9/14/85 from Wilsonville Run 249<br />
made with Wyoming (Clovis Point mine) subbituminous <strong>co</strong>al.<br />
ITSL bituminous distill<strong>at</strong>e - A <strong>co</strong>mposite <strong>of</strong> the 85OOF- distill<strong>at</strong>e portions <strong>of</strong><br />
fourteen daily samples taken between 9/12 and 12/9/84 <strong>of</strong> the recycle solvent<br />
(V-131B) from Wilsonville Run 247 made with Illinois 6 (Burning Star mine)<br />
bituminous <strong>co</strong>al.<br />
H-Coal subbituminous distill<strong>at</strong>e - the 1000°F- distill<strong>at</strong>e portion (96.1%) <strong>of</strong> a<br />
<strong>co</strong>mposite <strong>of</strong> seventeen daily samples taken between 9/1 and 9/17/80 <strong>of</strong> the<br />
Wean <strong>oil</strong>" (a <strong>co</strong>mponent <strong>of</strong> the recycle solvent) from H-Coal PDU Run 10 made<br />
with Wyoming (Wyodak mine) subbituminous <strong>co</strong>al.<br />
SRC-1 bituminous distill<strong>at</strong>e - the 1000°F- distill<strong>at</strong>e portion (95.6%) <strong>of</strong> a sample<br />
<strong>of</strong> recycle solvent taken 10/2/78 from Wilsonville Run 149 made with Kentucky<br />
#9 bituminous <strong>co</strong>al.<br />
ITSL subpituminous deasphalted resid - the hexane-solubles <strong>of</strong> a <strong>co</strong>mposite <strong>of</strong><br />
the 850OF resid portions <strong>of</strong> twenty-one daily samples taken between 5/6 and<br />
8/6/84 <strong>of</strong> the recycle solvent (V-1318) from Wilsonville Run 246 made with<br />
Wyoming (Clovis Point mine) subbituminous <strong>co</strong>al.<br />
DISCUSSION<br />
Results from dewaxing experiments using authentic <strong>co</strong>al liquefaction recycle <strong>oil</strong>s are<br />
discussed below. In this report, "wax" refers to the precipit<strong>at</strong>ed portion <strong>of</strong> the <strong>oil</strong><br />
obtained in the procedure,, "dewaxed <strong>oil</strong>" refers to the non-precipit<strong>at</strong>ed portion and<br />
"feed <strong>oil</strong>" refers to the original, untre<strong>at</strong>ed <strong>oil</strong>. It should be re<strong>co</strong>gnized th<strong>at</strong> the<br />
ketone process as <strong>co</strong>mmercially practiced is performed in .two stages called dewaxing<br />
and de-<strong>oil</strong>ing (11). Except in one case, these experiments were done in a single<br />
stage. TherefoZ, it is expected th<strong>at</strong> these results <strong>co</strong>uld be further improved.<br />
Experimental <strong>co</strong>nditions and yields are shown in Table 1. Results <strong>of</strong> microautoclave<br />
liquefaction tests are shown In Table 2. Analyses <strong>of</strong> the various <strong>oil</strong>s are shown In<br />
Table 3. 'H-NMR spectra and gas chrom<strong>at</strong>ograms <strong>of</strong> the feed <strong>oil</strong>, dewaxed <strong>oil</strong> and<br />
waxes from experiment 5 are shown in Figures 1 and 2.<br />
Wax Yields<br />
Wax yields ranged from 3 to 47 wt % (Table 1). Wax purity spanned a range as<br />
well. Generally, the gre<strong>at</strong>est yields <strong>of</strong> wax and the purest waxes were produced<br />
from <strong>oil</strong>s derived from subbituminous <strong>co</strong>al (feed <strong>oil</strong>s 81, 2 and 4). Those <strong>oil</strong>s were<br />
very paraffinic. as determined by the paraffinic hydrogen <strong>co</strong>ntent from 1H-NMR and<br />
by GC (Table 3) and would be expected to produce the most wax. This is <strong>co</strong>nsis-<br />
tent with the <strong>co</strong>ncept th<strong>at</strong> lower rank <strong>co</strong>als tend to be more paraffinic. One highly<br />
paraffinic <strong>oil</strong> produced from subbituminous <strong>co</strong>al (feed <strong>oil</strong> 12) yielded 47% <strong>of</strong> a<br />
reasonably pure wax (Tables 1 and 3).<br />
The wax yields obtained in these experiments would not be expected to be <strong>at</strong>tained<br />
<strong>at</strong> equilibrium in a liquefaction process employing dewaxing . The liquefaction<br />
processes from which these samples were taken all employed recycle and therefore,<br />
the total feed to these liquefaction processes included varying amounts <strong>of</strong> wax<br />
<strong>co</strong>mponents. If dewaxing <strong>of</strong> all or part <strong>of</strong> the recycle were used, the wax <strong>co</strong>ntent<br />
<strong>of</strong> the feed would be reduced thereby reducing the wax <strong>co</strong>ntent <strong>of</strong> the product. It<br />
may be possible to dewax only th<strong>at</strong> portion <strong>of</strong> the recycle solvent th<strong>at</strong> is necessary<br />
260
I<br />
to keep wax levels below some set point. Dewaxing only a portion <strong>of</strong> the recycle<br />
solvent <strong>co</strong>uld reduce both capital and oper<strong>at</strong>ing <strong>co</strong>sts <strong>of</strong> the dewaxing unit.<br />
Improvement in Donor Solvent Quality<br />
In all cases, donor solvent quality, as measured by microautoclave tests. increased<br />
upon dewaxing. In general, the improvement in solvent quality upon dewaxing, as<br />
measured by the difference in the microautoclave tests with the feed <strong>oil</strong> and the<br />
<strong>co</strong>rresponding dewaxed <strong>oil</strong>, increased with increasing wax yield. Thus, only 3%<br />
wax was removed in experiment 11 giving an improvement in donor solvent quality<br />
from 63 to 66%. whereas 47% wax was removed in experiment 14 giving an improve-<br />
ment in donor solvent quality from 71 to 87%. Clearly, the increase in donor<br />
solvent quality results from reducing the <strong>co</strong>ncentr<strong>at</strong>ion <strong>of</strong> paraffins and other<br />
s<strong>at</strong>ur<strong>at</strong>es which are non-donors and are generally <strong>co</strong>nsidered to be poor physical<br />
solvents for <strong>co</strong>al liquids. In fact, paraffins have been found to be Inimical to<br />
solvent quality in both microautoclave tests (2) and in the development <strong>of</strong> the<br />
CSF (4) and EDS (2) processes.<br />
In direct <strong>co</strong>al liquefaction processes, the quality <strong>of</strong> the recycle <strong>oil</strong> generally is fixed<br />
by the feed <strong>co</strong>al, the oper<strong>at</strong>ing <strong>co</strong>nditions and the characteristics <strong>of</strong> the process.<br />
Making changes in oper<strong>at</strong>ing <strong>co</strong>nditions to improve the product sl<strong>at</strong>e or to <strong>co</strong>mpen-<br />
s<strong>at</strong>e for c<strong>at</strong>alyst deactiv<strong>at</strong>ion can have the undesirable side effect <strong>of</strong> reducing<br />
solvent quality which, in turn, can affect product yields unexpectedly. Alter-<br />
n<strong>at</strong>ely, oper<strong>at</strong>ing <strong>co</strong>nditions th<strong>at</strong> provide a high quality recycle solvent may not be<br />
desirable from a product yield or product quality standpoint. Dewaxing provides a<br />
means <strong>of</strong> improving recycle solvent quality th<strong>at</strong> is independent <strong>of</strong> liquefaction<br />
<strong>co</strong>nditions and may permit simultaneous optimiz<strong>at</strong>ion <strong>of</strong> product and recycle-solvent<br />
qualities.<br />
It is interesting to note th<strong>at</strong> the three dewaxed distill<strong>at</strong>e ITSL recycle solvents all<br />
gave similar <strong>co</strong>al <strong>co</strong>nversions in the microautoclave tests, even though the non-<br />
dewaxed feed <strong>oil</strong>s gave significantly different results as shown below.<br />
Run No.<br />
5<br />
13<br />
14<br />
Feed Oil<br />
#1<br />
#3<br />
#2<br />
Coal Conversion, wt % MAF<br />
Feed Dewaxed<br />
80.9<br />
79.2<br />
70.8<br />
88.2<br />
86.2<br />
87.1<br />
This result would indic<strong>at</strong>e th<strong>at</strong> not only can donor solvent quality <strong>of</strong> recycle <strong>oil</strong>s be<br />
improved by dewaxing, but th<strong>at</strong> donor solvent quality can also be made more<br />
<strong>co</strong>nstant regardless <strong>of</strong> feed <strong>co</strong>al.<br />
Effect <strong>of</strong> Temper<strong>at</strong>ure on Ketone Dewaxing<br />
Experiments were performed with three feed <strong>oil</strong>s <strong>at</strong> both -20 and -5OOC. In each<br />
case, the lower temper<strong>at</strong>ure produced about twice as much wax (Table 1). The<br />
dewaxed <strong>oil</strong>s produced <strong>at</strong> -5OOC were better <strong>co</strong>al liquefaction donor solvents as<br />
measured by microautoclave tests (Table 2). This is <strong>co</strong>nsistent with the lower<br />
paraffinic <strong>co</strong>ntent <strong>of</strong> those dewaxed <strong>oil</strong>s (Table 3). However, the waxes produced<br />
<strong>at</strong> -5OOC were lower purity paraffins than those produced <strong>at</strong> -2OOC as evidenced by<br />
the increased arom<strong>at</strong>icities and carbon <strong>co</strong>ntents and decreased hydrogen and<br />
paraffinic hydrogen <strong>co</strong>ntents and by the GC results (Table 3). Even though the<br />
additional m<strong>at</strong>erial removed <strong>at</strong> -5OOC was largely not paraffinic, its removal further<br />
improved donor solvent quality. It is believed th<strong>at</strong> the additional m<strong>at</strong>erial removed<br />
<strong>at</strong> -5OOC largely <strong>co</strong>nsists <strong>of</strong> highly s<strong>at</strong>ur<strong>at</strong>ed and alkyl<strong>at</strong>ed <strong>co</strong>mpounds. Clearly,<br />
ketone dewaxing can be performed to maximize the improvement in solvent quality or<br />
to maximize the purity <strong>of</strong> the product wax depending on oper<strong>at</strong>ing temper<strong>at</strong>ure. It<br />
should be possible to optimize both fe<strong>at</strong>ures simultaneously by selecting appropri<strong>at</strong>e<br />
261
temper<strong>at</strong>ure, time and solvent power <strong>co</strong>nditlons. These experiments were all<br />
oper<strong>at</strong>ed with no hold time <strong>at</strong> temper<strong>at</strong>ure. Commercial petroleum dewaxing<br />
oper<strong>at</strong>ions tailor solvent power by using solutions <strong>of</strong> varying r<strong>at</strong>ios <strong>of</strong> ketone and<br />
toluene as the dewaxing solvent (1 1). Commercial petroleum oper<strong>at</strong>ions also improve<br />
the selectivity <strong>of</strong> the process bToper<strong>at</strong>ing in two stages in which the wax is<br />
separ<strong>at</strong>ed and then de-<strong>oil</strong>ed (XI.<br />
Ketone Dewaxing <strong>of</strong> Resid<br />
One deasphalted 850OF+ resid sample was dewaxed (experiment 12) yielding 7.9% <strong>of</strong><br />
a very hard wax. The results <strong>of</strong> 'H-NMR and elemental analyses (Table 3) indic<strong>at</strong>e<br />
the wax is reasonably pure paraffin. This wax was too high b<strong>oil</strong>ing for <strong>co</strong>mplete<br />
CC analysis, but the eluted <strong>co</strong>mponents were Predominantly n-paraffins <strong>co</strong>ntaining<br />
24 to over 40 carbon <strong>at</strong>oms. This wax was very hard and had a freezing point<br />
upon <strong>co</strong>oling <strong>of</strong> 54OC by differential scanning calorimetry. This experiment showed<br />
th<strong>at</strong> deasphalted resids can be dewaxed and may indic<strong>at</strong>e th<strong>at</strong> full-range <strong>co</strong>al lique-<br />
faction recycle <strong>oil</strong>s can be dewaxed providing th<strong>at</strong> they are solids-free and<br />
asphaltene-free. This may have applicability in ITSL and the developing C<strong>at</strong>alytic<br />
Two-Stage <strong>Liquefaction</strong> process in which the full-range recycle <strong>oil</strong> is typically<br />
solids-free and, when <strong>processing</strong> subbituminous <strong>co</strong>al, <strong>co</strong>ntains rel<strong>at</strong>ively low levels<br />
<strong>of</strong> asphaltenes (u),<br />
N<strong>at</strong>ure and Quality <strong>of</strong> Product Waxes<br />
The product waxes were predominantly s<strong>at</strong>ur<strong>at</strong>ed hydrocarbons. This is evidenced<br />
by their very low H-arom<strong>at</strong>icities (as low as 0.3% arom<strong>at</strong>icltotal hydrogen) and their<br />
elemental analyses (Table 3). Specifically, these s<strong>at</strong>ur<strong>at</strong>ed hydrocarbons were<br />
mostly paraffins as shown by the high <strong>co</strong>ncentr<strong>at</strong>ion <strong>of</strong> paraffinic protons from<br />
'H-NMR analysis (as high as 91% paraffinicltotal hydrogen). Even pure paraffins<br />
do not give 100% paraffinic protons in this 'H-NMR analysis because <strong>of</strong> spinning<br />
side bands. For example, pure n-tetra<strong>co</strong>sane gives 92.1% paraffinic protons in this<br />
analysis. n-Paraffins predomin<strong>at</strong>e in the gas chrom<strong>at</strong>ograms <strong>of</strong> the waxes (Table 3<br />
and Figure 3). ac<strong>co</strong>unting for as much as 60% <strong>of</strong> the wax. The carbon preference<br />
indices (14) <strong>of</strong> the waxes were near unity, averaging 1.03 with a standard devi<strong>at</strong>ion<br />
<strong>of</strong> 0.04 (range = 0.92 to 1.09).<br />
Dewaxing is most efficient for the higher molecular weight n-paraffins as seen by<br />
the GC d<strong>at</strong>a in Table 3. For all but one experiment, the n-paraffin <strong>of</strong> gre<strong>at</strong>est<br />
<strong>co</strong>ncentr<strong>at</strong>ion has the highest carbon number in the wax and the lowest carbon<br />
number in the dewaxed <strong>oil</strong>.<br />
Enough wax was produced in experiments 13 and 14 to test the effectiveness <strong>of</strong> the<br />
wax as a donor solvent. The waxes from these experiments were also two <strong>of</strong> the<br />
least pure waxes re<strong>co</strong>vered. As shown in Table 2, these waxes performed<br />
<strong>co</strong>nsiderably more poorly than the <strong>co</strong>rresponding feed <strong>oil</strong>s, though not as poorly as<br />
pure n-tetra<strong>co</strong>sane which gave 25.4% <strong>co</strong>al <strong>co</strong>nversion in the same test. It is<br />
expected th<strong>at</strong> the purer waxes would behave more similarly to the n-tetra<strong>co</strong>sane.<br />
ACKNOWLEDGEMENT<br />
This work was funded by the U.S. Department <strong>of</strong> Energy under Contract No.<br />
DE-ACZ2-84PC70018.<br />
262
REFERENCES<br />
1.<br />
2.<br />
3.<br />
4.<br />
5.<br />
6.<br />
7.<br />
8.<br />
9.<br />
10.<br />
11.<br />
12.<br />
13.<br />
14.<br />
Bartle, K. D., Jones, D. W. and Pakdel, H., "Separ<strong>at</strong>ion and Spectros<strong>co</strong>py <strong>of</strong><br />
Paraffinic Hydrocarbons from Coalt1 in "Analytical Methods for Coal and Coal<br />
Products", C. Karr, Jr., ed., Academic Press, NY, NY, 1978.<br />
Burke, F. P., Winschel, R. A. and Pochapsky, T.C., Fuel 1981, 60, 562.<br />
Winschel, R. A., Robbins, G. A. and Burke, F. P., Fuel 1986, 65(4), 526.<br />
Consolid<strong>at</strong>ion Coal Company, "Pilot Scale Development <strong>of</strong> the CSF Process",<br />
U.S. Department <strong>of</strong> Interior, OCR RED Rep. No. 39, Vol. 5, Book 3,<br />
November, 1971.<br />
Plumlee, K. W., Zaczepinski, S., and Hu, A. Y., Proceedings <strong>of</strong> the Seventh<br />
Annual EPRl Contractors' Conference on Coal <strong>Liquefaction</strong>, EPRl Report No.<br />
AP-2718, p. 12-1.<br />
Cronauer, D. C., Jewell, D. M., Shah, Y. T. and Kuesar, K. A., Ind. Eng.<br />
Chem. Fundam. 1978, 17, 291.<br />
Curran, C. P., Struck, R. T. and Corin, E., Ind. Eng. Chem.<br />
Dev. 1967, 6, 166.<br />
Clarke, J. W., Rantell, T. D. and Snape, C. E., Fuel 1984, 63, 1476.<br />
Proc. Des.<br />
Youtcheff, J. S., Given, P. H., Baset, Z. and Sundaram, M. S., Org.<br />
Ceochem. 1983, 5(3), 157.<br />
Chemical Marketing Reporter, December 23, 1985.<br />
Zurcher, P., "Dewaxing" in "Petroleum Refinery Engineering", Fourth Edition,<br />
W. L. Nelson, ed., McCraw-Hill Book Co., Inc., NY, NY, 1958.<br />
Burke, F. P., Winschel, R. A. and Robbins, C. A., "Recycle Slurry Oil<br />
Characteriz<strong>at</strong>ion - Final Report", DOE Contract No. DE-AC22-80PC30027,<br />
March, 1985.<br />
Winschel, R. A., Robbins, C. A. and Burke, F. P., "Coal <strong>Liquefaction</strong><br />
Process Solvent Characteriz<strong>at</strong>ion and Evalu<strong>at</strong>ion - Technical Progress Report<br />
for July 1, 1985, through September 30, 1985", DOE Contract No.<br />
DE-AC22-84PC70018, December, 1985.<br />
Maxwell, J. R., Pillinger, C. T. and Eglington, C., 4. Rev., Chem. SOC.<br />
1971, 25, 571.<br />
263
TABLE 1<br />
EXPERIMENTAL CONDITIONS AND PRODUCT YIELDS<br />
Conditions Yields, wt %<br />
Experiment Feed O i l Acetone/ Dewaxed Mass<br />
No. I.D. T, OC Oil, v/v Wax Oi 1 Balance<br />
---<br />
Urea Adduction Method<br />
2 #1<br />
Ketone Method<br />
4 #1<br />
5 #1<br />
Room 5.0 88.8 93.8<br />
-20 2.0 9.1 90.7 99.8<br />
-50 3.3 20.6 79.3 99.9<br />
14 #2 -50 3.0 47.4 52.0 99.4<br />
7 #3 -20 3.0 14.5 83.2 97.7<br />
13<br />
9<br />
#3(a)<br />
#4<br />
-50<br />
-20<br />
3.0<br />
3.0<br />
32.7<br />
5.2<br />
66.5<br />
93.9<br />
99.1<br />
99.1<br />
10<br />
11<br />
#4<br />
#5<br />
-50<br />
-50<br />
3.0<br />
3.0<br />
9.2<br />
3.2<br />
90.0<br />
96.3<br />
99.1<br />
99.5<br />
12<br />
#6 -50 (C) 7.9 90.7 98.6<br />
(a) Redistilled to 1000°F- immedi<strong>at</strong>ely before experiment (98.3% distill<strong>at</strong>e).<br />
(b) Urea phod used, see Experimental section.<br />
(c) 85OOF residual <strong>oil</strong> used as feed, see Experimental section.<br />
Experiment<br />
No.<br />
2<br />
4<br />
5<br />
14<br />
7<br />
13<br />
TABLE 2<br />
MICROAUTOCLAVE TEST RESULTS<br />
Coal Conversion, wt % MAF Waxes<br />
Feed Oil Dewaxed Oil -<br />
80.9<br />
80.9<br />
80.9<br />
84.9<br />
87.1<br />
88.2<br />
70.8<br />
80.1<br />
87.1<br />
83.7<br />
55.1<br />
-<br />
79.2<br />
55.7<br />
55.7<br />
63.3<br />
86.2<br />
55.4157.2<br />
60.5<br />
65.6<br />
62.5<br />
-<br />
264
Experiment<br />
No.<br />
2<br />
4<br />
5<br />
14<br />
7<br />
13<br />
9<br />
10<br />
11<br />
12<br />
Fraction<br />
Feed<br />
Dewaxed<br />
Wax<br />
Feed<br />
Dewaxed<br />
Wax<br />
Feed<br />
Dewaxed<br />
Wax<br />
Feed<br />
Dewaxed<br />
Wax<br />
Feed<br />
oewaxed<br />
Wax<br />
Feed<br />
Dewaxed<br />
Wax<br />
Feed<br />
Dewaxed<br />
Wax<br />
Feed<br />
Dewaxed<br />
Wax<br />
Feed<br />
Dewaxed<br />
Wax<br />
Feed<br />
Dewaxed<br />
Wax<br />
TABLE 3<br />
ANALYSES OF FEEDS AND PRODUCTS<br />
Hydrogen Types<br />
by 'H-NHR. %<br />
Arom<strong>at</strong>ic Paraffinic<br />
-~ - C - H --<br />
N S<br />
14.6<br />
17.0<br />
2.6<br />
14.6<br />
19.5<br />
2.2<br />
14.6<br />
19.3<br />
2.7<br />
9.3<br />
15.3<br />
4.1<br />
10.8<br />
12.5<br />
4.6<br />
10.8<br />
14.1<br />
5.0<br />
21.3<br />
22.9<br />
0.3<br />
21.3<br />
24.6<br />
1.9<br />
25.9<br />
26.9<br />
0.8<br />
20.7<br />
23.8<br />
2.8<br />
42.6<br />
34.2<br />
82.5<br />
42.6<br />
32.5<br />
88.4<br />
42.6<br />
28.8<br />
71.2<br />
49.4<br />
33.6<br />
62.0<br />
40.7<br />
38.4<br />
57.6<br />
42.5<br />
35.2<br />
55.4<br />
40.3<br />
35.6<br />
91.3<br />
40.3<br />
32.5<br />
90.9<br />
31.4<br />
30.1<br />
86.7<br />
33.8<br />
26.4<br />
76.9<br />
Elwnental Analysis,<br />
rt 8 (a)<br />
89.1 10.0 0.3 c0.1<br />
88.4 9.8 0.4
la<br />
Figure 1. lH-NMR spectra <strong>of</strong> samples from experiment 5. (a) feed <strong>oil</strong>. (b) wax,<br />
(c) dewaxed <strong>oil</strong>. TMS - tetramethylsilane internal reference, SSB -<br />
spinning side band.<br />
266
i<br />
2a<br />
t . 5 10 15 20 15 n I5 UI 45 0<br />
Irtentlo" TI-. .I".<br />
Figure 2. Gas chrom<strong>at</strong>ograms <strong>of</strong> samples from experiment 5. (a) feed <strong>oil</strong>, (b)<br />
wax, (c) dewaxed <strong>oil</strong>. (internal standard, n-decane)<br />
c<br />
267
PERFORMANCE OF THE LOW TEMPERATURE FIRST STAGE OF<br />
HRI 'S CATALYTIC TWO-STAGE LIQUEFACTION PROCESS<br />
J. B. McLean, A. G. Comolli and T. 0. Smith<br />
Hydrocarbon Research, Inc.<br />
(A Subsidiary <strong>of</strong> Dynalectron Corpor<strong>at</strong>ion)<br />
P. 0. Box 6047<br />
Lawrenceville, New Jersey 08648<br />
ABSTRACT<br />
Hydrocarbon Research, Inc. (HRI), under the sponsorship <strong>of</strong> the U. S. Department<br />
<strong>of</strong> Energy (DOE), is developing a c<strong>at</strong>alytic two-stage <strong>co</strong>al liquefaction process.<br />
The process <strong>co</strong>nsists <strong>of</strong> two direct-<strong>co</strong>upled ebull<strong>at</strong>ed-bed reactors in series, with<br />
the first stage oper<strong>at</strong>ed <strong>at</strong> lower temper<strong>at</strong>ures (t800'F) than typically used in<br />
direct liquefaction. Studies <strong>of</strong> both bituminous and sub-bituminous <strong>co</strong>als in a<br />
nominal 50 lb/day <strong>co</strong>ntinuous, integr<strong>at</strong>ed recycle bench unit have shown <strong>co</strong>n-<br />
siderable improvements in both distill<strong>at</strong>e yield and product quality over other<br />
processes. In order to better understand the chemistry <strong>of</strong> the unique first-stage<br />
reactor <strong>co</strong>nditions, a special on-line sampling system was added to the bench<br />
unit. Samples obtained over a wide range <strong>of</strong> oper<strong>at</strong>ing <strong>co</strong>nditions indic<strong>at</strong>e th<strong>at</strong><br />
the first stage is an efficient hydrogen<strong>at</strong>ion system, achieving balanced r<strong>at</strong>es <strong>of</strong><br />
<strong>co</strong>al <strong>co</strong>nversion and dissolution. solvent to <strong>co</strong>al hydrogen transfer, solvent<br />
regener<strong>at</strong>ion, and liquefaction product upgrading and stabiliz<strong>at</strong>ion. Differences<br />
in responses <strong>of</strong> the two <strong>co</strong>als studied are noted and discussed.<br />
INTRODUCTION<br />
Hydrocarbon Research, Inc. (HRI) has long been actively involved in the development<br />
<strong>of</strong> <strong>co</strong>al liquefaction process technology. The H-Coal@ Process, fe<strong>at</strong>uring a<br />
single-stage ebul l<strong>at</strong>ed-bed c<strong>at</strong>alytic reactor, was successfully developed and<br />
demonstr<strong>at</strong>ed through oper<strong>at</strong>ion <strong>of</strong> a 200 ton per day pilot plant <strong>at</strong> C<strong>at</strong>lettsburg.<br />
Kentucky, in the early 1980's.(l) In 1981-1982, HRI <strong>co</strong>nducted a series <strong>of</strong><br />
labor<strong>at</strong>ory investig<strong>at</strong>ions to evalu<strong>at</strong>e various two-stage liquefaction <strong>co</strong>ncepts<br />
which fe<strong>at</strong>ured a thermal first-stage re ctor followed by a closely <strong>co</strong>upled,<br />
ebull<strong>at</strong>ed bed, c<strong>at</strong>alytic se<strong>co</strong>nd stage.(2-6f The results <strong>of</strong> these programs formed<br />
the basis for the ctirrent C<strong>at</strong>alytic Two-Stage <strong>Liquefaction</strong> (CTSL) <strong>co</strong>ncept, which<br />
fe<strong>at</strong>ures two direct-<strong>co</strong>upled, ebull<strong>at</strong>ed-bed reactors in series. Under OOE<br />
sponsorship, HRI has been <strong>co</strong>nducting a development rogram for the CTSL Process<br />
Since 1983. Program results have been re~orted(7-~~) with C4-975OF distil l<strong>at</strong>e<br />
yields <strong>of</strong> 65 W I MAF <strong>co</strong>al achieved for both Illinois No. 6 and Wyodak <strong>co</strong>als. The<br />
process e<strong>co</strong>nomics have been shown to be favorable in c parison with other twostage<br />
approaches by an independent <strong>co</strong>ntractor's study(12?for both <strong>co</strong>als.<br />
268<br />
i
PROCESS FEATURES<br />
The salient fe<strong>at</strong>ures <strong>of</strong> the CTSL Process are listed in Table 1. The key fe<strong>at</strong>ure<br />
which distinguishes this from other single- or two-stage processes is the opera-<br />
tion Of a low temper<strong>at</strong>ure (
BENCH UNIT DESCRIPTION<br />
Process deve1opmnt studies have been <strong>co</strong>nducted in HRI ‘S <strong>co</strong>ntinuous two-stage<br />
Bench Unit 227, shown in Figure 1. It is necessary to study the process in a<br />
<strong>co</strong>ntinuous unit “4th self-sustained recycle solvent gener<strong>at</strong>ion in order to fully<br />
understand the<br />
and avoid the pitfalls <strong>of</strong> smaller b<strong>at</strong>ch or once-through<br />
unfts. The unit fe<strong>at</strong>ures two 200Occ ebull<strong>at</strong>ed-bed reactors in<br />
series. A special high-pressure. on-line sampling System was adapted to the<br />
first-stage reactor to obtain the d<strong>at</strong>a re uired to independently assess the<br />
effectiveness <strong>of</strong> the two reactor stages. hior to this, it was necessary to<br />
<strong>at</strong>tempt to interpret the effects <strong>of</strong> first-stage variables based on overall<br />
results only. Since the reactors are direct-<strong>co</strong>upled, and the desired sample<br />
quantities represent a significant fraction <strong>of</strong> the first-stage reactor inventory,<br />
the design and oper<strong>at</strong>ion <strong>of</strong> the sampling system is critical to obtain<br />
represent<strong>at</strong>ive samples while minimizing unit disruption. The d<strong>at</strong>a presented in<br />
this paper are based on analyses <strong>of</strong> the first-stage samples. A <strong>co</strong>ntinuous<br />
<strong>at</strong>mospheric still was also added to the unit during this program to provide<br />
accur<strong>at</strong>e <strong>co</strong>ntrol <strong>of</strong> recycle solvent cut points. The <strong>at</strong>mspheric still bottoms<br />
are subjected to further b<strong>at</strong>ch filtr<strong>at</strong>ion and/or vacuum distil l<strong>at</strong>ion oper<strong>at</strong>ions<br />
to study various recycle <strong>oil</strong> prepar<strong>at</strong>ion techniques. System inventories are<br />
minimized in order to provide a rapid lineout response to <strong>co</strong>ndition changes.<br />
PROGRAM HISTORY<br />
A sumnary <strong>of</strong> bench unit oper<strong>at</strong>ions <strong>co</strong>nducted through February 1986 is shown in<br />
Table 2. The first year <strong>of</strong> the program was dedic<strong>at</strong>ed to Illinois No. 6 <strong>co</strong>al, and<br />
the se<strong>co</strong>nd year to Wyodak sub-bituminous <strong>co</strong>al. Following renewal <strong>of</strong> the <strong>co</strong>ntract<br />
for two additional years in 1985, additional studies are being <strong>co</strong>nducted with<br />
Illinois No. 6 <strong>co</strong>al. Each <strong>of</strong> these <strong>co</strong>als has been studied in previous single-<br />
and two-stage oper<strong>at</strong>ions, so th<strong>at</strong> an extensive d<strong>at</strong>a base for canparison <strong>of</strong> CTSL<br />
results exists. The implement<strong>at</strong>ion <strong>of</strong> the first-stage sampling system, l<strong>at</strong>e in<br />
the, original Illinois <strong>co</strong>al program. gre<strong>at</strong>ly enhanced the understanding <strong>of</strong> the<br />
observed favorable performance, and first-stage sample analyses were used<br />
extensively in a1 1 subsequent work.<br />
FIRST-STAGE PERFORMANCE<br />
Coal Conversion R<strong>at</strong>e<br />
One <strong>of</strong> the primary benefits <strong>of</strong> the lower temper<strong>at</strong>ure liquefaction stage is th<strong>at</strong><br />
<strong>co</strong>al is <strong>co</strong>nverted <strong>at</strong> a <strong>co</strong>ntrolled r<strong>at</strong>e, allowing a balance between thennal and<br />
c<strong>at</strong>alytic reaction r<strong>at</strong>es to be maintained. Figures 2 and 3 show the rel<strong>at</strong>ionship<br />
between <strong>co</strong>al <strong>co</strong>nversion (to quinoline solubles) and both temper<strong>at</strong>ure and<br />
residence time for several sets <strong>of</strong> d<strong>at</strong>a for both <strong>co</strong>als. In each case. the<br />
<strong>co</strong>nnected d<strong>at</strong>a points represent studies where a1 1 other parameters (se<strong>co</strong>nd-stage<br />
<strong>co</strong>nditions, solvent/<strong>co</strong>al r<strong>at</strong>io, etc.) are held <strong>co</strong>nstant. Comparisons <strong>of</strong> non-<br />
<strong>co</strong>nnected p<strong>of</strong>nts should not be made since other parameters are different as well.<br />
Note th<strong>at</strong> increasing severity by both parameters always results in an increased<br />
<strong>co</strong>al <strong>co</strong>nversion, indic<strong>at</strong>ing kinetic r<strong>at</strong>e <strong>co</strong>ntrol. It should also be noted th<strong>at</strong><br />
2 70
overall process <strong>co</strong>nversions were in all cases substantially higher, and tended to<br />
<strong>co</strong>rrel<strong>at</strong>e with first-stage <strong>co</strong>nversions. In the case <strong>of</strong> the Illinois No. 6 <strong>co</strong>al,<br />
it appears th<strong>at</strong> "maximum" <strong>co</strong>al <strong>co</strong>nversions (95-96%, typically) are being<br />
approached <strong>at</strong> 750-775OF. while the Wyodak <strong>co</strong>al is much slower to <strong>co</strong>nvert and<br />
requires additional thermal severity (90-93% <strong>co</strong>nversion typically achieved in<br />
se<strong>co</strong>nd stage).<br />
Hydrogen Transfer Efficiency<br />
Figure 4 shows the <strong>at</strong>omic hydrogen/carbon r<strong>at</strong>io <strong>of</strong> THF insoluble ICY4 from both<br />
first- and se<strong>co</strong>nd-stage samples as a function <strong>of</strong> <strong>co</strong>al <strong>co</strong>nversion for Wyodak <strong>co</strong>al.<br />
Surprisingly, this r<strong>at</strong>io stays quite high (<strong>at</strong> or above'the original <strong>co</strong>al level)<br />
over a wide range <strong>of</strong> first-stage <strong>co</strong>nversions. It would be expected th<strong>at</strong> the most<br />
reactive <strong>co</strong>mponents <strong>of</strong> the <strong>co</strong>al would be the most hydrogen-rich. and would leave<br />
behind a residue <strong>of</strong> depleted hydrogen <strong>co</strong>ntent. This in fact does occur in higher<br />
temper<strong>at</strong>ure, thermal processes. However. the <strong>co</strong>ntrolled <strong>co</strong>nversion r<strong>at</strong>e in CTSL<br />
allows for efficient hydrogen transfer to the <strong>co</strong>al as it reacts. A similar<br />
rel<strong>at</strong>ionship has been noted for the Illinois No. 6 <strong>co</strong>al. Only <strong>at</strong> the more severe<br />
thermal <strong>co</strong>nditions <strong>of</strong> the se<strong>co</strong>nd stage does the hydrogen transfer appear to drop<br />
<strong>of</strong>f. as evidenced by the lower H/C r<strong>at</strong>ios for the high <strong>co</strong>nversion samples.<br />
No <strong>at</strong>tempt has been made here to distinguish "unreacted <strong>co</strong>al" from IOM formed by<br />
regressive reaction. However, the <strong>co</strong>mbin<strong>at</strong>ion <strong>of</strong> the observed kinetic response,<br />
residue analyses, and mild severity <strong>co</strong>nditions indic<strong>at</strong>e th<strong>at</strong> regressive reaction<br />
should be minimal in the first stage. While residue analyses are interesting,<br />
they are <strong>of</strong> limited utility, particularly since the overall <strong>co</strong>al <strong>co</strong>nversions<br />
achieved in CTSL are no better than in the single-stage H-Coals Process. Of more<br />
importance are the analyses <strong>of</strong> the liquids which are formed <strong>at</strong> first-stage<br />
<strong>co</strong>nditions, which are substantially different than those produced in other direct<br />
1 iquefaction processes.<br />
Sol vent Hydrogent<strong>at</strong>i on<br />
Since the <strong>co</strong>al is liquefied in the presence <strong>of</strong> a c<strong>at</strong>alyst <strong>at</strong> <strong>co</strong>nditions which<br />
favor hydrogen<strong>at</strong>ion. donor species present in the solvent can be regener<strong>at</strong>ively<br />
rehydrogen<strong>at</strong>ed. This is illustr<strong>at</strong>ed for a typical <strong>co</strong>ndition for each <strong>co</strong>al in<br />
Table 3, which <strong>co</strong>mpares properties <strong>of</strong> first-stage <strong>oil</strong> and pressure filter liquid<br />
(PFL), which is both the se<strong>co</strong>nd-stage <strong>oil</strong> and process recycle solvent. Note th<strong>at</strong><br />
even though substantial <strong>co</strong>al <strong>co</strong>nversion has occurred in the first stage in each<br />
case, there is no indic<strong>at</strong>ion <strong>of</strong> solvent quality deterior<strong>at</strong>ion - in fact, the<br />
solvent quality, as measured by standard microautoclave tests, has improved.<br />
This is due to simultaneous solvent hydrogen<strong>at</strong>ion, as indic<strong>at</strong>ed by the improved<br />
hydrogen <strong>co</strong>ntent and lower arom<strong>at</strong>ics level in the first-stage liquid. This is a<br />
key difference from other two-stage processes, where solvent quality is depleted<br />
in the liquefaction stage due to mre severe thermal <strong>co</strong>nditions and the lack <strong>of</strong><br />
an effective hydrogen<strong>at</strong>ion c<strong>at</strong>alyst. One positive benefit <strong>of</strong> this effect on the<br />
overall process is th<strong>at</strong> the feed solvent/<strong>co</strong>al r<strong>at</strong>io can be set <strong>at</strong> a minimum pum-<br />
pable level, without <strong>co</strong>ncern for available donor hydrogen levels. Bench unit<br />
oper<strong>at</strong>ions on Illinois No. 6 <strong>co</strong>al have been <strong>co</strong>nducted <strong>at</strong> feed slurry solvent/<strong>co</strong>al<br />
r<strong>at</strong>ios as low as 1.1, and still lower r<strong>at</strong>ios may well be possible on a larger<br />
scale. This has a large favorable impact on process e<strong>co</strong>nomics.<br />
271
Recycle Residuum Hydrogen<strong>at</strong>i On<br />
Residuum in the recycle solvent is upgraded by hydrogen<strong>at</strong>ion in the first stage,<br />
making it more reactive for cracking to lighter distill<strong>at</strong>es in the se<strong>co</strong>nd stage.<br />
This is indic<strong>at</strong>ed in Table 4, which shows net positive yields <strong>of</strong> residuum <strong>co</strong>m-<br />
ponents in the first stage, and net <strong>co</strong>nversion to distill<strong>at</strong>es in the se<strong>co</strong>nd<br />
stage. As a result, the overall 975'F+ yields are quite low, and the quality (as<br />
indic<strong>at</strong>ed by high <strong>oil</strong> and low preasphaltene <strong>co</strong>ntents) is also quite good.<br />
C<strong>at</strong>alytic Stabi liz<strong>at</strong>ion/Upgrading <strong>of</strong> Primary <strong>Liquefaction</strong> Products<br />
The discussion .above had highlighted the effect <strong>of</strong> first-stage <strong>co</strong>nditions on<br />
recycle solvent properties. In fact, the <strong>oil</strong> properties presented are for<br />
liquids which are a blend <strong>of</strong> recycle solvent and direct first-stage products.<br />
Depending on feed solvent/<strong>co</strong>al r<strong>at</strong>io and net first-stage reactions, the first-<br />
stage <strong>oil</strong> <strong>co</strong>ntent is estim<strong>at</strong>ed to be 20-508 directly produced fran <strong>co</strong>al, with the<br />
remainder derived from recycle solvent. (Of <strong>co</strong>urse, in an integr<strong>at</strong>ed oper<strong>at</strong>ion<br />
all <strong>of</strong> the m<strong>at</strong>erial is ultim<strong>at</strong>ely <strong>co</strong>al-derived; here the distinction is being<br />
made to specifically include m<strong>at</strong>erial which has not yet been exposed to se<strong>co</strong>nd-<br />
stage <strong>co</strong>nditions.) With this in mind, the level <strong>of</strong> hydrogen<strong>at</strong>ion is even more<br />
notable since the primary liquefaction products should be <strong>of</strong> substantially lower<br />
quality than the recycle solvent.<br />
COAL COMPARISON<br />
Evidence has been presented for both Illinois No. 6 and Wyodak <strong>co</strong>als which sup-<br />
port the process <strong>co</strong>ncept <strong>of</strong> first-stage hydrogen<strong>at</strong>ion, resulting in improved<br />
overall liquid yields and product qualities. However, the response <strong>of</strong> the two<br />
<strong>co</strong>als - and hence the optimum process <strong>co</strong>nditions for each - are quite different.<br />
As has been noted in Figure 2, the sub-bituminous <strong>co</strong>al is much slower to <strong>co</strong>nvert,<br />
and probably requires a first-stage temper<strong>at</strong>ure <strong>of</strong> <strong>at</strong> least 75OOF to achieve<br />
enough <strong>co</strong>al <strong>co</strong>nversion for the c<strong>at</strong>alytic tre<strong>at</strong>ment to be effective. The<br />
bituminous <strong>co</strong>al liquefies much more readily, but (as noted in Table 4) gives much<br />
higher net residuum yields. Work to d<strong>at</strong>e has indic<strong>at</strong>ed optimum performance <strong>at</strong><br />
750-775OF. but it is probable th<strong>at</strong> this can be reduced by the appropri<strong>at</strong>e<br />
<strong>co</strong>mbin<strong>at</strong>ion <strong>of</strong> c<strong>at</strong>alyst, space velocity, etc. This objective is being pursued in<br />
the present program. Other items being investig<strong>at</strong>ed include optimiz<strong>at</strong>ion <strong>of</strong><br />
liquid yield distribution, particularly the extinction <strong>co</strong>nversion <strong>of</strong> all 650DFt<br />
products, and oper<strong>at</strong>ion <strong>at</strong> lower se<strong>co</strong>nd-stage temper<strong>at</strong>ures to improve product<br />
quality and extend c<strong>at</strong>alyst life.<br />
ACKNOWLEDGEMENTS<br />
This work was supported by the United St<strong>at</strong>es Department <strong>of</strong> Energy under Contracts<br />
DE-AC22-83PC-60017 and DE-AC22-85PC80002. Analytical d<strong>at</strong>a provided by Drs. F. P.<br />
Burke and R. A. Winschel <strong>of</strong> the Cono<strong>co</strong> Coal Research Division under DOE Contract<br />
OE-AC22-84PC70018 have been quite helpful in our interpret<strong>at</strong>ion <strong>of</strong> process<br />
perf onnance .<br />
2 72
REFERENCES<br />
Tallb, A., 0. Gray, and M. Neuworth. Assessment <strong>of</strong> H-Coal@ Process<br />
Developments, MITRE Corpor<strong>at</strong>ion Report MTR-83W199. January 1984.<br />
"H-Coal@ Process Improvement Study - Bench Unit Baseline Run With<br />
prehe<strong>at</strong>er/Reactor", Hydrocarbon Research, hC., FE-10152-65, May 1981.<br />
"Bench Unit Prehe<strong>at</strong>er Study To Determine Optimum Prehe<strong>at</strong>er Conditions for<br />
High Coal Conversion", Hydrocarbon Research, Inc., FE-10152-86, October<br />
1982.<br />
"Two-Stage Bench Run on Illinois No. 6 Coal - Runs 227-2 and 227-3.<br />
Hydrocarbon Research, Inc., FE-10152-87. August 1982. (Not Issued)<br />
"Two-Stage <strong>Liquefaction</strong> <strong>of</strong> Kyodak Coal - Runs 227-5 and 227-6". Hydrocarbon<br />
Research, Inc., FE-10152-90, September 1982. (Not Issued)<br />
"Two-Stage <strong>Liquefaction</strong> Study Using Illinois No. 6 Coal and Equal Volume<br />
Reactors - Run 227-7". Hydrocarbon Research Inc.. FE-10152-92. September<br />
1982. (Not Issued)<br />
"New Technology Concept for Two-Stage <strong>Liquefaction</strong> <strong>of</strong> Coal - Illinois No. 6<br />
Coal", Hydrocarbon Research, Inc.. DE-60017-TOP-1, August 1985.<br />
"New Technology Concept for Two-Stage <strong>Liquefaction</strong> <strong>of</strong> Coal - Wyoming<br />
(Wyodak) Coal Study", Hydrocarbon Research, Inc.. DE-60017-TOP-3. February<br />
1986.<br />
"New Technology Concept for Two-Stage <strong>Liquefaction</strong> <strong>of</strong> Coal - Final Summary<br />
Report", Hydrocarbon Research, Inc., DE-60017-10, February 1986.<br />
"HRI's C<strong>at</strong>alytic Two-Stage <strong>Liquefaction</strong> Process - Performance Comparison For<br />
Illinois No. 6 and Wyodak Coals", McLean, J. E., A. G. Comolli, and J. 8.<br />
MacArthur. Presented <strong>at</strong> the American Chemical Society's Division <strong>of</strong><br />
Petroleum Chemistry Symposium. September 1985.<br />
"The C<strong>at</strong>alytic Two-Stage <strong>Liquefaction</strong> Process", McLean, J. 8. A. G.<br />
Comolli, J. E. Duddy ?nd T. 0. Smith. Department <strong>of</strong> Energy's Direct<br />
<strong>Liquefaction</strong> Contractors Conference, November 1985.<br />
"Technical and E<strong>co</strong>nomic Impacts <strong>of</strong> Staged <strong>Liquefaction</strong> Configur<strong>at</strong>ions".<br />
Mitre Corpor<strong>at</strong>ion. Contract No. 21-5262 (Final Report to be issued April<br />
1986).<br />
"Recent Developments With The Lumus-Crest Integr<strong>at</strong>ed Two-Stage <strong>Liquefaction</strong><br />
Process", M. Peluso. et al. Proceedings <strong>of</strong> 9th Annual EPRI Contractors'<br />
Conference on Coal <strong>Liquefaction</strong>, May 1984.<br />
"Process Studies <strong>of</strong> Integr<strong>at</strong>ed Two-Stage <strong>Liquefaction</strong> <strong>of</strong> Wilsonvi 1 le", M.<br />
J. Moniz, et al. Proceedings <strong>of</strong> 9th Annual EPRI Contractors' Conference on<br />
Coal <strong>Liquefaction</strong>. May 1984.<br />
273
FIRST STAGE<br />
HRI'S CATALYTIC TWO STAGE LIQUEFACTION PROCESS<br />
"Low" Temper<strong>at</strong>ure ( 8OO0F)<br />
Hydro<strong>co</strong>nversion C<strong>at</strong>alyst (e.9. Amoc<strong>at</strong> 1A. CoMo)<br />
Functions: Complete Coal Conversion (Thenal in an improved solvent envirorment)<br />
Residuum Conversion to Distill<strong>at</strong>e Products<br />
Hetero<strong>at</strong>om Removal<br />
Avoi d Dehy d r oge n <strong>at</strong> i on<br />
OTHER PROCESS FEATURES<br />
Reaction Stages are Oi rect-Coupled<br />
€bull<strong>at</strong>ed Bed Technology Scaleable Based on H-Coal@/H-Oil@ Experience<br />
Highest Conversion to Distill<strong>at</strong>es <strong>of</strong> any Direct <strong>Liquefaction</strong> Process<br />
More Aliph<strong>at</strong>ic/Petroleum-Like Products than other Direct <strong>Liquefaction</strong> Processes<br />
2 74
CATALYTIC TWO STAGE LIQUEFACTION PROCESS DEVELOPMENT<br />
History <strong>of</strong> Bench Unit Oper<strong>at</strong>ions (through February 1986)<br />
Number <strong>of</strong> First Stage<br />
Runs Conditions Samples<br />
Illinois No. 6 Coal (1983-1984) -<br />
Process Variable Studies 8 149 38<br />
First Stage Sampling 1 12 4 4<br />
Process Demonstr<strong>at</strong>ion --<br />
1 25 - 1 -<br />
Total Illinois No. 6<br />
( 1983- 1984 ) 10 186 43 4<br />
Wyodak Sub-bi tuminous Coal<br />
(1983-1985)<br />
Process Variable Studies<br />
Process Demonstr<strong>at</strong>ion<br />
Total Wyodak Coal<br />
3 80<br />
--<br />
2 44<br />
5 124<br />
25<br />
- 3<br />
28<br />
18<br />
8<br />
26<br />
-<br />
Illinois No. 6 Coal (1985-1986)<br />
Process Variable Studies 2 57 16 15<br />
275<br />
TABLE 2
FIRST-STAQ COAL CONVERSION (W I IIAF)<br />
VERSUS TEMPERATURE<br />
FIRST-STAGE TEMPERATURE, O F<br />
o Illinois No. 6 (Run 227-18)<br />
0 Illinois No. 6 (Run, 227-32)<br />
+ Wyodak, Run 227-22 (Conditions 4, 5 and 6)<br />
x Wyodak, Run 227-22 (Conditions 7 and 9)<br />
276<br />
FIGURE 2
A<br />
I I<br />
I<br />
I<br />
I<br />
I<br />
I<br />
I<br />
I<br />
I<br />
I<br />
I<br />
I<br />
9<br />
be<br />
2c<br />
z<br />
0<br />
w<br />
v,<br />
!A CL<br />
> z<br />
0<br />
V<br />
_1<br />
3<br />
V<br />
w<br />
cl<br />
2<br />
v, I<br />
I- v,<br />
5<br />
U<br />
I<br />
I<br />
I<br />
I<br />
I<br />
I<br />
I<br />
I<br />
I<br />
I<br />
I<br />
I<br />
I<br />
I<br />
I<br />
I<br />
I<br />
I<br />
V<br />
FIRST-STAGE COAL CONVERSION (W 2 MAF) VERSUS TIME<br />
ILLINOIS NO. 6<br />
I I<br />
+ Run 227-32, 775OF.<br />
o Run 227-30, 75OoF<br />
I<br />
I-<br />
i<br />
+ Run 227-22<br />
o Run 227-25<br />
I I<br />
+/+<br />
.5 I, 1.5 2.<br />
I<br />
WYODAK<br />
750°F<br />
750°F<br />
! !<br />
RELATIVE RESIDENCE TIME (RELATIVE SV-1)<br />
277<br />
FIGURE 3
REACTOR SOLIOS HTOROGEN/CARBON ATOllIC RATIO<br />
VERSUS COAL COWVERSION<br />
WYOOAK COAL<br />
l . [ . , . , 1 ! ' !<br />
L '<br />
.9 + +<br />
I ' I ~ l ' ! ' 7<br />
V \<br />
= -5 I I I I I I I I I<br />
0. 10. 20. 38. 48. 50. 60. 70. 80. 90. 100.<br />
COAL CONVERSION, W % MAF COAL<br />
+ First-Stage Solids<br />
o Se<strong>co</strong>nd-Stage Solids<br />
* Range <strong>of</strong> Analyses for Fresh Coal<br />
278<br />
FIGURE 4
COllpARISON OF FIRST-STAGE OIL AND PFL PROPERTIES<br />
Bench Unit Coal Conversion, W % MAF<br />
Microautoclave Sol vent Quality<br />
Test. W % THF Conversion<br />
HRI\Il<br />
Cono<strong>co</strong> (2) *<br />
H/C R<strong>at</strong>io - 650-850°F<br />
850-975'F<br />
975'F+<br />
Proton? NMR - X Arom<strong>at</strong>ics<br />
850°F- Di sti 1 l<strong>at</strong>e<br />
85OoF+ Residuum<br />
TABLE 3<br />
ILLINOIS NO. 6 COAL YYODAK COAL<br />
Run No. 227-18-12 Run No. 227-25-16<br />
< ___________ s T A GI s -----_------ ><br />
----<br />
FIRST SECOND FIRST SECOND<br />
OIL PFL OIL PFL<br />
--<br />
--<br />
87.1 92;7 73.6 91.6<br />
83.3<br />
82.9<br />
1.28<br />
1.19<br />
0.95<br />
14.6<br />
29.2<br />
(1) HRI procedure uses m<strong>at</strong>ched <strong>co</strong>al and solvent.<br />
(2) Cono<strong>co</strong> procedure uses Indiana V <strong>co</strong>al.<br />
D<strong>at</strong>a provided by CONOCO.<br />
W % 975"F+ In Oil<br />
97iV[+H;;p;erti es<br />
X Nitrogen<br />
% Oil<br />
% Asphal tenes<br />
% Preasphaltenes<br />
Estim<strong>at</strong>ed Net 9750Ft<br />
Yield - W % MAF Coal<br />
Oils<br />
Asphal tenes<br />
Pre-Asphal tenes<br />
TOTAL<br />
- 975OF' RESIDUUM PROPERTIES<br />
76.6 54.5 52.0<br />
79.9 64.5 64 .O<br />
1.25 1.40 1.40<br />
1.13 1.34 1.24<br />
0.91 1.06 0.98<br />
15.7 11 .o 10.6<br />
31.4 19.3 25.3<br />
ILLINOIS NO. 6 WYOOAK<br />
Run 227-32-9 Run 227-25-16<br />
< _______ S T A G S _______ ><br />
First Se<strong>co</strong>nd First Se<strong>co</strong>nd<br />
-<br />
39.5<br />
1.03<br />
0.65<br />
64.5<br />
28.6<br />
6.9<br />
9.6<br />
9.3<br />
3.3<br />
22.2<br />
2 79<br />
32.1<br />
0.99<br />
0.53<br />
71.9<br />
23.5<br />
4.6<br />
-3.2<br />
-7.2<br />
-2.9<br />
-13.3<br />
-<br />
12.1<br />
1.06<br />
0.79<br />
75.3<br />
24.1<br />
0.6<br />
2.4<br />
2.2<br />
0.1<br />
-<br />
4.7<br />
9.0<br />
0.98<br />
0.73<br />
84.7<br />
15.0<br />
0.3<br />
-0.2<br />
-1 .R<br />
-0.1<br />
-<br />
-2.1<br />
TABLE 4
TRANSPORTATION FUELS FROM TWO-STAGE LIQUEFACTION PRODUCTS<br />
Richard F. Sullivan<br />
Chevron Research Company, P. 0. Box 1627<br />
Richmond, California 94802-0627<br />
INTRODUCTION<br />
FOK several years, Chevron Research Company under a <strong>co</strong>ntract with<br />
the US Department <strong>of</strong> Energy has been studying the refining <strong>of</strong> <strong>co</strong>al<br />
liquids. Detailed results are given in a series <strong>of</strong> DOE Interim<br />
Reports (1). The earlier work emphasized upgrading <strong>of</strong> products from<br />
single-stage processes: SRC-11, H-Coal, and EDS. More recently, we<br />
have been studying products from two different two-stage processes:<br />
the Integr<strong>at</strong>ed Two-Stage <strong>Liquefaction</strong> (ITSL) Process and the C<strong>at</strong>alytic<br />
Two-Stage <strong>Liquefaction</strong> (CTSL) Process.<br />
The purpose <strong>of</strong> this paper is to <strong>co</strong>mpare results for syncrudes<br />
from single-stage and two-stage processes, from different two-stage<br />
processes, from different <strong>co</strong>als [Illinois No. 6 (bituminous) and<br />
wyodak (subbituminous)], and <strong>of</strong> different b<strong>oil</strong>ing ranges from a given<br />
<strong>co</strong>al and process.<br />
The ITSL process, developed by Cities Service and Lummus Crest,<br />
Inc., oper<strong>at</strong>es with a high temper<strong>at</strong>ure (over 80O0F) first stage with<br />
no added c<strong>at</strong>alyst. The product is then deashed, and sent to a<br />
se<strong>co</strong>nd-stage th<strong>at</strong> oper<strong>at</strong>es <strong>at</strong> lower temper<strong>at</strong>ures (typically below<br />
800OF) with an ebull<strong>at</strong>ed c<strong>at</strong>alyst bed (2).<br />
The CTSL process, developed by Hydrocarbon Research, Inc. (HRI),<br />
oper<strong>at</strong>es with two c<strong>at</strong>alytic ebull<strong>at</strong>ed-bed stages. In <strong>co</strong>ntrast to the<br />
ITSL process, the CTSL first stage oper<strong>at</strong>es <strong>at</strong> a lower temper<strong>at</strong>ure<br />
(below 8 OOOF) than the se<strong>co</strong>nd (!which oper<strong>at</strong>es above 800°F) (3).<br />
Depending upon how each liquefaction process is oper<strong>at</strong>ed, the end<br />
point (EP) <strong>of</strong> the net whole-liquid product will vary. Typically, part<br />
or all <strong>of</strong> the vacuum gas <strong>oil</strong> (VGO) made by the process is used as<br />
recycle solvent for the <strong>co</strong>al. Some or all <strong>of</strong> it is ultim<strong>at</strong>ely<br />
<strong>co</strong>nverted to lower b<strong>oil</strong>ing products. Thus, the net whole-liquid<br />
product can have an EP ranging from below 65OoF to over 85OOF. As we<br />
will see, the ease or difficulty <strong>of</strong> upgrading is affected to a large<br />
extent by product EP. The 650-850'F VGO is rel<strong>at</strong>ively difficult to<br />
upgrade, but is reported to be an excellent recycle solvent.<br />
Therefore, there may be both upstream and downstream advantages to<br />
recycling this VGO, as shown for example, by MacArthur et a1 (4).<br />
Ultim<strong>at</strong>ely, <strong>of</strong> <strong>co</strong>urse, the <strong>co</strong>sts and yields <strong>of</strong> both liquefaction and<br />
upgrading must be used to determine the optimum EP.<br />
In this paper, we will use results for upgrading products from<br />
the H-Coal process (1, 5) as our primary basis for <strong>co</strong>mparison with<br />
single-stage processes.<br />
FEEDSTOCKS<br />
Key factors th<strong>at</strong> determine how easy or difficult a particular<br />
syncrude is to refine are EP, b<strong>oil</strong>ing range, hydrogen <strong>co</strong>ntent, and<br />
hetero<strong>at</strong>om <strong>co</strong>ntent. Also, hot-heptane insoluble <strong>co</strong>mpounds (low-<br />
solubility polycyclic-arom<strong>at</strong>ic and polar <strong>co</strong>mpounds, asphaltenes, and<br />
ash) can make syncrudes difficult to processes.<br />
280
I<br />
Table I shows properties <strong>of</strong> pairs <strong>of</strong> H-Coal and ITSL syncrudes<br />
derived from Illinois <strong>co</strong>al. Table I1 shows properties <strong>of</strong> pairs <strong>of</strong><br />
H-Coal, CTSL, and ITSL syncrudes derived from wyodak <strong>co</strong>al. In each<br />
case, the syncrude identified as "A" had a higher EP than th<strong>at</strong><br />
i identified as "B". The A syncrudes were blended from <strong>co</strong>mponents<br />
supplied HRI and Lummus in r<strong>at</strong>ios re<strong>co</strong>mmended by DOE to represent, as<br />
nearly as possible, "net whole-liquid prOdUCtS" from these processes.<br />
[Note: the heavy fractions <strong>of</strong> Illinois ITSL A and Wyodak CTSL A,<br />
1 as-received, <strong>co</strong>ntained large quantities <strong>of</strong> hot-heptane insolubles and<br />
i<br />
Process<br />
Sample Identific<strong>at</strong>ion<br />
LV% <strong>of</strong> As-Received Oil<br />
Inspections<br />
Gravity, "API<br />
Sulfur, ppm<br />
Nitrogen, ppm<br />
Oxygen, ppm<br />
Hydrogen, Wt %<br />
Carbon, Wt %<br />
Hot-Heptane<br />
Insolubles, ppm<br />
TBP Distill<strong>at</strong>ion, OF<br />
(ASTM D2887)<br />
St/5<br />
10/30<br />
50<br />
70/90<br />
95/99<br />
B<strong>oil</strong>ing Range, LV%<br />
St-40O0F<br />
400-700'F<br />
700O~+<br />
Table I<br />
SYNCRUDES FROM ILLINOIS NO. 6 COAL<br />
A B<br />
100 87<br />
A B<br />
95 69<br />
25.8 28.1 13.6 17.6<br />
2000 1400 865 700<br />
4600 3300 1050 730<br />
18000 19600 2600 1800<br />
11.29 11.44 10.19 10.68<br />
86.25 86.13 89.35 88.99<br />
3500 54 375 290<br />
56/177 56/170 97/275 97/214<br />
213/333 200/310 375/532 314/471<br />
404 380 602 56 0<br />
476/588 440/508 665/745 609/676<br />
654/765 538/589 793/859 703/763<br />
49 57<br />
48 43<br />
3 0<br />
281<br />
12 18<br />
69 76<br />
19 6
Process<br />
Identific<strong>at</strong>ion<br />
LV% Of AS-<br />
Received Oil<br />
Inspections<br />
Gravity , OAPI<br />
Sulfur, ppm<br />
Nitrogen, ppm<br />
Oxygen, ppm<br />
Hydrogen, Wt %<br />
Carbon, Wt %<br />
Hot-Heptane<br />
Insol., ppm<br />
TBP Dist., OF<br />
(ASTM D2887)<br />
St/5<br />
10/3 0<br />
50<br />
70/90<br />
95/99<br />
B<strong>oil</strong>ing Range,<br />
LV%<br />
St-40ODF<br />
400-700°F<br />
700°F+<br />
Table I1<br />
SYNCRUDES FROM WYODAK COAL<br />
100 96 93 62 loo* 52*<br />
35.1<br />
410<br />
1700<br />
8500<br />
35.1<br />
250<br />
1500<br />
6700<br />
29.0<br />
140<br />
1230<br />
1500<br />
36.1<br />
88<br />
935<br />
1400<br />
8.8<br />
580<br />
1670<br />
4600<br />
15.8<br />
305<br />
1020<br />
3900<br />
12.74<br />
86.20<br />
12.97<br />
86.20<br />
12.14<br />
87.35<br />
12.65<br />
87.11<br />
9.35<br />
89.76<br />
10.48<br />
89.00<br />
680
I<br />
5<br />
I<br />
<strong>co</strong>mparable CTSL <strong>oil</strong>s. Probably, the higher hydrogen <strong>co</strong>ntent was a<br />
result Of the higher severity required for the single-stage process.<br />
In <strong>co</strong>ntrast, Figure 3 shows th<strong>at</strong> within a given b<strong>oil</strong>ing range, the<br />
two-stage products had higher hydrogen <strong>co</strong>ntents than <strong>co</strong>mparable H-Coal<br />
<strong>oil</strong>s. Together, these two sets <strong>of</strong> observ<strong>at</strong>ions may seem to present a<br />
paradox. However, the results are explained by the b<strong>oil</strong>ing<br />
distributions--the H-Coal <strong>oil</strong>s <strong>co</strong>ntained more <strong>of</strong> the <strong>co</strong>mpar<strong>at</strong>ively<br />
hydrogen-rich low-b<strong>oil</strong>ing <strong>co</strong>mponents than the two-stage <strong>oil</strong>s.<br />
HYDROTREATING PILOT PLANT TESTS<br />
Discussion. The major goals <strong>of</strong> the hydrotre<strong>at</strong>ing runs were<br />
either (1) to make specific<strong>at</strong>ion jet fuel or diesel fuel and a naphtha<br />
suitable for c<strong>at</strong>alytic reforming in a single hydrotre<strong>at</strong>ing step; or<br />
(2) to make a product suitable for hydrocracking in a se<strong>co</strong>nd step.<br />
To meet either goal, almost all <strong>of</strong> the hetero<strong>at</strong>om<br />
<strong>co</strong>ntaminants-sulfur, nitrogen, and oxygen--had to be removed by the<br />
hydrotre<strong>at</strong>ment. Typically, the <strong>co</strong>ntrol target for product nitrogen<br />
<strong>co</strong>ntent was 0.5 ppm or below. Sulfur is rel<strong>at</strong>ively easy to remove<br />
<strong>co</strong>mpared to nitrogen, and therefore was <strong>of</strong> little <strong>co</strong>ncern in this<br />
study. [Although sulfur is much easier to remove than nitrogen, the<br />
equilibrium <strong>co</strong>ncentr<strong>at</strong>ions <strong>of</strong> sulfur are somewh<strong>at</strong> higher than nitrogen<br />
in products hydrotre<strong>at</strong>ed in a single stage.] Oxygen-<strong>co</strong>ntaining<br />
<strong>co</strong>mpounds can be as hard or harder remove than nitrogen <strong>co</strong>mpounds.<br />
However, when the nitrogen was removed to 0.5 ppm, organic oxygen<br />
<strong>co</strong>ntent was also removed to less than 10 ppm (based on limited<br />
analytical results). Most <strong>of</strong> the reported 50-100 ppm oxygen in the<br />
products was dissolved w<strong>at</strong>er.<br />
In addition to removing the hetero<strong>at</strong>oms, it is necessary to<br />
hydrogen<strong>at</strong>e most <strong>of</strong> the arom<strong>at</strong>ics <strong>co</strong>mpounds in these fractions if<br />
finished jet fuel or diesel are to be the main products from a single<br />
hydrotre<strong>at</strong>ing step. One <strong>of</strong> the purposes <strong>of</strong> this work was to show the<br />
degree <strong>of</strong> arom<strong>at</strong>ics s<strong>at</strong>ur<strong>at</strong>ion needed for specific<strong>at</strong>ion diesel and jet<br />
fuel. The amount <strong>of</strong> hydrogen <strong>co</strong>nsumed will be determined by the<br />
hydrogen <strong>co</strong>ntents <strong>of</strong> the feed and products, and the amounts <strong>of</strong><br />
hetero<strong>at</strong>oms removed.<br />
If the hydrotre<strong>at</strong>ed product is to be hydrocracked, the<br />
hydrotre<strong>at</strong>ing severity can be somewh<strong>at</strong> less severe than if jet and<br />
diesel fuels are to be finished product. Additional hydrogen will be<br />
added in the se<strong>co</strong>nd-stage hydrocracker.<br />
283
Table I11<br />
HYDROTREATING TESTS WITH ICR 106 CATALYST<br />
Syncrudes listed in increasing Order <strong>of</strong> hydrotre<strong>at</strong>ing difficulty<br />
LHSV<br />
H~ Pressure, psia.<br />
wyodak CTSL B (EPx634OF)<br />
Temper<strong>at</strong>ure, OF<br />
H2 Consumption, SCF/B<br />
Product Nitrogen, ppm<br />
Product Arom<strong>at</strong>ics, LV%<br />
Wyodak H-Coal A (EP=603OF)*<br />
Illinois H-Coal B (EP=58g°F)<br />
Temper<strong>at</strong>ure, OF<br />
H2 Consumption, SCF/B<br />
Product Nitrogen, ppm<br />
Product Arom<strong>at</strong>ics, LV%<br />
Wyodak ITSL B (EP=731°F)<br />
Temper<strong>at</strong>ure, OF<br />
n2 Consumption, SCF/B<br />
Product Nitrogen, ppm<br />
Product Arom<strong>at</strong>ics, LV%<br />
wyodak H-Coal A (EP=785'F)<br />
Temper<strong>at</strong>ure, O F<br />
H2 Consumption, SCF/B<br />
Product Nitrogen, ppm<br />
Product Arom<strong>at</strong>ics, LV%<br />
Illinois ITSL B (EP=763OF)<br />
Temper<strong>at</strong>ure, OF<br />
n2 Consumption, SCF/B<br />
Product Nitrogen, ppm<br />
Product Arom<strong>at</strong>ics, LV%<br />
Illinois H-Coal A (EP=765OF)<br />
Temper<strong>at</strong>ure, OF<br />
H2 Consumption, SCF/B<br />
Product Nitrogen, ppm<br />
Product Arom<strong>at</strong>ics, LV%<br />
Wyodak CTSL A (EP=85E°F)<br />
Temper<strong>at</strong>ure , OF<br />
H2 Consumption, SCF/B<br />
Product Nitrogen, ppm<br />
Product Arom<strong>at</strong>ics, LV%<br />
Illinois ITSL A (EP=859'=F)*<br />
Wyodak ITSL A (EP=941°F)<br />
Temper<strong>at</strong>ure, OF<br />
n2 Consumption, SCF/B<br />
Product Nitrogen, ppm<br />
Product Arom<strong>at</strong>ics, Lv%<br />
0.5 1.0 1.5 1.5 1.5 1.5<br />
2300 2300 2300 1800 1400 1000<br />
680<br />
775<br />
0.5<br />
3<br />
6<br />
16<br />
Some generaliz<strong>at</strong>ions can be made, based on the ease <strong>of</strong><br />
hydrotre<strong>at</strong>ing and feed properties:<br />
(1) For a given b<strong>oil</strong>ing range, syncrudes from two-stage<br />
liquefaction are easier to upgrade than those made in one-stage--th<strong>at</strong><br />
is, lower hydrotre<strong>at</strong>ing severity is needed for a given product quality<br />
in upgrading. This result appears to be the effect <strong>of</strong> the lower<br />
hetero<strong>at</strong>om <strong>co</strong>ntents <strong>of</strong> two-stage syncrudes.<br />
(2) For syncrudes from a given liquefaction process, rel<strong>at</strong>ively<br />
small increases in EP can make the syncrudes much harder to upgrade.<br />
For example, a good <strong>co</strong>rrel<strong>at</strong>ion (roughly linear) was found between<br />
required c<strong>at</strong>alyst temper<strong>at</strong>ure and syncrude EP for a group <strong>of</strong> ITSL<br />
Oils, regardless <strong>of</strong> <strong>co</strong>al source. For example, Wyodak ITSL B (EP =<br />
634'F) <strong>co</strong>uld be hydrotre<strong>at</strong>ed <strong>at</strong> a temper<strong>at</strong>ure about 100DF lower than<br />
Wyodak ITSL A (EP = 94l0F) for the same degree <strong>of</strong> hetero<strong>at</strong>om removal.<br />
[See Figure 4, Reference 6.1<br />
Not surprisingly, the easiest <strong>oil</strong>s to process were the three<br />
syncrudes with EPs below 65O0F--Wyodak CTSL light <strong>oil</strong> (B), and the two<br />
redistilled H-Coal (B) <strong>oil</strong>s. The CTSL appears to be the easiest <strong>of</strong><br />
the three. Although it had a slightly higher EP than the others, it<br />
had the advantage <strong>of</strong> a lower hetero<strong>at</strong>om <strong>co</strong>ntent.<br />
Of the four <strong>oil</strong>s with EPs between 70OOF and 8OOoF, Wyodak ITSL<br />
<strong>oil</strong> B had the lowest EP and was easiest. Next is wyodak H-Coal B.<br />
Although it had a slightly higher EP than the <strong>oil</strong>s in this group, it<br />
had a much lower average b<strong>oil</strong>ing range. Illinois ITSL B ranked next.<br />
It was much easier than Illinois H-Coal A, which had about the same EP<br />
but a much higher hetero<strong>at</strong>om <strong>co</strong>ntent.<br />
Finally, <strong>of</strong> the three <strong>oil</strong>s with EPs above 800°F, Wyodak CTSL <strong>oil</strong><br />
A was clearly the easiest. Although its EP was about the same as<br />
Illinois ITSL A, it had a lower average b<strong>oil</strong>ing range and lower<br />
hetero<strong>at</strong>om <strong>co</strong>ntent. Of all the <strong>oil</strong>s, wyodak ITSL A was the most<br />
difficult to process. It <strong>co</strong>ntained the most 7OOoF+ m<strong>at</strong>erial <strong>of</strong> any <strong>of</strong><br />
the syncrudes, and had the highest EP (941OF).<br />
C<strong>at</strong>al st Stabilit . The length <strong>of</strong> specific tests varied from<br />
sever+ ays to +<br />
severa months. With one exception and within the<br />
limits <strong>of</strong> the tests, ICR-106 c<strong>at</strong>alyst appeared to stable for<br />
hetero<strong>at</strong>om removal during all <strong>of</strong> the tests shown in Table 111. The<br />
exception: With Illinois H-Coal A, the c<strong>at</strong>alyst lost about 20°F <strong>of</strong><br />
activity during 1100-hr <strong>at</strong> 1.5 LHSV and 1800 psia hydrogen partial<br />
pressure. In <strong>co</strong>ntrast, Illinois ITSL B (with about the same EP as<br />
Illinois H-Coal A) was stable during a 900-hr test <strong>at</strong> the same<br />
<strong>co</strong>nditions. The difference was probably due to the lower hetero<strong>at</strong>om<br />
and hot-heptane insolubles <strong>co</strong>ntents <strong>of</strong> the ITSL <strong>oil</strong>. [The higher EP<br />
<strong>oil</strong>s were not tested <strong>at</strong> this pressure, but would be expected to cause<br />
appreciable c<strong>at</strong>alyst deactiv<strong>at</strong>ion also.]<br />
YIELDS<br />
For syncrudes with EPs below 800°F, there was rel<strong>at</strong>ively little<br />
cracking during hydrotre<strong>at</strong>ing, and the feed b<strong>oil</strong>ing range determined<br />
I product b<strong>oil</strong>ing range (except for some EP reduction due to<br />
I hydrogen<strong>at</strong>ion). As an example, Table IV <strong>co</strong>ntrasts yields <strong>of</strong> products<br />
1 from Illinois H-Coal A and Illinois ITSL 8, two <strong>oil</strong>s th<strong>at</strong> have about<br />
i the same EP but widely different b<strong>oil</strong>ing ranges.<br />
1<br />
L<br />
A 285<br />
t<br />
1
Table IV<br />
HYDROTREATING TO 0.2 PPM NITROGEN (0.5 LHSV, 2300 psia H2)<br />
Syncrude H-Coal ITSL<br />
C<strong>at</strong>alyst Temper<strong>at</strong>ure, OF 750 710<br />
Yields, Based on Fresh Feed<br />
C1-C4, Wt % 0.3 0.2<br />
C5-250°F, LV % 20.4 7.0<br />
25O-35O0F, LV% 26.3 6.8<br />
350-550°F, LV% 57.7 53.6<br />
55OoF+, LV% 6.4 40.6<br />
Total C5+, Lv% 111 108<br />
Chemical H2 Consumption, SCF/B 2150<br />
Product Arom<strong>at</strong>ics, LV% 2<br />
1600<br />
12<br />
For higher EP syncrudes, higher hydrotre<strong>at</strong>ing severities were<br />
required and more cracking occurred. Still, Cl-C4 yields were low (2<br />
LV% OK below), indic<strong>at</strong>ing efficient use <strong>of</strong> the hydrogen.<br />
PRODUCT PROPERTIES<br />
General Comments. After hydrotre<strong>at</strong>ing, products <strong>of</strong> similar<br />
b<strong>oil</strong>ing ranges from the different liquefaction processes and different<br />
<strong>co</strong>als were quite similar. After removal <strong>of</strong> hetero<strong>at</strong>om-<strong>co</strong>ntaining<br />
<strong>co</strong>mpounds, the products mainly <strong>co</strong>nsisted <strong>of</strong> cyclic hydrocarbons. The<br />
severity <strong>of</strong> hydrotre<strong>at</strong>ing determined the amount <strong>of</strong> hydrogen<strong>at</strong>ion <strong>of</strong><br />
arom<strong>at</strong>ics to naphthenes. There were, however, some differences.<br />
Products from subbituminous <strong>co</strong>als <strong>co</strong>ntained more paraffins than those<br />
from bituminous <strong>co</strong>als.<br />
Naphtha. Hydrotre<strong>at</strong>ed and hydrocracked naphthas from <strong>co</strong>al<br />
liquids are excellent feeds for c<strong>at</strong>alytic reformers because <strong>of</strong> the<br />
high <strong>co</strong>ntent <strong>of</strong> cyclic <strong>co</strong>mpounds. Paraffin <strong>co</strong>ntents <strong>of</strong> all the<br />
hydrotre<strong>at</strong>ed naphthas were low, although the Wyodak naphthas <strong>co</strong>ntained<br />
somewh<strong>at</strong> more paraffins than those from Illinois <strong>co</strong>al as shown by<br />
Table V.<br />
PARAFFIN<br />
Table V<br />
CONTENTS OF TYPICAL 150-350'F HYDROTREATED NAPHTHAS<br />
--<br />
Feed Source Paraffins, e<br />
Wyodak H-Coal 23<br />
Wyodak ITSL 23<br />
Wyodak CTSL 18<br />
Illinois H-Coal 11<br />
Illinois ITSL 7<br />
At the higher hydrotre<strong>at</strong>ing severities, the cyclics in the<br />
naphthas were almost all hydrogen<strong>at</strong>ed. The naphthenes, however, <strong>co</strong>uld<br />
be dehydrogen<strong>at</strong>ed to high-octane arom<strong>at</strong>ics by c<strong>at</strong>alytic reforming <strong>at</strong><br />
rel<strong>at</strong>ively mild <strong>co</strong>nditions <strong>co</strong>mpared to those required for typical<br />
petroleum naphthas. Or, when reformed <strong>at</strong> higher severities, these<br />
naphthas would make extremely high octane products for gasoline<br />
blending OK for chemicals production (benzene, toluene, and xylene).<br />
In the reforming process, much <strong>of</strong> the hydrogen <strong>co</strong>nsumed during<br />
hydrotre<strong>at</strong>ing would be re<strong>co</strong>vered. [We have not performed c<strong>at</strong>alytic<br />
reforming studies on naphthas from the two-stage, processes, but
esults would be expected to be similar to those previously reported<br />
for naphthas from single stage processes (7).1<br />
Jet. To make jet fuel meeting the ASTM smoke point specific<strong>at</strong>ion<br />
<strong>of</strong> 20mm.(minimum), most <strong>of</strong> the arom<strong>at</strong>ics in the <strong>co</strong>al liquids had to<br />
be hydrogen<strong>at</strong>ed.<br />
Figure 4 is a plot <strong>of</strong> smoke point versus arom<strong>at</strong>ic <strong>co</strong>ntent <strong>of</strong><br />
kerosene jet fuels from the various syncrudes. The results fall into<br />
two rough groups, those from Wyodak <strong>co</strong>al and those from Illinois <strong>co</strong>al.<br />
At a given arom<strong>at</strong>ics <strong>co</strong>ntent, those from Wyodak <strong>co</strong>al had smoke points<br />
2-3 mm higher than those from Illinois <strong>co</strong>al, a <strong>co</strong>nsequence <strong>of</strong> the<br />
higher Wyodak paraffin <strong>co</strong>ntent. [The Wyodak jet <strong>co</strong>ntained about 10<br />
LV% paraffins; the Illinois jet, 1-3 LV%.] The Illinois jet fuels met<br />
the jet smoke specific<strong>at</strong>ion <strong>of</strong> 20 mm <strong>at</strong> 10% arom<strong>at</strong>ics or lower; the<br />
Wyodak jet fuels met the specific<strong>at</strong>ion <strong>at</strong> about 16 LV% arom<strong>at</strong>ics.<br />
[Some <strong>of</strong> the sc<strong>at</strong>ter in results for products from a given <strong>co</strong>al was due<br />
to different b<strong>oil</strong>ing distributions. Those jet fuels <strong>co</strong>ntaining more<br />
low b<strong>oil</strong>ing m<strong>at</strong>erial had somewh<strong>at</strong> higher smoke points.]<br />
Jet fuels from <strong>co</strong>al <strong>of</strong>fer some unique advantages over those from<br />
petroleum. Because they <strong>co</strong>ntain high <strong>co</strong>ncentr<strong>at</strong>ions <strong>of</strong> naphthenes,<br />
they are very dense and have high he<strong>at</strong>ing values by volume.<br />
Therefore, they <strong>co</strong>uld have specialized uses, such as for military<br />
fuels. FOK example, Figure 5 shows the densities <strong>of</strong> narrow b<strong>oil</strong>ing<br />
fractions <strong>of</strong> hydrotre<strong>at</strong>ed ITSL <strong>oil</strong>. Jet fuel <strong>of</strong> a desired density<br />
<strong>co</strong>uld be made by adjusting the b<strong>oil</strong>ing range. The ASTM specific<strong>at</strong>ion<br />
for jet fuel gravity is 37OAPI (minimum). However, this specific<strong>at</strong>ion<br />
is probably unnecessary for aircraft with modern flow <strong>co</strong>ntrollers, and<br />
lower gravity (higher density) fuels <strong>co</strong>uld be acceptable. Also, these<br />
jet fuels have unusually low freezing points, because <strong>of</strong> low normal<br />
paraffin <strong>co</strong>ntents.<br />
Diesel. Diesel products from both single-stage and two-stage<br />
procemet typical ASTM specific<strong>at</strong>ions. A rel<strong>at</strong>ively high degree<br />
<strong>of</strong> hydrogen<strong>at</strong>ion was needed to meet the cetane-number specific<strong>at</strong>ion <strong>of</strong><br />
40 (minimum).<br />
Figure 6 shows the rel<strong>at</strong>ionship for cetane number versus<br />
arom<strong>at</strong>ics <strong>co</strong>ntent for products from single-stage and two-stage<br />
processes. With the two-stage <strong>oil</strong>s, the specific<strong>at</strong>ion was met with an<br />
arom<strong>at</strong>ics <strong>co</strong>ntent <strong>of</strong> about 20 Lv%; with single-stage <strong>oil</strong>s, an arom<strong>at</strong>ic<br />
<strong>co</strong>ntent <strong>of</strong> less than 10 LV% was needed. These differences, however,<br />
were not necessarily the result <strong>of</strong> single-stage versus two-stage<br />
<strong>processing</strong>. R<strong>at</strong>her, they appear to be due to changes in b<strong>oil</strong>ing<br />
ranges <strong>of</strong> the diesels. For example, Table VI <strong>co</strong>mpares pairs <strong>of</strong><br />
samples <strong>of</strong> different b<strong>oil</strong>ing ranges. The arom<strong>at</strong>ics and paraffin<br />
<strong>co</strong>ntents within a given pair were about the same. Within each pair,<br />
the higher b<strong>oil</strong>ing sample had the higher cetane number. Also, in<br />
other <strong>co</strong>mparisons (1). the more paraffinic diesels had higher cetane<br />
numbers, when other properties were about equal.<br />
AS with the jet fuels described above, these <strong>co</strong>al-derived diesel<br />
fuels had excellent <strong>co</strong>ld we<strong>at</strong>her properties, and high volumetric<br />
energy <strong>co</strong>ntents.<br />
287
Table VI<br />
EFFECT OF BOILING RANGE ON CETANE NUMBER<br />
Source Initial, Midpoint, Cetane Arom<strong>at</strong>ics, Paraffins,<br />
O F (TBP) OF (TBP) No. LV, % LV%<br />
Wyodak CTSL 250 414 44.2 3.9 9.5<br />
350 454 48.7 4.6 7.7<br />
Illinois ITSL 250 520 43.1 9.8
th<strong>at</strong> the equilibrium was unfavorable for hydrogen<strong>at</strong>ion <strong>of</strong> some <strong>of</strong> the<br />
high-b<strong>oil</strong>ing polycyclic-arom<strong>at</strong>ic <strong>co</strong>mpounds <strong>at</strong> the run <strong>co</strong>nditions.<br />
Therefore, we tried an two-step approach: (1) Hydrotre<strong>at</strong> <strong>at</strong><br />
rel<strong>at</strong>ively high temper<strong>at</strong>ure (e.g., 750°F) to remove most <strong>of</strong> the<br />
hetero<strong>at</strong>oms. (2) Further hydrogen<strong>at</strong>e <strong>at</strong> lower temper<strong>at</strong>ures<br />
(e.g.,600-650°~) for further arom<strong>at</strong>ics s<strong>at</strong>ur<strong>at</strong>ion.<br />
In the next test, product from the initial experiment (75O0F, 0.5<br />
LHSV, 2300 psia H2), which <strong>co</strong>ntained 42 % arom<strong>at</strong>ics, was hydrotre<strong>at</strong>ed<br />
a se<strong>co</strong>nd time using the same c<strong>at</strong>alyst. The LHSV and pressure were<br />
kept the same, but the temper<strong>at</strong>ure decreased to 650°F--1000F lower<br />
than previously. Due to the more favorable equilibrium <strong>at</strong> 650°F.<br />
product arom<strong>at</strong>ics were reduced to 12%. The jet and diesel fractions,<br />
respectively, exceeded smoke point and cetane number specific<strong>at</strong>ions.<br />
Also, enough EP reduction was achieved so th<strong>at</strong> less than 5% <strong>of</strong> the<br />
product b<strong>oil</strong>ed above the diesel range.<br />
When,the temper<strong>at</strong>ure was further decreased to 6OO0F, the arom<strong>at</strong>ic<br />
<strong>co</strong>ntent <strong>of</strong> the product did not decrease further, but increased to 20%.<br />
[The r<strong>at</strong>e <strong>of</strong> hydrogen<strong>at</strong>ion was lower, although the equilibrium was<br />
even more favorable than <strong>at</strong> 65OOF.l The diesel fraction still met the<br />
cetane number specific<strong>at</strong>ion, however.<br />
The results show th<strong>at</strong> two-step hydrotre<strong>at</strong>ing [with the se<strong>co</strong>nd<br />
hydrotre<strong>at</strong>ment <strong>at</strong> a rel<strong>at</strong>ively low temper<strong>at</strong>ure] is an altern<strong>at</strong>ive to<br />
the hydrotre<strong>at</strong>ing/hydrocracking route for upgrading high EP syncrudes,<br />
provided diesel fuel is a desired product.<br />
CONCLUSIONS<br />
Coal liquids produced in the ITSL and CTSL processes with EPs<br />
from about 600°F to over 900°F were hydrotre<strong>at</strong>ed to make diesel and<br />
jet fuels, and naphthas suitable for c<strong>at</strong>alytic reforming to gasoline.<br />
Specific <strong>co</strong>nclusions are as follows:<br />
(1) Oils from two-stage processes were easier to upgrade than<br />
<strong>co</strong>mparable b<strong>oil</strong>ing-range products from single-stage processes, due to<br />
lower nitrogen and oxygen <strong>co</strong>ntents. However, as with products from<br />
single-stage processes, rel<strong>at</strong>ively small increases in EPs made the<br />
<strong>oil</strong>s much harder to upgrade.<br />
(2) Except for modest differences in paraffin <strong>co</strong>ntents,<br />
properties <strong>of</strong> finished products <strong>of</strong> given b<strong>oil</strong>ing ranges from both<br />
wyodak and Illinois <strong>co</strong>als, and both one- and two-stage processes<br />
studied were fairly similar, and mainly <strong>co</strong>nsisted <strong>of</strong> cyclic<br />
hydrocarbons. Products from Wyodak <strong>co</strong>al were somewh<strong>at</strong> more paraffinic<br />
than those from Illinois <strong>co</strong>al.<br />
(3) Product b<strong>oil</strong>ing ranges were different, depending upon the<br />
liquefaction process and the cut point used in th<strong>at</strong> process. The<br />
single-stage processes made more naphtha than the two-stage processes<br />
<strong>at</strong> a given cut point; the two-stage processes made more middle<br />
distill<strong>at</strong>e. The ITSL process made more middle distill<strong>at</strong>e than the<br />
CTSL process. Diesel products from two-stage processes had higher<br />
cetane numbers <strong>at</strong> a given arom<strong>at</strong>ic <strong>co</strong>ntent than those from<br />
single-stage processes. At least in part, this was due to product<br />
b<strong>oil</strong>ing range differences.<br />
289
(4) In all cases studied, the jet fuel and diesel products had<br />
high densities and, therefore, high volumetric-energy <strong>co</strong>ntents.<br />
(5) Wyodak CTSL light <strong>oil</strong> had a higher hydrogen <strong>co</strong>ntent and lower<br />
hetero<strong>at</strong>om <strong>co</strong>ntent than the other <strong>oil</strong>s. These factors, plus its low<br />
EP, made it easier to upgrade than the other syncrudes studied.<br />
(6) For high EP syncrudes, an <strong>at</strong>tractive upgrading route is a<br />
two-step process--hydrotre<strong>at</strong>ing to remove most <strong>of</strong> the hetero<strong>at</strong>oms,<br />
followed by low-temper<strong>at</strong>ure hydrogen<strong>at</strong>ion to s<strong>at</strong>ur<strong>at</strong>e arom<strong>at</strong>ics.<br />
ACKNOWLEDGMENT<br />
This work was funded by the US Department <strong>of</strong> Energy under DOE<br />
Contract DE-AC22-76ET10532. The feedstocks were provided by<br />
Hydrocarbon Research, Inc., and Lummus Crest, Inc. The analytical<br />
work was performed by the Research Services Department <strong>of</strong> Chevron<br />
Research Company.<br />
REFERENCES<br />
1. Chevron Research Co., Refining and Upgrading <strong>of</strong> Synfuels from<br />
Coal and Oil Shales by Advanced C<strong>at</strong>alytic Processes, DOE Reports<br />
DOE/ET/10532 Series, N<strong>at</strong>ional Technical Inform<strong>at</strong>ion Service, US<br />
Department <strong>of</strong> Commerce, Springfield, Virginia 22161. (See reference 6<br />
for a more detailed listing <strong>of</strong> interim reports.)<br />
2. R. S. Chillingworth, J. D. Potts, H. D.Schindler, J. M. Chen,<br />
M. Peluso, Preprints, Div. Petroleum Chem., ACS, 2 (4), Sept. 1982,<br />
pp. 859-876.<br />
3. J. B. MacArthur, J. E. Duddy, A. S. Ambegaonkar, A. V. Moomjy,<br />
"H-Coal Liquids - Upgrading Upstream or Downstream," AIChE 1983 Spring<br />
N<strong>at</strong>l. Meeting, Houston, March 27-31, 1983.<br />
4. J. B. McLean, A. G. Comolli, J. B. MacArthur, Preprints, Div.<br />
Petroleum Chem., ACS, 30 (3), Aug. 1985, p. 530. TAbstract only,<br />
Manuscript Available from Authors.]<br />
5. D. J. O'Rear, R. F. Sullivan, B. E. Stangeland, ACS Symposium<br />
Series 156, Edited,by R. F. Sullivan, American Chemical Society,<br />
Washington D. C. 1981, pp. 115-144.<br />
6. R. F. Sullivan and H. A. Frumkin, Preprints, Div. Fuel Chem.,<br />
(2), April 1986, pp. 325-339.<br />
7. R. C. Robinson, H. A. Frumkin, R. F Sullivan, Energy Progress, 3<br />
(3). Sept. 1983, pp. 163-172.<br />
8. R. F. Sullivan, D. J. O'Rear, 8. E. Stangeland, Preprints, Div.<br />
Petroleum Chem., ACS, 25 (3), Aug. 1980, pp. 583-607.<br />
9. R. F. Sullivan, Preprints, Div. Petroleum Chem., ACS, 0 13),<br />
Aug. 1985, pp. 503-512.<br />
290
J<br />
U<br />
0<br />
W<br />
80<br />
z<br />
291<br />
-0 0<br />
l-<br />
-m 0<br />
-W 0<br />
-0 U<br />
-0 (Y<br />
s<br />
2
292
FIGURE 5<br />
API GRAVITIES OF NARROW BOILING<br />
FRACTION OF HYDROTREATED ILLINOIS ITSL OIL<br />
Total Arom<strong>at</strong>ics, 11 LV VO<br />
0 Mldb<strong>oil</strong>lng Point<br />
-Bolllng Range <strong>of</strong><br />
Fractlon<br />
io0 200 300 400 500 600 700 800<br />
B<strong>oil</strong>ing Point, ‘F<br />
FIGURE 6<br />
EFFECT OF AROMATICS ON THE<br />
CETANE NUMBER OF DIESEL<br />
FUELS FROM HYDROTREATED COAL-DERIVED OILS<br />
50-<br />
30-<br />
A<br />
0 liiinols ITSL<br />
Wyodak ITSL<br />
A Wyodak CTSL<br />
0 Wyodak H-Coal<br />
A IlllnOIl H-Coal<br />
Solid Points = 2-Stage<br />
25 I I I I I I<br />
0 10 20 30 40 SO 60<br />
Arom<strong>at</strong>ic Contant, LV ah<br />
2 93
INTEGRATED TWO-STAGE LIQUEFACTION: THE LEGACY AND THE UNFINISHED WORK<br />
INTRODUCTION<br />
ENEO C. MORONI<br />
U.S. Department <strong>of</strong> Energy, FE-34 GTN, Washington, D.C. 20545<br />
The Integr<strong>at</strong>ed Two-Stage <strong>Liquefaction</strong> (ITSL) <strong>co</strong>ncept has received <strong>co</strong>nsiderable<br />
<strong>at</strong>tention by many labor<strong>at</strong>ories and has emerged as one <strong>of</strong> the most promising<br />
technology in direct <strong>co</strong>al liquefaction.<br />
DOE/Lummus/Cities Service, EPRI/Kerr McGee, the Wilsonville teams, Chevron and<br />
Exxon (1) have been in one way or another involved in processes <strong>co</strong>nforming to<br />
the ITSL <strong>co</strong>ncept, each one with a somewh<strong>at</strong> different approach or <strong>processing</strong><br />
scheme. Recently, HRI (2) has developed another staged liquefaction <strong>co</strong>ncept<br />
which recently has received DOE support for extended <strong>co</strong>ntinuous bench-scale<br />
testi ny.<br />
Major achievements were obtained during the development <strong>of</strong> the ITSL process <strong>at</strong><br />
Lummus, from May 1980 t o June 1985, which have changed substantially our<br />
approach to <strong>co</strong>al liquefaction techniques and inspired new thoughts in<br />
unraveling the mechanism <strong>of</strong> direct <strong>co</strong>al liquefaction <strong>at</strong> low severity opera-<br />
tions. These novel mechanistic <strong>co</strong>nsider<strong>at</strong>ions need to be supported with<br />
further studies using suitable model <strong>co</strong>mpounds, some <strong>of</strong> which have been<br />
recently proposed (3).<br />
Several papers have reported the early development <strong>of</strong> the ITSL process and<br />
rel<strong>at</strong>ed projects (4-8). This paper has the objective to divulge the most<br />
recent achievements <strong>of</strong> an evolved ITSL process, pointing out the unfinished<br />
work, and to expand the <strong>co</strong>ncept <strong>of</strong> the low-severity staged approach which<br />
resulted from the evolved ITSL process and emerged as the most desirable<br />
p<strong>at</strong>hway for the direct production <strong>of</strong> marketable liquid fuels from <strong>co</strong>als:<br />
The ultim<strong>at</strong>e objective is to interest the researchers dedic<strong>at</strong>ed to<br />
fundamentals <strong>of</strong> <strong>co</strong>al liquefaction toward the technological needs and the<br />
understanding <strong>of</strong> reaction mechanism, kinetics and thermodynamic limit<strong>at</strong>ions<br />
residing with the novel, low-severity staged <strong>co</strong>al liquefaction approach.<br />
THE ITSL LEGACY<br />
Major ac<strong>co</strong>mplishments obtained <strong>at</strong> low-severity oper<strong>at</strong>ions in a 3/4 ton/day<br />
ITSL process development unit <strong>at</strong> Lummus and from rel<strong>at</strong>ed bench-scale studies<br />
in various labor<strong>at</strong>ories, include:<br />
LOW severity produced extracts are low in hetero<strong>at</strong>oms and more<br />
easily bydrogen<strong>at</strong>abl e, <strong>co</strong>nsistently yielding excel 1 ent equi 1 i brium<br />
donor solvent (9).<br />
No high vis<strong>co</strong>sity gel region is apparent over the 28D-35OoC<br />
temper<strong>at</strong>ure range for a slurry <strong>of</strong> bituminous <strong>co</strong>al and ITSL<br />
solvent, as was the case for slurries prepared with the same<br />
<strong>co</strong>al and other types <strong>of</strong> solvent (10).<br />
294
Low severity <strong>processing</strong> forms mostly reactive low molecular-<br />
weight fragments. Conversely, single stage thermal and thermal/<br />
c<strong>at</strong>alytic <strong>processing</strong> produce high-molecular weight products<br />
thought to be actually <strong>co</strong>ndens<strong>at</strong>ion products <strong>of</strong> such smaller<br />
fragments and <strong>co</strong>nsequently, less reactive (11).<br />
Proton NMR analysis, modified to provide d<strong>at</strong>a on ITSL distill<strong>at</strong>e<br />
and non-distill<strong>at</strong>e fractions, has been shown to be useful in the<br />
development <strong>of</strong> a kinetic model for <strong>co</strong>al extract hydro<strong>processing</strong>,<br />
thus enabling us to distinguish c<strong>at</strong>alytic hydrogen<strong>at</strong>ion and<br />
cracking reactions, and to predict the solvent donor capability<br />
as well as the yield structure <strong>of</strong> the upgraded products (12).<br />
A mixture <strong>of</strong> <strong>co</strong>ndensed arom<strong>at</strong>ics, hydroarom<strong>at</strong>ics, paraffins and<br />
their respective hetero<strong>at</strong>om deriv<strong>at</strong>ives is produced during <strong>co</strong>al<br />
liquefaction. This mixture tends to be unstable because <strong>of</strong> the<br />
in<strong>co</strong>mp<strong>at</strong>ibility between polar hetero<strong>at</strong>om <strong>co</strong>mpounds and hydro-<br />
carbons. as well as between <strong>co</strong>ndensed arom<strong>at</strong>ics and paraffins.<br />
Condensed hydroarom<strong>at</strong>ics, having closer affinity for both<br />
arom<strong>at</strong>ics and paraffins, tend to keep them in solution, thus<br />
<strong>co</strong>ntributing to the stability <strong>of</strong> the <strong>co</strong>al extract. Low severity<br />
<strong>co</strong>al extraction yields a larger quantity <strong>of</strong> hydroarom<strong>at</strong>ics and<br />
small amounts <strong>of</strong> high hetero<strong>at</strong>om, <strong>co</strong>ndensed arom<strong>at</strong>ics and<br />
paraffins (12).<br />
Best c<strong>at</strong>alysts tested are those modified to suppress the<br />
hydrocracking activity and enhance hydrogen<strong>at</strong>ion functionality (9).<br />
Coal derived transport<strong>at</strong>ion fuels, produced by refining <strong>of</strong><br />
distill<strong>at</strong>e from low severity oper<strong>at</strong>ions, possess inherent high<br />
quality which is due mostly to their hydroarom<strong>at</strong>ic (naphthenic)<br />
n<strong>at</strong>ure. Coal derived naphthas <strong>co</strong>ntain large quantities <strong>of</strong> highly<br />
alkyl<strong>at</strong>ed cyclohexanes which, by reforming, <strong>co</strong>nvert to the<br />
<strong>co</strong>rresponding benzenes and in the process, re<strong>co</strong>ver a large<br />
portion <strong>of</strong> the hydrogen to make the overall <strong>co</strong>al liquefaction<br />
approach e<strong>co</strong>nomically more <strong>at</strong>tractive. A1 kyl<strong>at</strong>ed benzenes are<br />
the major <strong>co</strong>ntributors to the high octane gasoline thus formed.<br />
Coal derived middle distill<strong>at</strong>e is <strong>co</strong>nstituted mostly <strong>of</strong> di-and<br />
tri-hydroarom<strong>at</strong>ics and <strong>co</strong>rresponding arom<strong>at</strong>ics.<br />
Further refining has been successfully employed to <strong>co</strong>nvert some<br />
<strong>of</strong> the arom<strong>at</strong>ics to meet marketable jet and diesel specific<strong>at</strong>ions<br />
<strong>of</strong> smoke point and cetane number, respectively (13).<br />
From the oper<strong>at</strong>ion <strong>of</strong> the process development unit (PDU) <strong>at</strong> Lumnus (9) the<br />
following important results were obtained:<br />
Subbituminous <strong>co</strong>al was demonstr<strong>at</strong>ed to be an <strong>at</strong>tractive feed for<br />
direct liquefaction: The distill<strong>at</strong>e yield was slightly lower<br />
(2.9 bbl/ton <strong>of</strong> moisture-ash-free <strong>co</strong>al <strong>co</strong>mpared to 3.2 for<br />
bituminous <strong>co</strong>als) but its lower <strong>co</strong>st, higher reactivity in the<br />
se<strong>co</strong>nd stage and its ease in being <strong>co</strong>nverted to a lighter product<br />
295
are over-riding fe<strong>at</strong>ures in its favor. In addition, ITSL with<br />
Wyodak <strong>co</strong>al demonstr<strong>at</strong>ed good operability in both reaction stages<br />
and was easily deashed.<br />
When the deasher is placed after the LC-Fining reactor, the<br />
distill<strong>at</strong>e yield was increased seven percent. More importantly,<br />
the LC-Fining c<strong>at</strong>alyst was unaffected by ash feed and reactor<br />
volume is unchanged.<br />
Most <strong>of</strong> the LC-Fining reactor volume <strong>co</strong>uld be replaced by a fixed<br />
bed hydrocracking unit. This resulted in an equally good yield<br />
<strong>of</strong> -65OOF product with no loss in hydrogen efficiency. The<br />
-65OoF product <strong>co</strong>ntained less than 100 ppm sulfur and less than<br />
500 ppm nitrogen, making this a clean, light and environmentally<br />
acceptable product. Furthermore, this flow <strong>co</strong>nfigur<strong>at</strong>ion reduces<br />
the se<strong>co</strong>nd stage reactor volume by <strong>at</strong> least 18 percent and may<br />
also gre<strong>at</strong>ly simplify and reduce the <strong>co</strong>st <strong>of</strong> the deashing section.<br />
The SCT reaction was oper<strong>at</strong>ed <strong>at</strong> 500 and 1000 psig, with no<br />
adverse effect on yields or hydrogen usage. This leaves the LC-<br />
Fining as the only high pressure section <strong>of</strong> the process.<br />
Analysis <strong>of</strong> the results indic<strong>at</strong>ed th<strong>at</strong> a <strong>co</strong>mmercial SCT reactor<br />
can be designed to retain all the important fe<strong>at</strong>ures <strong>of</strong> the PDU.<br />
There is every reason to believe th<strong>at</strong> SCT can be scaled to<br />
<strong>co</strong>mmerci a1 size .<br />
Deasher bottoms were <strong>co</strong>ked to produce additional liquid products.<br />
The liquid yield was about 20 percent <strong>of</strong> the organic m<strong>at</strong>ter in<br />
the ash-rich feed.<br />
Low temper<strong>at</strong>ures <strong>of</strong> about 7OO0F, do not provide sufficient<br />
hydrogen<strong>at</strong>ion to replenish solvent quality, while <strong>at</strong> 80OoF the<br />
solvent <strong>co</strong>ntains insufficient transferable hydrogen. Therefore,<br />
the optimum temper<strong>at</strong>ure for both <strong>co</strong>nversion and regener<strong>at</strong>ion <strong>of</strong><br />
recycle solvent is about 724-750°F (9).<br />
In antisolvent deashing experiments, THF-insoluble/quinol ine-<br />
soluble preasphaltenes precipit<strong>at</strong>e <strong>co</strong>nsistently with the mineral<br />
m<strong>at</strong>ters, whereas the THF-soluble preasphaltenes do not. This<br />
indic<strong>at</strong>es th<strong>at</strong> THF-insoluble preasphaltenes may be the major cause<br />
<strong>of</strong> mineral m<strong>at</strong>ters agglomer<strong>at</strong>ion, increasing their diameter and<br />
causing the particles to settle faster (14).<br />
THE UNFINISHED WORK<br />
The mst significant achievements <strong>of</strong> the ITSL program came into focus during<br />
the last part <strong>of</strong> the ITSL project and <strong>of</strong> the rel<strong>at</strong>ed projects before they<br />
ceased oper<strong>at</strong>ion. In this particular period, those who closely monitored<br />
the overall program, g<strong>at</strong>hered the large set <strong>of</strong> d<strong>at</strong>a made available, and<br />
structured them for suitable process engineering and e<strong>co</strong>nomic evalu<strong>at</strong>ion,<br />
296
ecame aware <strong>of</strong> an evolutionary trend in <strong>co</strong>al liquefaction <strong>processing</strong>.<br />
The major factors <strong>co</strong>ntributing to this novel trend were: 1) the better<br />
understanding <strong>of</strong> the very sensitive interdependency between the stages <strong>of</strong> <strong>co</strong>al<br />
extraction, <strong>of</strong> the <strong>co</strong>al extract upgrading and hydrogen donor recycle solvent<br />
requirements. which emerged only from the d<strong>at</strong>a produced in a <strong>co</strong>ntinuous,<br />
integr<strong>at</strong>ed recycle mode <strong>of</strong> oper<strong>at</strong>ions, and 2) the more favorable results <strong>of</strong><br />
low-severity oper<strong>at</strong>ions, practically solving, in an easy and elegant manner,<br />
most if not all the problems en<strong>co</strong>untered by using high-severity oper<strong>at</strong>ions<br />
which were practiced in earlier processes. i.e. German, H-Coal, SRC-11, etc.<br />
But perhaps more important to the fundamental research <strong>co</strong>mmunity is the<br />
fact th<strong>at</strong> the large set <strong>of</strong> ITSL d<strong>at</strong>a under scrutiny for process development,<br />
lacks the fundamental d<strong>at</strong>a to support the pr<strong>of</strong>ound changes in the mechanism<br />
and kinetics occurring <strong>at</strong> low-severity <strong>co</strong>al liquefaction.<br />
Some <strong>of</strong> the <strong>co</strong>ncepts and technology needs are outlined below.<br />
Preserv<strong>at</strong>ion <strong>of</strong> highly reactive, small fragments in the <strong>co</strong>al extract<br />
is <strong>of</strong> utmost importance in producing an excellent donor solvent and<br />
high quality distill<strong>at</strong>e fuel products. For this purpose, the<br />
fragments should be withdrawn from the extraction reactor as soon<br />
as they are formed.<br />
The un<strong>co</strong>nverted <strong>co</strong>al can be further <strong>co</strong>nverted<br />
by recycling it with the preasphaltenes as part <strong>of</strong> the recycle<br />
solvent.<br />
Better preserv<strong>at</strong>ion <strong>of</strong> the reactive small fragments can be<br />
achieved by increasing the don<strong>at</strong>able hydrogen level and decreasing<br />
the hetero<strong>at</strong>om <strong>co</strong>ntent <strong>of</strong> the recycle solvent. It i s important<br />
for the superior hydrogen donor solvent to penetr<strong>at</strong>e the less<br />
reactive macerals. Consequently, it is advisable to allow for a<br />
thermal soaking tre<strong>at</strong>ment. i .e., <strong>at</strong> 250-350°C temper<strong>at</strong>ure range<br />
for 10-30 minutes, prior to the short <strong>co</strong>ntact time (SCT) reaction<br />
<strong>of</strong> rapid he<strong>at</strong>ing (two minutes) to the 45OoC exit temper<strong>at</strong>ure. It<br />
is evident th<strong>at</strong> all the above activities are interdependent and<br />
the improvements maximized in an integr<strong>at</strong>ed recyle process.<br />
It is extremely difficult to capture in research bench scale units<br />
the essence <strong>of</strong> the results produced in the integr<strong>at</strong>ed recycle<br />
process, because most <strong>of</strong> the key benefits, i.e., <strong>co</strong>al <strong>co</strong>nversion<br />
and enhanced donor solvent quality, are obtained only after<br />
several cycles <strong>of</strong> the integr<strong>at</strong>ed staged oper<strong>at</strong>ions. Bench scale<br />
researchers <strong>co</strong>uld avoid the long and tedious recycle oper<strong>at</strong>ions<br />
by applying the aforementioned kinetic model for <strong>co</strong>al extract<br />
hydro<strong>processing</strong> (12) and using proton-NMR d<strong>at</strong>a <strong>of</strong> the <strong>co</strong>al<br />
extract to predict solvent donor capability and yield structure<br />
<strong>of</strong> the upgraded products.<br />
Proton-NMR analysis is' rapid, requires small samples, is highly<br />
reliable and has excellent reproducibility.<br />
Removal <strong>of</strong> the hetero<strong>at</strong>oms in the early stage <strong>of</strong> <strong>co</strong>al extraction<br />
is desirable and ought to be sequential, removing first the more<br />
abundant oxygen and thus making easier the subsequent nitrogen<br />
removal.<br />
297
Complementary fundamental studies on C-0 and C-N bond scission<br />
should be emphasized over the current C-C bond cracking effort.<br />
Most <strong>of</strong> the sulfur is <strong>co</strong>nverted to hydrogen sulfide during the<br />
two above sequences, and the H S must be kept in the system as<br />
c<strong>at</strong>alyst itself and as "activa?or" <strong>of</strong> transition metal c<strong>at</strong>alysts.<br />
It is essential th<strong>at</strong> the ITSL technology be pursued to the<br />
<strong>co</strong>mpletion <strong>of</strong> the evolutionary trend which became almost dormant<br />
with the termin<strong>at</strong>ion <strong>of</strong> most <strong>of</strong> the ITSL projects. It is up to<br />
the fundamental research <strong>co</strong>mmunity to fill-up the gap <strong>of</strong><br />
supportive fundamental research through studies <strong>of</strong> thermodynamics,<br />
kinetics and reaction mechanisms involved in low-severity<br />
oper<strong>at</strong>ions which are part <strong>of</strong> the ITSL process. Of particular<br />
interest would be the m<strong>at</strong>ching <strong>of</strong> reaction kinetics <strong>of</strong><br />
dehydrogen<strong>at</strong>ion <strong>of</strong> the hydrogen donor solvent with the hydrogen<br />
acceptancy <strong>of</strong> <strong>co</strong>al extracts.<br />
Those <strong>of</strong> us involved in these efforts are optimistic about the<br />
future <strong>of</strong> low-severity direct <strong>co</strong>al liquefaction and the quite<br />
similar <strong>co</strong>al/<strong>oil</strong> <strong>co</strong>-<strong>processing</strong> as the practical approaches in<br />
helping to allevi<strong>at</strong>e an increasingly energy-deficient world.<br />
REFERENCES<br />
1.<br />
2.<br />
3.<br />
4.<br />
5.<br />
6.<br />
7.<br />
8.<br />
S. Zaczepinski, Exxon ER&E Co., Priv<strong>at</strong>e <strong>co</strong>mmunic<strong>at</strong>ion, April 1983.<br />
A.G. Comolli, E. J. Hippo and E. S. Johanson, "Two-Stage Direct<br />
<strong>Liquefaction</strong>," McGraw-Hill Science and Technology <strong>of</strong> Synfuels 11,<br />
New York City, NY. February 2-4, 1983.<br />
E. C. Moroni. F. B. Burke. R. A. Winschel and B. W. Wilson. "Inteqr<strong>at</strong>ed<br />
Two-Stage <strong>Liquefaction</strong> Process--Sol<br />
Meeting, Se<strong>at</strong>tle, WA, March 20-25,<br />
M. Peluso, A. N. Schiffer and H. D.<br />
<strong>Liquefaction</strong> Process (ITSL)". Coal<br />
Texas, November 1981.<br />
H. D. Schindler, J. M. Chen. M. Pel<br />
"The Integr<strong>at</strong>ed Two-Stage Liquefact<br />
Meeting, New Orleans, LA, November<br />
ent Quality Effects," ACS N<strong>at</strong>ional<br />
983.<br />
Schindl er, "The Two-Stage<br />
Technology '81 Meeting, Houston,<br />
so, E. C. Moroni and J. D. Potts,<br />
on Process (ITSL) ," A1Ch.E Annual<br />
981.<br />
M. 8. Neuworth, "Advanced in Two-Stage <strong>Liquefaction</strong>" 9th Energy<br />
Technology Conference, Washington, D.C., March 1982.<br />
E. C. Moroni, "Future Development for the ITSL Concept," 7th Annual<br />
EPRI Contractors' Conference on Coal <strong>Liquefaction</strong>, Palo Alto, CA.<br />
May 11-13, 1983.<br />
M. B. Neuworth and E. C. Moroni, "Development <strong>of</strong> an Integr<strong>at</strong>ed Two-Stage<br />
Coal <strong>Liquefaction</strong> Process ," Fuel Processing Technology, 8, (1984)<br />
231-239. Elsevier Science Pub1 ishers.<br />
298
9. H. D. Schindler. J. M. Chen and J. 0. Potts. "Integr<strong>at</strong>ed Two-Stage<br />
<strong>Liquefaction</strong>", Final Technical Report - DOE Contract No. PC 50021-911,<br />
July 1985.<br />
10. B. R. Rodgers. Oak Ridge N<strong>at</strong>ional Labor<strong>at</strong>ory, Priv<strong>at</strong>e Communic<strong>at</strong>ion,<br />
1983.<br />
11. J. H. Shinn, "From Coal Single-Stage and Two-Stage Products:<br />
Model <strong>of</strong> Coal Structure, Fuel, Vol. 63, 1187, 1984.<br />
12. J. M. Chen and H. D. Schindler, "A Lumped Kinetic Model for<br />
Hydro<strong>processing</strong> Coal Extract," AIChE Spring N<strong>at</strong>ional Meeting, Houston,<br />
TX, April 1985.<br />
13.<br />
A Reactive<br />
R. F. Sullivan, "Two-Stage Hydrocracking <strong>of</strong> ITSL Oil for Jet Fyel and<br />
Naphtha." Proceedings <strong>of</strong> Direct Coal <strong>Liquefaction</strong> Contractors Review<br />
Meeting, Albuquerque, NM, Page 238, October 1984.<br />
14. R. A. Winschel and F. P. Burke, "Recycle Slurry Oil Characteriz<strong>at</strong>ion"<br />
Se<strong>co</strong>nd Annual Report, October 1, 1981 through September 30, 1982,<br />
DOE Contract PC 30027-39, August 1983.<br />
299
Compar<strong>at</strong>ive E<strong>co</strong>nomics <strong>of</strong> Two-Stage <strong>Liquefaction</strong> Processes<br />
David Gray, Glen Tomllnson, Abdel El Sawy and Abu Talib<br />
The MITRE Corpor<strong>at</strong>ion. 1820 Dolley Madison Blvd. Mclean, Va. 22102<br />
Background<br />
It has been re<strong>co</strong>gnized in research and development carried out<br />
by Lummus-Crest (1). th<strong>at</strong> two stage liquefaction provides an<br />
<strong>at</strong>tractive route to <strong>co</strong>al liquefaction by optimizing the discrete<br />
stages in <strong>co</strong>nversion <strong>of</strong> solid <strong>co</strong>al to distill<strong>at</strong>e. While<br />
improvements in process efficiency, product yield and quality have<br />
been demonstr<strong>at</strong>ed, the current limited kowledge <strong>of</strong> <strong>co</strong>al structure<br />
and liquefaction chemistry still necessit<strong>at</strong>es empirical testing <strong>of</strong><br />
process altern<strong>at</strong>ives.<br />
A number <strong>of</strong> promising process altern<strong>at</strong>ives have been developed<br />
and are under current investig<strong>at</strong>ion <strong>at</strong> a bench or process<br />
development unit (PDU) scale by a number <strong>of</strong> <strong>co</strong>ntractors under DOE<br />
sponsorship. The <strong>co</strong>ntractors include Lummus-Crest, Southern<br />
Services (Wilsonville) (2), Hydrocarbon Research Inc., (3) and Amo<strong>co</strong><br />
(4). The process vari<strong>at</strong>ions under current investig<strong>at</strong>ion are as<br />
follows:<br />
0<br />
Production <strong>of</strong> a major part <strong>of</strong> the distill<strong>at</strong>e product in<br />
Stage 1 versus Stage 2.<br />
C<strong>at</strong>alytic first and se<strong>co</strong>nd stage versus thermal first<br />
stage and c<strong>at</strong>alytic se<strong>co</strong>nd stage.<br />
Critical Solvent Deashing versus Anti-Solvent Deashing.<br />
Direct <strong>co</strong>upling <strong>of</strong> Stages 1 and 2 without intermedi<strong>at</strong>e<br />
deashing.<br />
DOE requested th<strong>at</strong> MITRE undertake a <strong>co</strong>mprehensive technical<br />
and e<strong>co</strong>nomic analysis <strong>of</strong> all the two-stage <strong>co</strong>al liquefaction<br />
<strong>co</strong>nfigur<strong>at</strong>ions currently under development in order to quantify the<br />
improvements made in the production <strong>of</strong> high quality distill<strong>at</strong>es from<br />
<strong>co</strong>al. Table 1 lists the processes th<strong>at</strong> were analyzed in this task.<br />
The methodology used to perform this analysis was as follows:<br />
0<br />
0<br />
0<br />
0<br />
Review te<strong>at</strong> d<strong>at</strong>a. Select most represent<strong>at</strong>ive run for each<br />
process using both Illinois #6 and Wyoming <strong>co</strong>als.<br />
Scale test d<strong>at</strong>a to develop m<strong>at</strong>erial balances for<br />
<strong>co</strong>nceptual <strong>co</strong>mmercial plants <strong>processing</strong> 30,000 tons per<br />
day <strong>of</strong> moisture free <strong>co</strong>al to the liquefaction units.<br />
Identify unit oper<strong>at</strong>ions for <strong>co</strong>mmercial plants.<br />
Compute <strong>co</strong>al and energy requirements for plant balance.<br />
Estim<strong>at</strong>e capital and oper<strong>at</strong>ing <strong>co</strong>sts for <strong>co</strong>mmerciai plants.<br />
300<br />
i
Compute annual revenue requirements based on <strong>co</strong>nsistent<br />
e<strong>co</strong>nomic assumptions.<br />
Compute product <strong>co</strong>sts required to s<strong>at</strong>isfy revenue required.<br />
Results and Discussion<br />
Table 2 shows the characteristics <strong>of</strong> the <strong>co</strong>mmercial-scale<br />
plants for both Illinois No. 6 and Wyoming <strong>co</strong>als. Plants with high<br />
yields and/or high hydrogen <strong>co</strong>nsumption require large quantities <strong>of</strong><br />
additional <strong>co</strong>al for steam and hydrogen production.<br />
The <strong>co</strong>nstruction <strong>co</strong>sts <strong>of</strong> the <strong>co</strong>nceptual <strong>co</strong>mmercial plants were<br />
estim<strong>at</strong>ed using an 1981 UOP/SDC (5) <strong>co</strong>mmercial design <strong>of</strong> the Lummus<br />
Integr<strong>at</strong>ed Two-Stage <strong>Liquefaction</strong> (ITSL) plant as a basis for <strong>co</strong>sts<br />
<strong>of</strong> unit oper<strong>at</strong>ions, where possible. Costs <strong>of</strong> unit oper<strong>at</strong>ions not<br />
addressed in this report were obtained from other sources.<br />
Oper<strong>at</strong>ing and maintenance <strong>co</strong>sts were estim<strong>at</strong>ed using a standard<br />
procedure developed by UOP/SnC.<br />
these plants were then calcul<strong>at</strong>ed based on the capital re<strong>co</strong>very and<br />
oper<strong>at</strong>ing <strong>co</strong>sts developed from use <strong>of</strong> <strong>co</strong>nsistent e<strong>co</strong>nomic<br />
assumptions.<br />
Since each process <strong>co</strong>nfigur<strong>at</strong>ion produces a syncrude having a<br />
different quality, it was decided to ac<strong>co</strong>unt for this product<br />
quality difference. In order to do this, MITRE has calcul<strong>at</strong>ed the<br />
hydrogen requirements and volume gain which occur when the<br />
Cq-850°F raw output <strong>of</strong> each plant is hydrotre<strong>at</strong>ed to produce a<br />
hetero<strong>at</strong>om-free, 35OAP1, 13-percent hydrogen product. The <strong>co</strong>st <strong>of</strong><br />
this hydrotre<strong>at</strong>ment is calcul<strong>at</strong>ed based on the assumption th<strong>at</strong> the<br />
<strong>co</strong>st <strong>of</strong> hydrogen production and addition is $l.OO/pound.<br />
Product <strong>co</strong>sts were then <strong>co</strong>mputed to s<strong>at</strong>isfy the annual revenue<br />
requirements based on the following assumptions.<br />
The annual revenue requirements for<br />
It was assumed<br />
th<strong>at</strong> heavy products (i.e., 850°F+ products) were valued <strong>at</strong><br />
one-half <strong>of</strong> the value <strong>of</strong> a barrel <strong>of</strong> C4-85O0F liquid product.<br />
For the hydrocarbon gases (C1-C3), it was assumed th<strong>at</strong> 12~10~<br />
Btus were equivalent in value to one barrel <strong>of</strong> C4-850°F product.<br />
Figure 1 shows <strong>co</strong>mparisons <strong>of</strong> the annual revenue requirement,<br />
equivalent product yield, and required selling price <strong>of</strong> products<br />
from the <strong>co</strong>nceptual <strong>co</strong>mmercial plants when oper<strong>at</strong>ed with Illinois<br />
No. 6 <strong>co</strong>al. All values are shown as percentages <strong>of</strong> the Lummus<br />
Integr<strong>at</strong>ed Two-Stage <strong>Liquefaction</strong> (ITSL) base case. For <strong>co</strong>mpar<strong>at</strong>ive<br />
purposes two <strong>co</strong>nceptual plants based on single-stage processes<br />
(H-Coal (6) and EDS (7)) are included.<br />
The bars depicting annual revenue requirements are divided into<br />
four sections, to illustr<strong>at</strong>e the rel<strong>at</strong>ive <strong>co</strong>ntribution <strong>of</strong> capital<br />
re<strong>co</strong>very, <strong>co</strong>al, oper<strong>at</strong>ing and hydrotre<strong>at</strong>ing <strong>co</strong>sta. The capital<br />
re<strong>co</strong>very <strong>co</strong>sts for the two-stage plants vary by about 2 percent,<br />
indic<strong>at</strong>ing a similarly small vari<strong>at</strong>ion in the capital <strong>co</strong>sts <strong>of</strong> the<br />
plants. Capital <strong>co</strong>stebf the single-stage plants are 5.4- and<br />
9.5-percent lower than the Integr<strong>at</strong>ed Two-Stage <strong>Liquefaction</strong> (ITSL)<br />
base case for €I-Coal and EDS, respectively.<br />
301
The vari<strong>at</strong>ion in hydrotre<strong>at</strong>ing <strong>co</strong>sts reflects vari<strong>at</strong>ion in both<br />
the quality <strong>of</strong> the raw product and the quantity <strong>of</strong> the C4-850°F<br />
fraction. Hydrotre<strong>at</strong>ing <strong>co</strong>sts <strong>of</strong> EDS are lower than the other<br />
systems, because <strong>of</strong> the rel<strong>at</strong>ively low yield and high API quality <strong>of</strong><br />
the raw EDS product. The total annual revenue requirements vary<br />
from a low <strong>of</strong> 89.7 percent <strong>of</strong> base for EDS, to a high <strong>of</strong> 107.5<br />
percent <strong>of</strong> base for CTSL.<br />
The equivalent barrels <strong>of</strong> yield show a much wider vari<strong>at</strong>ion<br />
than the annual revenue requirements. EDS yield is lowest <strong>at</strong> 84.6<br />
percent <strong>of</strong> base, while CTSL is highest <strong>at</strong> 115.7 percent.<br />
The lower portion <strong>of</strong> Figure 1 <strong>co</strong>mpares the required selling<br />
price <strong>of</strong> hydrotre<strong>at</strong>ed products from the <strong>co</strong>nceptual plants. The<br />
prices vary from 105.9 percent <strong>of</strong> base for EDS to a low <strong>of</strong> 92.9<br />
percent <strong>of</strong> base for CTSL, a spread <strong>of</strong> 13 percent. The most advanced<br />
systems, e.g., modified Lummus, Wilsonville RITSL, and CTSL, <strong>of</strong>fer<br />
the lowest product prices. All these systems c<strong>at</strong>alytically tre<strong>at</strong> an<br />
ash-<strong>co</strong>ntaining extract. It is doubtful th<strong>at</strong> the one-percent<br />
difference between CTSL and RITSL is significant. However, the<br />
slight superiority <strong>of</strong> these systems rel<strong>at</strong>ive to the modified Lummus<br />
is believed to be significant and is traceable to the higher<br />
rejection <strong>of</strong> soluble m<strong>at</strong>erial which is inherent in the deashing<br />
system employed <strong>at</strong> Lummus. Lummus has suggested th<strong>at</strong> the additional<br />
liquids in the deashed overflow <strong>co</strong>uld be re<strong>co</strong>vered by <strong>co</strong>king.<br />
Figure 2 shows e<strong>co</strong>nomic <strong>co</strong>mparisons for plants oper<strong>at</strong>ed with<br />
Wyoming <strong>co</strong>al. Capital re<strong>co</strong>very <strong>co</strong>st vari<strong>at</strong>ions between the plants<br />
are very similar to those observed in the Illinois No. 6 plants.<br />
The total revenue requirements vary from 93.8 to 100 percent <strong>of</strong> base<br />
for the two-stage plants, but are much lower <strong>at</strong> 89 and 83.3 percent<br />
<strong>of</strong> base for the single-stage H-Coal and EDS plants, respectively.<br />
Plant yields show a much gre<strong>at</strong>er vari<strong>at</strong>ion than was observed in<br />
the plants <strong>processing</strong> Illinois No. 6 <strong>co</strong>al. Yields vary from a low<br />
<strong>of</strong> 91.6 percent <strong>of</strong> base for EDS to a high <strong>of</strong> 141.5 percent <strong>of</strong> base<br />
for CTSL.<br />
The required selling prices also show a wide vari<strong>at</strong>ion. The<br />
Lummus ITSL shows the highest selling price <strong>at</strong> 100 percent <strong>of</strong> base,<br />
while CTSL <strong>of</strong>fers the lowest price <strong>at</strong> 68.9 percent <strong>of</strong> base. The<br />
single-stage H-Coal and Wilsonville DITSL processes <strong>of</strong>fer similar<br />
prices <strong>of</strong> 80.3 and 80.7 percent <strong>of</strong> base, respectively.<br />
The results with both Illinois No. 6 and Wyoming <strong>co</strong>als indic<strong>at</strong>e<br />
th<strong>at</strong> the additional <strong>co</strong>st and <strong>co</strong>mplexity <strong>of</strong> two-stage <strong>processing</strong> is<br />
justified by the increases in yield and product quality which can be<br />
obtained.<br />
302
REFERENCES<br />
1. Integr<strong>at</strong>ed Two-Stage <strong>Liquefaction</strong>, Final Technical Report,<br />
Volume I, prepared for DOE by the Lummus Crest Company<br />
Engineering Development Center, Bloomfield, New Jersey,<br />
DOE Report No. PC50021-Ql1, July 1985.<br />
2. C<strong>at</strong>alytic, Inc., Wilsonville, Alabama, Technical Progress<br />
Report: Run 247 with Illinois No. 6 Coal, The Wilsonville<br />
Advanced Coal <strong>Liquefaction</strong> Research and Development<br />
Facility, prepared for the U.S. Department <strong>of</strong> Energy and<br />
Electric Power Research Institute, Document No.<br />
WE/PC/50041 (UC-Sod), 1985.<br />
3. Comolli, A. G., et al., New Technology Concept for<br />
Two-Stage <strong>Liquefaction</strong> <strong>of</strong> Coal, Topical Report prepared by<br />
HRI under DOE Contract No. DE-AC22-83-PC60017, August 1985.<br />
4. Hydrocarbon Research, Inc., AMOCO Continuous Ebull<strong>at</strong>ed-Bed<br />
Bench Unit Study, report prepared under DOE Contract No.<br />
DE-AC22-81PC400009, July 1985.<br />
5. Schachtschneider, A. B., R. N. Dinapoli, C. S. Yin, W. F.<br />
Charba, and J. R. Schulze, Conceptual Design <strong>of</strong> Commercial<br />
Integr<strong>at</strong>ed Two-Stage Coal <strong>Liquefaction</strong> Facilit , Report<br />
No. TR-82/014-003, prepared for DOE by UOP/SDCf June 1981.<br />
6. Talib, A., D. Gray, and M. B. Neuworth, Assessment <strong>of</strong><br />
€I-Coal Process Developments, MITRE Technical Report No.<br />
MTR-83W199, January 1984 (published a8 DOE Report No.<br />
DOE/ET/13800-5 January 1984).<br />
7. Exxon Research and Engineering Company, EDS Coal<br />
<strong>Liquefaction</strong> Process Development, Phase V, Annual<br />
Technical Progress Report for July 1, 1980 - June 30,<br />
- 1981, DOE/ET/10069-T12 (Volume l), December 1981.<br />
303
FIGURE 1: COMPARISONS OF ILLINOIS NO. 6 COAL PLANTS<br />
130<br />
120<br />
Annual Revenue Requirement<br />
-<br />
110 Yields in Barrels<br />
P.MI no<br />
o(n1u (00<br />
90<br />
0<br />
(0<br />
¶a<br />
&met no<br />
o(8.u (00<br />
D<br />
0<br />
EDS AMOCO M a l Lummur Wilson Modilied Rm<strong>co</strong>n. MRi<br />
Lummur Wl(r0n ClSL<br />
EDS AMOCO H-Coai Lummur Wilson Modified Re<strong>co</strong>n. MRI<br />
Lummur Wilson ClSL<br />
304<br />
1
Penant<br />
<strong>of</strong> Base<br />
FIGURE 2: ECONOMIC COMPARISONS OF WYOMING COAL PLANTS<br />
120<br />
110<br />
Annual Revenue Requirement<br />
Lurnrnuo Wilson Modified H.Coal<br />
Wilson<br />
97<br />
HRI<br />
CTSL<br />
150<br />
140<br />
130<br />
Pmrcml 120<br />
0' 110<br />
iw<br />
sa<br />
M Lummur EDS Wilson Modillad HCoal HUI<br />
Wllron CISL<br />
130<br />
120 Required Selling Price<br />
tin<br />
hunt um<br />
Of BBU 90<br />
M<br />
70<br />
60 Lummur EDS Wilaon Moditid HCoal nus<br />
wuwn ClSL<br />
305<br />
Hydrotmmting<br />
<strong>co</strong>st<br />
Opnt1ng<br />
<strong>co</strong>st
Process<br />
TABLE 1: TWO-STAGE PROCESSES<br />
--<br />
Scale Stage I<br />
Lumrnus-Crest<br />
Integr<strong>at</strong>ed TwWStage 600<br />
<strong>Liquefaction</strong> (ITSL)<br />
Wilsonvllle Two-Stage<br />
LblDay Thermal<br />
<strong>Liquefaction</strong> 3 TlDay Thermal'<br />
Hydrocarbon Research. Inc.<br />
C<strong>at</strong>alytic Two-Stage 50-100<br />
<strong>Liquefaction</strong> (CTSL) LblDay C<strong>at</strong>alytic<br />
AMOCO ThermallC<strong>at</strong>alylic<br />
Two-Stage 50-100<br />
<strong>Liquefaction</strong> LblDay Thermal<br />
*Sometimer Slurry C<strong>at</strong>alyst Used<br />
306<br />
Deashlng Stage I1<br />
AntI.Solven1 C<strong>at</strong>alytic<br />
Critical C<strong>at</strong>alytic<br />
Solvent Deashing<br />
No Deashlng C<strong>at</strong>alytic<br />
Between Stages<br />
No Deashing C<strong>at</strong>alytic<br />
Between Stages
-<br />
TABLE 2: CONCEPTUAL COMMERCIAL PLANT SUMMARIES<br />
Rouu:<br />
input<br />
Slam hi. TPD (UF)<br />
Garifl~r Cod. TPD (MF)<br />
TOW PIml W, TPD (MF)<br />
- OUlpUl<br />
SNQ, MMSCFD<br />
Raw 4 + Uguld, BPSD<br />
Total Llquld Vkld<br />
aflw Hydroln<strong>at</strong>menl, BPSD<br />
Process:<br />
ILLINOIS NO. 6 COAL FEED<br />
WYOMING COAL FEED<br />
Lummus Wilsonville Wilsonville<br />
ITSL ITSL DIEL CTSL<br />
----<br />
- Input<br />
Steam Coal, TPD (MF)<br />
Gaslfier Coal, TPD (MF)<br />
3,000<br />
0<br />
2,000<br />
3.000<br />
2,000<br />
4.000<br />
4,000<br />
s,m<br />
Total Plant Coal, TPD (MF) 33,000 36.000 36,OOO 43,OOO<br />
Output<br />
SNG, MMSCFD<br />
Raw C4 + Liquld, BPSD<br />
33<br />
70,000<br />
26<br />
00.000<br />
6<br />
97,000<br />
3<br />
114,000<br />
Total Liquid Yield<br />
after Hydrotre<strong>at</strong>ment. BPSD 80,OOO 94,OOO 96,OOO 119,OOO<br />
307
Abstract<br />
Temper<strong>at</strong>ure-Staged C<strong>at</strong>alytic Coal <strong>Liquefaction</strong><br />
Frank Derbyshire, Alan Davis, Mike Epstein, and Peter Stansberry<br />
College <strong>of</strong> Earth and Mineral Sciences<br />
The Pennsylvania St<strong>at</strong>e University<br />
University Park, PA 16802, USA<br />
Coal liquefaction has been investig<strong>at</strong>ed under <strong>co</strong>nditions where reaction is<br />
<strong>co</strong>nducted in successive stages <strong>of</strong> increasing temper<strong>at</strong>ure and in the presence <strong>of</strong> a<br />
dispersed sulfided Mo c<strong>at</strong>alyst. This sequence leads not only to high <strong>co</strong>nversions<br />
but also gre<strong>at</strong>ly increases the selectivity <strong>of</strong> the products to <strong>oil</strong>s <strong>at</strong> the expense <strong>of</strong><br />
asphaltenes, with only marginal increase in gas make. The product distribution is<br />
strongly influenced by the solvent <strong>co</strong>mposition and the reaction <strong>co</strong>nditions in the<br />
two stages.<br />
Examin<strong>at</strong>ion <strong>of</strong> the liquefaction residues from the liquefaction <strong>of</strong> a<br />
bituminous and a subbituminous <strong>co</strong>al has provided supporting evidence to show th<strong>at</strong><br />
the temper<strong>at</strong>ure-staged reaction sequence favors hydrogen<strong>at</strong>ive processes. Moreover,<br />
the choice <strong>of</strong> reaction <strong>co</strong>nditions for optimum performance is rank-dependent; for<br />
example, low-rank <strong>co</strong>als appear to require a lower first stage temper<strong>at</strong>ure than<br />
bituminous <strong>co</strong>als in order to minimize the potential for regressive reactions.<br />
Introduction<br />
In some earlier reported research (1,2) a bituminous and a subbituminous <strong>co</strong>al<br />
were pretre<strong>at</strong>ed by dry c<strong>at</strong>alytic hydrogen<strong>at</strong>ion, using a molybdenum c<strong>at</strong>alyst <strong>at</strong> 35OoC<br />
for 1 h, following which they were mixed with naphthalene (2:l solvent to <strong>co</strong>al<br />
r<strong>at</strong>io) and reacted <strong>at</strong> 425OC for 10 min. The results showed th<strong>at</strong> the low-temper<strong>at</strong>ure<br />
pretre<strong>at</strong>ment improved both the net <strong>co</strong>al <strong>co</strong>nversion, based upon solubility in<br />
tetrahydr<strong>of</strong>uran, and the product distribution. Notably, the <strong>oil</strong> to asphaltene r<strong>at</strong>io<br />
was substantially increased with only marginal additional gas make.<br />
Based upon these findings, further research has been directed to investig<strong>at</strong>ing<br />
the chemistry and the potential <strong>of</strong> temper<strong>at</strong>ure-staged <strong>co</strong>al liquefaction. The<br />
results <strong>of</strong> this research are presented in this paper. Similar studies are being<br />
<strong>co</strong>nducted on a larger scale by Hydrocarbon Research Inc. (3).<br />
Experimental<br />
Coal Prepar<strong>at</strong>ion<br />
Samples <strong>of</strong> bituminous and subbituminous <strong>co</strong>al were provided by the Penn St<strong>at</strong>e<br />
Coal Sample Bank for use in this research. The <strong>co</strong>als were obtained undried and in<br />
lump form about 12 mm size and were crushed in a glove box under oxygen-free<br />
nitrogen to 0.8 mm top size. The crushed <strong>co</strong>als were subdivided by riffling into a<br />
number <strong>of</strong> 10 g represent<strong>at</strong>ive samples and sealed in vials under nitrogen.<br />
Properties <strong>of</strong> the <strong>co</strong>als are shown in Table 1.<br />
The <strong>co</strong>als were impregn<strong>at</strong>ed with Mo c<strong>at</strong>alyst by slurrying with an aqueous<br />
solution <strong>of</strong> ammonium tetr<strong>at</strong>hiomolybd<strong>at</strong>e in the <strong>co</strong>ncentr<strong>at</strong>ion necessary to <strong>at</strong>tain a<br />
loading <strong>of</strong> 1% wt Mo on a dmmf basis. The quantity <strong>of</strong> <strong>co</strong>al impregn<strong>at</strong>ed was<br />
sufficient for a <strong>co</strong>mplete series <strong>of</strong> experiments. After slurrying, the excess w<strong>at</strong>er<br />
was removed by vacuum freeze-drying.<br />
<strong>Liquefaction</strong><br />
The impregn<strong>at</strong>ed <strong>co</strong>al was mixed in a r<strong>at</strong>io <strong>of</strong> 1:2 with liquefaction solvent. In<br />
most <strong>of</strong> the experiments, naphthalene was selected as the solvent because, <strong>at</strong> least<br />
308<br />
I<br />
' I<br />
1
<strong>at</strong> the onset <strong>of</strong> reaction, no H-donor would be present, which would allow the effects<br />
<strong>of</strong> added c<strong>at</strong>alyst and low-temper<strong>at</strong>ure <strong>co</strong>al pretre<strong>at</strong>ment to be more clearly<br />
discerned. Approxim<strong>at</strong>ely 0.1 g <strong>of</strong> CS was added to the reaction mixture to ensure<br />
2<br />
th<strong>at</strong> the molybdenum was maintained in the fully sulphided st<strong>at</strong>e.<br />
Reactions were carried out in tubing bomb reactors <strong>of</strong> about 30 cm3 capacity<br />
which were he<strong>at</strong>ed by immersion in a fluidized sandb<strong>at</strong>h. More detailed descriptions<br />
<strong>of</strong> the experimental procedures have been given elsewhere (4). Reactions were<br />
<strong>co</strong>nducted either <strong>at</strong> 425OC for 10 min or under these same <strong>co</strong>nditions after first<br />
pretre<strong>at</strong>ing <strong>at</strong> 35OoC for 60 min. The initial hydrogen pressure (<strong>co</strong>ld) for both<br />
pretre<strong>at</strong>ment and the higher temper<strong>at</strong>ure reaction was 7 MPa.<br />
When the low-temper<strong>at</strong>ure pretre<strong>at</strong>ment was carried out, the bombs were quenched<br />
to room temper<strong>at</strong>ure <strong>at</strong> the end <strong>of</strong> the reaction period and the yields <strong>of</strong> light gases<br />
were determined by volumetric measurement and gas chrom<strong>at</strong>ographic analysis. The<br />
main purpose <strong>of</strong> the <strong>co</strong>oling and venting procedure was to ensure th<strong>at</strong> there was a<br />
high partial pressure <strong>of</strong> hydrogen in the reactor <strong>at</strong> the beginning <strong>of</strong> the higher<br />
temper<strong>at</strong>ure stage. The r<strong>at</strong>ionale for choosing a short reaction time for the<br />
high-temper<strong>at</strong>ure stage (10 min <strong>at</strong> 425OC) was the same as th<strong>at</strong> used for solvent<br />
selection; namely to accentu<strong>at</strong>e the effects which would be caused by the<br />
low-temper<strong>at</strong>ure reaction.<br />
Following high-temper<strong>at</strong>ure reaction, the gas yield and <strong>co</strong>mposition were<br />
determined and the solid and liquid products were worked-up to obtain the yields <strong>of</strong><br />
insoluble residue (tetrahydr<strong>of</strong>uran, THF) asphaltenes (hexane-insoluble, THF-soluble)<br />
and <strong>oil</strong>s (hexane-soluble). In these calcul<strong>at</strong>ions, it was assumed th<strong>at</strong> the<br />
naphthalene reported to the hexane-solubles. Despite extensive precautions, some <strong>of</strong><br />
the lighter liquefaction products were lost during product work-up and especially<br />
during the removal <strong>of</strong> solvents. Oil yields are therefore calcul<strong>at</strong>ed from the mass<br />
balance assuming th<strong>at</strong> most <strong>of</strong> the mass balance deficit is <strong>at</strong>tributable to the loss<br />
<strong>of</strong> light ends.<br />
A further factor, which is not ac<strong>co</strong>unted in the product distribution, is the<br />
yield <strong>of</strong> w<strong>at</strong>er produced by reaction. It is not anticip<strong>at</strong>ed th<strong>at</strong> this will<br />
<strong>co</strong>nstitute more than a few percent <strong>of</strong> the liquid yields, even with the subbituminous<br />
<strong>co</strong>al, although firm estim<strong>at</strong>es have not yet been made. Because <strong>of</strong> the method <strong>of</strong><br />
calcul<strong>at</strong>ion <strong>of</strong> the <strong>oil</strong> yield, any w<strong>at</strong>er which is produced is <strong>co</strong>nsidered as <strong>oil</strong>.<br />
Consequently, the actual <strong>oil</strong> yield will be somewh<strong>at</strong> lower than reported.<br />
A few experiments were <strong>co</strong>nducted to explore the effects <strong>of</strong> extended reaction<br />
time <strong>at</strong> 425OC and the influence <strong>of</strong> a more reactive solvent than naphthalene. In the<br />
l<strong>at</strong>ter instance, the solvent was a process-derived recycle solvent fraction (454+Oc)<br />
obtained from the Lummus Integr<strong>at</strong>ed Two-Stage <strong>Liquefaction</strong> (ITSL) process, when<br />
oper<strong>at</strong>ing on Wyodak subbituminous <strong>co</strong>al.<br />
In determining the product distribution, quantities <strong>of</strong> <strong>oil</strong> and asphaltene,<br />
equivalent to those present in the original solvent, were subtracted from the<br />
product yields in order to obtain the net yields <strong>at</strong>tributable to <strong>co</strong>al.<br />
Residue Micros<strong>co</strong>py<br />
The dried liquefaction residues (THF insolubles) were embedded in epoxy resin<br />
and polished with a series <strong>of</strong> alumina,slurries. Examin<strong>at</strong>ion was undertaken with a<br />
polarizing ref lected-light microsope under <strong>oil</strong> immersion <strong>at</strong> a magnific<strong>at</strong>ion <strong>of</strong> 625;<br />
a rot<strong>at</strong>able <strong>co</strong>mpens<strong>at</strong>ion pl<strong>at</strong>e was used as an aid in distinguishing between<br />
isotropic and anisotropic m<strong>at</strong>erials. some observ<strong>at</strong>ions were made in blue-light<br />
irradi<strong>at</strong>ion, in order to observe the proportions <strong>of</strong> liptinite macerals present.<br />
309
Results and Discussion<br />
Reactions in Naphthalene<br />
The <strong>co</strong>nversions and product distributions obtained by the liquefaction <strong>of</strong> the<br />
subbituminous and bituminous <strong>co</strong>als under various <strong>co</strong>mbin<strong>at</strong>ions <strong>of</strong> pretre<strong>at</strong>ment and<br />
liquefacti-on reactions are summarised in Tables 2 and 3. Similar trends are<br />
apparent for both <strong>of</strong> the <strong>co</strong>als. Reaction in the presence <strong>of</strong> the c<strong>at</strong>alyst produced<br />
higher net <strong>co</strong>nversions than the 'thermal' experiments, as would be expected.<br />
However, the <strong>co</strong>mbin<strong>at</strong>ion <strong>of</strong> low-temper<strong>at</strong>ure c<strong>at</strong>alytic pretre<strong>at</strong>ment followed by the<br />
higher temper<strong>at</strong>ure c<strong>at</strong>alytic reaction had the gre<strong>at</strong>est influence in improving the<br />
product selectivity <strong>co</strong>n<strong>co</strong>mitant with <strong>at</strong>taining the highest <strong>co</strong>nversion. In<br />
particular, the highest <strong>oil</strong> yields were obtained without any <strong>at</strong>tendant increase in<br />
the production <strong>of</strong> light hydrocarbon gases.<br />
An examin<strong>at</strong>ion <strong>of</strong> the liquefaction products by gas chrom<strong>at</strong>ography showed th<strong>at</strong><br />
there was no significant <strong>co</strong>nversion <strong>of</strong> naphthalene to tetralin (less than 1%) in any<br />
<strong>of</strong> these experiments. While this finding does not exclude the possibility th<strong>at</strong> the<br />
c<strong>at</strong>alyst may promote liquefaction through the successive gener<strong>at</strong>ion and<br />
dehydrogen<strong>at</strong>ion <strong>of</strong> donor solvent, it does suggest th<strong>at</strong> other reaction p<strong>at</strong>hways are<br />
oper<strong>at</strong>ive and may be more important.<br />
The addition <strong>of</strong> c<strong>at</strong>alyst, without pretre<strong>at</strong>ment, significantly increased the<br />
<strong>co</strong>nversion <strong>of</strong> both <strong>co</strong>als over th<strong>at</strong> obtained in the thermal experiments. At the same<br />
time, these <strong>co</strong>nditions produced the lowest <strong>oil</strong> yields and the lowest r<strong>at</strong>ios <strong>of</strong> <strong>oil</strong>s<br />
to asphaltenes. The-pretre<strong>at</strong>ment evidently allows the c<strong>at</strong>alyst to perform certain<br />
functions which ultim<strong>at</strong>ely lead to higher <strong>oil</strong> yields and these functions appear to<br />
be not as readily gerformed during a short c<strong>at</strong>alytic reaction <strong>at</strong> the higher<br />
temper<strong>at</strong>ure <strong>of</strong> 425 C.<br />
Micros<strong>co</strong>pic Examin<strong>at</strong>ion <strong>of</strong> <strong>Liquefaction</strong> Residues<br />
There were notable differences in the appearance <strong>of</strong> the residues from the<br />
bituminous and subbituminous <strong>co</strong>als. The bituminous <strong>co</strong>als hydrogen<strong>at</strong>ed in the<br />
absence <strong>of</strong> c<strong>at</strong>alyst showed clear evidence <strong>of</strong> the development <strong>of</strong> plasticity. i.e.,<br />
rounded particle outlines and the form<strong>at</strong>ion <strong>of</strong> spheres <strong>of</strong> vitroplast.<br />
Vitroplast is<br />
a low-reflecting, isotropic, pitch-like m<strong>at</strong>erial. usually derived from vitrinite.<br />
th<strong>at</strong> occurs as spheres and agglomer<strong>at</strong>es (5). The vitroplast observed in this study<br />
is the type which Shibaoka (6) has referred to as a primary vitroplast, being<br />
derived directly by s<strong>of</strong>tening <strong>of</strong> vitrinite.<br />
In <strong>co</strong>ntrast, the residues derived from the c<strong>at</strong>alytically hydrogen<strong>at</strong>ed<br />
bituminous <strong>co</strong>als had apparently undergone more extensive reaction. There was no<br />
evidence <strong>of</strong> simple melting, and the vitrinite-derived m<strong>at</strong>erial was <strong>co</strong>nsiderably<br />
reduced in volumetric proportion rel<strong>at</strong>ive to th<strong>at</strong> <strong>of</strong> other macerals. The<br />
reflectance <strong>of</strong> this vitrinite-derived m<strong>at</strong>erial was lower than either th<strong>at</strong> <strong>of</strong> the<br />
vitrinite in the feed <strong>co</strong>al or th<strong>at</strong> <strong>of</strong> the vitroplast in the residues <strong>of</strong> unc<strong>at</strong>alyzed<br />
runs. These observetions are <strong>co</strong>nsistent with the action <strong>of</strong> the c<strong>at</strong>alyst being<br />
instrumental in the hydrogen<strong>at</strong>ion and breakdown <strong>of</strong> the vitrinite structure. An<br />
unexpected fe<strong>at</strong>ure <strong>of</strong> the residues was the large proportion <strong>of</strong> remaining, although<br />
not necessarily unchanged, liptinite (sporinite and cutinite) present in samples<br />
from the c<strong>at</strong>alysed experiments with bituminous <strong>co</strong>al.<br />
None <strong>of</strong> the residues from the subbituminous <strong>co</strong>al <strong>co</strong>ntained vitroplast or showed<br />
other evidence <strong>of</strong> plasticity during tre<strong>at</strong>ment. R<strong>at</strong>her, the residues <strong>of</strong> the<br />
vitrinite (huminite) <strong>co</strong>nsisted <strong>of</strong> t<strong>at</strong>tered skeletons <strong>of</strong> the structures present in<br />
the original <strong>co</strong>al. However, the vitrinite reflectance was significantly higher in<br />
the residues than in the parent <strong>co</strong>al; th<strong>at</strong> <strong>of</strong> the residue from the c<strong>at</strong>alysed and<br />
pretre<strong>at</strong>ed <strong>co</strong>al was judged to be somewh<strong>at</strong> lower than th<strong>at</strong> <strong>of</strong> other residues. This<br />
310
un also resulted in more particle disintegr<strong>at</strong>ion than was observed in the other<br />
residues.<br />
The micros<strong>co</strong>pic studies <strong>of</strong> the liquefaction residues reflect the trends shown<br />
by the yield d<strong>at</strong>a in <strong>co</strong>nfirming th<strong>at</strong> the staged c<strong>at</strong>alytic liquefaction produced the<br />
Conditions most <strong>co</strong>nducive to <strong>co</strong>al hydrogen<strong>at</strong>ion and liquefaction.<br />
From the residue analysis for the subbituminous <strong>co</strong>al, it appears th<strong>at</strong> the<br />
temper<strong>at</strong>ure selected for the low-temper<strong>at</strong>ure stage was too high as shown by the<br />
increase in reflectance <strong>of</strong> the vitrinite-derived m<strong>at</strong>erials in the residues rel<strong>at</strong>ive<br />
to the vitrinite (huminite) in the untre<strong>at</strong>ed <strong>co</strong>al. In <strong>co</strong>ntrast, the residues from<br />
c<strong>at</strong>alysed bituminous <strong>co</strong>al hydrogen<strong>at</strong>ion display the predominance <strong>of</strong> hydrogen<strong>at</strong>ion<br />
reactions as evidenced by the lower vitrinite reflectance <strong>co</strong>mpared to the parent<br />
<strong>co</strong>al. Without c<strong>at</strong>alyst, the very obvious development <strong>of</strong> plasticity indic<strong>at</strong>es the<br />
domin<strong>at</strong>ing effect <strong>of</strong> thermal tre<strong>at</strong>ment.<br />
Effect <strong>of</strong> Other Reaction Conditions<br />
The d<strong>at</strong>a presented above have illustr<strong>at</strong>ed the potential advantages to be<br />
derived by liquefying <strong>co</strong>als in stages <strong>of</strong> increasing temper<strong>at</strong>ure and in the presence<br />
<strong>of</strong> a c<strong>at</strong>alyst. Verific<strong>at</strong>ion <strong>of</strong> these phenomena has been demonstr<strong>at</strong>ed more<br />
<strong>co</strong>mprehensively and on a larger scale by Hydrocarbon Research Inc. (3).<br />
In the labor<strong>at</strong>ory scale studies, no system<strong>at</strong>ic <strong>at</strong>tempt has yet been made to<br />
investig<strong>at</strong>e how independent variables such as the reaction <strong>co</strong>nditions in the first<br />
and se<strong>co</strong>nd stages, the solvent <strong>co</strong>mposition and the c<strong>at</strong>alyst type and <strong>co</strong>ncentr<strong>at</strong>ion<br />
affect the performance <strong>at</strong>tainable in such a reaction sequence. Some preliminary<br />
d<strong>at</strong>a sre presented in Figure 1 which show the <strong>co</strong>mpar<strong>at</strong>ive affects on the product<br />
distribution for the subbituminous <strong>co</strong>al (PSOC-1401) due to (i) increasing the high<br />
temper<strong>at</strong>ure residence time from 10 to 45 min while employing naphthalene as solvent<br />
and (ii) using the more reactive process solvent and the 45 min high-temper<strong>at</strong>ure<br />
residence time.<br />
With naphthalene, increasing the reaction time <strong>at</strong> high-temper<strong>at</strong>ure is evidently<br />
advantageous in promoting further inter<strong>co</strong>nversion <strong>of</strong> <strong>oil</strong>s to asphaltenes (the <strong>oil</strong> to<br />
asphaltene r<strong>at</strong>io increased from 0.8 to 2:l) with some simultaneous increase in gas<br />
make; the CO yield increased from 7.9 to 9.8% and the yield <strong>of</strong> C -C hydrocarbons<br />
increased frh 0.8 to 3.0%. As in the other experiments using nabhthalene as<br />
solvent, there was no significant <strong>co</strong>nversion <strong>of</strong> naphthalene to tetralin.<br />
A much more dram<strong>at</strong>ic change in product selectivity was achieved by using the<br />
process solvent when the <strong>oil</strong> to asphaltene r<strong>at</strong>io increased to approxim<strong>at</strong>ely 14:l.<br />
To <strong>of</strong>fset this gain there was a more significant increase in gas make; the CO and<br />
C -c yields being 12.2 and 4.7%. respectively. Quite evidently, the <strong>co</strong>mposition <strong>of</strong><br />
4<br />
the solvent is an important parameter even in the presence <strong>of</strong> an active c<strong>at</strong>alyst.<br />
Acknowledgements<br />
Three <strong>of</strong> the authors, Derbyshire, Epstein. and Stansberry, wish to acknowledge<br />
the financial support <strong>of</strong> the Department <strong>of</strong> Energy, Grant No. DE-FE22-83PC60811 and<br />
Contract No. DE-FG-22-84PC7003. Frank Derbyshire wishes to scknowledge Dr. Frank<br />
Burke <strong>of</strong> Cono<strong>co</strong> Coal Research for his helpful discussions and <strong>co</strong>ntributions and Dr.<br />
Eneo Moroni for his steadfast support.<br />
References<br />
1. Derbyshire, F. J., Davis, A., Lin. R., Stansberry. P. G., and Terrer, 14.-T.,<br />
Accepted for public<strong>at</strong>ion in Fuel Proc. Tech.<br />
310 A
2.<br />
3.<br />
4.<br />
5.<br />
6.<br />
Derbyshire, F. J. and Luckie, P. T.. Quarterly Report to U.S. DOE for the<br />
Period July 1st-September 30th, 1985, Contract No. DE-FG-22-84-PC7003.<br />
Comolli, A. G., MacArthur, J. B., and McLean, J. B.. Proc. Tenth Annual EPRI<br />
Contractors' Conference on Clean Liquid and Solid Fuels, 1985, p. 2-1.<br />
Derbyshire, F. J., Davis, A., Lin, R., Stansberry, P. G., and Terrer, M.-T..<br />
ACS Fuel Division Preprints, 1985, 30. No. 4, 326.<br />
Mitchell, G. D., Davis, A.. and Spackman, W., in R. T. Ellington, ed., "Liquid<br />
Fuels from Coal." pp. 245-270. Academic Press, 1977.<br />
Shibaoka. M., Fuel, 60. 240-246, 1981.<br />
311<br />
al<br />
a<br />
e<br />
Y<br />
u<br />
0 4<br />
I(<br />
s<br />
i:<br />
3<br />
ij<br />
a<br />
Q<br />
Y U<br />
n<br />
t<br />
.I<br />
m<br />
4<br />
Q<br />
P)<br />
a<br />
Y 9<br />
al a<br />
al<br />
Y<br />
8<br />
4<br />
x<br />
t"
C<br />
r(<br />
Y U<br />
I<br />
0<br />
P<br />
cl<br />
u<br />
1 Q<br />
Y<br />
u<br />
O<br />
L Y<br />
? ? ? m<br />
0 0 0 6<br />
h ? ? ?<br />
I - ~ W I O<br />
? Y ? 1<br />
0 0 0 4<br />
rl 0<br />
I . I -<br />
I 0 1 0<br />
n<br />
I . Y<br />
II-IIO<br />
N 0<br />
I . I .<br />
I 0 1 0<br />
. . . .<br />
h m m -<br />
u m r<br />
v<br />
l<br />
u<br />
~ ~<br />
- * A h<br />
Q P V W<br />
312<br />
h<br />
1 W<br />
N<br />
v<br />
m<br />
-0<br />
rl<br />
O<br />
r(<br />
r<br />
u V<br />
W<br />
e<br />
&<br />
Q<br />
0<br />
.A<br />
Y<br />
W .I<br />
5<br />
I .J<br />
I 4<br />
I N<br />
8<br />
I<br />
8<br />
I<br />
0"<br />
3<br />
I N<br />
8<br />
I<br />
8<br />
l<br />
4 0<br />
0"<br />
0<br />
O<br />
rl Y<br />
9<br />
c<br />
T<br />
C<br />
.A<br />
m<br />
Y<br />
><br />
0<br />
e<br />
4<br />
Y U<br />
w PI<br />
w<br />
cl r(<br />
u<br />
C<br />
B<br />
Y 9<br />
2<br />
o<br />
L Y<br />
U<br />
V ? ? ?<br />
d o 0 0<br />
0 1 1 ?<br />
d o 0 0<br />
YI IO<br />
- A h -<br />
9 1 V W
I<br />
LL<br />
a<br />
rJJW ., .. ;><br />
0 i s W 8 a<br />
313
TWO-STAGE COAL LIQUEFACTION<br />
WITHOUT GAS-PHASE HYDROGEN<br />
H. P. Stephens<br />
Sandia N<strong>at</strong>ional Labor<strong>at</strong>ories<br />
Albuquerque, New Mexi<strong>co</strong> 87185<br />
INTRODUCTION<br />
Current two-stage direct <strong>co</strong>al liquefaction processes require the use<br />
<strong>of</strong> high-pressure purified hydrogen to hydrogen<strong>at</strong>e either the solvent<br />
or a <strong>co</strong>al/solvent slurry. This paper describes techniques to<br />
elimin<strong>at</strong>e the direct use <strong>of</strong> hydrogen gas in the solvent production<br />
and primary <strong>co</strong>al liquefaction stages. The approach employs the<br />
w<strong>at</strong>er-gas shift (WGS) reaction to gener<strong>at</strong>e liquefaction hydrogendonor<br />
solvents <strong>at</strong> low temper<strong>at</strong>ures and pressures in a c<strong>at</strong>alytic<br />
solvent production stage, followed by reaction <strong>of</strong> the solvent with<br />
<strong>co</strong>al, in the absence <strong>of</strong> hydrogen, in a thermal primary liquefaction<br />
stage.<br />
Previous researchers (1,2) have used mixtures <strong>of</strong> carbon monoxide and<br />
steam to <strong>co</strong>nverg <strong>co</strong>al .in single-stage processes oper<strong>at</strong>ed <strong>at</strong> high<br />
temper<strong>at</strong>ures (380 to 475 C) and pressures (to 5000 psi). Although<br />
pilot tests with high-reactivity, low-rank <strong>co</strong>als achieved moder<strong>at</strong>e<br />
<strong>co</strong>nversions to benzene-soluble products, yields <strong>of</strong> distill<strong>at</strong>e <strong>oil</strong>s<br />
were low (3).<br />
In an initial portion <strong>of</strong> this study it was proposed (4) th<strong>at</strong> a<br />
significant improvement in <strong>co</strong>al liquefaction using CO/H 0 mixtures<br />
may be realized by separ<strong>at</strong>ing, or staging, the &GS-solvent<br />
production and <strong>co</strong>al liquefaction reactions, allowing each to be<br />
performed <strong>at</strong> an optimum temper<strong>at</strong>ure. Results <strong>of</strong> thermodynamic<br />
calcul<strong>at</strong>ions and greliminary experiments proved th<strong>at</strong> use <strong>of</strong> low<br />
temper<strong>at</strong>ures (
M<strong>at</strong>erials<br />
Feeds to the WGS-solvent production reactor <strong>co</strong>nsisted <strong>of</strong> carbon<br />
monoxide, deionized w<strong>at</strong>er and a nearly s<strong>at</strong>ur<strong>at</strong>ed mesitylene solution<br />
<strong>of</strong> polynuclear arom<strong>at</strong>ic hydrocarbons (weight basis PAHIS: 11.6%<br />
phenanthrene, 12.0% pyrene and 16.7% fluoranthene). Mesitylene was<br />
chosen as the solvent for the gAH's because <strong>of</strong> its rel<strong>at</strong>ively low<br />
vapor pressure (75 psi <strong>at</strong> 240 C), its ability to dissolve large<br />
amounts <strong>of</strong> PAHIS <strong>at</strong> room temper<strong>at</strong>ure, and its stability under high<br />
temper<strong>at</strong>ure <strong>co</strong>al liquefaction <strong>co</strong>nditions. Extrud<strong>at</strong>es (0.8 mm<br />
diameter by 4 m length) <strong>of</strong> Shell 324M, a 2.8 wt. % Ni, 12.4 wt. %<br />
Mo on alumina c<strong>at</strong>alyst, were used in the WGS-solvent production<br />
reactor. Prior to use, the c<strong>at</strong>alyst was presukfided, in-situ for<br />
six hours, with 10 mole % H2S in H2 <strong>at</strong> 385 C and <strong>at</strong>mospheric<br />
pressure.<br />
<strong>Liquefaction</strong> reactions were performed with a bituminous <strong>co</strong>al,<br />
Illinois #6 (Burning Star Mine--proxim<strong>at</strong>e analysis: 3.78 moisture,<br />
9.4% ash, 34.5% vol<strong>at</strong>ile and 52.4% fixed carbon: dry basis ultim<strong>at</strong>e<br />
analysis: 72.5% C, 4.7% H, 1.0% N, 0.1% C1, 2.9% S, 9.8% ash, and<br />
9.0% 0 by difference; mineral m<strong>at</strong>ter <strong>co</strong>ntent: 13.7%).<br />
Appar<strong>at</strong>us and Procedure<br />
WGS-solvent production was performed in a <strong>co</strong>ncurrent flow trickle-<br />
bed reactor <strong>co</strong>nsisting <strong>of</strong> six 1.0 cm ID by 15 cm long c<strong>at</strong>alyst-<br />
filled stainless steel tubes <strong>co</strong>nnected in series. Each tube was<br />
filled with 10.5 g <strong>of</strong> c<strong>at</strong>alyst. The reactor was <strong>co</strong>ntained in a<br />
forced-air <strong>co</strong>nvection oven thermost<strong>at</strong>ted to 51. Oo C. Reactor<br />
pressure was <strong>co</strong>ntrolled with a precision back-pressure regul<strong>at</strong>or and<br />
gas and liquid products were sampled subsequent to pressure letdown.<br />
After pressuriging to 508 psig with CO, the reactor temper<strong>at</strong>ure was<br />
ramped to 240 C <strong>at</strong> 10 C/min and w<strong>at</strong>er flow was initi<strong>at</strong>ed. Upon<br />
detection <strong>of</strong> <strong>co</strong>nversion <strong>of</strong> CO/H 0 to CO /H , PAH solution flow was<br />
started. Carbon monoxide, w<strong>at</strong>er ind the hH2solution were delivered<br />
to the reactor <strong>at</strong> weight hourly space velocities <strong>of</strong> 0.124, 0.079,<br />
and 0.48 g-feed/hr/g-c<strong>at</strong>alyst, respectively. For the WGS reaction,<br />
the amount <strong>of</strong> w<strong>at</strong>er delivered was one percent in excess <strong>of</strong> th<strong>at</strong><br />
required by stoichiometry to ensure th<strong>at</strong> <strong>co</strong>nversion was limited only<br />
by thermodynamic equilibrium. It was estim<strong>at</strong>ed from the reactor<br />
void volume and fluid flow r<strong>at</strong>es th<strong>at</strong> the residence time <strong>of</strong> the gas<br />
was two minutes and th<strong>at</strong> <strong>of</strong> the liquid phase was approxim<strong>at</strong>ely sixty<br />
minutes.<br />
Prior to use for the liquefaction reactions, the solvent produced by<br />
the flow reactor was <strong>co</strong>ncentr<strong>at</strong>ed by nearly a factor <strong>of</strong> two by<br />
evapor<strong>at</strong>ion <strong>of</strong> mesitylene under vacuum. This higher <strong>co</strong>ncentr<strong>at</strong>ion,<br />
which allowed the use <strong>of</strong> lower solvent to <strong>co</strong>al r<strong>at</strong>ios for the<br />
liquefaction reactions, <strong>co</strong>uld be achieved because <strong>of</strong> the increased<br />
solubility <strong>of</strong> the hydroarom<strong>at</strong>ics formed in the flow reactor.<br />
<strong>co</strong>al liquefaction reactions were gerformed in b<strong>at</strong>ch micrqautoclaves<br />
with slurry capacities <strong>of</strong> 8 cm and gas volumes 35 cm (5). Four<br />
reactors <strong>co</strong>uld be oper<strong>at</strong>ed simultaneously. After the reactors were<br />
charged with <strong>co</strong>al and solvent, they were gressurized to 450 psig<br />
with nitrogen. They were then he<strong>at</strong>ed to 445 C for 36 min (time <strong>at</strong><br />
temper<strong>at</strong>ure) in a fluidized sand b<strong>at</strong>h while being agit<strong>at</strong>ed with a<br />
wrist-action shaker <strong>at</strong> 200 cycles/min. Following the he<strong>at</strong>ing<br />
315
period, the reaction vessels were quenched in w<strong>at</strong>er, the final<br />
temper<strong>at</strong>ures and pressures were re<strong>co</strong>rded, a gas sample was taken,<br />
and the product slurry was quantit<strong>at</strong>ively removed for analysis. All<br />
experimental variables for both the flow and b<strong>at</strong>ch reactors were<br />
monitored and re<strong>co</strong>rded with a <strong>co</strong>mputer-<strong>co</strong>ntrolled d<strong>at</strong>a acquisition<br />
system.<br />
Four <strong>co</strong>al liquefaction reactions were performed. To test the impact<br />
<strong>of</strong> amount <strong>of</strong> don<strong>at</strong>able solvent hydrogen on <strong>co</strong>al <strong>co</strong>nversion, three<br />
reactions were performed with WGS-produced solvent to <strong>co</strong>al r<strong>at</strong>ios <strong>of</strong><br />
2:1, 3:1, and 4:l. A <strong>co</strong>ntrol experiment, without don<strong>at</strong>able<br />
hydrogen, was performed with a portion <strong>of</strong> the flow reactor PAH feed<br />
solution, which <strong>co</strong>ntained no hydroarom<strong>at</strong>ics. The <strong>co</strong>ntrol experiment<br />
had a l*solventl* to <strong>co</strong>al r<strong>at</strong>io <strong>of</strong> 3:l.<br />
Product Analvses<br />
On-line analyses for the partial pressures <strong>of</strong> CO and CO in the gas<br />
stream from the flow reactor were performed with a Hewfett-Packard<br />
5710A gas chrom<strong>at</strong>ograph. Prior to analysis, residual w<strong>at</strong>er vapor<br />
was elimin<strong>at</strong>ed from the gas sample with a <strong>co</strong>ld trap. The partial<br />
pressure <strong>of</strong> hydrogen was obtained by the difference between the sum<br />
<strong>of</strong> the CO and CO pressures and the sample pressure. Gas samples<br />
from the liquefac8ion reactions were analyzed for N , H , CO, CO ,<br />
and C -C hydrocarbons with a Carle series 500 gal chlom<strong>at</strong>ogra6h<br />
with a'hy8rogen transfer system.<br />
The amounts <strong>of</strong> PAHIS and hydroarom<strong>at</strong>ics in the flow reactor feed and<br />
liquid product samples were determined with a Hewlett-Packard 5890<br />
capillary <strong>co</strong>lumn-equipped gas liquid chrom<strong>at</strong>ograph. Coupled gas<br />
chrom<strong>at</strong>ography/mass spectrometry techniques were used to identify<br />
the order <strong>of</strong> elution <strong>of</strong> the PAHIS and hydrogen<strong>at</strong>ed PAH's.<br />
Conversion <strong>of</strong> <strong>co</strong>al to products was quantified by tetrahydr<strong>of</strong>uran<br />
(THF) and n-heptane (C ) solubility. Dry, mineral m<strong>at</strong>ter free<br />
(dmmf) basis <strong>co</strong>nversions Zere calcul<strong>at</strong>ed from the difference between<br />
the weight <strong>of</strong> organic <strong>co</strong>al and the insoluble organic m<strong>at</strong>ter<br />
resulting from THF or C extraction <strong>of</strong> the product. In addition,<br />
the C soluble m<strong>at</strong>erials , 'which <strong>co</strong>ntained the post-reaction solvent<br />
<strong>co</strong>mponhs, were examined by capillary <strong>co</strong>lumn chrom<strong>at</strong>ography to<br />
determine the extent <strong>of</strong> dehydrogen<strong>at</strong>ion <strong>of</strong> solvent hydroarom<strong>at</strong>ics.<br />
RESULTS AND DISCUSSION<br />
WGS Solvent Hvdroaen<strong>at</strong>ion<br />
The performance <strong>of</strong> the WGS-solvent production reactor can be<br />
evalu<strong>at</strong>ed in terms <strong>of</strong> <strong>co</strong>nversion <strong>of</strong> CO/H20 to C02/H2, and the extent<br />
<strong>of</strong> hydrogen<strong>at</strong>ion <strong>of</strong> the PAHIS.<br />
From the gaseous product analyses, the <strong>co</strong>nversion <strong>of</strong> CO/H 0 to<br />
CO /H was observed to be 97%. This is significantly gre<strong>at</strong>er'than<br />
thg vhue <strong>of</strong> 92% calcul<strong>at</strong>ed from the initial partial pressutjes <strong>of</strong> CO<br />
and steam and the pressure equilibrium <strong>co</strong>nstant ( 6) for 240 C. The<br />
observed larger <strong>co</strong>nversion results from removal <strong>of</strong> hydrogen due to<br />
hydrogen<strong>at</strong>ion <strong>of</strong> the PAHIS, which causes an additional shift to<br />
316
products. Thus, <strong>co</strong>upling the WGS and solvent hydrogen<strong>at</strong>ion<br />
reactions promotes efficiency for the WGS reaction.<br />
The extent <strong>of</strong> hydrogen<strong>at</strong>ion <strong>of</strong> the PAH's can be seen in Figure 1,<br />
which shows a <strong>co</strong>mparison <strong>of</strong> the chrom<strong>at</strong>ogram <strong>of</strong> the feed solution to<br />
th<strong>at</strong> <strong>of</strong> the product. Analysis <strong>of</strong> the product solution showed th<strong>at</strong><br />
31% <strong>of</strong> the phenanthrene, 49% <strong>of</strong> the pyrene and 92% <strong>of</strong> the<br />
fluoranthene were <strong>co</strong>nverted to hydroarom<strong>at</strong>ics. From the amount and,<br />
distribution <strong>of</strong> the hydroarom<strong>at</strong>ics and the extent <strong>of</strong> the WGS<br />
reaction, it was calcul<strong>at</strong>ed th<strong>at</strong> 30% <strong>of</strong> the hydrogen gener<strong>at</strong>ed was<br />
used to produce hydroarom<strong>at</strong>ics. The liquid product was found to<br />
<strong>co</strong>ntain 0.52 wt. 8 don<strong>at</strong>able hydroarom<strong>at</strong>ic hydrogen. The solvent<br />
for the <strong>co</strong>al liquefaction reactions, <strong>co</strong>ncentr<strong>at</strong>ed by removal <strong>of</strong><br />
mesitylene from the flow reactor product, <strong>co</strong>ntained 0.87 wt. %<br />
don<strong>at</strong>able hydrogen, a high value by current process standards.<br />
It is notable th<strong>at</strong> almost <strong>co</strong>mplete <strong>co</strong>nversion <strong>of</strong> fluoranthene to<br />
hydr<strong>of</strong>luoranthenes (primarily tetrahydr<strong>of</strong>luoranthene, which<br />
ac<strong>co</strong>unted for half <strong>of</strong> the don<strong>at</strong>able hydrogen) was achieved, while<br />
only half <strong>of</strong> the pyrene and a third <strong>of</strong> the phenanthrene were<br />
hydrogen<strong>at</strong>ed. For pyrene (Py), the limit<strong>at</strong>ion for <strong>co</strong>nversion to<br />
dihydropyrene (H Py) is a thermodynamic one. The equilibrium r<strong>at</strong>io<br />
<strong>of</strong> [H Py]/[Py] lay be calcul<strong>at</strong>ed from the reactor outlet hydrogen<br />
partia.f pressure (155 psia) and the pressure equilibrium <strong>co</strong>nstant<br />
(7) <strong>at</strong> 240 c, 0.0042/psia. The calcul<strong>at</strong>ed value <strong>of</strong> 0.65 is in<br />
agreement with the observed value <strong>of</strong> 0.66, indic<strong>at</strong>ing th<strong>at</strong> the<br />
<strong>co</strong>ncentr<strong>at</strong>ion <strong>of</strong> dihydropyrene was limited by thermodynamics, r<strong>at</strong>her<br />
than kinetics. The production <strong>of</strong> hydrophenanthrenes may also be<br />
thermodynamically limited, though no thermodynamic d<strong>at</strong>a are<br />
available for <strong>co</strong>mparison. Although the WGS-solvent production<br />
reactor yielded high <strong>co</strong>ncentr<strong>at</strong>ions <strong>of</strong> hydroarom<strong>at</strong>ics, previously<br />
reported work (7) indic<strong>at</strong>es th<strong>at</strong> even better performance can be<br />
achieved with a more active c<strong>at</strong>alyst <strong>at</strong> lower temper<strong>at</strong>ures, where<br />
form<strong>at</strong>ion <strong>of</strong> hydroarom<strong>at</strong>ics is favored.<br />
Coal Liauefaction<br />
The effectiveness <strong>of</strong> the <strong>co</strong>al liquefaction reactions performed<br />
without gas phase hydrogen can be judged by the <strong>co</strong>nversion <strong>of</strong> the<br />
. <strong>co</strong>al to THF and C soluble products, and to C -C hydrocarbons: by<br />
the amount <strong>of</strong> hyarogen transferred from h4dr8arom<strong>at</strong>ic hydrogen<br />
donors to the <strong>co</strong>al; and by the percentage <strong>of</strong> hydrogen lost from the<br />
solvent to the gas phase.<br />
Table 1 presents a summary <strong>of</strong> the results <strong>of</strong> the liquefaction<br />
experiments. As can be seen, the <strong>co</strong>nversion <strong>of</strong> <strong>co</strong>al was dependent<br />
on solvent hydrogen availability. For the <strong>co</strong>ntrol experiment (No.<br />
l), <strong>co</strong>ntaining no don<strong>at</strong>able hydrogen, the THF and C <strong>co</strong>nversions<br />
were very low: 30% and 17%, respectively. Howevzr, all the<br />
experiments with WGS-produced solvent, <strong>co</strong>ntaining hydroarom<strong>at</strong>ics,<br />
yielded much higher <strong>co</strong>nversions, which increased with increasing<br />
solvent to <strong>co</strong>al r<strong>at</strong>io. The 4:l solvent to <strong>co</strong>al experiment (No. 4),<br />
resulted in the highest THF and C <strong>co</strong>nversions, 98% and 482,<br />
respectively. The C -C hydr~carbon~gas make for the experiments<br />
with the WGS-produced QolSent (Nos. 2-4) were low, nominally 3%.<br />
317
The f<strong>at</strong>e <strong>of</strong> the don<strong>at</strong>able hydroarom<strong>at</strong>ic hydrogen in the solvent was<br />
determined from the amounts in the solvent before reaction with <strong>co</strong>al<br />
H , th<strong>at</strong> remaining in the solvent after reaction with <strong>co</strong>al H , and<br />
th<strong>at</strong> The percentage don<strong>at</strong>ed eo the<br />
<strong>co</strong>al H, can be calcul<strong>at</strong>ed by differsnce:<br />
transfered to the gas phase H .<br />
From the values for H in Table 2 it can be seen th<strong>at</strong> the<br />
utiliz<strong>at</strong>ion <strong>of</strong> hydrogen gas efficient, as only 10% <strong>of</strong> the don<strong>at</strong>able<br />
hydrogen was lost to the gas phase, the balance being don<strong>at</strong>ed to the<br />
<strong>co</strong>al or remaining with the solvent. It is also noted from the<br />
values <strong>of</strong> H th<strong>at</strong> nearly all o'f the don<strong>at</strong>able hydrogen was depleted<br />
from the s8lvent. In fact, experiment No. 2 with a 2:l solvent to<br />
<strong>co</strong>al r<strong>at</strong>io was cfearly hydrogen starved, resulting in the lowest THF<br />
and C <strong>co</strong>nversions for the experiments with WGS-produced solvent.<br />
In <strong>co</strong>hrast, experiment No. 4, with a solvent to <strong>co</strong>al r<strong>at</strong>io <strong>of</strong> 4:l<br />
had sufficient don<strong>at</strong>able hydrogen to achieve high <strong>co</strong>nversions, as<br />
evidenced by the 20% don<strong>at</strong>able hydrogen remaining after <strong>co</strong>mpletion<br />
<strong>of</strong> the reaction.<br />
PROCESS IMPLICATIONS<br />
The results <strong>of</strong> the experiments presented in this paper clearly<br />
demonstr<strong>at</strong>e th<strong>at</strong> <strong>co</strong>al can be effectively liquified without the use<br />
<strong>of</strong> high-pressure purified hydrogen feed gas. This suggests th<strong>at</strong><br />
substantial e<strong>co</strong>nomic improvements in direct <strong>co</strong>al liquefaction can be<br />
achieved. Figure 2 shows a schem<strong>at</strong>ic flow diagram for a two-stage<br />
liquefaction process proposed on the basis <strong>of</strong> these results.<br />
Notable differences between this and current two-stage processes<br />
are: 1) elimin<strong>at</strong>ion <strong>of</strong> high-pressure purified hydrogen for solvent<br />
production; 2) use <strong>of</strong> low temper<strong>at</strong>ure in the solvent production<br />
reactor; 3) elimin<strong>at</strong>ion <strong>of</strong> gas-phase hydrogen and high pressures in<br />
the thermal liquefaction reactor; and 4) selective recycle <strong>of</strong><br />
solvent <strong>co</strong>mponents (primarily PAHIS). Use <strong>of</strong> this process would<br />
elimin<strong>at</strong>e the requirements for a separ<strong>at</strong>e WGS reactor and gas<br />
separ<strong>at</strong>ion units for hydrogen production, and high pressure<br />
equipment for solvent production and liquefaction reactors. Because<br />
these units ac<strong>co</strong>unt for approxim<strong>at</strong>ely.ha1f <strong>of</strong> the estim<strong>at</strong>ed $1.5<br />
billion capital investment <strong>of</strong> a 50,000 barrel/day plant, this<br />
process would result in substantial savings in capital <strong>co</strong>sts.<br />
Oper<strong>at</strong>ing <strong>co</strong>sts such as those for <strong>co</strong>mpression <strong>of</strong> gases would also be<br />
significantly lower.<br />
REFERENCES<br />
1. F. Fisher, and H. Schrader, Brennst<strong>of</strong>f-Chem. 2, 257 (1921).<br />
2. H. R. Appell, I. Wender, and R. D. Miller, Chem. Ind. 47,<br />
1703 (1969).<br />
3. P. Nowacki, Coal Limefaction Processes, Noyes D<strong>at</strong>a Corp.,<br />
Park Ridge, N.J. (1979)<br />
4. H. P. Stephens, Proceedinas <strong>of</strong> the 1985 Intern<strong>at</strong>ional<br />
Conference on Coal Science, 327 (1985).<br />
318
5. R. J. Xottenstette, Sandia N<strong>at</strong>ional Labor<strong>at</strong>ories ReDort,<br />
SAND82-2495, 6 (1983).<br />
6. H. E. Benson, Chemistry <strong>of</strong> Coal Utiliz<strong>at</strong>ion. 2nd SuvDl.<br />
vol.. Chap. 25, John Wiley & Sons, New York (1981).<br />
7. H. P. Stephens and R. J. Kottenstette, Am. Chern. SOC. Fuel<br />
Div. Preprints, 30, 345 (1985).<br />
ACKNOWLEDGEMENT<br />
This work supported by the U.S. Department <strong>of</strong> Energy <strong>at</strong> Sandia<br />
N<strong>at</strong>ional Labor<strong>at</strong>ories under Contract DE-AC04-76DP00789.<br />
Figure 1. Comporison <strong>of</strong> high resolution gas<br />
liquid chrom<strong>at</strong>ograms <strong>of</strong> the flow reactor feed<br />
and product solutions.<br />
Flow Reactor Feed<br />
Phenanthrene (Ph) Fluoranthene (FI) Pyrene (Py)<br />
Flow Reactor Product<br />
Ph H , FI<br />
319
Figure 2. Schem<strong>at</strong>ic flow diagram for a two-stage<br />
liquefaction process via WGS-solvent production.<br />
TABLE 1<br />
Results <strong>of</strong> Coal <strong>Liquefaction</strong> Experiments a<br />
Exp. Solvent:<br />
Conversions<br />
(% dmmf basis)<br />
No. Coal THF C, C,- C,<br />
-----<br />
l b 3:l 30 17 1.6<br />
2c 2:l 91 21 2.9<br />
3' 3:l 97 42 3.1<br />
4' 4:l 98 48 3.1<br />
a)<br />
b)<br />
C) Performed with WGS-produced solvent.<br />
Products<br />
Solvent Hydrogen Balance<br />
(X don<strong>at</strong>able hydrogen)<br />
To Coal To Gas Remaining<br />
--<br />
90 9 1<br />
04 11 5<br />
69 11 20<br />
Reaction <strong>co</strong>nditions for all experiments: 445 C, 36 min,<br />
450 psig <strong>co</strong>ld chorge nitrogen. No gas-phase hydrogen used.<br />
Performed with flow reactor feed solution. PAH's only;<br />
no don<strong>at</strong>able solvent hydrogen.
ENHANCED COAL LIQUEFACTION WITH STEAM PRETREATMENT<br />
P.R. Bienkowski, R. Narayan, R.A. Greenkorn and K.C. Chm'<br />
School <strong>of</strong> Chemical Engineering<br />
Purdue University<br />
West Lafayette, Indiana 47907<br />
SUMMARY<br />
A two step process for the liquefaction <strong>of</strong> <strong>co</strong>al, in a semi-Eow micro reactor, was investig<strong>at</strong>ed. The<br />
process <strong>co</strong>nsisted <strong>of</strong> pretre<strong>at</strong>ing <strong>co</strong>al with low temper<strong>at</strong>ure steam, followed by tre<strong>at</strong>ment with supercriti-<br />
cal steam. The maximum observed <strong>co</strong>nversion <strong>of</strong> a Wyodak subbituminous <strong>co</strong>al, using this two step pro-<br />
cess, was 40 wt% on a moisture and ash free basis (MAF). The 240 ' c pretre<strong>at</strong>ment step resulted io a<br />
32% increase over the <strong>co</strong>nversion observed with just a 400 c tre<strong>at</strong>ment. The <strong>co</strong>al liquid obtained has<br />
a number average molecular weight <strong>of</strong> 325 and a mass average molecular weight <strong>of</strong> 373, with a narrow<br />
molecular weight distribution. The hydrogen and oxygen <strong>co</strong>ntent <strong>of</strong> the extract is increased, a significant<br />
amount <strong>of</strong> the oxygen is present as dihydroxyl arom<strong>at</strong>ics. A highly <strong>co</strong>ndensed residue <strong>of</strong> lower hydrogen<br />
and oxygen <strong>co</strong>ntent is obtained which can be <strong>of</strong> value as a solid fuel.<br />
INTRODUCTION<br />
Recent investig<strong>at</strong>ions have led to the observ<strong>at</strong>ion (1) th<strong>at</strong> in its n<strong>at</strong>ive unwe<strong>at</strong>hered st<strong>at</strong>e <strong>co</strong>al is a<br />
reactive m<strong>at</strong>erial, far from being the inert solid th<strong>at</strong> it is <strong>co</strong>mmonly regarded. Extensive hydrogen bonds<br />
<strong>co</strong>nnect the poly nuclear arom<strong>at</strong>ic cluster to form a semi-permanent macro molecular structure. The<br />
structure is particularly fragile in subbituminous <strong>co</strong>als, and may le subject to rupture with mild tre<strong>at</strong><br />
ment to dissoci<strong>at</strong>e the hydrogen bonds. GraU and Brandes (2) found th<strong>at</strong> carbon <strong>co</strong>nversion to liquids in<br />
pyroiysls <strong>at</strong> 940 c was raised from 23% to over 50% if the <strong>co</strong>al was exposed to steam for times less<br />
than 30 minutes <strong>at</strong> temper<strong>at</strong>ures between about 320 and 360 O c. Both pretre<strong>at</strong>ment and pyrolysis were<br />
<strong>co</strong>nducted in 50 <strong>at</strong>m <strong>of</strong> steam. This result suggests th<strong>at</strong> <strong>co</strong>al is partially depolymerized by the steam<br />
pretre<strong>at</strong>ment, perhaps by the removal <strong>of</strong> oxygen linkages. If this is indeed the case, improved yields<br />
and/or lighter liquids should result if the pretre<strong>at</strong>ed <strong>co</strong>al is liquefied instead <strong>of</strong> pyrolyzed.<br />
321
EXPERIMENTAL<br />
Wyodak <strong>co</strong>al was pretre<strong>at</strong>ed and tre<strong>at</strong>ed with steam in a semi-Row micro reactor <strong>at</strong> <strong>co</strong>ntrolled <strong>co</strong>ndi-<br />
tions to exclude oxygen. The appar<strong>at</strong>us is depicted in Figure 1. The main <strong>co</strong>mponents are a Milton Roy<br />
metering pump (29 to 290cm3/hr) which provides a <strong>co</strong>ntinuous and <strong>co</strong>nstant Row <strong>of</strong> distilled deoxy-<br />
gen<strong>at</strong>ed w<strong>at</strong>er to the re<strong>at</strong>or, a Tecam fluidized sand b<strong>at</strong>h (model SBS-4) <strong>co</strong>ntrolled with a Leeds and<br />
Northrup <strong>co</strong>ntroller (Electromax Ill), a Heli<strong>co</strong>id pressure gauge (0 to 3000 PSIG), an autoclave micro<br />
metering valve and a Picro reactor equipped with a she<strong>at</strong>hed thermo<strong>co</strong>uple (see Figure 2).<br />
The feed m<strong>at</strong>erial was an unwe<strong>at</strong>hered subbituminous Wyodak <strong>co</strong>al provided by EPRI in a w<strong>at</strong>er<br />
slurry kept in a sealed air-tight barrel (#4171). Table 1 shows the elemental analysis <strong>of</strong> samples taken<br />
from this barrel. Samples A and B were taken from the top and middle <strong>of</strong> the barrel and were suction<br />
dried for use. These were the main feed m<strong>at</strong>erial. About a 50 gm sampled <strong>of</strong> this m<strong>at</strong>erial was removed<br />
from the barrel and vacuum dried; a sample <strong>of</strong> this m<strong>at</strong>erial is repoted as sample C. A tew reactions<br />
were carried out with this m<strong>at</strong>erial.<br />
The suction dried <strong>co</strong>al was prepared in a Buchner filter, by applying suction to it tor about one hour.<br />
The top portion <strong>of</strong> the m<strong>at</strong>erial was removed from the filter and thoroughly mixed to insure a homogeneous<br />
sample for reaction. The moisture <strong>co</strong>ntent <strong>of</strong> this m<strong>at</strong>erial (z 30 wt %) was determined by weighing<br />
a sample before and after further drying in a vacuum oven <strong>at</strong> 45 O c for 6 hours. Duplic<strong>at</strong>e samples<br />
were used for moisture determin<strong>at</strong>ions.<br />
Suction dried Wyodak <strong>co</strong>al is paste-like, and is messy to bandle. A free-Rowing <strong>co</strong>al powder was<br />
obtained for reaction experiments by drying the <strong>co</strong>al in vacuum under mild he<strong>at</strong>ing. The vacuum dried<br />
<strong>co</strong>al wan prepared by suction drying in a Buchner funnel over nitrogen; the m<strong>at</strong>erial was then transferred<br />
in a jar 611ed with nitrogen, to a vacuum oven and dried for 72 hours <strong>at</strong> 45 O c. The experimental reac-<br />
tion procedure for vacuum dried <strong>co</strong>al was similar to suction dried <strong>co</strong>al except th<strong>at</strong> the separ<strong>at</strong>e moisture<br />
determin<strong>at</strong>ion <strong>of</strong> the dried <strong>co</strong>al was no longer necessary.<br />
To prepare for reaction the oven dried micro reactor (see Figure 2) was weighed and then filled with<br />
about 4 gm ot wet suction dried <strong>co</strong>al or 3 gm <strong>of</strong> vacuum dried <strong>co</strong>al and reweighed for an accur<strong>at</strong>e deter-<br />
min<strong>at</strong>ion <strong>of</strong> the quantity <strong>of</strong> m<strong>at</strong>erial charged to the reactor. The charged reactor was fitted into the<br />
appar<strong>at</strong>ns upon <strong>co</strong>nnecting tubings and fittings. The system was purged <strong>of</strong> air by Rushing with nitrogen.<br />
Enough w<strong>at</strong>er was pumped from the buret into the reactor to raise the pressure to SO pia, about 10 to<br />
nCm3 being required. The reactor was lowered into the sand b<strong>at</strong>h, the pressure adjusted to 750 psia<br />
with the aid <strong>of</strong> the micro metering valve, as w<strong>at</strong>er was pumped into the system <strong>at</strong> 0.4cm3/min. The<br />
steam gener<strong>at</strong>ed in the he<strong>at</strong>ing <strong>co</strong>il passed through the reactor, was <strong>co</strong>ndensed, and <strong>co</strong>llected in an Erlen-<br />
meyer Bask.<br />
At the termin<strong>at</strong>ion <strong>of</strong> an experiment the reactor WBB removed trom the aand b<strong>at</strong>h, and placed in a<br />
vacuum oven over night <strong>at</strong> 45 O c to remove any w<strong>at</strong>er. The reactor was then wekhed, the <strong>co</strong>ntents<br />
were removed and placed in a predried and weighed thimble. The reactor was then reweighed, the<br />
322
change in weight being <strong>co</strong>mpared to the weight gain <strong>of</strong> the thimble.<br />
The <strong>co</strong>al in the thimble was extracted with toluene for 4 to 6 hours in a Soxhlet appar<strong>at</strong>us. The<br />
thimble was dried over night in a vacuum oven and reweighed to detemined the amount <strong>of</strong> <strong>co</strong>al<br />
extracted. On selected runs the effluent <strong>co</strong>llected in the Erlenmeyer Bask was extracted with chlor<strong>of</strong>orm<br />
and sent to analysis.<br />
The only time the <strong>co</strong>al sample was exposed to oxygen <strong>of</strong> the air was during suction drying <strong>at</strong> ambient<br />
temper<strong>at</strong>ure. Oxyged was carefully excluded from the reaction system. Feed w<strong>at</strong>er to the reactor was<br />
deoxygen<strong>at</strong>ed by blowing it with nitrogen. >From the time <strong>of</strong> steam pretre<strong>at</strong>ment the reactor remained<br />
tightly closed and <strong>co</strong>mpletely isol<strong>at</strong>ed from air until the reactor was <strong>co</strong>oled down after steam tre<strong>at</strong>ment.<br />
The only <strong>co</strong>al th<strong>at</strong> was he<strong>at</strong>ed before reaction was th<strong>at</strong> which was vacuum dried <strong>at</strong> 45 c. The suction<br />
dried <strong>co</strong>al was never he<strong>at</strong>ed.<br />
EXPERIMENTAL RESULTS<br />
Table 2 shows the <strong>co</strong>nversion <strong>of</strong> Wyodak <strong>co</strong>al upon pretre<strong>at</strong>ment and tre<strong>at</strong>ment with steam <strong>at</strong> 750<br />
pia. Tre<strong>at</strong>ment and pretre<strong>at</strong>ment (if used) steps each lasted 30 minutes. Conversion expresses the frac-<br />
tion <strong>of</strong> <strong>co</strong>al th<strong>at</strong> was extracted by the steam from the reactor plus the fraction th<strong>at</strong> was extracted by<br />
toluene from the Soxhlet appar<strong>at</strong>us. The toluene extraction was small, ranging from 0% to less than 3%<br />
<strong>of</strong> the total reported <strong>co</strong>nversion. Toluene extraction was in<strong>co</strong>rpor<strong>at</strong>ed in our experimental procedure in<br />
order to put our experimental results on the same basis with those <strong>of</strong> other investig<strong>at</strong>ors (3,4,5,6) who<br />
wash their <strong>co</strong>al residue with toluene.<br />
With suction dried <strong>co</strong>al, when not pretre<strong>at</strong>ed, tre<strong>at</strong>ment with steam <strong>at</strong> 200 c gives a practically<br />
zero (2.2%) <strong>co</strong>nversion. Raising the tre<strong>at</strong>ment temper<strong>at</strong>ure to 400 * c raisea the <strong>co</strong>nversion to 30.5%.<br />
Pretre<strong>at</strong>ment with steam <strong>at</strong> 200 c further raises the <strong>co</strong>nversion to 38.5%. Raising the pretre<strong>at</strong>ment<br />
temper<strong>at</strong>ure to 240 ' c raises the <strong>co</strong>nversion to 40.3%. This is the highest <strong>co</strong>nversion observed in this<br />
work, for, upon raking the pretre<strong>at</strong>ment temper<strong>at</strong>ure to 320 e c <strong>co</strong>nersion is lowered to 33.8%. Raising<br />
the tre<strong>at</strong>ment temper<strong>at</strong>ure to 430 c further lowers the Conversion to 34.2%.<br />
The <strong>co</strong>nversion <strong>of</strong> vacuum dried <strong>co</strong>al is reported in the se<strong>co</strong>nd part <strong>of</strong> Table 2, and is generally lower<br />
than the <strong>co</strong>rresponding results for suction dried <strong>co</strong>al. Even the mild he<strong>at</strong>ing <strong>at</strong> 45 O c during vacuum<br />
drying made the <strong>co</strong>al more refractory. Comparison <strong>of</strong> the results obtained with the two diRerent <strong>co</strong>al<br />
samples <strong>co</strong>nvinced us to stop using vacuum dried <strong>co</strong>al. All other experiments reported here used suction<br />
dried <strong>co</strong>al.<br />
The <strong>co</strong>nversions reported in Table 2 were obtained with 750 psia steam for both the pretre<strong>at</strong>ment snd<br />
tn<strong>at</strong>ment. The eUect <strong>of</strong> steam pressure <strong>at</strong> the tre<strong>at</strong>ment stage was investig<strong>at</strong>ed, the results are reported<br />
in Table 3. Holding the pretre<strong>at</strong>ment pressure <strong>co</strong>nstant <strong>at</strong> 750 psi, an increase in tre<strong>at</strong>ment preasure<br />
from 750 tO 260 psia, produced a slight reduction in the observed <strong>co</strong>nversion. Higher pressure<br />
apparently increm the r<strong>at</strong>e <strong>of</strong> retrograde reactions. This eRect more than <strong>co</strong>mpens<strong>at</strong>es for any increase<br />
323
in solvent power <strong>of</strong> the steam <strong>at</strong> a higher pressure, leading to reduced extraction.<br />
Twenty five experiments were performed in all. The <strong>co</strong>nversions in Tables 3 and 3 represent average<br />
values for experiments <strong>at</strong> the same <strong>co</strong>nditions.<br />
CHEMISTRY<br />
Extract and residue samples from two experiments, Runs 5 and 27, were analyzed by infrared spec-<br />
tros<strong>co</strong>py, NMR and'FIMS. Run 5 <strong>co</strong>nsisted <strong>of</strong> a 320 ' c pretre<strong>at</strong>ment followed by a 430 * C tre<strong>at</strong>-<br />
ment, while Run 27 was <strong>at</strong> 200 ' c and 400 ' c.<br />
Table 4 shows the elemental analyses <strong>of</strong> the extract and the residue <strong>of</strong> Run 27. The analysis <strong>of</strong> the<br />
feed <strong>co</strong>al is also shown for <strong>co</strong>mparhon. The extract has a higher H/C r<strong>at</strong>io (1.28) a8 <strong>co</strong>mpared to the<br />
feed <strong>co</strong>al (0.95), whereas the residue has a lower r<strong>at</strong>io (0.68). The O/C r<strong>at</strong>io follows a similar p<strong>at</strong>tern,<br />
being higher in the extract (0.32) than in the feed (0.28), and lower in the residue (0.13).<br />
The steam pretre<strong>at</strong>ment/extraction process produces a hydrogen-rich extract which <strong>co</strong>ntains oxy-<br />
gen<strong>at</strong>ed <strong>co</strong>mpounds and hetero<strong>at</strong>omic species <strong>of</strong> the original <strong>co</strong>al, leaving behind a more <strong>co</strong>ndensed<br />
arom<strong>at</strong>ic residue.<br />
The same trend is indic<strong>at</strong>ed lor nitrogen and sulfur, being enriched in the extract and reduced in the<br />
residue. But here because <strong>of</strong> the small.amounts, especially <strong>of</strong> nitrogen, accuracy in low.<br />
The infrared spectrum <strong>of</strong> the steam extract or Run 27 is shown in Figure 3. The IR spectrum is dom-<br />
in<strong>at</strong>ed by broad, strong -OH stretching vibr<strong>at</strong>ions in the 3400-3100 cm-' region. The presence <strong>of</strong> sharp<br />
aliph<strong>at</strong>ic -OH hands just below 3000 cm-' suggests th<strong>at</strong> the extract <strong>co</strong>ntains aliph<strong>at</strong>ic m<strong>at</strong>erial, and<br />
reinforces the observed enrichment <strong>of</strong> H and 0 in the extract.<br />
Further <strong>co</strong>nfirm<strong>at</strong>ion is provided by the 'H NMR spectrum <strong>of</strong> the steam extract from Run 5 shown in<br />
Figure 4. The r<strong>at</strong>io 01 H,,/H,,, is 1:30. 43% <strong>of</strong> the H,,, hydrogens are associ<strong>at</strong>ed with methylene,<br />
methine, or methyl groups which are not directly bonded to arom<strong>at</strong>ic nuclei. Another 20% 01 the K<strong>at</strong><br />
appearing as a group <strong>of</strong> signals in the 2-3 ppm region are associ<strong>at</strong>ed with hydroarom<strong>at</strong>ic structures or<br />
associ<strong>at</strong>ed with methyl, methylene and methine groups directly <strong>at</strong>tached to an arom<strong>at</strong>ic nucleus. The<br />
dominant sharp signal <strong>at</strong> 1.2 ppm is characteristic <strong>of</strong> long chain polymethylene groups. Thus, one <strong>of</strong> the<br />
major <strong>co</strong>nstituents <strong>of</strong> the hydrogen rich extract is aliph<strong>at</strong>ic8 present primarily as long chain<br />
polymethylenes, either as free species or <strong>at</strong>tached to arom<strong>at</strong>ic/hydroarom<strong>at</strong>ic ring systems.<br />
The NMR spectrum (Figure 5) is domin<strong>at</strong>ed by a number <strong>of</strong> well resolved lines riding on a spec-<br />
tral envelope in the 15-80 ppm region which in the normal chemical shift region for aliph<strong>at</strong>ic <strong>co</strong>mpounds,<br />
supporting the presence <strong>of</strong> signitlcant quantities <strong>of</strong> aliph<strong>at</strong>ic m<strong>at</strong>erials in the steam extract. The most<br />
intense signal is <strong>at</strong> 30.2 which is generally assigned to the internal methylene carbons<br />
(c~-c~-c~-cn-) <strong>of</strong> straight chain alkanes <strong>co</strong>rrobor<strong>at</strong>ing the presence <strong>of</strong> long chain polymethylene<br />
groups (minimum average carbon chain length; nc+?-lO) which may or may not be <strong>at</strong>tached to an<br />
324
I<br />
I<br />
arom<strong>at</strong>ic ring.<br />
The group <strong>of</strong> signals in the 1520 ppm region is probably due to the methylene carbons <strong>at</strong>tached to<br />
arom<strong>at</strong>ic rings. The presence <strong>of</strong> a broad spectral envelope in addition to the sharp alkane lines demon-<br />
str<strong>at</strong>e the extract's <strong>co</strong>mplexity. The spectral <strong>co</strong>mplexity is due to the presence <strong>of</strong> small amounts <strong>of</strong><br />
polymethylene type <strong>co</strong>mpounds. The <strong>co</strong>mplex band <strong>of</strong> carbon signals in the 120-130 ppm region is due<br />
to the arom<strong>at</strong>ic and polycyclic arom<strong>at</strong>ic species. Interestingly, a small, but distinctive signal occurs <strong>at</strong><br />
179 ppm which is whpre the carbonyl carbon <strong>of</strong> a -COOH group appears suggesting the presense <strong>of</strong> some<br />
carboxylic acids in the extract.<br />
Field ioniz<strong>at</strong>ion mass spectomety (FIMS) M a mass spectometry technique which uses a s<strong>of</strong>t ioniza-<br />
tion mode and allows most molecules to be observed as unfragmented molecular ions. The method can<br />
provide a true molecular weight pr<strong>of</strong>ile for any given <strong>co</strong>mplex mixture. Figure 6 represents the field ioni-<br />
z<strong>at</strong>ion mass spectrum <strong>of</strong> the extract. The extract has a very narrow molecular weight distribution witb<br />
number average (M,) and weight average (M,) molecular weight <strong>of</strong> 315 and 373, respectively. Since<br />
75% <strong>of</strong> the m<strong>at</strong>erial was vol<strong>at</strong>ilized in the FIMS probe the observed molecular weights are a true<br />
represent<strong>at</strong>ion <strong>of</strong> the extract and the extract is <strong>co</strong>mposed <strong>of</strong> low molecular weight <strong>co</strong>mpounds. The most<br />
prominent peaks in the spectrum appear <strong>at</strong> m/z 110, 124, and 138 and can be assigned to dihydroxyl<br />
benzene and its methyl and ethyl analogs, respectively. Suprisingly no prominent peaks due to monohy-<br />
droxyl benzene (phenol) or its C-1 or C-2 analogs are found. The oxygen<strong>at</strong>ed <strong>co</strong>mpounds present in tbe<br />
extract are best represented by the clabs <strong>of</strong> dihydroxyl benzenes and other dihydroxyl arom<strong>at</strong>ics. There<br />
are a number <strong>of</strong> other prominent peaks in the higher molecular weight range which, in all probability,<br />
arise from the polymethylenes <strong>at</strong>tached to an arom<strong>at</strong>ic ring (identified by NMR) but the FIMS analysis<br />
does not allow ready identific<strong>at</strong>ion <strong>of</strong> these <strong>co</strong>mpounds.<br />
Tbe presence <strong>of</strong> reactive <strong>co</strong>mponents like the polymethylene species, the dihydroxyl benzenes, and tbe<br />
low molecular weight pr<strong>of</strong>ile <strong>of</strong> the extract sugkests th<strong>at</strong> the <strong>co</strong>al is very reactive and not a highly <strong>co</strong>n-<br />
densed, very large molecular weight, intractable molecule. Self <strong>co</strong>ndens<strong>at</strong>ion and crosslinking reactions<br />
<strong>of</strong> the dihydroxyl arom<strong>at</strong>ics, alkyl<strong>at</strong>ion <strong>of</strong> the activ<strong>at</strong>ed arom<strong>at</strong>ic rings by the polymethylene species in<br />
the <strong>co</strong>al , are some <strong>of</strong> the retrogressive reactions th<strong>at</strong> these <strong>co</strong>als can undergo, under the severe process-<br />
ing <strong>co</strong>nditions generally employed.<br />
Pretre<strong>at</strong>ment witb steam, <strong>at</strong> lower temper<strong>at</strong>ures, allows the breaking <strong>of</strong> hydrogen bonds, loosening up<br />
the <strong>co</strong>al m<strong>at</strong>rix, and stabilizing some <strong>of</strong> the reactive <strong>co</strong>mponents in the <strong>co</strong>al. When the temper<strong>at</strong>ure<br />
increases during the supercritical extraction step, many <strong>of</strong> these reactive molecules can be steam vol<strong>at</strong>il-<br />
ized or steam extracted, escaping the loosened <strong>co</strong>al m<strong>at</strong>rix structure before undergoing retrogressive reac-<br />
tions. This explan<strong>at</strong>ion is supported since the introduction <strong>of</strong> a low temper<strong>at</strong>ure pretre<strong>at</strong>ment step<br />
before the supercritical steam extraction leads to a 32% increase in <strong>co</strong>nversion.<br />
The presence <strong>of</strong> reactive dihydroxyl benzenes in the extract is also supporting evidence. Dihydroxyl<br />
arom<strong>at</strong>ics have never been reported M occuring in <strong>co</strong>al liquids obtained under normal <strong>co</strong>al <strong>processing</strong><br />
325
<strong>co</strong>nditions generally employed. They cannot survive the severe <strong>processing</strong> <strong>co</strong>nditions. Small amounts <strong>of</strong><br />
dihydroxyl arom<strong>at</strong>ics have been obtained in Bash or fsst pyrolysis <strong>co</strong>nditions. The very rapid he<strong>at</strong>ing<br />
allows the dihydroxyl arom<strong>at</strong>ics to escape the <strong>co</strong>al m<strong>at</strong>rix before they can undergo retrogrrssive reac-<br />
tions.<br />
CONCLUSIONS<br />
The steam pretre<strong>at</strong>mentextraction process produces enhanced extractan yields. The extract has a<br />
high H/C r<strong>at</strong>io due to the presence <strong>of</strong> long chain polymethylene <strong>co</strong>mpounds which may or may not be<br />
<strong>at</strong>tached to arom<strong>at</strong>ic rins. The extract <strong>co</strong>ntains significant amounts <strong>of</strong> oxygen<strong>at</strong>ed <strong>co</strong>mpounds, some <strong>of</strong><br />
which are present as dihydroxyl arom<strong>at</strong>ics. A highly <strong>co</strong>ndensed residue (low H/C r<strong>at</strong>io) is obtained<br />
which can be <strong>at</strong>tractive a8 a solid fuel lor <strong>co</strong>mbustion.<br />
ACKNOWLEDGEMENTS<br />
This work has been supported by the Electric Power Research Institute through research project<br />
2147-9. Shu-Yen Huang performed the IR and NMR analysis and prepared the FlMS samples. We<br />
thank Linda F. Atberton for helpful discussions.<br />
LITERATURE CITED<br />
1. Atherton, L.F., Kulik, C.J. "Advanced Coal <strong>Liquefaction</strong>" paper presented <strong>at</strong> AIChE Annual Meet<br />
ing, Los Angeles, CA, November 1982.<br />
2. Coal Technology Report Vol. 2, No. 11, p.1, May 28, 1984.<br />
3. Kushaw, J.R.; Bagnell, L.J. ACS Div. Fuel Chem. 1985,30(3), 101.<br />
4. Kenhaw, J.R.; Jezko, J.; Separ<strong>at</strong>ion Science and Technolou 1982, 17(1), 151.<br />
5. Kershaw J.R.; Fuel Processing Technology 1982, 5, 241.<br />
6. Jezko, J.; Gray, D.; Kershaw, J.R.; Fuel Proces. Tech. 1982, 5, 229.<br />
326<br />
!
T,\llI.l< I<br />
ASAI,YSIS OP \\YOl)Al\: (;O.\l,<br />
s.wri.i; A 11 c<br />
\Vet Wct Vnruum<br />
Drird<br />
Carhn. 59.51 59.72 58.17<br />
Ilydrqcn 4.70 4.13 4.35<br />
OXYP 17.90 2?.44 18.9-1<br />
Nitrryrn<br />
Chlorine<br />
0.70<br />
-<br />
0.76 0.62<br />
Sulfur 1.m 1.55 3.K<br />
Ash I3.W 13.47 16.48<br />
TOTAL 90.55 103.07 101.02<br />
BTU/lb 10,814 - 10.I41<br />
An.- .I q L r mu pwfarnwd by I!uUmu Labor<strong>at</strong>ory.<br />
Sham. pi.<br />
750 38.5<br />
1500 a0<br />
?GOO 36.0<br />
32 7<br />
TABLE 4<br />
EIemeabl h dysh d ExCut, Raidn,<br />
A ~4<br />
~(RU n<br />
Wt% Extract R d u Fwd<br />
C<br />
n<br />
51.21<br />
6.77<br />
u.n 60.71<br />
3.69 4.!3<br />
0<br />
N<br />
23.42<br />
4.00<br />
ILIO<br />
1.01<br />
22.44<br />
0.70<br />
S<br />
kh<br />
4-<br />
8.04<br />
1.71<br />
1T.U<br />
2.s<br />
13.47<br />
Atomic<br />
Ralw<br />
II/C 1.28 0.08 0.05<br />
O/C 0.32 0.13 0.28
Orll.1<br />
t
00 3000 2000 le00 1600 1400 1200 IO00 800 SO0<br />
WAVENUMBER CM"<br />
rlgure 3: Infrared Spect- <strong>of</strong> Stem Eltract <strong>of</strong> Run 27<br />
From. 10.00 100.50 PPM<br />
Imte9r.I. 100.00<br />
329<br />
cn, vw &rYity<br />
k Aromolic
200<br />
150 100<br />
FFM<br />
Cn In. 4.5.6 ... I<br />
'2 TMS<br />
I<br />
50 0<br />
,,B.-.<br />
I<br />
.- - .. . . .... . ,... . . . . . . ._ . .<br />
N AV YW. 315 WT 4V W.373<br />
MASS IYIZI<br />
Flgure 6: FlllS <strong>of</strong> Stem Extract 01 Run 5<br />
330
THE EFFECT OF REACTION CONDITIONS ON SOLVENT LOSS<br />
DURING COAL LIQUEFACTION<br />
Bruce R. Utz and Sidney Friedman<br />
U.S. Department <strong>of</strong> Energy<br />
Pittsburgh Energy Technology Center<br />
P.O. Box 10940<br />
Pittsburgh, PA 15236<br />
INTRODUCTION<br />
The f<strong>at</strong>e <strong>of</strong> a <strong>co</strong>al-liquefaction recycle solvent during Integr<strong>at</strong>ed Two-Stage<br />
<strong>Liquefaction</strong> (ITSL) and other direct liquefaction processes is a major <strong>co</strong>ncern.<br />
If the solvent has an inadequ<strong>at</strong>e <strong>co</strong>ncentr<strong>at</strong>ion <strong>of</strong> hydrogen donors or other solvent<br />
<strong>co</strong>mponents th<strong>at</strong> might enhance liquefaction, then the quality <strong>of</strong> the solvent will<br />
be degraded and <strong>co</strong>nversion to liquid products will be adversely affected. Studies<br />
using the solvents phenol (l), tetrahydroquinoline (2-4), quinoline (5), and<br />
pyridine (6) have shown th<strong>at</strong> these <strong>co</strong>mpounds are partially in<strong>co</strong>rpor<strong>at</strong>ed by<br />
<strong>co</strong>valent bonds, but a rel<strong>at</strong>ively large amount is h drogen bonded to <strong>co</strong>al-derived<br />
products. In other studies using pure "C- and '*C-labeled arom<strong>at</strong>ic or hydro-<br />
arom<strong>at</strong>ic <strong>co</strong>mpounds as solvents (3,6-8), the amount <strong>of</strong> adduction was determined.<br />
Although adduction and hydrogen bonding <strong>of</strong> solvent <strong>co</strong>mponents ac<strong>co</strong>unt for solvent<br />
loss, other degrad<strong>at</strong>ive reactions also ac<strong>co</strong>unt for loss <strong>of</strong> original solvent and<br />
solvent quality. Ring <strong>co</strong>ntraction <strong>of</strong> tetralin (9), and octahydrophenan-<br />
threne (8,lO) under <strong>co</strong>al-liquefaction <strong>co</strong>nditions reduces the hydrogen-donor <strong>co</strong>n-<br />
centr<strong>at</strong>ion and therefore the solvent quality.<br />
The use <strong>of</strong> a one-<strong>co</strong>mponent solvent system can be misleading because reactions<br />
with <strong>co</strong>al may be occurring th<strong>at</strong> normally would not occur if other <strong>co</strong>mponents <strong>of</strong> a<br />
solvent were present. The objective <strong>of</strong> this study was to examine a multi<strong>co</strong>mponent<br />
synthetic solvent and determine if solvent <strong>co</strong>mponents are preferentially lost or<br />
degraded during short-<strong>co</strong>ntact-time liquefaction (SCTL) , and how solvent loss in<br />
SCTL <strong>co</strong>mpares with other liquefaction <strong>co</strong>nditions th<strong>at</strong> involve gradual he<strong>at</strong>-up.<br />
EXPERIMENTAL<br />
Experiments were performed in a microreactor assembly (11) <strong>co</strong>nsisting <strong>of</strong> a<br />
316 stainless steel 1/2-in. union tee with two end caps. Five grams <strong>of</strong> a<br />
synthetic solvent and one gram <strong>of</strong> moisture-free Western Kentucky 9/14 <strong>co</strong>al were<br />
added to the microreactor, which was assembled and pressurized to 1100 psig with<br />
Hz. The synthetic solvent <strong>co</strong>nsisted <strong>of</strong> 4% quinoline, 13% m-cresol, 20% tetralin,<br />
33% 1-methylnaphthalene, 20% phenanthrene, and 10% pyrene. Experiments were <strong>co</strong>n-<br />
ducted with one- and two-<strong>co</strong>mponent solvents and <strong>co</strong>al in order to identify unique<br />
products from each <strong>of</strong> the simple solvent systems. The two-<strong>co</strong>mponent solvent <strong>co</strong>n-<br />
sisted <strong>of</strong> 20% pyrene and 80% tetralin, or 20% phenanthrene and 80% tetralin. One-<br />
<strong>co</strong>mponent solvents <strong>of</strong> 1-methylnaphthalene, tetralin, and m-cresol were also used.<br />
The microreactor assembly was <strong>at</strong>tached to a wrist-action shaker th<strong>at</strong> moved the<br />
reactor through a small arc for adequ<strong>at</strong>e mixing <strong>of</strong> the reactor <strong>co</strong>ntents. The<br />
shaker was positioned above a vertically moving pl<strong>at</strong>form th<strong>at</strong> supported a<br />
fluidized sand b<strong>at</strong>h (11).<br />
For SCTL experiments, the b<strong>at</strong>h was he<strong>at</strong>ed to an initial temper<strong>at</strong>ure <strong>of</strong> 46OOC.<br />
When the experiment was initi<strong>at</strong>ed, the temper<strong>at</strong>ure set point was changed to 425OC<br />
and the pl<strong>at</strong>form (with b<strong>at</strong>h) was raised, immersing the microreactor. He<strong>at</strong>-up to<br />
425oC took approxim<strong>at</strong>ely 1 min. Reaction time was 3 min <strong>at</strong> 425OC. For experi-<br />
ments <strong>co</strong>nducted under traditional or severe <strong>co</strong>nditions, the reactor was immersed<br />
in the sand b<strong>at</strong>h, which was <strong>at</strong> room temper<strong>at</strong>ure, gradually he<strong>at</strong>ed (55 min) to<br />
425oC, and held <strong>at</strong> reaction temper<strong>at</strong>ure for 30 min or 6 hours. At the end <strong>of</strong> the<br />
331
eaction, the pl<strong>at</strong>form was lowered and the microreactor was <strong>co</strong>oled with a stream<br />
<strong>of</strong> room-temper<strong>at</strong>ure air. The outside <strong>of</strong> the <strong>co</strong>oled microreactor assembly was<br />
cleaned (to remove sand) with <strong>co</strong>mpressed air. Gases were vented, and the micro-<br />
reactor was disassembled.<br />
The reacted suspension was pipetted into a 50-mL volumetric flask <strong>co</strong>ntaining<br />
0.100 gm durene and 0.100 gm fluorenone, which were the internal standards used<br />
for quantit<strong>at</strong>ive capillary gas chrom<strong>at</strong>ography, The remaining residue and<br />
synthetic solvent in the microreactor were removed with tetrahydr<strong>of</strong>uran (THF) and<br />
added to the flask. Aliquots were then analyzed using a 50-meter highly cross-<br />
linked phenylmethylsili<strong>co</strong>ne capillary <strong>co</strong>lumn. Flame ioniz<strong>at</strong>ion was used to detect<br />
individual <strong>co</strong>mponents <strong>of</strong> the tre<strong>at</strong>ed synthetic solvent, although in certain<br />
instances a mass-selective detector was also used to assist in the identific<strong>at</strong>ion<br />
<strong>of</strong> products.<br />
Solvent loss <strong>of</strong> each <strong>co</strong>mponent was determined by calcul<strong>at</strong>ing the difference<br />
between the original amount <strong>of</strong> the <strong>co</strong>mponent and the re<strong>co</strong>vered amount <strong>of</strong> the <strong>co</strong>m-<br />
ponent and its reaction by-products. Solvent th<strong>at</strong> was lost and unac<strong>co</strong>unted for<br />
represented adducted or polymerized m<strong>at</strong>erial. A l l percentages will be discussed<br />
on an absolute basis; therefore, the loss <strong>of</strong> 10% tetralin would represent 10% <strong>of</strong><br />
the synthetic solvent and not 10% <strong>of</strong> the 20% tetralin present in the synthetic<br />
solvent. The solvent losses would be five times larger if they were based on the<br />
weight <strong>of</strong> the <strong>co</strong>al sample, since the so1vent:<strong>co</strong>al r<strong>at</strong>io was 5:1, i.e., loss <strong>of</strong> 3%<br />
<strong>of</strong> the solvent by adduction would represent a 15% addition to the weight <strong>of</strong> the<br />
<strong>co</strong>al.<br />
RESULTS AND DISCUSSION<br />
The effect <strong>of</strong> reaction <strong>co</strong>nditions on solvent loss, and specifically on pref-<br />
erential loss <strong>of</strong> <strong>co</strong>mponents <strong>of</strong> the solvent, was examined <strong>at</strong> three sets <strong>of</strong> reaction<br />
<strong>co</strong>nditions. Experiments were <strong>co</strong>nducted under SCTL <strong>co</strong>nditions (rapid he<strong>at</strong>-up, 3<br />
min <strong>at</strong> 425OC). traditional <strong>co</strong>nditions (gradual he<strong>at</strong>-up, 30 min <strong>at</strong> 425OC), and<br />
severe <strong>co</strong>nditions (gradual he<strong>at</strong>-up, 6 hours <strong>at</strong> 425OC). The advantages in using<br />
this synthetic solvent are th<strong>at</strong> all <strong>co</strong>mponents <strong>of</strong> the solvent are known, its<br />
elemental <strong>co</strong>mposition is similar to a <strong>co</strong>al-derived recycle solvent, and the multi-<br />
<strong>co</strong>mponent solvent simul<strong>at</strong>es a recycle solvent better than a one-<strong>co</strong>mponent solvent<br />
does. The use <strong>of</strong> a synthetic solvent also allows the study <strong>of</strong> individual <strong>co</strong>m-<br />
ponents in a more realistic environment.<br />
The objective was to examine solvent re<strong>co</strong>very with increasing severity <strong>of</strong><br />
<strong>co</strong>al-liquefaction reaction <strong>co</strong>nditions, The SCTL stage <strong>of</strong> an ITSL process involves<br />
rapid he<strong>at</strong>-up, followed by a short residence time <strong>at</strong> reaction temper<strong>at</strong>ure. During<br />
rapid he<strong>at</strong>-up, the r<strong>at</strong>e <strong>of</strong> free-radical production should increase significantly<br />
in a SCTL stage. An increase in free-radical <strong>co</strong>ncentr<strong>at</strong>ion was hypothesized,<br />
since the demand for hydrogen, with increased free-radical production, would<br />
increase and would be less likely s<strong>at</strong>isfied by hydrogen donors and gaseous<br />
hydrogen. With an increase in free-radical <strong>co</strong>ncentr<strong>at</strong>ion, a <strong>co</strong>n<strong>co</strong>mitant increase<br />
in solvent adduction was hypothesized, since free-radical addition, arom<strong>at</strong>ic sub-<br />
stitution, and polymeriz<strong>at</strong>ion reactions involving solvent and <strong>co</strong>al free-radicals<br />
xould be likely.<br />
In <strong>co</strong>mparing results obtained under SCTL <strong>co</strong>nditions (Table 1) with results<br />
obtained from experiments <strong>co</strong>nducted under traditional liquefaction <strong>co</strong>nditions<br />
(gradual he<strong>at</strong>-up to 425OC, 30 min <strong>at</strong> 425OC), solvent loss due to adduction should<br />
also be occurring under traditional <strong>co</strong>nditions. As the severity <strong>of</strong> the lique-<br />
faction <strong>co</strong>nditions is increased, adducted solvent should undergo cleavage (crack-<br />
ing, hydrogenolysis, etc. ) and re-form solvent-like products, resulting in<br />
improved solvent balance. While the original <strong>co</strong>mponents may not be re<strong>co</strong>vered,<br />
products having similar structures and chemical properties should be. For<br />
example, adduction <strong>of</strong> phenanthrene with a <strong>co</strong>al-derived benzylic radical may occur<br />
332
and may ultiontely form methyl-substituted phenanthrene (Figure 1). Baaed on this<br />
hypothesis, severe solvent loss uas expected uith a SCTL process <strong>co</strong>mpared to<br />
moder<strong>at</strong>e solvent loss under traditional liquefaction <strong>co</strong>nditions, and if the <strong>co</strong>n-<br />
ditions were severe enough, solvent th<strong>at</strong> was initially adducted, or solvent-like<br />
products, <strong>co</strong>uld be re<strong>co</strong>vered.<br />
Results from SCTL reactions demonstr<strong>at</strong>ed th<strong>at</strong> <strong>co</strong>mponents <strong>of</strong> the synthetic<br />
solvent were not adducted, degraded, or lost. While 755-855 <strong>of</strong> the <strong>co</strong>al was <strong>co</strong>n-<br />
verted to THF-soluble m<strong>at</strong>erial, only 2.61 tetralin underwent dehydrogen<strong>at</strong>ion. No<br />
adduction <strong>of</strong> m-cresol or quinoline uas observed. Results wing a synthetic<br />
solvent demonstr<strong>at</strong>e th<strong>at</strong> SCTL is a favorable process because solvent balance with<br />
little solvent degrad<strong>at</strong>ion can be achieved. These results were unexpected and<br />
suggest th<strong>at</strong> an increased <strong>co</strong>ncentr<strong>at</strong>ion <strong>of</strong> free radicals in a short period <strong>of</strong> time<br />
does not cause solvent loss or degrad<strong>at</strong>ion uhen sufficient readily donable<br />
hydrogen is present. The results also imply th<strong>at</strong> free radical production does not<br />
play an important role in solvent loss via adduction.<br />
It should also be understood th<strong>at</strong> little, if any, <strong>of</strong> the re<strong>co</strong>vered, original<br />
solvent <strong>co</strong>mponents are <strong>co</strong>al-derived. Based on <strong>co</strong>al experiments performed uith<br />
one- and two-<strong>co</strong>mponent solvents, negligible quantities <strong>of</strong> quinoline, m-cresol, and<br />
1-methylnaphthalene <strong>co</strong>uld be <strong>co</strong>nsidered <strong>co</strong>al-derived, while a maximum <strong>of</strong> 0.2%<br />
tetralin, 0.3% naphthalene, 0.2% pyrene, and 0.1% phenanthrene were <strong>co</strong>al-derived.<br />
Experiments <strong>co</strong>nducted using traditional <strong>co</strong>al liquefaction reaction <strong>co</strong>nditions<br />
shoued th<strong>at</strong> solvent balance <strong>co</strong>uld still be achieved, but degrad<strong>at</strong>ion <strong>of</strong> the<br />
solvent had started to occur. Results from experiments performed in the absence<br />
<strong>of</strong> <strong>co</strong>al showed th<strong>at</strong> little, if any, demethyl<strong>at</strong>ion and de<strong>co</strong>mposition <strong>of</strong> 1-<br />
methylnaphthalene had occurred. Reactions in the presence <strong>of</strong> <strong>co</strong>al showed th<strong>at</strong><br />
approxim<strong>at</strong>ely 3.21 l-mthylnaphthalene had undergone demethyl<strong>at</strong>ion and de<strong>co</strong>mpo-<br />
sition. Some <strong>of</strong> the demethyl<strong>at</strong>ed product (2.2%) <strong>co</strong>uld be ac<strong>co</strong>unted for by the<br />
increased amounts <strong>of</strong> naphthalene. Tetralin reactions included dehydrogen<strong>at</strong>ion to<br />
naphthalene (6. l%), rearrangement to 1-methylindane (0.5%), and de<strong>co</strong>mposition to<br />
butylbenzene (0.1%). The total amount <strong>of</strong> tetralin and its reaction products is<br />
22.2%; therefore, the additional amount (based on 20% tetralin in the synthetic<br />
solvent) <strong>co</strong>uld be ac<strong>co</strong>unted for by demethyl<strong>at</strong>ion <strong>of</strong> 1-methylnaphthalene. It is<br />
possible th<strong>at</strong> more than 2.2% 1-methylnaphthalene underwent demethyl<strong>at</strong>ion to<br />
naphthalene if tetralin was being lost via unidentified reactions and not via<br />
dehydrogen<strong>at</strong>ion to naphthalene. If this occurred, then gre<strong>at</strong>er amounts <strong>of</strong><br />
re<strong>co</strong>vered naphthalene <strong>co</strong>uld be <strong>at</strong>tributed to the demethyl<strong>at</strong>ion <strong>of</strong> 1-<br />
methylnaphthalene and not to the dehydrogen<strong>at</strong>ion <strong>of</strong> tetralin. Methyl<strong>at</strong>ion was<br />
also occurring, and 0.4% dimethylnaphthalene and 0.3% dimethylphenol were pro-<br />
duced. Hydrogen<strong>at</strong>ion <strong>of</strong> the arom<strong>at</strong>ic <strong>co</strong>mponents was also occurring, producing<br />
1.03 methyltetralin, 1.7% dihydrophenanthrene, 0.1% tetrahydrophenanthrene, 0.8%<br />
dihydropyrene, and 1.3% tetrahydroquinoline. The production <strong>of</strong> methyltetralin is<br />
most likely occurring via the hydrogen<strong>at</strong>ion <strong>of</strong> 1-methylnaphthalene, since no<br />
methyl<strong>at</strong>ion <strong>of</strong> tetralin was observed when blank experiments using tetralin as a<br />
solvent were <strong>co</strong>nducted. The major degrad<strong>at</strong>ion reactions occurring were rearrange-<br />
ment <strong>of</strong> tetralin to 0.5% 1-methylindane (Equ<strong>at</strong>ion 1 ) and cracking to butylbenzene<br />
(0.1%).<br />
H HU H<br />
333
While some <strong>of</strong> the quinoline was undergoing hydrogen<strong>at</strong>ion to tetrahydroquinoline<br />
(1.3%), 0.3% <strong>co</strong>uld not be ac<strong>co</strong>unted for. Solvent balance under traditional lique-<br />
faction <strong>co</strong>nditions was quite good. While methyl<strong>at</strong>ion, demethyl<strong>at</strong>ion, rearrange-<br />
ment, and cracking reactions have be<strong>co</strong>me apparent, 98.6% <strong>of</strong> the solvent can still<br />
be ac<strong>co</strong>unted for (less possible <strong>co</strong>al-derived products) and had not been lost<br />
because <strong>of</strong> adduction with <strong>co</strong>al-derived products.<br />
4<br />
Tre<strong>at</strong>ment <strong>of</strong> <strong>co</strong>al and solvent <strong>at</strong> more severe <strong>co</strong>nditions resulted in gre<strong>at</strong>er<br />
rearrangement and degrad<strong>at</strong>ion <strong>of</strong> the individual solvent <strong>co</strong>mponents, although the<br />
solvent balance was approxim<strong>at</strong>ely 97.5%. As much as 16.5% <strong>of</strong> 1-methylnaphthalene<br />
had undergone reaction. At least 12% 1-methylnaphthalene <strong>co</strong>uld be ac<strong>co</strong>unted for<br />
because <strong>of</strong> demethyl<strong>at</strong>ion, methyl<strong>at</strong>ion, and hydrogen<strong>at</strong>ion. Extensive dehydrogen<strong>at</strong>ion<br />
<strong>of</strong> tetralin was expected, and only 6.3% tetralin was re<strong>co</strong>vered.<br />
Rearrangement reactions were significant and again were a major reason for the<br />
decrease in solvent quality. Approxim<strong>at</strong>ely 2.1% tetralin had rearranged to 1-<br />
methylindane. Some loss <strong>of</strong> most <strong>co</strong>mponents <strong>of</strong> the solvent had occurred. I<br />
Methyl<strong>at</strong>ion and demethyl<strong>at</strong>ion <strong>of</strong> m-cresol was gre<strong>at</strong>er than observed under<br />
3<br />
traditional liquefaction <strong>co</strong>nditions, and approxim<strong>at</strong>ely 11.1% was re<strong>co</strong>vered. While<br />
quinoline represented the <strong>co</strong>mponent th<strong>at</strong> was present in the smallest amount, it<br />
was preferentially lost, and only 1.8% <strong>of</strong> quinoline and tetrahydroquinoline <strong>co</strong>uld<br />
be ac<strong>co</strong>unted for. While most <strong>of</strong> the solvent <strong>co</strong>uld be ac<strong>co</strong>unted for (approxim<strong>at</strong>ely<br />
97.511, only 61.1% was re<strong>co</strong>vered as original solvent and represented a solvent <strong>of</strong><br />
much poorer quality.<br />
Experiments <strong>co</strong>nducted under these severe reaction <strong>co</strong>nditions have shown th<strong>at</strong><br />
the synthetic solvent has undergone many reactions. Degrad<strong>at</strong>ion reactions were<br />
evident; and gre<strong>at</strong>er, not lesser, quantities <strong>of</strong> solvent were lost. Even under<br />
these severe <strong>co</strong>nditions, the overall solvent balance was better than expected.<br />
SUMMARY<br />
The effect <strong>of</strong> reaction <strong>co</strong>nditions on solvent loss was determined. Solvent<br />
re<strong>co</strong>very and solvent balance were better than expected for SCTL and traditional<br />
liquefaction <strong>co</strong>nditions. Surprisingly, very little adduction <strong>of</strong> solvent <strong>co</strong>mponents<br />
was observed. Increased severity <strong>of</strong> reaction <strong>co</strong>nditions caused an<br />
increase in degrad<strong>at</strong>ion <strong>of</strong> the synthetic solvent and an increase in adduction 'd'<br />
(approxim<strong>at</strong>ely 2.5%). From these results, the extent <strong>of</strong> solvent adduction under<br />
most liquefaction <strong>co</strong>nditions is minimal, and the solvent quality is most affected<br />
because <strong>of</strong> increased degrad<strong>at</strong>ion with increasing severity.<br />
ACKNOWLEDGMENTS<br />
The authors would like to thank John Siciliano, K<strong>at</strong>herine Lew, and<br />
Joseph Sharrow for their technical assistance in the labor<strong>at</strong>ory. These<br />
individuals were part <strong>of</strong> the Oak Ridge Associ<strong>at</strong>ed Universities (ORAU) Student<br />
Particip<strong>at</strong>ion Program. The authors would also like to thank Tom Williams, whose<br />
meticulous efforts in the labor<strong>at</strong>ory resulted in excellent precision.<br />
REFERENCES<br />
1. Larsen, J.W., Sams, T.L., and Rogers, B.R. Fuel 1981, 60, 335.<br />
2. Bruecker, R., and Koelling, G. Brennst<strong>of</strong>f-Chemie 1965, 46, 41.<br />
3.<br />
WcNeil, R.I., Young, D.C., and Cronauer, D.C. Fuel 1983, 62, 806.<br />
4. Hellgeth, J.W., Taylor, L.T., and Squires, A.M. Int. Conf. Coal Science<br />
i9a3, 172.<br />
.. ,<br />
334 I
!<br />
5. Narain, N.K., Utz, B.R., Appell, H.A., and Blaustein, B.D. Fuel, 1983, 62,<br />
1417.<br />
6. Collings, C.J.,<br />
359.<br />
Haggman, E.W., Jones, R.M., and Raeen, V.F. Fuel 1981, 60,<br />
7. Cronauer, D.C., McNeil, R.I., Danner, D.A., Wieland, J.H., and Ablchandanl,<br />
J.S. Prepr. Am. Chem. SOC. Div. Fuel Chem., 28(5), 40.<br />
8. Cronauer, D.C., Jewell, D.M., Shah, Y.T., Modl, R.J., and Seshadri, K.S.<br />
Ind. Eng. Chem., Fundam. 1979, 18(4), 368.<br />
9. Franz, J.A.,<br />
26(1), 105.<br />
and Camaioni, D.M. Prepr. Am. Chem. SOC. Div. Fuel Chem. 1981,<br />
10. Ruberto, R.G. Fuel Proc. Tech. 1980, 3,7.<br />
11. Utz, B.R.,<br />
Fuel.<br />
Appell, H.A., and Blaustein, B.D. Accepted for publfc<strong>at</strong>lon in<br />
335
4<br />
a<br />
4<br />
><br />
%<br />
m<br />
><br />
0<br />
p:<br />
U<br />
0)<br />
4<br />
v)<br />
W -1<br />
m<br />
4<br />
c<br />
m --<br />
-- 9"-<br />
. I I . . I I I . . l . I l ... I I<br />
m I I00 I I IO0 IN I I mo-fo I I<br />
m a N<br />
mi 1 0 1 I I io1 irni 1 0 1<br />
ml I N 1 I I IN1 1 - 1 1 - I 3 I I<br />
336<br />
W<br />
W<br />
m L<br />
C S<br />
W Y<br />
L E<br />
cY C m 4<br />
Gal 0<br />
mums c<br />
c c n m<br />
a m 0<br />
L S L<br />
snu4-1<br />
U 0 % O h<br />
c L S 4s4 m<br />
m o m m u 0 c<br />
ChLLaISO)<br />
W S Y V e w L<br />
c.4 0) I .-s ><br />
anc ~ n a a<br />
1
4<br />
(u<br />
I<br />
t<br />
+<br />
9<br />
(u<br />
I<br />
337<br />
-<br />
Q<br />
0