Kompletní sborník ke stažení (60 MB) - Fakulta chemická - Vysoké ...
Kompletní sborník ke stažení (60 MB) - Fakulta chemická - Vysoké ...
Kompletní sborník ke stažení (60 MB) - Fakulta chemická - Vysoké ...

You also want an ePaper? Increase the reach of your titles
YUMPU automatically turns print PDFs into web optimized ePapers that Google loves.
<strong>Vysoké</strong> učení technické v Brně<br />
<strong>Fakulta</strong> <strong>chemická</strong><br />
Sborník příspěvků<br />
soutěže Studentské tvůrčí činnosti „STUDENT 2006”<br />
a doktorské soutěže „O cenu děkana 2005 a 2006”<br />
Brno 2006
© <strong>Vysoké</strong> učení technické v Brně, <strong>Fakulta</strong> <strong>chemická</strong>, 2006<br />
ISBN: 80-214-3321-3<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
strana 2
OBSAH:<br />
Předmluva...........................................................................................................................................................7<br />
PRÁCE STUDENTŮ MAGISTERSKÝCH STUDIJNÍCH PROGRAMŮ<br />
SOUTĚŽE STUDENTSKÉ TVŮRČÍ ČINNOSTI<br />
SEKCE STČ 2006<br />
VLIV PROVOZNÍCH PARAMETRŮ FLOTACE NA SEPARAČNÍ ÚČINNOST ÚPRAVNY VODY MOSTIŠTĚ<br />
Jana Burianová..................................................................................................................................................9<br />
SYNTHESIS, CHARACTERIZATION AND ECOTOXICOLOGICAL ASSESSMENT OF NEW FLEXIBLE<br />
POLYURETHANE FOAMS WITH BIODEGRADABLE FILLERS<br />
Jan David..........................................................................................................................................................15<br />
NOVÝ SORPČNÍ GEL NA BÁZI SPHERON-OXINU PRO UŽITÍ V TECHNICE DGT<br />
Michaela Gregušová.........................................................................................................................................20<br />
MOLECULAR DYNAMICS SIMULATION OF POLYMER CHAIN IN THE VICINITY OF<br />
NANOPARTICLE<br />
Kateřina Hynštová............................................................................................................................................24<br />
APPLIKÁCIA NOVEJ EURÓPSKEJ NORNY NA STANOVENIE Cr(VI) V CEMENTE<br />
Tomáš Ifka.........................................................................................................................................................28<br />
DISSOLUTION OF HUMIC SUBSTANCES IN UREA IN LIGHT OF SUPRAMOLECULAR THEORY<br />
Jiří Kislinger.....................................................................................................................................................34<br />
WETTABILITY OF PLASMA POLYMERIZED VINYLTRIETHOXYSILANE FILM<br />
Soňa Lichovníková............................................................................................................................................39<br />
URČOVANIE DISTRIBÚCIE DOMÉN V SPIN CROSSOVER SYSTÉMOCH POMOCOU ANALYTIC-<br />
KEJ FUNKCIE<br />
Ján Pavlik.........................................................................................................................................................44<br />
A STUDY OF INTERFACIAL ASPECTS OF EPOXY-BASED HYBRID COMPOSITES REINFORCED<br />
WITH BASALT, CARBON AND CERAMIC FIBRES BY DMA ANALYZIS<br />
Lukáš Recman...................................................................................................................................................48<br />
ŠTÚDIUM ANTIOXIDANTOV A ICH ÚČINKOV PRI STABILIZÁCII POLYMÉROV<br />
Ján Rimarčík.....................................................................................................................................................54<br />
HOŘLAVOST A MECHANICKÉ VLASNOTSTI NANOKOMPOZITŮ EVA/Mg(OH) 2<br />
Jiří Sadílek........................................................................................................................................................59<br />
STUDIUM MECHANISMU KOORDINAČNÍ POLYMERACE HEXA-1,5-DIENU KATALYZOVANÉ<br />
FENOXYIMINOVÝM KOMPLEXEM TITANU A METHYLALUMINOXANEM<br />
Jan Ševčík.........................................................................................................................................................64<br />
MOLEKULOVÁ DYNAMIKA C-GLYKOZYLNITROMETÁNOV<br />
Stanislava Šoralová..........................................................................................................................................67<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
strana 3
STUDIUM DEGRADACE TISKU NA TENKÝCH POLYMERNÍCH VRSTVÁCH<br />
Jiří Stančík.......................................................................................................................................................71<br />
ZMĚNY OBSAHU AMINOKYSELIN V BRA<strong>MB</strong>ORÁCH V ZÁVISLOSTI NA HNOJENÍ DUSÍKEM<br />
Petra Valová.....................................................................................................................................................77<br />
PRECIPITATION OF DL – VALINE FROM AQUEOUS ISOPROPANOL SOLUTIONS<br />
Miroslav Variny................................................................................................................................................81<br />
VÝVOJ MIKROŠTRUKTÚRY SODNO-BORITO-KREMIČITÝCH SKIEL S PRÍDAVKOM TIO 2<br />
Oto Vojtechovský..............................................................................................................................................86<br />
VÝZNAM VOĽNÝCH RADIKÁLOV PRI POŠKODENÍ PAPIERA (EPR ŠTÚDIUM)<br />
Zuzana Vrecková..............................................................................................................................................91<br />
PRÁCE STUDENTŮ DOKTORSKÝCH STUDIJNÍCH PROGRAMŮ<br />
SOUTĚŽE O CENU DĚKANA 2005<br />
SEKCE DSP 2005<br />
CHITOSAN - A NEW TYPE OF POLYMER COAGULANT<br />
Marcela Borovičková.......................................................................................................................................97<br />
PREPARATION OF SELF-CLEANING PHOTOCATALYTICALLY ACTIVE SURFACES<br />
Jana Chomoucká............................................................................................................................................101<br />
RHEOLOGICAL, VISCOUS, AND SURFACE AACTIVE BEHAVIOR OF POLYSACCHARIDES IN<br />
AQUEOUS SOLUTIONS<br />
Martin Chytil..................................................................................................................................................106<br />
STUDY OF PHYSICOCHEMICAL AND ANTIOXIDATIVE PROPERTIES OF YEAST ME<strong>MB</strong>RANE<br />
COMPONENTS<br />
Michaela Drabkova........................................................................................................................................112<br />
DETERMINATION OF YEAST VIABILITY BY MEANS OF FLUORESCENCE MICROSCOPY AND<br />
IMAGE ANALYSIS<br />
Petra Jeřábková..............................................................................................................................................115<br />
FLOW BEHAVIOUR OF DILUTED SUSPENSIONS IN CARBOXYMETHYLCELLULOSE-WATER<br />
SOLUTION<br />
Michal Klimovič.............................................................................................................................................119<br />
USE OF VARIOUS POLYACRYLAMIDE DIFFUSIVE GELS FOR ASSESSMENT OF METAL AVAIL-<br />
ABILITY IN SOILS<br />
Vladěna Kovaříková.......................................................................................................................................123<br />
SPECTROPHOTOMETRIC STUDY OF SOME BIOLOGICALBUFFERS SOLUTIONS<br />
Miroslava Krčmová........................................................................................................................................128<br />
PHYSIOLOGICAL CHARACTERISATION OF XYLOSE-UTILISING SACCHAROMYCES CEREVI-<br />
SIAE STRAINS<br />
Jitka Kubešová................................................................................................................................................133<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
strana 4
THE EFFECT OF STERILIZATION HEATING ON NUTRITION QUALITY OF PROCESSED CHEESE<br />
Blanka Loupancová........................................................................................................................................137<br />
INVESTIGATION OF ENVIRONMENTAL AND VARIETAL INFLUENCES ON DISTRIBUTION OF<br />
STARCH GRANULES IN BARLEY KERNELS BY GFFF AND LASER PARTICLE SIZE ANALYSIS<br />
Karel Mazanec................................................................................................................................................142<br />
FIBER ASPECT RATIO INFLUENCE ON FRACTURE IMPACT TOUGNESS OF POLYMETHYL<br />
METHACRYLATE/POLYVINYL ALCOHOL COMPOSITES<br />
Vladimír Pavelka............................................................................................................................................147<br />
DIAGNOSTIC OF THE DIAPHRAGM DISCHARGE IN WATER SOLUTIONS BY OPTICAL<br />
EMISSION SPECTROSCOPY<br />
Jana Procházková..........................................................................................................................................151<br />
SENSORIC PROPERTIES OF SOLUBLE PHTHALOCYANINES<br />
Kateřina Severová..........................................................................................................................................155<br />
INFLUENCE OF TRIETHYLALUMINIUM COCATALYST ON INITIAL KINETIC OF PROPENE<br />
POLYMERIZATION<br />
Miroslav Skoumal..........................................................................................................................................159<br />
PHYSICO-CHEMICAL PROPERTIES OF PLASMA-POLYMERIZED TETRAVINYLSILANE<br />
Jan Studýnka...................................................................................................................................................164<br />
PULSED 1 H-NMR OF IMPACT COPOLYMERS OF PROPYLENE (ICP)<br />
Ladislav Vilč...................................................................................................................................................169<br />
SPECIATION OF FIVE SELENIUM COMPOUNDS BY HPLC-HEATING-UV-HG-AFS<br />
Eva Vitoulová.................................................................................................................................................175<br />
EPITAXIAL GROWTH OF Ni AND Co ON (111) METALLIC SUBSTRATES<br />
Martin Zelený.................................................................................................................................................182<br />
ACETYLCHOLINESTERASE INHIBITOR FROM NOSTOC SLIZ. KOL.<br />
Petr Zelík........................................................................................................................................................187<br />
HUMIC ACIDS IDENTIFICATION BY PY-GCMS TECHNIQUE<br />
Pavla Žbánková..............................................................................................................................................191<br />
PRÁCE STUDENTŮ DOKTORSKÝCH STUDIJNÍCH PROGRAMŮ<br />
SOUTĚŽE O CENU DĚKANA 2006<br />
SEKCE DSP 2006<br />
PRODUCTION OF SELECTED SECONDARY METABOLITES IN TRANSFORMED BACTERIAL<br />
CELLS<br />
Jana Hrdličková.............................................................................................................................................197<br />
THE SIMPLE METHOD FOR THE RECOGNITION OF REDUCING AND NONREDUCING NEUTRAL<br />
CARBOHYDRATES BY MALDI-TOF MS<br />
Markéta Laštovičková....................................................................................................................................201<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
strana 5
MINIATURE METALLIC DEVICE FOR COLLECTION OF HYDRIDE FORMING ELEMENTS<br />
Pavel Krejčí....................................................................................................................................................206<br />
INFLUENCE OF POLYUNSATURATED FATTY ACIDS INTAKE ON LIPID METABOLISM IN<br />
PATIENTS WITH HYPERLIPIDAEMIA<br />
Simona Macuchová........................................................................................................................................210<br />
HYDROPHOBIZED SODIUM HYALURONATE IN AQUEOUS SOLUTION - A FLUORESCENCE<br />
STUDY<br />
Filip Mravec...................................................................................................................................................213<br />
CHARACTERIZATION AND DEGRADATION BEHAVIOUR OF TRIBLOCK COPOLYMER<br />
Ludmila Nová.................................................................................................................................................218<br />
STUDY OF THE COPPER(II) IONS NON–STATIONARY DIFFUSION IN HUMIC GEL<br />
Petr Sedláček..................................................................................................................................................223<br />
LIVING RADICAL POLYMERIZATIONS INITIATED WITH SILSESQUIOXANES<br />
Ondřej Smrtka................................................................................................................................................228<br />
MEASUREMENT OF THERMOPHYSICAL PROPERTIES OF PMMA BY PULSE TRANSIENT METHOD<br />
Pavla Štefková................................................................................................................................................233<br />
THE ANALYSIS OF GAMMA LINOLENIC ACID IN EVENING PRIMROSE OIL<br />
Hana Štoudková.............................................................................................................................................238<br />
IMPROVED FLUORIMETRIC DETERMINATION OF Al, Ga AND In BY MICELLE ENHANCED<br />
8-HYDROXYQUINOLINE -5-SULPHONIC ACID COMPLEX<br />
Šimon Vojta.....................................................................................................................................................243<br />
PHYSICAL-CHEMICAL ASPECTS OF PAINT FILMS DEGRADATION<br />
Lucie Wolfová.................................................................................................................................................249<br />
USING LOW-MOLECULAR MASS PI MARKERS IN PROTEOMIC STAINING-FREE METHOD<br />
FOR STUDY OF POSTTRANSLATIONALLY MODIFIED PROTEINS<br />
Karel Mazanec, Karel Šlais and Josef Chmelík............................................................................................254<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
strana 6
PŘEDMLUVA<br />
Tak jako v minulých letech, i letos se v měsíci říjnu na půdě Fakulty chemické VUT uskutečnila<br />
soutěž posluchačů bakalářských a magisterských studijních programů Student 2006, jakož<br />
i soutěž posluchačů doktorských studijních programů. V doktorských studijních programech<br />
proběhla tato soutěž již po deváté, soutěž Student 2006 se konala po šesté. Jako člen hodnotících<br />
komisí soutěže doktorandů v posledních letech mohu konstatovat významný kvalitativní růst<br />
její úrovně nejen po stránce odborné, ale také po stránce kvality prezentace příspěvků v anglickém<br />
jazyku. Rovněž hodnotící komise soutěže Student FCH 2006 konstatovala velmi dobrou<br />
úroveň prezentovaných prací. Předkládaný soubor příspěvků, zaslaných do obou soutěží,<br />
doplněný o některé příspěvky ze soutěže v minulém roce, dokumentuje tento trend. Stává se<br />
již tradicí, že se do studentské soutěže přihlašují také posluchači mimobrněnských chemických<br />
fakult, především Fakulty chemické a potravinářské technologie STU v Bratislavě. V tomto<br />
roce se však poprvé představil také zástupce UTB ve Zlíně. I v příštích ročnících bychom v této<br />
tradici chtěli pokračovat. Předpokládá se, že nejlepší účastníci soutěže z FCH VUT budou<br />
naši fakultu reprezentovat na podobných soutěžích v Bratislavě a Zlíně. Právě ve výsledcích<br />
vzájemné konfrontace s posluchači jiných fakult spatřuje vedení Fakulty chemické důležitý<br />
prvek, směřující <strong>ke</strong> zvýšení kvality pedagogického a vědecko-výzkumného procesu. Proto<br />
bude akce podobného typu i v budoucnosti všestranně podporovat.<br />
prof. Ing. Ladislav Omelka, DrSc.<br />
proděkan pro tvůrčí činnost<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
strana 7
Příspěvky soutěže<br />
Studentské tvůrčí činnosti<br />
„STUDENT 2006”<br />
(Sekce STČ 2006)
VLIV PROVOZNÍCH PARAMETRŮ FLOTACE NA SEPARAČNÍ<br />
ÚČINNOST ÚPRAVNY VODY MOSTIŠTĚ<br />
Jana Burianová, 5.ročník<br />
vedoucí práce: doc. Ing. Petr Dolejš, CSc.<br />
konzultant práce: Ing. Pavel Dobiáš<br />
<strong>Vysoké</strong> učení technické v Brně, <strong>Fakulta</strong> <strong>chemická</strong>,<br />
Ústav chemie a technologie ochrany životního prostředí, Purkyňova 118, 612 00 Brno,<br />
e-mail: xcburianova@fch.vutbr.cz<br />
ÚVOD<br />
Práce se zabývá vlivem intenzity míchání v agregačním reaktoru flotace a délky a<br />
frekvence intervalu shrabování kalu na kvalitu upravené vody vycházející ze zařízení flotace<br />
rozpuštěným vzduchem (DAF - dissolved air flotation) na úpravně vody Mostiště. Tyto dvě<br />
studované proměnné mohou mít vliv jak na kvalitu upravené vody a také na ekonomiku<br />
provozu flotační jednotky a je proto nezbytné zjistit jejich optimální nastavení v provozu.<br />
TEORETICKÁ ČÁST<br />
Flotace rozpuštěným vzduchem je založena na snížení specifické hmotnosti vloček vzniklé<br />
suspenze vytvořením agregátů mezi vločkami a vzduchovými mikrobublinkami. Pak je<br />
hustota těchto agregátů nižší než vody. Agregáty tedy překonávají gravitaci a stoupají ve vodě<br />
směrem k hladině.<br />
Vlastní reaktor DAF se skládá ze dvou částí, ve kterých probíhají dva základní procesy<br />
(obr. 1). Do první části, zvané kontaktní zóna, vstupuje voda obsahující vyvločkované<br />
nečistoty. Do této vody je zaváděn proud kapaliny nasycené pod tla<strong>ke</strong>m rozpuštěným<br />
vzduchem a z něj vzniká velké množství vzduchových mikrobublin. Mikrobublinky vznikají<br />
při vstupu nasycené recirkulační vody ve speciálně upravených tryskách. Poté dochází<br />
k agregaci vloček a těchto mikrobublin. Kontaktní zóna tedy zajišťuje kontakt a připoutání<br />
mikrobublin a vloček [1]. Náhlým snížením tlaku dochází k uvolnění vzduchových<br />
mikrobublin a jejich proud je míchán s hlavním proudem vody. Vyloučené mikrobublinky<br />
vzduchu způsobují „mléčný zákal“ vody, a proto se této vodě také říká „bílá voda“ („white<br />
water“). Vzduchem nasycená recirkulační voda je vytvářena v saturátoru, do kterého proudí<br />
část již upravené vody. Obvykle to bývá 6 – 12 % upravovaného proudu vody. Voda je<br />
sycena pod tla<strong>ke</strong>m 0,5 – 0,6 MPa.<br />
Suspenze agregátů vločka-mikrobublina vytvořená v kontaktní zóně pak vstupuje do druhé<br />
části reaktoru, zvané separační zóna. Zde dochází k dalšímu růstu agregátů vločka-bublina a<br />
jejich pohybu k hladině. Tam tvoří souvislou vrstvu kalu, která je z hladiny shrabována<br />
mechanickým zařízením a odchází do kanálu pro odtah kalu. Upravená voda je odváděna ze<br />
dna separační zóny nádrže na další stupeň úpravny vody - filtraci.<br />
ÚPRAVNA VODY MOSTIŠTĚ<br />
Úpravna vody (ÚV) Mostiště má kapacitu 220 l/s a je z ní zásobován skupinový vodovod<br />
pro oblast Velké Meziříčí, Velká Bíteš, Olší nad Oslavou a Měřín, přivaděč do Třebíče a<br />
přivaděč do Žďáru nad Sázavou. Zásobováno je cca 75 000 obyvatel.<br />
Z vodní nádrže Mostiště je surová voda přiváděna na provzdušňovací kaskádu. Do odtoku<br />
z kaskády je dávkován koagulant. Na ÚV Mostiště se používá jako koagulant 41% roztok<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 9
Fe2(SO4)3. Dále následuje hydraulický mísič. Odtud vzniklá suspenze pokračuje do DAF<br />
zařízení s celkovou kapacitou 110 l/s. DAF zařízení je tvořeno dvěma flotačními jednotkami.<br />
Souběžně s DAF zařízením mohou ještě fungovat dvě jednotky původních čiřičů v případě<br />
potřeby vyšší produkce upravené vody. Voda je dále vedena na pískové filtry. Desinfekce<br />
vody je prováděna pomocí Cl2 a ClO2 a pH je upraveno vápennou vodou.<br />
Flotace rozpuštěným vzduchem je na ÚV Mostiště novou technologií. Zařízení bylo<br />
uvedeno do provozu 28.11.2005. Důvodem zařazení nové technologie do procesu úpravy<br />
vody bylo zhoršení kvality surové vody vlivem havarijního stavu zdroje surové vody<br />
(přehrady nádrže Mostiště) a následných stavebních prací a s tím spojené nevyhovující jakosti<br />
surové vody. Stalo se prvním procesem flotace v zemích střední a východní Evropy [2,3].<br />
Obr. 1. Schematické znázornění úpravy vody flotací rozpuštěným vzduchem<br />
EXPERIMENTÁLNÍ ČÁST<br />
Jako ukazatele kvality vody byla brána velikostní distribuce částic měřená pomocí<br />
analyzátoru velikostní distribuce částic (Water Particle Counter WPC-21), hmotnostní<br />
koncentrace Fe 3+ stanovovaná rhodanidem a absorbance při vlnové délce 387 nm měřené na<br />
spektrometru SPEKOL 11. Dále byly zaznamenávány hodnoty pH, absorbance při vlnové<br />
délce 254 nm a teplota vody měřené provozními měřícími přístroji ÚV Mostiště.<br />
Analyzátor částic: voda z výtoku flotace je přiváděna do analyzátoru z odtoku upravené<br />
vody na konci flotační jednotky. Analyzátor počítá částice o velikosti 2, 5, 7, 10, 15, 25, 50 a<br />
100 μm. Neustále je měřen počet částic o velikosti 2 μm, druhá hodnota udává postupně<br />
v určených intervalech počet částic ostatních velikostí.<br />
Použité metody stanovení a materiály<br />
Stanovení hmotnostní koncentrace Fe 3+ : připraví se zásobní roztok (NH4)2Fe(SO4)2.6H2O<br />
(0,7022 g do 1000 ml) okyselený 2 ml koncentrované H2SO4. Z něho se připraví pracovní<br />
roztok I (0,01 mg/ml) a roztok II (0,001 mg/ml). Z nich se připraví série roztoků pro sestrojení<br />
kalibrační křivky. Pracovní roztok se pipetuje do 25 ml odměrné baňky, přidá se 0,5 ml HCl<br />
(1:1) a 0,5 ml 3% H2O2. Po 5 min. se vzorek doplní asi na 15 – 20 ml, přidá se 2,5 ml KSCN a<br />
doplní se po rysku. Stejně se připraví roztoky neznámých vzorků. Absorbance se měří při<br />
vlnové délce 480 nm.<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 10
Obr.2. Analyzátor částic<br />
VÝSLEDKY MĚŘENÍ A DISKUSE<br />
Změna intenzity míchání<br />
Výsledky jsou uvedeny pouze pro nádrž 2, ve které byla intenzita míchání měněna. Nádrž<br />
1 fungovala jako referenční, tj. se stálým provozním nastavením a výsledky naměřené pro<br />
nádrž 2 byly srovnány s hodnotami pro nádrž 1. Podmínky a výsledky měření při různé<br />
rychlosti míchání znázorňuje tabulka 1. Ve dvou případech byla použita stejná rychlost<br />
míchání, ale došlo <strong>ke</strong> změně průtoku.<br />
Tabulka 1. Podmínky a výsledky měření při různé rychlosti míchání<br />
č. měř.<br />
doba<br />
t (°C)<br />
intenzita<br />
míchání %<br />
rychlost míchání<br />
ot./min.<br />
D (mg/l) zbyková koncentrace Fe (mg/l)<br />
měření<br />
(2<br />
hod.)<br />
teplota<br />
vody<br />
M1 M2 M1 M2<br />
dávka<br />
síranu<br />
zákal<br />
(NTU)<br />
nádrž<br />
1<br />
nádrž<br />
2<br />
rozdíl<br />
koncentrací<br />
1 17,10 <strong>60</strong> 40 4,25 2,67 38,87 0,301 0,328<br />
2 17,10 <strong>60</strong> 0 4,25 0,00 38,87 0,317 0,401 0,321 0,080<br />
3 17,10 100 80 7,08 5,33 38,87 0,361 0,344 0,191 0,153<br />
4 17,00 100 <strong>60</strong> 7,08 4,00 38,81 0,335 0,486 0,234 0,252<br />
5 17,10 100 <strong>60</strong> 7,08 4,00 39,70 0,243 0,108 0,281 0,141<br />
6 17,20 100 40 7,08 2,67 38,64 0,247 0,449 0,362 0,087<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 11
počet částic/ml<br />
120<br />
100<br />
80<br />
<strong>60</strong><br />
40<br />
20<br />
0<br />
Porovnání počtu částic/ml při různé rychlosti míchání<br />
2 5 7 10 15 25 50 100<br />
rozmezí velikosti částic (μm)<br />
4,25; 2,67<br />
4,25; 0,00<br />
7,08; 5,33<br />
7,08; 4,00<br />
7,08; 4,00<br />
7,08; 2,67<br />
Obr. 3. výsledky měření analyzátorem částic při různé rychlosti míchání<br />
V legendě je vždy nejprve rychlost pro první míchadlo ve flotaci, kde je míchání rychlé a<br />
poté následuje rychlost pro druhé míchadlo, kde je míchání pomalé. Z porovnání počtu<br />
částic/ml je vidět, že nejvhodnější nastavení intenzity míchání, z těch, které jsem použila, je<br />
nastavení 7,08 ot/min (100 %) pro první míchadlo a 2,67 ot/min (40 %) pro druhé míchadlo.<br />
Změna intervalu shrabování kalu<br />
Podmínky a výsledky měření pro různé intervaly shrabování jsou uvedeny v tabulce 2. I<br />
v tomto případě je nádrž 1 referenční.<br />
Tabulka 2. Podmínky a výsledky měření při různých intervalech shrabování kalu<br />
č.měř. t ( °C) shrabování<br />
doba<br />
měření<br />
(2<br />
hod.)<br />
teplota<br />
vody<br />
prodleva<br />
(min.)<br />
hrabání<br />
(min.)<br />
rychlost<br />
shrabování<br />
%<br />
rychlost<br />
(cm/min.)<br />
D<br />
(mg/l)<br />
dávka<br />
síranu<br />
zákal<br />
(NTU)<br />
zbyková koncentrace Fe<br />
nádrž<br />
1<br />
(mg/l)<br />
nádrž<br />
2<br />
rozdíl<br />
koncentrací<br />
1 17,00 20 1 30 61,0 37,41 0,247 0,524 0,373 0,151<br />
2 17,00 15 1 30 61,0 37,41 0,250 0,424 0,363 0,061<br />
3 17,00 10 1 30 61,0 38,33 0,237 0,405 0,356 0,049<br />
4 17,00 5 1 30 61,0 37,65 0,249 0,539 0,490 0,049<br />
5 17,00 2 1 30 61,0 37,65 0,251 0,586 0,574 0,012<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 12
počet částic/ml<br />
<strong>60</strong><br />
50<br />
40<br />
30<br />
20<br />
10<br />
0<br />
Porovnání počtu částic/ml pro různé intervaly shrabování<br />
2 5 7 10 15 25 50 100<br />
rozmezí velikosti částic (μm)<br />
20 + 1 min.<br />
15 + 1 min.<br />
10 + 1 min.<br />
5 + 1 min.<br />
2 + 1 min.<br />
Obr. 4. výsledky měření analyzátorem částic při různých intervalech shrabování kalu<br />
Při porovnání výsledků analyzátoru částic pro různé intervaly shrabování jsem zjistila, že<br />
nejvhodnější by byl interval 10 min. stání a 1 min. hrabání. Při nižších intervalech se počet<br />
částic zvýšil. To platí i pro zákal. Opět je tomu jinak pro koncentraci zbytkového železa, kde<br />
nejvhodnější interval je 20 min. + 1 min. Jako vhodný interval bych tedy zvolila 10 min. + 1<br />
min., protože stanovení ukázala dobrou kvalitu vody. Při nižších intervalech došlo pouze k<br />
nepatrnému zhoršení, ale zvýšila by se spotřeba energie. Při vyšších intervalech není také<br />
zhoršení příliš velké, ale později docházelo <strong>ke</strong> vzniku vrstvy kalu vysoké asi 20 cm, která byla<br />
velmi hustá a její váha způsobovala prohýbání lopatek hrabáku a nakonec jeho úplné<br />
zastavení. Kal musel být shrábnut ručně. Na intervalu 10 min. + 1 min. jsem se shodla i s<br />
obsluhou zařízení.<br />
ZÁVĚR<br />
Vlivy rychlosti míchání, délky intervalu a frekvence shrabování se projevily na kvalitě<br />
upravené vody. Větší vliv má změna intenzity míchání. Jako nejvhodnější se ukázalo<br />
nastavení, kdy první míchadlo má se otáčí rychlostí 7,08 ot./min. a druhé míchadlo 2,67<br />
ot./min. Při změně intervalu shrabování se ukázalo nejlepší nastavení 10 min. stání a 1 min.<br />
shrabování. Přihlédnutím k těmto výsledkům při provozu úpravny by tedy mohla být zvýšena<br />
kvalita upravené vod vycházející z flotace a tedy i vody dodávané <strong>ke</strong> spotřebiteli.<br />
Optimalizací studovaných provozních parametrů dochází především <strong>ke</strong> snížení počtu částic<br />
obsažených ve vodě z flotace, což má příznivý vliv na další stupně úpravy. Prodlužuje se doba<br />
provozu pískových filtrů, které nevyžadují tak časté praní a snižuje se i možnost průniku<br />
částic filtrem.<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 13
CITOVANÁ LITERATURA<br />
[1] Haarhoff, J. and Edzwald, J.K.: Dissolved air flotation modelling: insights and<br />
shortcomings. Journal of Water Supply: Reseach and Technology-AQUA, 2004, vol. 53, pp.<br />
127-150.<br />
[2] Dolejš P., Dobiáš P., Mazel L.: Provozní výsledky první vodárenské flotace v ČR<br />
realizované na ÚV Mostiště. Sborník konference Vodárenská biologie 2006, s. 92-97. Praha<br />
31.1.-2.2.2006. VŠCHT Praha a Ekomonitor, s.r.o. Chrudim, Praha 2006.<br />
[3] Dolejš P.: Flotace rozpuštěným vzduchem (DAF) pro úpravu pitné vody a její první<br />
provozní realizace v ČR. Vodní hospodářství 56, č. 4, s. 99-101 (2006).<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 14
SYNTHESIS, CHARACTERIZATION AND ECOTOXICOLOGICAL<br />
ASSESSMENT OF NEW FLEXIBLE POLYURETHANE FOAMS WITH<br />
BIODEGRADABLE FILLERS<br />
Jan DAVID 1 , 5. year<br />
Advisor: Prof. Milada VÁVROVÁ 1 ; Consultant: Dr. Lucy VOJTOVÁ 2<br />
1 Institute of Chemistry and Technology of Environmental Protection<br />
2 Institute of Materials Science<br />
Faculty of Chemistry, Brno University of Technology, Faculty of Chemistry,<br />
Purkynova 118, 612 00 Brno<br />
e-mail: xcdavid@fch.vutbr.cz<br />
ABSTRACT<br />
This work deals with the synthesis of new flexible biodegradable polyurethane foams<br />
(BIO-PU), in which non-degradable polyol polyether was partly substituted by bio-polyol,<br />
mainly the cellulose or starch derivatives. The bio-polyol replacements are expected to<br />
provide the PU foam cheaper, easily biodegradable and less toxic.<br />
Samples of the BIO-PU foams were characterized by means of Fourier Transform – Infra<br />
Red Spectroscopy (FTIR) and Thermogravimetric Analysis (TGA) methods and consequently<br />
hydrolyzed under reflux in freshwater. The ecotoxicological aspects of the foam leaches were<br />
determined by microbiotest screening toxkit „Thamnotoxkit F TM “. Values of toxicity were<br />
expressed as percentage mortality of the II-III larvae instars hatched from the cysts of<br />
freshwater fairy shrimps Thamnocephalus platyurus dependence on the effect criterion of the<br />
respective assay.<br />
INTRODUCTION<br />
Polyurethanes (PU) are polymers made by addition polymerizations of multi-functional<br />
isocyanates and multi-functional alcohols (polyols), e.g. polyether or polyester 1 , which<br />
functionality determine on linear or cross-lin<strong>ke</strong>d structure. Various methods can be used to<br />
produce polyurethanes but for the PU foam production is often used the one-shot process,<br />
where direct mixing of polyols, catalysts, blowing agent and isocyanates are used. Foams<br />
(expanded plastics) are microcellular structures produced by gas (CO2) bubbles during the<br />
polyurethane preparation. PU foams properties are affected mainly by the raw materials and<br />
can be modified by a wide variety of additives, such as stabilizers, fillers, cross-linking agents<br />
and chain extenders 2,3 . PU foams are materials with a broad variety of applications li<strong>ke</strong><br />
furniture, automotive and shoe industry, building construction and packaging, agriculture and<br />
medicine. Due to the recycling complications the PU foams are discarded after use, which<br />
represents contamination problems because of their difficult disintegration and incorporation<br />
to the environment. 2 Solution of this problem is including a natural material in the PU foam<br />
network in order to evo<strong>ke</strong> easier biodegradation and lower ecotoxicity. Degradable natural<br />
additives containing –OH groups might be used as –OH providers to modify the PU<br />
properties and structure. 4 Various compounds of natural origin can be used instead of<br />
commercial polyol polyethers li<strong>ke</strong> starches, celluloses and their derivatives, soy flour, bark,<br />
castor oil and palm oil, wattle tannin or oil palm empty fruit bunch 2,3,4,5,6,7,8 .<br />
This work is focused on the use of carboxymethyl cellulose sodium salt (CMC-Na),<br />
acetylated potato starch (AS), cellulose acetate (CA), 2-hydroxyethyl cellulose (2-HEC) and<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 15
wheat protein /gluten/ (WP-G) as bio polyols. Recently, the biodegradation of these flexible<br />
modified PU foams by thermophille bacteria Thermophillus sp. was published by our group of<br />
researchers from FCH BUT 9 . However, the levels of toxic substances must not exceed the<br />
permissible limit.<br />
The interest in biological testing is growing rapidly and toxicity testing is now gradually<br />
incorporated in environmental legislation in many countries. The most worldwide known and<br />
useful alternative toxicity biotests are Toxkits owing to their miniaturization, simplicity, quick<br />
availability, high sensitivity and reproducibility and low cost. The Toxkits tests are<br />
particularly suited for routine toxicity testing of chemicals, solid waste leaches or effluents<br />
released in aquatic environment 10 . The sensitivity of these microbiotests is equal to that of<br />
conventional bioassays, and the new alternative assays have already been validated in various<br />
studies 11,12,13,14 .<br />
In this proposed work, BIO-PU foams modified by AS, AC, CMC-Na,2-HEC and WP-G<br />
were characterized by FTIR, TGA, extracted in freshwater and used for ecotoxicological<br />
evaluation via Thamnotoxkit F.<br />
EXPERIMENTAL<br />
Raw materials<br />
Polyol polyether (hydroxyl number OH = 47) (PEP), tolylene diisocyanate 80/20 (TDI),<br />
tin-bis(2-ethylhexanoate) surfactant, and mixed catalyst were obtained from Gumotex, a.s.,<br />
carboxymethyl cellulose sodium salt (CMC-Na), Mn = 250 000, cellulose acetate (CA),<br />
Mn = 30 000, and 2-hydroxyethyl cellulose (2-HEC), Mn = 90 000 were purchased from<br />
Aldrich, acetylated starch AMISOL HS (AS) was obtained from Starch and Potato Products<br />
Research Laboratory, Poland and wheat protein (gluten) (WP-G) was received from<br />
Amylon, a.s.<br />
Synthesis of BIO-PU foams and preparation of leaches<br />
The foams were synthesized according to a standard industrial recipe for furniture industry<br />
flexible foams, except of the partial PEP substitution (from 1 up to 30 w%) by starch or<br />
cellulose derivates or gluten, which were pre-mixed with the original PEP. Consequently the<br />
catalyst, surfactant and TDI were added, mixed up to cream time and quickly poured into a<br />
mould. BIO-PU foams were cured for 48 hours.<br />
0,5 g pieces of cured PU foams were cut to a smaller pieces and boiled under reflux with<br />
25 ml of standard freshwater (for Thamnocephalus platyurus) for 8 hours.<br />
Ecotoxicological assessment<br />
Alternative crustacean toxicity screening test Thamnotoxkit F based on the mortality of<br />
the instars II-III larvae of freshwater fairy shrimps Thamnocephalus platyurus hatched from<br />
the cysts after 24 hours exposure was applied to different concentrations of the PU foams<br />
freshwater leaches. Standard freshwater was prepared from solutions supplied with<br />
Thamnotoxkit F. The toxicity tests were performed according to the standard operational<br />
procedure manual of the Thamnotoxkit F producer 15 Shortly, cysts of Thamnocephalus<br />
platyurus were hatched for 23 hours before the use by pre-hydrating with diluted freshwater<br />
for 30 minutes and incubating at 25°C for 22 hours under continuous illumination. Then the<br />
hatched larvae were transferred to the test wells containing the toxicant dilution and incubated<br />
at 25°C in darkness for 24 hours. Then the immobilized/dead individuals of larvae were<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 16
counted and toxicity data were expressed as percentage mortality of Thamnocephalus<br />
platyurus depending on the effect criterion of the respective assay.<br />
Characterization of foams<br />
Small pieces of cured PU foams were frozen by liquid N2 and pulverized. FTIR spectra<br />
were measured using Nicolet Impact 400D FTIR spectrometer with common KBr technique.<br />
The spectra analyses were performed on Omnic and MS Excel software.<br />
Thermogravimetric analyses were carried out via Perkin Elmer TGA 6, where 7 mg of<br />
samples were heated from 100°C to 500°C at a heating rate of 10°C.min −1 under N2<br />
atmosphere. The curve analyses were performed on PE Pyris 6 and MS Excel software.<br />
RESULTS AND DISCUSSION<br />
Synthesis and characterization of PU foams<br />
Five types of bio-polyols AS, AC, 2-HEC, CMC-Na and WP-G were successfully included<br />
in the 3D PU network in order to substitute as much of commercial polyether polyol as<br />
possible. It was found to substitute only 10 % of PEP by AC and 2-HEC, but 20 % by AS and<br />
even 30 % by CMC-Na. Resulting modified flexible foams were not distinguished at first<br />
sight from the respective CONTROL PU foam as for the size, shape, color, polymer network<br />
and flexibility, except the WP-G foam, where the wheat protein replaced only 5 % of<br />
commercial PEP. It was probably caused due to the side reactions of amino acids with the raw<br />
materials during the PU foam preparation resulting in collapsed hard foam.<br />
The chemical structure of the modified PU foams was characterized in terms of FTIR<br />
proving the natural polymers incorporation into the modified PU foam network (see Fig.1).<br />
The interesting point of view is shown at the FTIR spectra in the range of 2250 – 2400 cm −1<br />
where the peak at 23<strong>60</strong> cm −1 was suppressed and on the other hand the peak at 2280 cm −1 was<br />
highlighted at modified PU foams evidential of forming new bonds between the filler and PU<br />
network. Other peaks corresponded to the urethane linkages.<br />
From the literature and previous experiments 2,3 it is known that modifying of PU foam<br />
with bio-polymer filler up to 10 % has no significant influence on thermal properties of the<br />
modified foam, which was confirmed. The TGA showed that the influence rises with the<br />
increasing bio-polymer filler amount in the foam when is higher than 10 %.<br />
Ecotoxicology<br />
Ecotoxicity of modified and control PU foams leaches was evaluated using alternative<br />
crustacean toxicity test Thamnotoxkit F. Values of toxicity were expressed as percentage<br />
mortality of Thamnocephalus platyurus dependence on the effect criterion of the respective<br />
assay. Dilution series of 100 %, 80 %, <strong>60</strong> %, 40 % and 20 % of the leaches were prepared<br />
according to the procedure of Thamnotoxkit F manual 15 . As for 100 % (non-diluted)<br />
leaches made of 10 % substituted foams, the lowest toxicity (80 %) performed the PU foam<br />
with 10 % CMC-Na. All <strong>60</strong> % diluted leaches showed lower toxicity than the CONTROL PU<br />
foam, except the leach made of 5 % WP-G PU foam. As for other dilution series, also the<br />
most toxic leaches were the CONTROL PU and 2-HEC leaches (see Fig.2).<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 17
% of mortality<br />
4000,0<br />
120,00<br />
100,00<br />
80,00<br />
<strong>60</strong>,00<br />
40,00<br />
20,00<br />
0,00<br />
3500,0<br />
46,67<br />
3000,0<br />
SUBSTITUTED FOAMS - COMPARE ALL<br />
2500,0<br />
Control PUR<br />
10 % CMC-Na<br />
10 % ACS<br />
10 % CA<br />
10 % 2-HEC<br />
1 % WP(G)<br />
2000,0<br />
wavenumber [cm –1 ]<br />
1500,0<br />
Fig.1: FTIR spectra of modified PU foams.<br />
1000,0<br />
500,0<br />
Mortality graph - <strong>60</strong> % (diluted) leachates of various PU foams<br />
43,33<br />
50,00<br />
23,33<br />
56,67<br />
100,00<br />
10 % CMC-Na PU 10 % AS PU 10 % CA PU 10 % 2-HEC PU CONTROL PU 5 % WP-G PU<br />
foam sample<br />
Fig. 2: Percentage mortality graphs of diluted leaches of various modified PU foams.<br />
100,0<br />
95,0<br />
90,0<br />
85,0<br />
80,0<br />
75,0<br />
70,0<br />
65,0<br />
<strong>60</strong>,0<br />
0,0<br />
CONCLUSION<br />
Flexible modified PU foams with 10 % of PEP substituted by CMC-Na, AS, CA and<br />
2-HEC were successfully synthesized. The most suitable bio-polymer fillers for both physical<br />
and ecotoxicological properties were found to be CMC-Na and AS, which wor<strong>ke</strong>d up to 30 %<br />
and 20 % of substitution, respectively. The worst properties presented the PU foam filled with<br />
WP-G.<br />
Moreover, based on our last research 9 the AS PU foam indicates also good<br />
biodegradability.<br />
Further ecotoxicological assessments and new TGA experiments of the modified PU foams<br />
are needed and planned as well as the analysis of the PU foams leaches on Gel Permeation<br />
Chromatography and High Pressure Liquid Chromatography in order to identify the products<br />
of hydrolysis of the foams for better understanding the ecotoxicological assessment.<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 18<br />
transmittance [1]
ACKNOWLEDGEMENT<br />
I would li<strong>ke</strong> to thank to both of my tutors, Dr. Lucy Vojtová and Prof. Milada Vávrová.<br />
Special thanks are going to Dr. Karel Bednařík who gave me good advice and also to the<br />
manager of this research project Prof. Josef Jančář.<br />
This research was supported by the Ministry of Education, Youth and Physical Training of<br />
the Czech Republic under the research project MSM 0021630501.<br />
REFERENCES<br />
1<br />
Schyzer, M.: Schyzer's Handbook of Polyurethanes. CRC Press.<br />
2<br />
Vojtová, L., Zdilna, P., Jancar, J.: Degradace a recyklace polyuretanovych pen.[.doc<br />
document]. Brno: FCH Brno University of Technology, 2004 [cited 07.10.2006].<br />
3<br />
Rivera-Armenta, J. L., Heinze, T., Mendoza-Martínez, A. M.: New polyurethane foams modified with<br />
cellulose derivatives. European Polymer Journal, 2004, vol. 40, pp. 2803-2812. ISSN 0014-3057/$<br />
4<br />
Chang, L.-C., Xue, Y., Hsieh F.-H.: Comparative study of physical properties of water-blown rigid<br />
polyurethane foams with commercial soy flours. Journal of Apllied Polymer Science, 2001, vol. 80 (2001), pp.<br />
10-19.<br />
5<br />
Ge, J., Zhong, W., Guo, Z., Li, W., Sakai, K. : Biodegradable polyurethane materials from bark and starch I.<br />
Highly resilient foams. Journal of Applied Polymer Science, 2000, vol. 77 (2000), pp. 2575-2580.<br />
6<br />
Ge, J., Shi, X., Cai, M., Wu, R., Wang, M.: A novel biodegradable antimicrobial PU foam from wattle<br />
tannin. Journal of Applied Polymer Science, 2003, vol. 90, pp. 2756-2763.<br />
7<br />
Alfani, R., Iannace, S., Nicolais, L.: Synthesis and characterization of Starch-Based Polyurethane Foams.<br />
Journal of Applied Polymer Science, 1998, vol. 78, pp. 739-745.<br />
8<br />
Badri, K. H., Othman, Z. B., Razali I. M.: Mechanical properties of polyurethane composites from oil palm<br />
resources. Iranian Polymer Journal, 2005, vol. 14, no. 5, pp. 441-448.<br />
9<br />
Marova, I., Babak, L., Vavrova, M., Vojtova, L., Jancar, J.: Use of thermophillic bacteria to biodegradation<br />
of modified polyurethane foams, paper presented at The Sixth European Meeting on Environmental Chemistry,<br />
Belgrade, 6.-9. December (2005).<br />
10 th<br />
MicroBioTests Inc.: Background and general information [online]. Last update 30 of March 2006 [cited<br />
2006-10-07]. Available online at: .<br />
11<br />
Persoone, G., Van de Vel, A.: Report EUR 11342 EN, Commission of the European Communities (1987).<br />
12<br />
Latif, M., Licek, E.: Environmental toxicology, 19, 302 (2004).<br />
13 th<br />
MicroBioTests Inc.: Publications about microbiotests [online]. Last update 30 of March 2006<br />
[cited 2006-10-07]. Available online at: .<br />
14<br />
Torokne, A.: Sensitivity evaluation of the Daphtoxkit and Thamnotoxkit microbiotests on blind samples.<br />
Journal of Applied Toxicology, 2004, vol. 24, no. 5, pp. 323-326.<br />
15 TM<br />
MicroBioTests Inc., Belgium. (Ed.): Thamnotoxkit F . Standard Operation Procedure V310303.<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 19
NOVÝ SORPČNÍ GEL NA BÁZI SPHERON-OXINU PRO UŽITÍ<br />
V TECHNICE DGT<br />
Michaela Gregušová, 5. ročník<br />
Vedoucí práce: prof. RNDr. Hana Dočekalová, CSc.<br />
<strong>Vysoké</strong> učení technické v Brně, <strong>Fakulta</strong> <strong>chemická</strong>, ústav chemie a technologie ochrany<br />
životního prostředí, Purkyňova 118, 612 00 Brno, e-mail: xcgregusova@fch.vutbr.cz<br />
ÚVOD<br />
Kvantitativní stanovení chemických forem kovů (specií) přítomných v přírodních vodních<br />
systémech je stále nedořešeným problémem environmentálních chemiků. V průběhu odběru<br />
vzorku a jeho zpracování dochází k fyzikálně-chemickým dějům, které mají za následek<br />
změny v rozdělení specií. Skutečnou situaci odráží některé in situ prekoncentrační postupy.<br />
Mezi tyto postupy patří technika difúzního gradientu v tenkém filmu (DGT), která byla<br />
poprvé popsána v roce 1994 [1]. Technika DGT umožňuje stanovení stopových koncentrací<br />
kovů, radionuklidů a rovněž některých anionů (fosfátů, sulfidů) [2,3] ve vodném prostředí,<br />
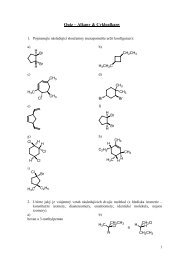
půdách a sedimentech. Technika DGT využívá vzorkovací jednotku tvaru pístu, ve které jsou<br />
uzavřeny dvě vrstvy polyakrylamidového gelu (APA) a membránový filtr (obr.1). Horní<br />
vrstvu tvoří polyakrylamidový hydrogel, přes který difundují ionty analytu. Ve spodní vrstvě<br />
PAM gelu se zakomponovaným iontoměničem se sorbuje analyt.<br />
Obr.1 Řez vzorkovací jednotkou DGT<br />
Technika DGT je založena na Fickových zákonech difúze. Po ponoření vzorkovací<br />
jednotky difundují mobilní ionty kovů přes difúzní gel známé tloušťky Δg k sorpčnímu gelu,<br />
kde se zachytávají na specifických funkčních skupinách sorbentu. V difúzním gelu se ve<br />
velmi krátké době ustaví lineární koncentrační gradient. Jestliže koncentrační gradient zůstává<br />
během měření konstantní (míchané vodné systémy), lze dle prvního Fickova zákona difúze<br />
vypočítat tok analytu F vrstvou Δg dle vztahu:<br />
M D ⋅ c<br />
F = =<br />
(1)<br />
A ⋅ t Δg<br />
kde M je množství kovu navázané na iontoměnič, A je plocha difúzního gelu, D je difúzní<br />
koeficient kovu a c je koncentrace kovu v měřeném roztoku.<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 20
Kovy zachycené na sorbentu jsou eluovány kyselinou dusičnou a stanoveny metodami<br />
atomové spektrometrie. Při stanovení specií kovů technikou DGT je klíčové nalézt vhodný<br />
sorbent a difúzní gel. Technika DGT nejčastěji využívá iontoměnič Chelex 100 s vázanými<br />
funkčními skupinami kyseliny iminodioctové a polyakrylamidový difúzní gel.<br />
Tato práce se zabývá studiem vlastností nového sorpčního gelu s navázanými skupinami<br />
8-hydroxychinolinu (Spheron-Oxin) [4], které mají silné chelatační schopnosti. Sorpční gel<br />
se Spheron-Oxinem přináší nové možnosti pro speciaci labilních i méně labilních komplexů<br />
technikou DGT.<br />
EXPERIMENTÁLNÍ ČÁST<br />
Příprava gelů a jednotky DGT. Difúzní gel i sorpční gel se Spheron-Oxinem byly<br />
připraveny z gelového roztoku, který obsahoval 15%obj akrylamidu (Merck), 0,3%obj<br />
agarosového síťovadla (DGT Research Ltd., Lancaster) v ultračisté vodě. Směs 10 μl<br />
katalyzátoru polymerace TEMED (tetramethylendiamin) zředěného 1:1 ultračistou vodou,<br />
20 μl čerstvě připraveného 8%-ního roztoku peroxosíranu amonného, iniciátoru polymerace, a<br />
2 ml gelového roztoku byla promíchána a nadávkována mezi dvě skla oddělená distanční<br />
teflonovou fólií. Do směsi na přípravu sorpčního gelu s menším obsahem katalyzátoru a<br />
iniciátoru polymerace bylo přidáno 0,4 g Spheron-Oxinu 1000 (Lachema Brno, 40–63 μm).<br />
Směs byla ponechána mezi skly v sušárně po dobu 45 min, kde při teplotě 42 ± 2 °C<br />
zpolymerovala. Sesítěné gely byly hydratovány v ultračisté vodě 24 hodin, kde nabobtnaly<br />
do stabilní tloušťky 0,8 mm u difúzního gelu a 0,4 mm u sorpčního gelu. Několikanásobná<br />
výměna ultračisté vody během hydratace zajišťuje odstranění zbytků polymeračních činidel<br />
z gelů. Po nabobtnání byly z gelů vykrájeny disky o průměru 25 mm. Disky pak byly<br />
uchovávány v ledničce v roztoku 0,01M dusičnanu sodného v případě difúzního gelu<br />
a v ultračisté vodě v případě sorpčního gelu.<br />
Při sestavování jednotky DGT se na píst (obr.1) vložil sorpční gel, který byl pak překryt<br />
difúzním gelem, a nakonec membránovým filtrem (Supor®-450) k ochraně gelů.<br />
Příprava roztoků, testování DGT. Pro přípravu modelových roztoků byly použity<br />
standardní roztoky kovů Astasol®-Pb, Cd, Cu, Ni (Analytika, Praha), dusičnan sodný p.a.<br />
99,8% (Lachema Brno), hydroxid sodný čistý (Onex Rožnov p. R.) a ultračistá voda<br />
připravená přístrojem Milli-Q Academic firmy Millipore. Roztoky byly připraveny zředěním<br />
standardů na příslušné koncentrace. Do plastové nádoby s 2 L ultračisté vody bylo přidáno<br />
20 ml 1M dusičnanu sodného a příslušné množství standardů kovů. Roztok byl neutralizován<br />
0,1M hydroxidem sodným a následně 24 hodin míchán, aby se ustavila rovnováha v roztoku.<br />
Po vnoření DGT jednotek do modelového roztoku bylo odebráno malé množství roztoku<br />
k analýze. Po uplynutí doby expozice byl opět odebrán vzorek roztoku, vyjmuty jednotky<br />
DGT a rozebrány. Sorpční gel byl loužen v 1M kyselině dusičné (Suprapur® Merck) po dobu<br />
24 hodin. Metodou atomové absorpční spektrometrie s elektrotermickou atomizací<br />
(AAnalyst <strong>60</strong>0, Perkin Elmer Analytical Instruments, USA) byly v eluátu, vstupním a<br />
konečném roztoku stanoveny Cd, Cu, Ni, Pb při vlnových délkách 228,8; 324,8; 232,0<br />
a 283,3 nm za teplotního programu doporučeného výrobcem.<br />
Rychlost ustanovení rovnováhy eluce. DGT jednotky byly exponovány 6 hodin v roztoku<br />
o koncentraci 40 ppb každého z kovů Cd, Cu, Ni, Pb. Sorpční gely se Spheron-Oxinem byly<br />
eluovány 1ml 1M kyseliny dusičné v časových intervalech 0,5; 1; 2; 4; 8 a 16 hod.<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 21
Eluční faktor. Ionty kovů byly opakovaně eluovány 1M kyselinou dusičnou ze sorpčních<br />
gelů z jednotek DGT exponovaných po dobu 24 h v roztoku kovů o koncentraci 10 ppb. Mezi<br />
jednotlivými po sobě následujícími elucemi byly gely pro odstranění vnitřního roztoku<br />
louženy ultračistou vodou.<br />
Sorpční kapacita. Z pentahydrátu síranu měďnatého p.a., Lachema Brno byly připraveny<br />
roztoky o koncentraci 25, 51, 146, 279 a 787 mg/l odpovídající 10, 20, <strong>60</strong>, 100 a 300%<br />
teoretické kapacity. Do 2 ml roztoků mědi byly vloženy gely se Spheron-Oxinem po dobu<br />
24 hodin a občas promíchány. Sorbované množství mědi bylo určeno z rozdílů koncentrace<br />
připravených roztoků a roztoků s exponovanými sorpčními gely. Koncentrace mědi byla<br />
stanovena plamenovou atomovou absorpční spektrometrií na přístroji Perkin Elmer 3110 AA.<br />
Vliv pH. V plastové nádobě s 2 L ultračisté vody, dusičnanem sodným a 30 ppb kovů bylo<br />
upravováno pH pomocí kyseliny octové Suprapur® 96% (Merck) a hydroxidu sodného<br />
Suprapur® 30% (Merck). Nejprve bylo pH roztoku sníženo kyselinou, ustalováno po dobu<br />
24 h a nakonec měřeno. V následující periodě 22 h byly v roztoku exponovány jednotky<br />
DGT. Po jejich vyjmutí bylo pH roztoku změřeno a dále postupně zvyšováno přídav<strong>ke</strong>m<br />
hydroxidu sodného a celý proces záchytu kovů v jednotkách DGT byl opakován.<br />
VÝSLEDKY<br />
Při přípravě sorpčního gelu se Spheron-Oxinem bylo nejprve optimalizováno množství<br />
sorbentu v gelovém roztoku. Ideální množství Spheron-Oxinu mělo být co největší z důvodu<br />
dostatečné kapacity disků, ale zároveň takové aby byla možná příprava homogenního gelu.<br />
Nalezené optimální množství činilo 0,4 g Spheron-Oxinu na 2 ml gelového roztoku.<br />
Při přípravě sorpčního gelu se částice Spheron-Oxinu usazovaly <strong>ke</strong> spodní straně odlévací<br />
skleněné formy, což vedlo k nehomogennímu rozdělení sorbentu v gelu. Bobtnání<br />
Spheron-Oxinu v gelovém roztoku po dobu minimálně tří dnů před přípravou gelu vedlo<br />
k potlačení tohoto negativního jevu. Pro přípravu homogenního gelu je rovněž důležitá<br />
důkladná homogenizace gelu po přidání iniciátoru polymerace a rychlé vlití polymerující<br />
směsi mezi skla formy.<br />
Pro vysoký obsah mědi a niklu v sorbentu bylo nutné Spheron-Oxin před použitím<br />
přečistit. Z důvodu možné kontaminace gelů během jejich přípravy byly proto vykrájené<br />
disky sorpčního gelu opakovaně několikrát eluovány v 1M kyselině dusičné (Suprapur®<br />
Merck) a následně několikrát hydratovány v ultračisté vodě, aby se zbytky kyseliny odstranily<br />
z vnitřního roztoku gelu.<br />
U připravených disků sorpčního gelu byl stanoven mrtvý objem a zároveň tloušťka gelu<br />
dvěma způsoby. Z úbytku hmotnosti gelů při sušení, který činil 0,19 g, a z průměru disků gelu<br />
25 mm byla odvozena tloušťka 0,4 mm. Přímo byla mikrometrem změřena tloušťka gelu,<br />
vloženého mezi dvě mikroskopická sklíčka. Přímé odečítání tloušťky bylo však<br />
komplikováno obtížností nastavení příslušného přítlaku na hlavičkách mikrometrického<br />
šroubu, při kterém již nedocházelo k nežádoucí deformaci (stlačování) gelu. Mikrometrem<br />
byla odečtena tloušťka 0,4 mm.<br />
Dalším zkoumaným parametrem byla rychlost ustanovení rovnováhy při eluci. Kinetika<br />
eluce kovů kyselinou dusičnou je procesem velmi rychlým. Během půl hodiny dochází<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 22
k vyloužení cca 90% nasorbovaných kovů. Z toho vyplývá, že i velmi krátká doba eluce<br />
30 min postačuje k ustanovení rovnováhy.<br />
Eluční faktor vyjadřuje jaké množství (M1) z celkového sorbovaného množství (ΣM) se<br />
vyluhuje v jednom elučním kroku do roztoku 1M kyseliny dusičné:<br />
M1<br />
fe = (2)<br />
M + M + M + M<br />
1<br />
2<br />
3<br />
4<br />
Stanovení elučního faktoru bylo nezbytné pro následné ověření funkce techniky DGT<br />
s novým sorpčním gelem. Eluční faktor nabývá hodnot pro Ni 0,86; Cd 0,98; Pb 0,93 a Cu<br />
0,71.<br />
Sorpční kapacita má spíše informační charakter pro analytickou praxi. Slouží k odhadu<br />
přípustného zatížení a doby expozice. Sorpční kapacita disků Spheron-Oxinu pro měď činila<br />
3,2 μmol/disk. Tato hodnota je nižší oproti teoretické hodnotě 8 μmol/disk. Kapacita je však<br />
dostatečně vysoká pro několikatýdenní až několikaměsíční vystavení sorpčního gelu do<br />
přírodních vodních systémů.<br />
Vliv kyselosti vnějšího roztoku na záchyt Cd, Ni a Cu byl sledován v rozsahu hodnot<br />
pH 3-8. Z výsledků vyplývá, že sorpční gel se Spheron-Oxinem váže silně sledované kovy,<br />
což může být při srovnání s tradičním sorpčním gelem Chelex 100 s výhodou využito<br />
k charakterizaci labilních a méně labilních komplexů kovů v přírodních vodních systémech.<br />
Po nalezení všech pomocných parametrů jako jsou tloušťka sorpčního gelu, eluční faktor,<br />
sorpční kapacita a vliv pH mohla být ověřena funkce nového sorpčního gelu pro techniku<br />
DGT výpočtem koncentrace vnějšího roztoku z množství kovů získaných elucí sorpčního<br />
gelu. Nalezené hodnoty technikou DGT odpovídaly pro Ni 92 ± 4 % a pro Cd 98 ± 4 %<br />
očekávaných hodnot. Z toho lze usoudit, že technika DGT s využitím sorpčního gelu na bázi<br />
sorbentu Spheron-Oxin může poskytovat spolehlivé výsledky.<br />
LITERATURA<br />
Davison W., Zhang H., Nature 367, 546 (1994)<br />
Zhang H., Davison W., Gadi R., Kobayashi T., Anal. Chim. Acta, 370, 29-38 (1998)<br />
Teasdale, Hayward S., Davison W., Anal. Chem. 71, 2186-2191 (1999)<br />
Slovák Z., Bulletin n.p. Lachema Brno, 30-34 (1979)<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 23
MOLECULAR DYNAMICS SIMULATION OF POLYMER CHAIN<br />
IN THE VICINITY OF NANOPARTICLE<br />
Kateřina Hynštová 1<br />
Advisor: prof. RNDr. Josef Jančář, CSc. 1 , Consultant: Mgr. Jan Žídek, Ph.D. 1<br />
1 Institute of Materials Science, School of Chemistry, Brno University of Technology,<br />
Purkynova 118, Brno, Czech Republic, e-mail: xchynstova@fch.vutbr.cz<br />
KEYWORDS Molecular dynamics, Rouse, Smoluchowski equation, viscoelasticity, model.<br />
ABSTRACT<br />
Molecular simulation of single chain in the vicinity of nanoparticle in comparison with pure<br />
system is presented. According to the Rouse theory, chains were considered as a sequence of<br />
beads connected together by harmonic springs. The motion of atoms was supported by thermal<br />
energy and retarded by the resistance of surrounding. New atom position, in given time, was<br />
determined by the Smoluchowski equation, that consists of two terms: first one includes the<br />
influence of the inter-atomic collisions, the sterical obstacles and the strong intermolecular<br />
interactions in friction coefficient, second one express the energy field aggregated from<br />
potentials of all atoms. Sinusoidal shear stress was applied to the chain. The output of the model<br />
was energy as a function of time. The energy course was also sinusoidal but shifted according<br />
to the deformation. The amplitudes and phase shifts were analyzed for the chains under<br />
different conditions .The chains were subjected to the model first as the standalone objects.<br />
Then, barrier was defined and chains placed in the vicinity of it. The barrier acted as a volume<br />
excluded hindrance. This type of chain molecular dynamics could be used as a stand-alone<br />
model or it could be suitable component for complex models, for example network model of<br />
polymer nanocomposite.<br />
INTRODUCTION<br />
The need for new materials has driven an increased interest in understanding the changes in<br />
structural, dynamic and melt properties caused by fillers and more recently, nanofillers. A<br />
considerable effort was devoted to understanding of viscoelastic properties of the materials.<br />
Phenomenological approach is not sufficient any longer; therefore the studying of the materials<br />
on multiple length scales is useful [1].<br />
Many efforts have been made on description of polymer behavior at a molecular level. Even<br />
though instrumental methods have improved, there are still problems with exact measurement<br />
of the structural and mechanical properties at this level. Hence, computer simulations ta<strong>ke</strong><br />
place in this area being very powerful tool that enable direct insight into investigated system<br />
[2].<br />
To define single chain motion in viscous surrounding the Rouse model is widely used [3,4].<br />
Atoms or atom groups are considered as rigid beads connected together by harmonic springs.<br />
The diffusion coefficient of surroundings and the potential of the atom group determine its<br />
position.<br />
The simulation progressed from chains in polymer melt that are sufficiently far from filler<br />
across the glassy polymer to the chains in the interphase layer vicinity of particle. Two factors<br />
influenced the relation between the particle and chain. The first factor was excluded volume<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 24
which caused immobilization of chain. This factor was observed rather in nanocomposites than<br />
in the microcomposites. For example, adding 1% by weight of ultra-fine, synthetic mica<br />
(30-nm diameter disks) to nylon gave super tough nylon, while adding the same amount of<br />
traditional mica (micron sized talc) gave only a slight improvement in toughness over unfilled<br />
polymer. Second factor was an acting of interactions, which were based on both physical and<br />
chemical mechanisms and their presence could cause diametrically different polymer response<br />
[5].<br />
SIMULATION<br />
An application of ROUSE model was used to model viscoelastic response of the polymer<br />
chain. The chain was composed of single atoms which are distributed in space. Each atom<br />
position was limited by position of the neighbor atoms. System tried to achieve thermodynamic<br />
equilibrium, but its motion was retarded. To trace the position of atoms in time the Newtonian<br />
and Smoluchowski equations were solved. For non-interacting particle undergoing the thermal<br />
motion, the probability for finding the particle at x and t as ψ ( x, t)<br />
follows from<br />
∂ψ<br />
∂ 1 ⎛ ∂ψ<br />
∂V<br />
⎞<br />
= ⎜kT<br />
+ ψ ⎟<br />
(1)<br />
∂t<br />
∂x<br />
ς ⎝ ∂x<br />
∂x<br />
⎠<br />
where resistance of surroundings is expressed in first member, energy influence including<br />
dihedral, bending and bond stretch potential are involved by latter one[8]. Potential functions<br />
and parameters adopted are proposed by Ryckaert-Bellemans for 4-body dihedral (torsion)<br />
potential V(φ) [6] and by by for 2-body bond length potential V(b) and 3-body bond angle<br />
potential V(θ), respectively [7], as shown in equations with parameters listed below in Table 1.<br />
5<br />
1<br />
2<br />
1<br />
2<br />
i<br />
V ( b)<br />
= kb<br />
( b − b0<br />
) V ( θ ) = kθ<br />
( θ −θ<br />
0 ) V ( ϕ)<br />
= Ci<br />
cos ( ϕ)<br />
(2,3,4)<br />
2<br />
2<br />
Table 1: Parameters of Ryckaert-Bellemans multinominal [6] and Smit, Karaborni and<br />
Siepmann equations [7] for the polyethylene chain<br />
Parameter Value Units<br />
kb (bond) 3.3475 10 5 kJmol -1<br />
nm -2<br />
b0 0.153 Nm<br />
kθ(bending) 519.6 kJ mol -1 rad -2<br />
θ0 114 Deg<br />
C0(torsion) 9.2789 kJ mol -1<br />
C1<br />
C2<br />
C3<br />
C4<br />
C5<br />
RESULTS AND DISCUSSION<br />
12.156<br />
-13.120<br />
-3.0597<br />
26.24<br />
-31.495<br />
A set of 10 polyethylene chains with 50 segments was generated. A sinusoidal shear<br />
deformation was applied to each chain and its energy response was analyzed. The amplitude of<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 25<br />
∑<br />
i=<br />
0
the deformation was 5% of the chain length and the frequency was 1,5,10 and 20 Hz<br />
respectively. Temperature was 298.15 K. The friction coefficient was 0.05. Parameters of the<br />
polymer chain were ta<strong>ke</strong>n from Table 1. Deformation response is shown in the Figure 1. The<br />
amplitudes were ta<strong>ke</strong>n as the height of energy peak and the phase shift was a shift between<br />
course of deformation and the energy. The results are shown in Table 2. In the second part, the<br />
half space barrier was defined. The chains were set in the vicinity of the barrier so its centers of<br />
gravity were in the distance 0.2, 0.3, and 0.4 nm from the surface (Figure 2). The chains were<br />
deformed with frequency 5 Hz. The calculated data were compared with measured data for the<br />
polyethylene and polyethylene/starch composites [8].<br />
Deformation(nm)<br />
1,0640<br />
1,0635<br />
1,0630<br />
1,0625<br />
1,0620<br />
1,0615<br />
1,0610<br />
1,0<strong>60</strong>5<br />
0,0 0,1 0,2 0,3 0,4<br />
Time(s)<br />
0,0045<br />
0,0040<br />
0,0035<br />
0,0030<br />
0,0025<br />
Figure 1: Deformation and energy course of sinusoidal deformation of set of 50 segments<br />
chains with frequency 5Hz, amplitude 0.05, T=298,15K, friction 0.05<br />
Amplitude of Energy (10 -15 J)<br />
1,4<br />
1,2<br />
1,0<br />
0,8<br />
0,6<br />
0,2 0,3 0,4 0,5 0,6<br />
Distance of chain from barrier (nm)<br />
Figure 2: Influence of barrier on the mechanical response of single chain. The<br />
mechanical response was represented by energy amplitude and phase shift.<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 26<br />
0,6<br />
0,5<br />
0,4<br />
0,3<br />
0,2<br />
0,1<br />
0,0<br />
tg(δ)<br />
Energy(pJ)
Table 2: Amplitudes and phase shifts of the energy course for different frequencies;<br />
M – results from our model; E – experimental values from the ref [9].<br />
Frequency (Hz) Amplitude E (pJ) Phase shift: tg δ<br />
1 M– 50 segment PE chain 0.00150 ± 0.00002 0.063 ± 0.006<br />
5 M 0.00085 ± 0.00002 0.161 ± 0.01<br />
10 M 0.00139 ± 0.00004 0.203 ± 0.012<br />
20 M 0.00109 ± 0.00001 0.042 ± 0.002<br />
1 E- (LLDPE) - 0.126<br />
1 E- (composite LLDPE/Starch <strong>60</strong>/40 wt) - 0.100<br />
CONCLUSIONS<br />
Sinusoidal deformation of single chain was modeled. Energy response of the model was<br />
analyzed. Amplitudes of the energy and phase shifts were ta<strong>ke</strong>n from average energy course of<br />
ten chains ensemble. We found that the energy was shifted to the deformation of the model<br />
chain, as it was observed in real polymer. In comparison to real polyethylene, our calculated<br />
phase shift by the same frequency was still slightly small, but comparable. The phase shift<br />
increased by the frequencies 5-10 Hz. It was because not all of many parameters were in their<br />
confidence intervals. The correct viscoelastic response might be in a short interval of each<br />
parameter and its right combination should be determined.<br />
The impact of barrier in the vicinity of the chain was detectable. The energy amplitude in the<br />
vicinity of barrier increased by 50 percent from the amplitude of untreated chain, but in reality<br />
the modulus of the polymer increases orderly when it is adsorbed on the solid surface. The<br />
calculated phase shift of one sample increased and of another sample it decreased in<br />
comparison to the chain without the barrier. In reality, the phase shift of the composite is<br />
slightly lower than the phase shift of the pure polymer. The models reasonably described the<br />
behavior of real polymer chain. However, further improvement has been desirable. Moreover,<br />
the properties depend on the supermolecular structure, which would be introduced to the<br />
model.<br />
ACKNOWLEDGEMENT<br />
This research was supported by Ministry of Education of the Czech Republic under research<br />
project MSM 0021630501<br />
REFERENCES<br />
[1] F.W. Starr and S. C. Glotzer: Mat. Res. Soc. Symp. Proc. 661 (2001), p. KK4.1.1.<br />
[2] D. Fren<strong>ke</strong>l, B. Smit: Understanding Molecular Simulation (Computational Science Series,<br />
Vol 1) Academic Press; 2 nd edition, USA (2001).<br />
[3] P.E. Rouse: J. Chem. Phys. 21 (1953) p.1273.<br />
[4] Yn-Hwang L.: Polymer viscoelasticity: Basics, molecular theories and experiments,<br />
World Scientific Publishing Co. Pte. Ltd., Singapore (2003).<br />
[5] F.W. Starr, T.B. Schrødr and S. C. Glotzer: Macromolecules 35 (2002), p. 4481.<br />
[6] J.P. Ryckaert and A. Bellemans: Mol. Phys. 44 (2001) p. 68.<br />
[7] B. Smit, S. Karaborni and J. I. Siepmann: J. Chem. Phys. 2126 (1995) p. 102.<br />
[8] A.G. Pedroso and D.S. Rosa: Carbohydrate Polymers 59 (2005) p. 1.<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 27
APPLIKÁCIA NOVEJ EURÓPSKEJ NORNY NA STANOVENIE Cr(VI)<br />
V CEMENTE<br />
Tomáš Ifka, 4. ročník<br />
Vedúci práce: Doc. Dr. Ing. Martin Palou<br />
Oddelenie <strong>ke</strong>ramiky, skla a cementu, Ústav anorganic<strong>ke</strong>j chémie, technológie a materiálov,<br />
FCHPT STU Bratislava, Radlinského 9, 812 37 Bratislava, Slovenská Republika,<br />
e-mail: ifkatomas@centrum.sk<br />
ÚVOD<br />
Chróm je neodstrániteľný stopový prvok surovinového materiálu používaného vo výrobe<br />
cementárs<strong>ke</strong>ho slinku. Vyskytuje sa v prírodných surovinách (íloch, vápencoch a najmä<br />
železitých prísadách) vo forme Cr(III) a vo vymurovkách cementárs<strong>ke</strong>j rotačnej pece. V<br />
cementárskych peciach sa však pri vyso<strong>ke</strong>j teplote, pri oxidačnej atmosfére a alkalic<strong>ke</strong>j<br />
podmien<strong>ke</strong> mení neškodný trojmocný chróm na šesťmocnú formu.<br />
Problematika obsahu chrómu v cemente je dnes vysoko aktuálna. Mnohé štáty, ako napríklad<br />
Nemecko či škandinávs<strong>ke</strong> krajiny, sprísňujú hygienické požiadavky na cementy najmä z<br />
hľadiska obsahu vodorozpustného chrómu. Škandinávska receptúra a Nemecké pravidlá pre<br />
nebezpečné materiály považujú 2 ppm Cr VI za maximálnu bezpečnú hodnotu na zabránenie<br />
výskytu ekzémov u murárov.<br />
Vedecké štúdie taktiež ukázali, že zlúčeniny Cr VI v cemente majú vysokú rozpustnosť vo<br />
vode a tak môžu ľahko prísť do kontaktu s pokožkou murárov. V samotnom Nemecku, 400<br />
murárov malo poškodenú pokožku zapríčinenú chrómanom (1997). Šesťmocný chróm, v<br />
podobe vodorozpustných chrómanov, môže spôsobiť precitlivené reakcie a alergické ekzémy<br />
pri priamom kontakte s pokožkou ľudí. Nebezpečenstvo hrozí aj z ich toxických a potenciálne<br />
karcinogénnych účinkov. Každé použitie cementu so sebou nesie riziko priameho dlhšieho<br />
styku s ľudskou pokožkou s výnimkou kontrolovaných uzatvorených a úplne<br />
automatizovaných procesov. Na predchádzanie kontaktu cementu s pokožkou sú potrebné, ale<br />
nie postačujúce, individuálne ochranné opatrenia. Z toho vyplýva, že na ochranu zdravia ľudí<br />
je potrebné obmedziť používanie cementu s vysokým obsahom vodorozpustného Cr(VI).<br />
Malo by sa obmedziť najmä uvádzanie na trh a používanie cementu alebo cementových<br />
stavebných materiáloch, ktoré obsahujú viac ako 2 ppm šesťmocného chrómu v prípade<br />
činností, kde existuje možnosť kontaktu s pokožkou. Preto podľa nariadenia Európs<strong>ke</strong>ho<br />
parlamentu platného od septembra 2001 je nevyhnutné označením na obale výrobku<br />
upozorniť zákazníkov kupujúcich cementy, resp. cement obsahujúce výrobky (suché<br />
omietkové zmesi, malty, betónové zmesi, cementové lepidlá, tmely a podobne) s obsahom<br />
vodorozpustného Cr(VI) nad 2 ppm, že ide o zdraviu škodlivý výrobok. Pri kontrolovaných<br />
uzavretých alebo úplne automatizovaných procesoch to nie je potrebné a preto im môže byť<br />
udelená výnimka.<br />
V rámci opatrení mnohé cementárs<strong>ke</strong> firmy vyvíjali a používajú činidlá na redukciu<br />
Cr(VI) v cemente. Medzi ne patria heptahydrát síranu železnatého, monosal, siderox alebo<br />
zlúčeniny na báze zeolitu. Cieľom použitia týchto činidiel je potlačiť obsah Cr(VI) na<br />
hodnotu pod 2 ppm. Intenzita redukcie môže závisieť od typu cementu, od činidla a jeho<br />
koncentrácií. Redukčné činidlá by sa mali použiť podľa možnosti čo najskôr, t.j. v mieste<br />
výroby cementu.<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 28
Cieľom projektu je:<br />
• aplikovať nový harmonizovaný európsky postup na stanovenie Cr(VI) v cemente<br />
• skúmať účinok redukčných činidiel na cementoch pripravených v závode<br />
Rohožník (Holcim Slovensko a.s.)<br />
• dlhodobý monitoring obsahu Cr(VI) v cementoch (Rohožník Holcim Slovensko<br />
a.s.) pred a po použití redukčných činidiel<br />
• zistiť začiatok účinkovania redukčných činidiel<br />
• zistiť dobu starnutia redukčných činidiel.<br />
Na dosiahnutie vytýčených cieľov boli použité dve redukčné činidlá ( heptahydrát síranu<br />
železnatého a monohydrát síranu železnatého -monosal) a cementy pripravené v závode<br />
Rohožník Holcim Slovensko a. s. (CEM I 42, 5 R, CEM II 32,5/ B-S)<br />
Eliminácia toxických účinkov Cr (VI) obsiahnutých v cemente<br />
Problematikou imobilizácie toxických látok, zameranú na látky obsahujúce zlúčeniny<br />
Cr(VI) sa už dlhšiu dobu zaoberajú pracovníci OKSC FCHPT STU. Tieto látky sa považujú<br />
za príčinu kožných ekzémov a rozpustným zlúčeninám sa pripisujú aj karcinogénne účinky.<br />
Problematika zvýšeného obsahu Cr(VI) v cementoch sa rieši dvoma zásadnými<br />
postupmi<br />
1) je snaha znížiť celkový obsah chrómu už vo vstupnej surovinovej zmesi, čím sa<br />
dosiahne primárne zníženie obsahu chrómu vo výslednom produkte.<br />
2) sekundárne zníženie obsahu zlúčenín Cr(VI) v cemente možno dosiahnuť prídavkom<br />
aktivovaných železnatých solí, ktoré zabezpečujú redukciu vylúhovateľných škodlivých<br />
zlúčenín Cr(VI) na prakticky vo vode nerozpustné a zdraviu neškodné zlúčeniny Cr(III),<br />
pevne zabudované do matrice zhydratovaného cementu.<br />
Síran železnatý ako redukčné činidlo Cr VI<br />
Síran železnatý kryštalizuje z vodných roztokov zvyčajne so siedmimi molekulami vody<br />
ako ,,zelená skalica“ - FeSO4 . 7 H2O . Tvorí svetlozelené jednoklonné kryštály hustoty 1,88<br />
g.cm -3 , ktoré na vzduchu pomaly zvetrávajú a súčasne podliehajú oxidácii na žltohnedý<br />
zásaditý síran železitý. Pri zahrievaní ľahko stráca zelená skalica šesť molekúl vody; omnoho<br />
ťažšie sa oddeľuje posledná molekula. Pri ešte väčšom zahrievaní dochádza k rozkladu s<br />
odštepovaním oxidu siričitého:<br />
2Fe II SO4 = (Fe III O)2SO4 + SO2<br />
Technicky sa pripravuje zelená skalica buď rozpúšťaním železa v zriedenej kyseline<br />
sírovej alebo sa železný kyz nechá vetrať na vzduchu za častého vlhčenia.<br />
Zelená skalica sa získava ako vedľajší produkt pri výrobe kremenca chromitého. Používa<br />
sa pri výrobe atramentu, berlíns<strong>ke</strong>j modrej, vo farbiarstve, na konzervovanie dreva, na ničenie<br />
buriny, tiež príležitostne v lekárstve. Čistí sa zrážaním z vodných roztokov liehom.<br />
V prírode sa zelená skalica vyskytuje v malých množstvách v podobe výkvetu na železnej<br />
rude (pyrite).<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 29
Princíp redukcie<br />
Cr 6+ + Fe 2+ → Cr 3+ + Fe 3+ .<br />
Cr 6+ + 3e - → Cr 3+<br />
3Fe 2+ - 3e - → 3Fe 3+<br />
Cr 6+ + 3Fe 2+ → Cr 3+ + 3Fe 3+<br />
EXPERIMENTÁLNA ČASŤ<br />
Príprava vzoriek<br />
- Priemyselné vzorky. Vzorky boli pripravené v závode s prídavkom 0,3 %<br />
heptahydrátu síranu železnatého.<br />
- Laboratórne vzorky. Vzorky boli pripravené prídavkom redukčných činidiel – monosal<br />
a heptahydrát síranu železnatého v koncentráciách uvedených v tabuľ<strong>ke</strong> 1. 500g vzorky<br />
s príslušnou koncentráciou boli dokonale homogenizované.<br />
- Príprava vzoriek na stanovenie Cr(VI):<br />
Na stanovenie vodorozpustného Cr(VI) v cemente sme postupovali podľa Európs<strong>ke</strong>j normy<br />
prEN196-10:2004. Paralelne sme pripravovali vzorky bez štandardného piesku.<br />
Stanovenie bolo realizované pomocou UV – VIS Spektrofometer HEλIOS.<br />
VÝSLEDKY A DISKUSIA<br />
1. Účinok monosalu a heptahydrátu síranu železnatého pri rôznych koncentráciách na<br />
CEM I 42,5 R.<br />
Hlavným cieľom je, aby obsah vodorozpustného Cr(VI) v cemente bol pod 2ppm. Ak<br />
porovnáme účinok monosalu a heptahydrátu síranu železnatého pri rovna<strong>ke</strong>j koncentrácii (tab.<br />
č.1), zistíme že s použitím heptahydrátu síranu železnatého je obsah Cr (VI) vo vzor<strong>ke</strong><br />
cementu nižší ako v prípade monosalu. Pri koncentrácii 0,1 hm. % monosalu bol obsah Cr<br />
(VI) v danej vzor<strong>ke</strong> takmer ekvivalentný množstvu Cr(VI) stanovenému vo vzor<strong>ke</strong> bez<br />
redukčného činidla. Účinok monosalu po týždni pri tejto koncentrácii je zanedbateľný.<br />
Z hľadiska dlhodobého účinku redukčného činidla ako aj z ekonomického hľadiska je podľa<br />
uvedených výsledkov (tab. č.1) vhodné používať na redukciu vodorozpustného Cr (VI)<br />
heptahydrát síranu železnatého pri koncentrácii 0,3 hm. %.<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 30
Tab. č.1: Výsledky analýzy Cr(VI) v CEM 42,5 R pri aplikácii redukčných činidiel s rôznou<br />
koncentráciou<br />
CEM I 42,5R<br />
Chemické zloženie [ hm. %]<br />
CaO SiO2 Al2O3 Fe2O3 MgO SO3 Na2O K2O Troska<br />
62,52 22,01 4,95 2,96 2,48 2,74 0,4 0,75 6,6<br />
Stanovenie → 1 týždeň od prípravy vzoriek<br />
pH1 pH2 c [mg/l] A K [ppm] Kkor [ppm]<br />
Neredukovaný 12,92 2,39 0,4782 0,377 2,3910 2,5<strong>60</strong>0<br />
Red. 0,1% FeSO4.7H2O 12,94 2,29 0,3292 0,259 1,64<strong>60</strong> 1,7623<br />
Red. 0,3% FeSO4.7H2O 12,63 2,29 0,0801 0,063 0,4005 0,4288<br />
Red. 0,5% FeSO4.7H2O 12,86 2,28 - - - -<br />
Red. 0,1% monosal 12,70 2,49 0,4187 0,330 2,0935 2,2414<br />
Red. 0,3% monosal 13,20 2,42 0,2139 0,169 1,0695 1,1451<br />
Red. 0,5% monosal 13,02 2,37 0,1413 0,111 0,7065 0,7564<br />
pH1 - pred prídavkom HCl, pH2 - po prídavku HCl, c - koncentrácia Cr(VI) z kalibračnej<br />
krivky v mg/l, A – absorbancia, K - obsah vodorozpustného Cr(VI) v cemente s obsahom<br />
trosky, Kkor - obsah vodorozpustného Cr(VI) v čistom cemente<br />
2. Začiatok pôsobenia redukčného činidla na Cr(VI)<br />
Na zistenie začiatku účinkovania redukčného činidla bol použitý heptahydrát síranu<br />
železnatého pri dvoch koncentráciách 0,3 a 0,5 hm. %. Stanovoval som obsah<br />
vodorozpustného Cr(VI) po dobu 1, 4 a 24 hodinách. Výsledky sú uvedené v grafe č.1, kde je<br />
evidentné, že po 1 jednej hodine sa obsah Cr(VI) redukoval na minimálnu hodnotu. To<br />
znamená, že heptahydrát síranu železnatého pri koncentrácii 0, 3 a 0,5 hm. % začína pôsobiť<br />
už po jednej hodine aplikácie.<br />
Tab. č.2: Chemické zloženie vzorky CEM II/ B-S 32,5R<br />
CEM II/ B-S 32,5R<br />
Chemické zloženie [ hm.%]<br />
CaO SiO2 Al2O3 Fe2O3 MgO SO3 Na2O K2O Troska<br />
56,20 27,44 6,02 2,57 3,64 2,77 0,45 0,77 28,1<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 31
K [ppm]<br />
4<br />
3.5<br />
3<br />
2.5<br />
2<br />
1.5<br />
1<br />
0.5<br />
0<br />
CEM II / B-S 32,5R<br />
1 4 24<br />
t [hod]<br />
nered.<br />
red. 0,3% hepta<br />
red. 0,5% hepta<br />
Graf č.1: Začiatok pôsobenia redukčného činidla na Cr(VI)<br />
3 Doba starnutia redukčného činidla<br />
Na určenie doby pôsobenia redukčného činidla som použil cement CEM II/B-S 32,5R, do<br />
ktorého som pridal 0,5 hm. % heptahydrátu síranu železnatého. Po stanovení<br />
vodorozpustného Cr (VI) v tejto vzor<strong>ke</strong> cementu som zistil, že pri danej koncentrácii<br />
redukčného činidla je obsah Cr (VI) vo vzor<strong>ke</strong> veľmi nízky a to aj po 65. dňoch. Pretože<br />
obsah stanoveného Cr(VI) v cemente je veľmi nízky, kolísanie výsledkov je pravdepodobne<br />
spôsobené chybou pri meraní. 44 dní od prípravy danej vzorky sa začína Cr (III) spätne<br />
oxidovať na Cr(VI).<br />
Tab. č.3: Chemické zloženie vzorky cementu CEM I 42,5R<br />
CEM II/ B-S 32,5R<br />
Chemické zloženie [ hm. %]<br />
CaO SiO2 Al2O3 Fe2O3 MgO SO3 Na2O K2O Troska<br />
55,98 27,16 5,82 2,32 4,27 2,55 0,44 0,78 29,4<br />
K [ppm]<br />
3<br />
2.5<br />
2<br />
1.5<br />
1<br />
0.5<br />
0<br />
CEM II / B-S 32,5R<br />
9 16 44<br />
t[dni]<br />
51 65<br />
nered.<br />
red. 0,5% hepta<br />
Graf č.2: Monitorovanie doby starnutia redukčného činidla za obdobie 65 dní<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 32
ZÁVER<br />
Podľa smernice 76/769/EHS Európs<strong>ke</strong>j Rady, každý balený cement musí obsahovať<br />
Cr(VI) pod 2 ppm. Z toho vyplýva, že cementársky priemysel musí vyvíjať metódy redukcie<br />
a kontroly obsahu Cr(VI) vo svojich výrobkoch. Aplikácia novej normy na stanovenie<br />
Cr(VI) v cemente bola na konktrétnych cementoch vyrábaných v závode Rohožník Holcim<br />
Slovensko, a.s. V práci som použil dve redukčné činidlá (heptahydrát a monohydrát síranu<br />
železnatého) a sledoval som ich účinok na Cr(VI) podľa koncentrácie (0,1; 0,3; 0,5 hm. %)<br />
a doby starnutia do troch mesiacov.<br />
Z laboratórnych výsledkov stanovenia Cr(VI) v cemente vyplýva:<br />
- Monosal začína účinkovať až pri (redukovať Cr(VI) pod 2 ppm) koncentrácii 0,3 hm. %.<br />
Pri koncentrácii 0,1 hm. % očakávaný efekt nebol dosiahnutý.<br />
- Heptahydrát síranu železnatého účinkuje už pri koncentrácii 0,1 hm. %<br />
- Výrazný účinok obidvoch redukčných činidiel je pri koncentrácii 0,5 hm. %<br />
- Redukčné činidlo začína pôsobiť už do jednej hodiny.<br />
- Po 45. dňoch začína spätná oxidácia redukovaného Cr(III) na Cr(VI). Obsah<br />
vodorozpustného Cr(VI) rastie s časom veľmi pomaly, takže sa nepredpokladá<br />
dosiahnutie kritic<strong>ke</strong>j hodnoty 2 ppm.<br />
- Monitorovanie obsahu Cr(VI) v cemente pred a po pridaní redukčného činidla prinieslo<br />
tieto výsledky:<br />
- Obsah Cr(VI) v cemente sa bez redukčného činidla menil podľa dátumu výroby.<br />
- Účinky redukčného činidla neboli v niektorých prípadoch uspokojivé, lebo obsah Cr(VI)<br />
nebol pod 2 ppm.<br />
- Aj v prípade vzoriek cementov zo závodu som zistil starnutie redukčného činidla<br />
(spätná oxidácia Cr(III) na Cr(VI)).<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 33
DISSOLUTION OF HUMIC SUBSTANCES IN UREA IN LIGHT OF<br />
SUPRAMOLECULAR THEORY<br />
Jiří Kislinger, 5 th year<br />
Supervisor: Prof. Alessandro Piccolo*<br />
Brno University of Technology, Faculty of Chemistry, Institute of Physical and Applied<br />
Chemistry, Purkyňova 118, 612 00, Brno, e-mail: xckislinger@fch.vutbr.cz<br />
* Università degli Studi di Napoli Federico II, Facoltà di Agraria, Dipartimento di Scienze del<br />
Suolo, della Pianta e dell’Ambiente, Via Università 100, 800 55, Portici, Italy<br />
INTRODUCTION<br />
Humic substances (HS) are natural organic compounds present in all terrestrial and aquatic<br />
systems. Processes of their accumulation and decomposition in different environmental<br />
domains are still largely unclear [1]. Nevertheless, the characteristics and quantity of HS<br />
greatly affect the environmental fate of organic pollutants in soils and natural waters [2].<br />
Molecular properties of dissolved HS have recently been recognized to influence removal of<br />
synthetic organic compounds from municipal and industrial wastewaters [3], and give rise to<br />
binding and environmental transport of both nonpolar and slightly polar organic contaminants<br />
[4].<br />
Despite their prominent environmental role, the chemical structure of HS is still far from<br />
clear mainly because of their chemical heterogeneities and spatial variabilities. As a<br />
consequence the conformational structures of HS are also ill-defined. There is a general<br />
acceptance of the concept that HS are polydisperse, long-chain, randomly coiled molecules<br />
that may have a slight degree of crosslinking [5]. Negative charges arising from ionization of<br />
carboxyl groups are responsible for mutual repulsion and expansion of the coils [6]. The<br />
random coil model depicts HS as most densely coiled at high concentrations, low pH and high<br />
ionic strength, and as linear colloids at neutral pH, low ionic strength and low concentrations<br />
[7]. This model has helped several investigators to explain results obtained by gel permeation<br />
chromatography [8], or by diffusion through ultrafiltration membranes [9]. A common<br />
observation was that elution at larger permeation volumes was retarded considerably when the<br />
ionic strength of the mobile phase was increased. The changes were attributed to coiling of<br />
humic molecules with consequent decrease of hydrodynamic radius and enhanced diffusion<br />
through smaller gel pores. The same phenomenon was also observed by varying the ionic<br />
strength of the mobile phase in High Pressure Size Exclusion Chromatography (HPSEC)<br />
experiments with organic colloids from natural waters [10]. Another conformational model<br />
still represents humic materials as polymers but aggregated in micelle-li<strong>ke</strong> or membrane-li<strong>ke</strong><br />
structures [11]. In this description, humic molecules would have inner hydrophobic domains<br />
with structural voids that could entrap hydrophobic organic compounds [12] and quench their<br />
fluorescence activity [13].<br />
Recent results, mainly from low-pressure size-exclusion chromatography experiments,<br />
have indicated that humic aggregates are composed of relatively small subunits weakly held<br />
together by hydrophobic forces [4, 14]. The aggregation of humic material in high molecular<br />
dimensions is considered not to be permanent but found to be able to be reversibly disrupted<br />
by the presence of an organic acid in a rapid change from acidic to alkaline pH. These<br />
findings have suggested that HS in solution would appear to consist of randomly selfassembling<br />
supramolecular associations rather than polymeric coils, as in biological<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 34
macromolecules [15]. This innovative explanation of the conformational behavior of HS had<br />
to be verified in other experimental conditions and HPSEC was utilized for further<br />
investigations. For a number of reasons, HPSEC is more valid chromatographic system than<br />
low-pressure gel permeation [16].<br />
The evidence that humic molecules are suprastructural associations in aqueous solutions is<br />
given by the elution patterns of HS in HPSEC experiments in 8 M urea. Concentrated urea<br />
was successfully employed to disrupt protein-protein interactions and to solubilize<br />
aggregating hydrophobic proteins for their further separation [17]. Thus the aim of this work<br />
was to confirm and deepen the knowledge of behavior of HS in solutions in the term of<br />
supramolecular approach.<br />
EXPERIMENTAL PART<br />
Humic acids were obtained from 3 agricultural soils from: Denmark (Haplic Luvisol,<br />
Roskilde) – DK; Germany (Eutric Luvisol, Munich) – DE; and Italy (Eutric Regosol, Caserta)<br />
– IT. These humic acids were extracted by standard procedures [5] and were subsequently<br />
suspended in distilled water and titrated for 3 hours to pH 7 using CO2–free solution of 0.5 M<br />
NaOH in an atmosphere of N2. The resulting sodium-humates were than filtered through a<br />
Millipore 0.45 μm membrane, and freeze-dried.<br />
The HPSEC system used consists of a Shimadzu LC–10ADVP solvent pump and two<br />
detectors in series, a Perkin–Elmer LC–295 UV/VIS detector set at 280 nm and a refractive<br />
index (RI) detector (Refractomonitor IV, Fisons Instruments). A Rheodyne rotary injector<br />
equipped with a 100 μL sample loop was used to charge the humic solutions. A Biosep SEC–<br />
S–2000 column (300 by 7.8 mm i.d.) was adopted in these experiments. Humate solutions for<br />
HPSEC analysis were prepared by dissolving 15 mg of each sample in 25 mL of the eluent<br />
(8 M urea) to reach concentration 0.6 g×L –1 .<br />
In the UV/VIS spectroscopy experiments the Na-humates were redissolved in 8 M urea to<br />
reach concentration 0.2 g×L –1 . Such solutions were diluted 20 times in 8 M urea. These were<br />
scanned for the light absorbance from 200 to 800 nm in a Perkin–Elmer Lambda 25 UV/VIS<br />
spectrophotometer using quartz cuvettes of 1 cm pathlength against the solvent. Two<br />
replicates for each sample were run to obtain absorbance curves.<br />
RESULTS AND DISCUSSION<br />
The elution profiles of three different purified HS dissolved in 8 M urea and eluted in the<br />
same solution are shown in Figs. 1, 2, and 3. The experiment is illustrative of the capacity of<br />
urea to separate the hydrophobic from the hydrophilic components of HS. The<br />
chromatograms show a net separation of humic matter: a high-molecular-size fraction at the<br />
void volume visible only by the UV detector and a low-molecular-size fraction eluting at the<br />
total volume detected only by the RI detector. The humic hydrophobic molecules are strongly<br />
associated through a hydrophobic effect induced by the highly concentrated urea that interacts<br />
more favorably than hydrophobic molecules with the network of the water structure.<br />
Differences among HS can also be noted. The humic acid from Italian soil was the only<br />
one to show two well-separated peaks at the RI detector system, whereas only one very<br />
intense peak (>1000 mV) at the column void volume was shown by the UV detector (Fig. 1).<br />
This indicates that while light-absorbing chromophores (280 nm) with a high molar<br />
absorptivity were excluded rapidly from the column, their concentration in the humic sample<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 35
was relevant and of similar order of magnitude to the nonabsorbing hydrophilic, probably<br />
ionized, compounds detected by the RI detector at the total exclusion volume.<br />
Fig. 1: UV- and RI-detected HPSEC chromatogram of humic acid from the Italian soil<br />
The HA from the Danish soil gave similar complete separation between the large-size<br />
hydrophobic chromophore fraction and the small-size ionized and hydrophilic components<br />
shown by the RI detector (Fig. 2). However, while the molar absorptivity of the excluding<br />
chromophores was still high (as suggested by an absorption intensity >500 mV), the lack of<br />
corresponding peaks at the RI detector indicated that their concentration in the sample was<br />
much less than that in the hydrophilic components excluded at large elution volumes.<br />
Fig. 2: UV- and RI-detected HPSEC chromatogram of humic acid from the Danish soil<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 36
Identical behavior was shown by the humic acid extracted from the German soil (Fig. 3).<br />
Also for this sample, the chromophore association excluded at the column void volume was<br />
hardly representative of the mass of humic sample since the corresponding peak at the RI<br />
detector was almost irrelevant in comparison to the signal of the small-sized nonabsorbing<br />
hydrophilic constituents.<br />
Fig. 3: UV- and RI-detected HPSEC chromatogram of humic acid from the German soil<br />
All 3 HA samples diluted in 8 M urea were scanned for the light absorbance to gain some<br />
information about the optical properties. In all cases the absorption spectra (Fig. 4) decreased<br />
in an approximately exponential fashion with increasing wavelength and exhibited no distinct<br />
absorption bands. The differences in light absorption of HAs are related to their specific<br />
molecular composition. For the preceding HPSEC experiments the wavelength 280 nm has<br />
verified to be the optimal to monitor the separation of humic matter to excluding moieties.<br />
Fig. 4: Absorbance curves of soil humic acids dissolved in 8 M urea<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 37
CONCLUSION<br />
These findings indicate that humic substances, rather than being polymeric macromolecules,<br />
may be more simply regarded as supramolecular associations of relatively small<br />
heterogeneous molecules stabilized mainly by dispersive forces. The model of dissolved<br />
humic substances based on the self-association of small components rather than on the<br />
polymeric random coil model should contribute to further understanding of the reactivity of<br />
humic substances in many ecological and environmental processes in which they are<br />
involved.<br />
ACKNOWLEDGEMENT<br />
Encouragement for work, help and useful comments of Prof. A. Piccolo and my Italian<br />
friends and colleagues Drs. P. Conte and R. Spaccini during my residence in Naples are<br />
gratefully acknowledged. Great thanks go also to Dr. Jiří Kučerík for his everlasting support.<br />
REFERENCES<br />
[1] Piccolo, A. 1996. Humus and soil conservation, p. 225-264 in Humic substances in<br />
terrestrial ecosystems. Elsevier, Amsterdam.<br />
[2] Carter, C.W., Suffet, I.H. 1982. Environ. Sci. Technol. 16: 735-740<br />
[3] Karanfil, T., Kilduff, J.E., Schlautman, J.A., Weber, W.J. Jr. 1996. Environ. Sci.<br />
Technol. 30: 2187-2201<br />
[4] Piccolo, A., Nardi, S., Concheri, G. 1996. Chemosphere 33: 595-<strong>60</strong>2<br />
[5] Stevenson, F.J. 1994. Humus chemistry: Genesis, Composition, Reactions. 2 nd ed.<br />
Wiley-Intersci., New York.<br />
[6] Cameron, R.S., Swift, R.S., Thornton, B.K., Posner, A.M. 1972. J. Soil Sci. 23: 395-408<br />
[7] Ghosh, K., Schnitzer, M. 1980. Soil Sci. 129: 266-276<br />
[8] De Haan, H., Jones, R.I., Salonen, K. 1987. Freshwater Biol. 17: 453-459<br />
[9] Cornel, P.K., Summers, S.R., Roberts, P.V. 1986. J. Colloid Interface Sci. 110: 149-163<br />
[10] Chin, Yu-P., Gschwend, P.M. 1991. Geochim. Cosmochim. Acta 55: 1309-1317<br />
[11] Wershaw, R.L. 1993. Environ. Sci. Technol. 27: 814-816<br />
[12] Schlautman, M.A., Morgan, J.J. 1993. Environ. Sci. Technol. 27: 961-969<br />
[13] Engebretson, R., von Wandruszka, R. 1997. Org. Geochem. 26: 759-767<br />
[14] Piccolo, A., Conte, P. 2000. Adv. Environ. Res. 3: 508-521<br />
[15] Cantor, C.R., Schimmel, P.R. 1980. Biophysical chemistry, Part I: The conformation of<br />
biological macromolecules. Freeman, New York.<br />
[16] Conte, P., Piccolo, A. 1999. Chemosphere 38: 517-528<br />
[17] Hjelmeland, L.M. 1990. Solubilization of native membrane proteins. In Guide to<br />
Protein Purification. Methods of Enzymology, Vol. 182: 253-264. Academic Press, San<br />
Diego.<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 38
WETTABILITY OF PLASMA POLYMERIZED<br />
VINYLTRIETHOXYSILANE FILM<br />
Soňa Lichovníková, 5 nd year<br />
Supervisor: Doc. RNDr. Vladimír Čech, PhD.<br />
Institute of Material Science, Faculty of Chemistry, Brno University of Technology,<br />
Purkyňova 118, 612 00 Brno, Czech Republic, e-mail: xclichovnikova@fch.vutbr.cz<br />
INTRODUCTION<br />
This paper deals with wettability of thin polymer films prepared by Plasma Enhanced<br />
Chemical Vapor Deposition (PE CVD) from vinyltriethoxysilane (VTES) monomer deposited<br />
on planar substrates. The sessile drop method was used to measure the contact angles and the<br />
surface free energy was evaluated using the approach of Owens-Wendt geometrical mean<br />
method, Wu harmonic mean method and the acid-base method. Changes in the surface<br />
structure were investigated by Fourier Transform Infrared Spectroscopy (FTIR) measurement,<br />
X-Ray Photoelectron Spectroscopy (XPS) measurement and Rutherford Backscattering<br />
Spectrometry (RBS). A relation between wettability and process parameters of the plasma<br />
deposition were discussed as well. [1]<br />
EXPERIMENTAL<br />
PLASMA POLYMERIZATION<br />
Plasma polymer films of VTES CH2-CH-Si(-O-CH2-CH3)3 (purity ≥ 98 %, Fluka) were<br />
deposited on glass substrates (special microscope slides without flaws, 1.0 × 26 × 76 mm 3 ,<br />
Knittel Gläser, Germany, for surface free energy determination) and on infrared-transparent<br />
silicon wafers (0.8 × 10 × 10 mm 3 , Terosil Co., for film analyses by FTIR, XPS and RBS),<br />
using a helical coupling plasma system (13.56 MHz) in a vacuum tubular chamber (40 mm in<br />
diameter, made of Pyrex glass). Details of the apparatus are described in Ref. [2].<br />
The vacuum system was evacuated to a pressure of 5 × 10 -3 Pa and then flushed with argon<br />
gas (10 sccm) for 10 min. A basic pressure of 2 × 10 -3 Pa was established in the deposition<br />
chamber after flushing. Monomer vapor at a mass flow rate of 0.45 sccm was introduced into<br />
the deposition chamber using a dose valve and the plasma then was ignited at a selected<br />
effective power of 0.1, 0.5, 2.5, 5.0, 10.0 and 25 W, using pulsed plasma with ton= 1 ms and a<br />
total power of 50 W. Deposition pressure was about 1.3 Pa and the film thickness (about 400<br />
nm) was controlled by deposition time ranging from 5-100 min. When the deposition process<br />
was finished, all the apparatus was flushed with argon gas and after 30 min the chamber was<br />
flooded by air to atmospheric pressure. Prepared samples were transported from the chamber<br />
into a desiccator to avoid contamination before measurements. [3]<br />
THIN FILM ANALYSIS<br />
The sessile drop method (tangent method) employing an OCA 10 goniometer<br />
(DataPhysics) with Software SCA 20 for Windows 9x/NT was used to measure the contact<br />
angles. The surface free energy was evaluated using the approach of Owens-Wendt<br />
geometrical mean method, Wu harmonic mean method and acid-base method [1]. The surface<br />
free energy, as well as the dispersive and polar components, the Lewis electron acceptor (γs (+) )<br />
and Lewis electron donor (γs (-) ) components of VTES film were evaluated from contact angle<br />
measurements using distilled water, ethylene glycol, glycerol and methylene iodide. A small<br />
drop of liquid was carefully placed on the surface of substrate (glass slide). The contact angle<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 39
is obtained by measuring the angle between the tangent to the profile at the point of contact<br />
with the solid surface. Software SCA 20 offers the calculation of the surface free energy of<br />
solids and their components. It also contains database of liquids and their necessary<br />
parameters. Owens-Wendt and Wu methods express the surface tension as a sum of<br />
components based on dispersion forces (γ d ) and polar forces (γ p ): γ=γ d +γ p . Van Oss, Good<br />
and Chaudhury´s acid-base theory is more detailed in the surface free energy specification and<br />
leads from Lewis theory, where Lewis acid is an electron-pair acceptor, Lewis base is an<br />
electron-pair donor. The surface tension is a sum of two components: the Lifshitz-van der<br />
Waals component (γs LW ) and acid-base component (γs AB ),<br />
γ +<br />
LW AB<br />
= γ γ . γs LW reflects the<br />
long-range interactions (to 100 Å), including dispersive interaction, the dipole-dipole and the<br />
dipole induced dipole interaction and it influences the total surface free energy at most. γs AB is<br />
associated with the transfer of electrons between an electron donor (γs (-) ) and an electron<br />
acceptor (γs (+) AB + −<br />
) in short range of < 3 Å and γ s = 2 γ s γ s . γs AB is relatively small and not so<br />
important when compared to γs LW . Values of the surface free energy parameters of probe<br />
liquids were ta<strong>ke</strong>n according to different authors. Distilled water and diiodomethane were<br />
ta<strong>ke</strong>n according to Erbil, formamide and glycerol according to Van Oss et al. Three different<br />
probe liquids have to be used for calculation by acid-base approach, averaged results of the<br />
surface tension calculation from the water-diiodomethane-formamide and waterdiiodomethane-glycerol<br />
triplets were used. The contact angle data of two different testing<br />
liquids are needed for the determination of γ d and γ p components according Owens-Wendt<br />
and Wu methods: water (author Strom et al.) and diiodomethane (author Janczuk et.al.).<br />
Several standard conditions have to be met to measure reproducible contact angles: drop<br />
volume (chec<strong>ke</strong>d by micrometer screw), time (10 seconds) and laboratory temperature. More<br />
information about equations for surface tension calculation and contact angle measurements<br />
can be found in Ref. [1,6]<br />
The elemental composition (C, Si, O) in the surface region (top 6-8 nm) of the deposited<br />
layers was determined by XPS on an ADES 400 VG Scientific photoelectron spectrometer<br />
using MgKα (1253.6 eV) photon beams. XPS is sensitive surface selective method which<br />
provides information about chemical composition and electron structure of the surface of<br />
solids. The elemental composition has been also studied by conventional and resonant RBS<br />
and Elastic Recoil Detection Analysis (ERDA) methods using Van de Graaf generator with a<br />
linear electrostatic accelerator. The RBS spectrum contains information about elemental<br />
composition in depth levels of sample. [4,5]<br />
Infrared measurements were carried out using a Nicolet Impact 400 Fourier transform<br />
infrared (FTIR) spectrophotometer and the chemical bonds in thin films were determined by<br />
interpreting the infrared absorption spectrum. Samples were measured without the protective<br />
atmosphere.<br />
RESULTS AND DISCUSSION<br />
Plasma-polymerized VTES films were deposited on planar glass substrates at different<br />
effective powers (0.1-25 W) at conditions described above. Sessile drop measurements were<br />
employed to analyze the surface free energy of the film. Measured contact angles of the probe<br />
liquids (water, diiodomethane, formamide, glycerol) were changed according to various<br />
effective power (W). In Figure 1, trend of decreasing contact angles with the magnitude of the<br />
input RF power can be seen. Contact angles of all liquids are slightly decreasing before<br />
reaching 5 W effective power. Maximum decrement of contact angle can be seen between 2.5<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 40
W and 5 W. The wettability of liquids generally increases with increasing effective power,<br />
except diiodomethane, where the maximum wettability is during 5 W effective power. In<br />
Figure 2, there can be seen the trend of increasing surface free energy (mJ.m -2 , average of<br />
both triplets) with the magnitude of the input RF power. The largest increase is during 5 W<br />
effective power. When the surface free energy increases, wettability is also increased. Total<br />
surface free energy has maximum during 25 W. As can be seen, the most important<br />
component which influences the total surface free energy is γs LW . The other part of the surface<br />
free energy is the γs AB divided in electron acceptor γs (+) (acid) and donor γs (-) (basic)<br />
components. The γs AB component is in comparison with the γs LW component small. γs (-)<br />
component is much higher than γs (+) with values near zero.<br />
Contact angle [deg.]<br />
90<br />
80<br />
70<br />
<strong>60</strong><br />
50<br />
40<br />
30<br />
20<br />
10<br />
0<br />
water<br />
diiodomethane<br />
formamide<br />
glycerol<br />
0,1 1 10<br />
W eff [Watt]<br />
Surface free energy [mJ/m 2 ]<br />
50<br />
45<br />
40<br />
35<br />
30<br />
25<br />
20<br />
15<br />
10<br />
5<br />
0<br />
0,1 1 10<br />
W eff [Watt]<br />
Figure 1, 2: Dependence of the contact angle of all testing liquids and dependence of the<br />
surface free energy components (acid-base approach) on the magnitude of the input RF<br />
power<br />
In Figure 3 can be seen calculated total surface free energy and its components (in mJ.m -2 )<br />
for set of samples, by Owens-Wendt geometric mean (O-W) and Wu harmonic mean (Wu)<br />
methods, depending on the effective<br />
Surface free energy [mJ/m 2 ]<br />
50<br />
45<br />
40<br />
35<br />
30<br />
25<br />
20<br />
15<br />
10<br />
5<br />
0<br />
0,1 1 10<br />
W eff [Watt]<br />
γ TOT (O-W)<br />
γ TOT (Wu)<br />
(d) (O-W)<br />
γ<br />
s<br />
(d)<br />
γ (Wu)<br />
s<br />
(p)<br />
γ (O-W)<br />
s<br />
(p)<br />
γ (Wu)<br />
s<br />
Figure 3: Dependence of the surface free<br />
energy components (Owen-Wendt and Wu<br />
approach) on effective power<br />
γ TOT<br />
γ LW<br />
γ +<br />
γ −<br />
power. Total surface free energy is sum<br />
of γ d and γ p components. Values<br />
obtained by Wu method are not exactly<br />
equal to the values obtained by O-W<br />
method. It is caused by different γ d and<br />
γ p values of probe liquid diiodomethane.<br />
Total surface free energy (of both<br />
methods) and wettability increase with<br />
increasing effective power. The largest<br />
increase of surface free energy is during<br />
5 W. Surface free energy can be<br />
considered almost constant during higher<br />
effective powers. Dispersion component<br />
influences the total surface free energy at<br />
most, but influence of polar component<br />
can’t be neglected. Both components increase with increasing effective power. Total surface<br />
free energy of O-W, Wu and acid-base approaches is quite the same. The difference is caused<br />
by interpretation of the surface free energy components. Surface free energy values obtained<br />
by acid-base approach are closer to values at O-W method than Wu method. Wettability of<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 41
pp-VTES thin films could be well controlled by process parameters, especially effective<br />
powers.<br />
To investigate relationship between wettability and chemical status of the thin VTES films,<br />
the data from FTIR, XPS and RBS measurements were used. In Figure 4 and 5 can be seen<br />
comparison of FTIR spectra of three samples with different effective powers. We can see<br />
some changes in chemical structure between the high-power and low-power deposited<br />
polymers. The absorption bands at 2973 cm -1 and 2924 cm -1 were assigned to CH3 and CH2<br />
stretching vibrations. With increasing of effective power, intensity of bands decreases as well<br />
as bands 1070 cm -1 assigned to Si-O-C stretching vibrations, 885 cm -1 assigned to Si-C, 790<br />
cm -1 assigned to Si-O, 11<strong>60</strong> cm -1 and 965 cm -1 assigned to Si-O-C2H5. The dominant<br />
absorption bands in all spectra can be found at a range 1000-1200 cm -1 because the plasma<br />
polymer of VTES was formed by the Si-O-C network. The greatest decrease of peak area was<br />
measured in Si-O-C group. This group is only one from Si-groups which is apparent at 25 W<br />
effective power. The pp-VTES films seemed to be free of vinyl groups as there were no<br />
absorption bands at positions mar<strong>ke</strong>d by dashed line (Fig. 4). With increasing power a new<br />
absorption band can be seen at 3480 cm -1 assigned to the OH stretching vibrations (strong<br />
polar group) and also at 1700 cm -1 corresponding to the C=O carbonyl stretching vibrations<br />
(medium polar group), so the presence of polar substances on the surface increases. It comes<br />
to that wettability also increases with increasing power which can be also connected with<br />
decreasing of stretch band intensity of apolar substances (especially CH3) and decreasing of<br />
inorganic Si-O-C substances. At lower effective powers (under 5 W), it seems that apolar<br />
groups (CH3, Si-Et, Si-C, Si-H) are lin<strong>ke</strong>d to Si-O-C network and create more apolar surface<br />
in comparison with samples at higher effective powers where polar groups li<strong>ke</strong> OH and C=O<br />
are lin<strong>ke</strong>d to the carbon network.<br />
Absorbance (a.u.)<br />
OH<br />
CH 3<br />
CH 2<br />
CH 2<br />
CH 3<br />
Si-H<br />
0.05 W<br />
5 W<br />
25 W<br />
Si-O-C<br />
Si-Et<br />
C=O CH2 Si-Et<br />
vinyl vinyl vinyl<br />
Si-O<br />
Si-C<br />
4000 3500 3000 2500 2000 1500 1000 500<br />
Wavenumber (cm -1 )<br />
Figure 4, 5: Comparison of VTES FTIR spectra of three different effective powers (W)<br />
and dependence of the peak area (a.u.) from VTES FTIR spectra on the effective power (W)<br />
In Figure 6 and 7 can be seen the results from XPS and RBS measurements – element<br />
abundances of C, O, Si (at. %) and elements ratio which also depend on effective power. With<br />
increasing of effective power, C concentration increases, O and Si concentration decrease.<br />
Ratios C/Si, O/Si, C/O at a higher power of 25 W are very similar to those of the monomer<br />
molecule. As can be seen, dependences of atomic concentration and element ratio on film<br />
thickness could be neglected. C concentration increases with increasing power but it seems<br />
that in high powers C create cage and polar groups OH, C=O are lin<strong>ke</strong>d to cage and stick out.<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 42
Hence wettability is higher. O and Si concentrations decrease with increasing power, but in<br />
lower powers Si-O-C groups created polymer network, apolar groups are lin<strong>ke</strong>d to network<br />
and stick out. Wettability is than smaller. This implies that wettability of VTES is influenced<br />
especially by groups which stick out the network and it depends if groups are polar or apolar.<br />
This conclusion corresponds to investigation in Ref. [7].<br />
Atomic concentration (at%)<br />
80<br />
70<br />
<strong>60</strong><br />
50<br />
40<br />
30<br />
20<br />
10<br />
0<br />
Film thickness: ~400 nm<br />
XPS<br />
RBS<br />
Carbon<br />
Oxygen<br />
Silicon<br />
0,1 1 10<br />
Effective power (W)<br />
Effective power: 5 W<br />
Carbon<br />
Oxygen<br />
Silicon<br />
XPS<br />
RBS<br />
200 500 1000<br />
Film thickness (nm)<br />
80<br />
70<br />
<strong>60</strong><br />
50<br />
40<br />
30<br />
20<br />
10<br />
0<br />
Element ratio<br />
10<br />
9<br />
8<br />
7<br />
6<br />
5<br />
4<br />
3<br />
2<br />
1<br />
Film thickness: ~400 nm<br />
C/Si (monomer)<br />
C/Si<br />
O/Si (monomer)<br />
O/Si C/O<br />
C/O (monomer)<br />
0<br />
0,1 1 10<br />
Effective power (W)<br />
Effective power: 5 W<br />
200 500 1000<br />
Film thickness (nm)<br />
Figure 6, 7: Comparison of RBS and XPS measurements: dependence of the atomic<br />
concentration (at %) on effective power (W) and dependence of the elements ratio on effective<br />
power (W)<br />
CONCLUSION<br />
Plasma-polymerized thin films of VTES were deposited on planar glass substrates and<br />
silicon wafers and investigated by contact angle measurements, FTIR, XPS and RBS<br />
methods. Process parameters, especially effective power can influence/control the chemical<br />
structure, elemental composition and wettability. Wettability increases with increasing<br />
effective power and it seems that wettability is influenced mainly by groups sticking out of<br />
the network and by polar groups.<br />
ACKNOWLEDGEMENTS<br />
This work was supported by the Czech Science Foundation, grant no.104/06/0437, and the<br />
Czech Ministry of Education, grant no. 1P05OC087 (COSTP12). The author would li<strong>ke</strong> to<br />
thank Dr. J. Zemek (XPS), Dr. V. Perina (RBS/ERDA) and J. Studynka (FTIR) for<br />
spectroscopic measurements and analyses.<br />
REFERENCES<br />
[1] F. Garbassi, M. Morra, E. Occhiello, Polymer Surfaces, John Wiley & Sons Ltd, New<br />
York, 1998.<br />
[2] R. Prikryl, O. Salyk, J. Vanek, V. Cech, Czech. J. Phys. 52 (2002), D816.<br />
[3] V. Cech, N. Inagaki, J. Vanek, R. Prikryl, A. Grycova, J. Zemek, Thin Solid Films 502<br />
(2006) 181-187.<br />
[4] Jiří Sova, Diploma Thesis, Brno University of Technology, Brno, 2005.<br />
[5] J.F. Watts, J. Wolstenholme, An Introduction to Surface Analysis by XPS and AES, John<br />
Wiley & Sons Ltd, London, 2003.<br />
[6] S. Wu, Polymer Interface and Adhesion, Marcel Dek<strong>ke</strong>r, Inc., New York, 1982.<br />
[7] V. Cech, J. Vanek, V. Perina, J. Zemek, Chemical properties of plasma-polymerized<br />
vinyltriethoxysilane, ISPC-17, Toronto, August 7-12, 2005, p.1-5.<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 43<br />
C/Si<br />
C/O<br />
O/Si<br />
10<br />
9<br />
8<br />
7<br />
6<br />
5<br />
4<br />
3<br />
2<br />
1<br />
0
URČOVANIE DISTRIBÚCIE DOMÉN V SPIN CROSSOVER<br />
SYSTÉMOCH POMOCOU ANALYTICKEJ FUNKCIE<br />
Ján Pavlik, 4. ročník<br />
vedúci práce: Prof. Ing. Roman Boča, DrSc.<br />
Slovenská technická univerzita v Bratislave, <strong>Fakulta</strong> chemic<strong>ke</strong>j a potravinárs<strong>ke</strong>j technológie,<br />
Oddelenie anorganic<strong>ke</strong>j chémie, Radlinského 9, 812 37 Bratislava,<br />
email: jan.pavlyk@gmail.com<br />
ÚVOD<br />
Niektoré komplexy prechodných kovov sa môžu predovšetkým v závislosti od teploty (ale<br />
napríklad aj tlaku, magnetického poľa a pôsobenia svetla) vyskytovať buď v nízkospinovom<br />
(LS) alebo vysokospinovom (HS) stave. Tieto dva stavy majú rozdielne optické, magnetické a<br />
objemové vlastnosti. Jav prechodu medzi týmito stavmi sa označuje ako spin crossover. Jeho<br />
možnosť je nutne podmienená tým, aby základným energetickým stavom molekuly v kryštáli<br />
bol nízkospinový. Ďalšou podmienkou je, aby entropia HS stavu bola vyššia ako LS. Pri<br />
splnení týchto podmienok bude existovať teplota (tzv. teplota prechodu), pri ktorej bude<br />
pozorovateľný prechod medzi nimi [6]. Spin crossover sa zaznamenáva najčastejšie formou<br />
závislosti zastúpenia HS molekúl od teploty.<br />
MODELY ISINGOVHO TYPU (MIKROSKOPICKÉ MODELY)<br />
Spoločnou črtou týchto modelov je predpoklad nerovnakého vzájomného pôsobenia<br />
susediacich molekúl, ktoré sú v rôznom spinovom stave a parametrizovanie tejto interakcie<br />
empirickou konštantou. Konkrétne sa pre spin crossover systém predpokladá hamiltonián<br />
nasledujúceho typu [9]:<br />
Hˆ= ( Δ ˆ ˆ<br />
0 /2) σ − J σ σ<br />
(1.1)<br />
kde Δ0 je energetický rozdiel molekuly v stave HS a LS, J je konštanta kooperativity, teda<br />
miera vplyvu stavov okolitých molekúl na stav sledovanej molekuly a ˆ σ je operátor tzv.<br />
fiktívneho spinu, ktorý rozlišuje medzi LS a HS stavom definičným vzťahom:<br />
ˆ σ LS = -1 LS (1.2)<br />
ˆ σ HS = +1 HS (1.3)<br />
Ďalej σ značí teplotný priemer fiktívneho spinu, teda<br />
∑<br />
( E kT )<br />
( −E<br />
kT )<br />
σ =<br />
σ iexp − i /<br />
i<br />
∑ exp i /<br />
i<br />
(1.4)<br />
Tento model sa nazýva aj dvojhladinový model Isingovho typu, pretože hovorí o dvoch<br />
energetických stavoch molekuly, ktoré sú ďalej modulované priemerným stavom okolitých<br />
molekúl. Vlastné energie molekuly budú<br />
E ( σ =− 1) =− Δ 2+<br />
J σ<br />
(1.5 a)<br />
1 0<br />
E2( σ =+ 1) = Δ0 2−<br />
J σ<br />
(1.5 b)<br />
Fyzikálne oprávnenejšie modely Isingovho typu sa získajú predstavou kryštálu zloženého z<br />
oblastí, v ktorých sa nachádzajú iba molekuly jedného stavu tzv. domény [2].<br />
FORC A FORC DISTRIBÚCIA<br />
Jednou z novších perspektívnych metód umožňujúcich stanoviť niektoré parametre<br />
spomínaných modelov je meranie tzv. FORC (first-order reversal curve). Postup merania je<br />
nasledujúci: vzorka pri teplote kde je úplne v HS stave sa pomaly ochladzuje a stanovuje sa<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 44
zastúpenie molekúl alebo domén v tomto stave. Pri určitej teplote (teplote obratu TA) sa<br />
ochladzovanie zastaví a začne sa znova s ohrevom až kým nie je vzorka opäť kompletne v<br />
pôvodnom stave. Zastúpenie HS stavu sa posudzuje ako funkcia TA a aktuálnej teploty TB.<br />
Vychádzať sa môže aj z LS stavu, <strong>ke</strong>dy hovoríme o ohrevnom móde narozdiel od skôr<br />
uvedeného postupu, ktorý sa označuje ako chladiaci mód. Príklad typic<strong>ke</strong>j FORC v<br />
chladiacom móde uvádza obr. 1.<br />
Obr. 1. FORC v ohrevnom móde pre [Fe0.6Zn0.4(btr)2(NCS)2]H2O získaná<br />
pomocou optických metód.<br />
Dôležitý pojem spájajúci sa s FORC je tzv. FORC distribúcia [4] definovaná vzťahom<br />
2<br />
1 ∂ nHS( TA, TB)<br />
ρ(<br />
TA, TB)<br />
=−<br />
(2.1)<br />
2 ∂TA∂TB Existujú pádne dôvody pre stotožnenie takto definovanej veličiny s distribúciou domén v<br />
kryštáli.<br />
URČOVANIE FORC DISTRIBÚCIE<br />
V súčasnosti sa FORC distribúcia určuje numericky, čo znamená, že diferencie vo vzťahu<br />
(2.1) sa nahradia malými zmenami daných veličín. Ich hodnoty sú samozrejme determinované<br />
hustotou bodov v skúmanej FORC a presnosť takto určenej distribúcie je teda veľmi závislá<br />
na hustote merania. Ako alternatíva sa ponúka možnosť aproximovať namerané FORC<br />
vhodne zvolenou funkciou derivovateľnou do druhého stupňa a FORC distribúciu získať jej<br />
analytickým derivovaním, čím sa aj pri redších meraniach dosiahne interpolácia chýbajúcich<br />
bodov a hladšia FORC distribúcia.<br />
Obr. 2. FORC s rôznymi osami pre TA resp. TB<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 45
Obr. 2. názorne uvádza FORC vo vyobrazení vhodnom pre predstavu o tvare ta<strong>ke</strong>jto funkcie.<br />
Ako vidieť, závislosť je vzhľadom na TB v celom rozsahu TA približne sigmoidálna, čo sa dá<br />
vystihnúť všeobecným vzťahom<br />
1<br />
xHS = f ( TA, TB) + g( TA, T<br />
C<br />
B)<br />
(3.1)<br />
⎛ ⎛ TB−a ⎞⎞<br />
1<br />
⎜1+ exp⎜−<br />
⎟⎟<br />
⎝ ⎝ b1<br />
⎠⎠<br />
kde a1, b1 a c sú parametre. Závislosť však bolo možné opísať tým lepšie, čím viac<br />
susediacich sigmoíd bolo použitých, v takom prípade vzťah (3.1) má tvar<br />
n 1 n<br />
xHS = f ( TA, TB) ∑ + g( TA, T<br />
C<br />
B)<br />
(3.2)<br />
i=<br />
1 ⎛ ⎛ TB − a ⎞⎞<br />
1i<br />
⎜1+ exp⎜−<br />
⎟⎟<br />
⎝ ⎝ b1<br />
⎠⎠<br />
Priebeh funkcie pri konštantnom TB sa ukázalo výhodné opisovať závislosťou gaussovského<br />
typu, čo viedlo k nasledujúcim výrazom<br />
( )<br />
( )<br />
2. D<br />
⎛ TA−a ⎞<br />
2<br />
f TA, TB<br />
= exp⎜−<br />
⎟<br />
(3.3)<br />
⎜ b ⎟<br />
⎝ 2 ⎠<br />
( )<br />
( )<br />
2. D<br />
⎛ TA−a ⎞<br />
2<br />
g TA, TB<br />
= 1−exp⎜− ⎟<br />
(3.4)<br />
⎜ b ⎟<br />
⎝ 2 ⎠<br />
a výsledná závislosť nadobudla tvar<br />
( ) 2.<br />
⎧ ⎫<br />
⎪ ⎪<br />
D<br />
n ⎪ 1 n<br />
⎪ ⎛ TA−a ⎞<br />
2<br />
xHS<br />
= 1+ ⎨∑ −1 exp<br />
C ⎬ ⎜− ⎟ (3.5)<br />
⎪ i=<br />
1 ⎛ ⎛ T b2<br />
B − a ⎞⎞<br />
⎜ ⎟<br />
1i<br />
⎪<br />
1+ exp −<br />
⎝ ⎠<br />
⎪ ⎜ ⎜ ⎟⎟<br />
b<br />
⎪<br />
⎩ ⎝ ⎝ 1 ⎠⎠<br />
⎭<br />
s voľnými parametrami a2, b1, b2, C, Da n parametrami a 1i , pričom D je celé číslo. Použitím<br />
vzťahu (3.2.6) dostávame pre FORC distribúciu analytický vzťah<br />
2D<br />
−C−1 ⎛ ( TA−a2) TB −a ⎞⎛ ⎛ 1i TB −a<br />
⎞⎞<br />
1i<br />
2D−1 CDexp⎜− − ⎟⎜1+ exp<br />
( TA−a2) n ⎜ b2 b ⎟ ⎜ ⎟⎟<br />
1 ⎝ ⎝ b1<br />
⎠⎠<br />
ρ ( TA, TB)<br />
=<br />
⎝ ⎠<br />
∑ (3.6)<br />
i=<br />
1 nb1b2 kde symboly majú už uvedený význam.<br />
ZÁVER<br />
Ako vidieť na obr. 3., existuje stále kvalitatívny rozdiel medzi navrhovanou analytickou<br />
závislosťou (3.6) a simulačne získanou FORC. V tomto ohľade tu zostáva priestor na vývoj<br />
uvedených vzťahov. FORC distribúcia získaná výpočtom zo vzťahu (3.6) však nadobúda tvar<br />
blízky očakávanému (obr. 4.). Na tomto mieste treba upozorniť na obmedzenú univerzálnosť<br />
navrhovaných vzťahov, nakoľko boli vyvíjané na základe dát získaných simuláciou správania<br />
sa Isingovho modelu pre jednojadrové komplexy, ktorý dobre opisuje síce podstatnú, nie však<br />
úplnú časť pozorovaných typov závislostí [7].<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 46
Obr. 3. Rozdiel hodnôt navrhovanej funkcie a FORC v percentách<br />
Obr. 4. Experimentálna FORC distribúcia k obr. 1 (vľavo) a vypočítaná podľa vzťahu (3.6)<br />
(vpravo)<br />
LITERATÚRA<br />
[1] Slichter CP, Drickamer HG (1972) J Chem Phys 56: 2142<br />
[2] Sorai M, Seki S. (1974) J Phys Chem Solids 35: 555<br />
[3] Boča R, Boča M, Dlháň Ľ, Fakl K, Fuess H, Haase H, Jaroščiak R, Papánková B, Renz F,<br />
Vrbová M, Werner R (2000) Inorg Chem 40: 3025<br />
[4] Enachescu C, Tanasa R, Stancu A, Codjovi E, Linares J, Varret F (2004) Physica B 343:<br />
15<br />
[5] Cambi L, Cagnaso A (1931) Atti Accad Naz Lintei 13: 809<br />
[6] Kahn O (1993) Molecular Magnetism. VCH, New York<br />
[7] Boča R, Linert W (2003) Monats Chem 134: 199<br />
[8] Zimmermann R, König E (1977) J Chem Phys Solids 38: 779<br />
[9] Boča R (1999) Theoretical Foundations of Molecular Magnetism. Elsevier, Amsterdam<br />
[10] Real J, Bolvin H, Bousseksou A, Dworkin A, Kahn O, Varret F, Zarembowitch J (1992)<br />
J Am Chem Soc 114: 4650<br />
[11] Nasser JA, Boukheddaden K, Linares J (2004) Eur Phys J B 39: 219<br />
[12] Herchel R, Boča R, Gembický M, Kožíšek J, Renz F (2004) Inorg Chem 43: 4103<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 47
A STUDY OF INTERFACIAL ASPECTS OF EPOXY-BASED<br />
HYBRID COMPOSITES REINFORCED WITH BASALT,<br />
CARBON AND CERAMIC FIBRES BY DMA ANALYZIS.<br />
Lukáš Recman, 5. ročník<br />
Vedoucí práce prof. RNDr. Josef Jančář, CSc.<br />
Konzultant ing. Petr Poláček<br />
<strong>Vysoké</strong> učení technické v Brně, <strong>Fakulta</strong> <strong>chemická</strong>, ústav chemie materiálů,<br />
Purkyňova118, 612 00 Brno, e-mail: xcrecman@fch.vutbr.cz<br />
ABSTRACT<br />
This article introduces the recent knowledge on continious fibre reinforced epoxy<br />
based composites. The following topics are included: (i) theoretical and experimental<br />
studies of continious fibers – epoxy matrix interphase (ii) preparation of dual-fiber<br />
hybrid composites DFHC (iii) DMA measurement of DFHC.<br />
INTRODUCTION<br />
Interfacial properties and matrix-reinforcement (communication) adhesion plays a<br />
vital role in composite´s aplications, properties and his behaviour under stress<br />
conditions. According to this expression it is usefull to ensure carefull interphase<br />
connection hence the expression about the wea<strong>ke</strong>st link. The appropriate adhesion is<br />
defined by various physical and chemical parameters. [1-2] But it will be unreliable<br />
ma<strong>ke</strong> ourselves satisfied that strong bonds between matrix and the reinforcement<br />
conquer all. So let discuss this phenomon detaily.<br />
Matrix and reinforcement can be connected by several types of bonding li<strong>ke</strong><br />
mechanical bonding also known as <strong>ke</strong>ying, electrostatic bonding, interdiffusion<br />
bonding and finally chemical bonding. [1] When none of this types of connection can<br />
be established another parameter rises – a wettability. Wettability is the ability of a<br />
liquid to be spread over a solid surface. But for an epoxy based composites this is the<br />
<strong>ke</strong>y-word, the stumbling block of this phenomon. The epoxy resins are extremely<br />
viscous and hydrofobic liquids. It is not easy to ensure the appropriate connection<br />
between epoxy matrix and the reinforcement.<br />
Matrix transmits the applied load from its surface to its volume and then to the<br />
reinforcement. [2] Therefor a well-calculated and right connection has to be made. This<br />
matrix-fibre bond transmit the applied load to the stiffer and stronger reinforcement<br />
where is being gathered. After reaching a critical value of absorbed energy a several<br />
modes of its dissipation occures: Reinforcement breakage, pull out, interphacial<br />
debonding and reinforcement „elongation“. The better the connection is the advanced<br />
material properties are.<br />
Matrix – reinforcement adhesion is a complicated manner, which can not be<br />
designed and described easily. It depends on wide physical and chemical parameters.<br />
But also another parameters play vital role: li<strong>ke</strong> homogenity, uniderectional<br />
orientation, avoiding fibre – fibre contact. All these parameters mentioned above<br />
influence composite´s parameters.<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 48
Let answer the question from the beginning of the chapter. Is strong bond always a<br />
desired manner? Sometimes yes and sometimes no. First it is necessary to know that<br />
the bond breakage serves as an energy dissipating process. Second it all depends on the<br />
applied stress. When the material is being stressed with increasing force during a long<br />
time period it retains a greather value of the force, then stress during a short time<br />
interval. A strong bonding for this case is a mortal habit. So the conclusion is materials<br />
are designed for one purpose only. When designing material for concrete purpose then<br />
confining his field of application! It is called tailor-made or tailoring.<br />
EXPERIMENTAL<br />
Materials<br />
Basalt, Carbon and Ceramic fibres.<br />
The basalt fibre was made from naturally occuring basalt rock from Charberec<br />
location. The chemical composition of the fibre is determinated by the native basalt<br />
rock, which was used as a raw material. The main composition is SiO2 43%, Al2O3<br />
14%, CaO 10%, MgO 9%, Na2O 4%, TiO2 2%, Fe2O3/FeO 12%, K2O 1%. Material<br />
was melted in alumina-mullite crucialbe at 1400 °C.<br />
The HS Carbon Celion fibres were obtrained form Union Carbide Company.<br />
The Nextel 440 and Nextel 550 fibres were obtained form 3MNextelIndustrial<br />
fibers & 3MNextelComposite fibres. The composition and mechanical data are<br />
included in table 1.<br />
Table 1. Composition and selected properties of used fibres and resin<br />
Property Units Nextel Nextel HS- Basalt Atlac 430<br />
440 550 Carbon<br />
composition % Al2O3 Al2O3 - -<br />
SiO2 (28)<br />
B2O3 (2)<br />
SiO2 (27)<br />
diameter μm 10-12 10-12 - - -<br />
Young´s<br />
modulus (E)<br />
GPa 190 193 230 89 2<br />
Density (ρ) g.cm -3 3.05 3,03 1,75 2,7 10<strong>60</strong><br />
Tensile<br />
strenght (σ)<br />
GPa 2 2 3,4 4,8 2<br />
Polymer matrix<br />
An epoxy resin ATLAC 430 was obtained from DSM corporate. It is based on<br />
unsaturated polyesters. It is a solution of linear unsaturated polyesters, containing a<br />
reactive doble-bond in polymerizable solution – styrene. Some useful data are<br />
mentioned in table 1.<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 49
METHODS<br />
DMA measurement<br />
To gain detailed informations about DFHC properties a complex DMA<br />
measurement on Dynamic Mechanical Analyser 2980 (TA Instruments) was designed.<br />
From a wide range of DMA modes a DMA single-cantilever multifrequency mode was<br />
selected (DMA-SC-MF). DMA-SC-MF is an application of a constant amplitude as a<br />
function of time, temperature and frequency of oscillation. The set parameters:<br />
Frequency 10Hz, equilibrate at 40 °C, ramp 3 °C/min to 180 °C and amplitude 2μm.<br />
Loss modulus storage modulus and their mutual ration tanδ were obtained.<br />
Preparation of dual-fibre hybrid composites (DFHC) specimens.<br />
The dimensions of the specimen was 2mm thick, 40mm long and 10mm wide. The<br />
fibres were embedded in flexibilized epoxy resin in a rectangular-shaped (Lukopren)<br />
silicon-rubber mold. They were than heated at 65 °C for 2h, because of gasses and as a<br />
bubbles prevention. The specimens were than post-cured at 90 °C for 6h in a kiln.<br />
To obtain rigid epoxy specimens with DBP, the resin and curing agent 1wt% DBP<br />
was thoroughly mixed and degassed in a vacuum oven to remove air. The mixture was<br />
poured slowly in the mold and first continious fibres were put in, then another layer of<br />
resin was appplied and so the fibres until complete amount of fibres has been used.<br />
After taking the specimens out of the mold (when finished curing process) a standard<br />
metallographic techniques were used to obtain a smooth surface, when necessary. The<br />
two best specimens of each species and ratios were chosen and performed a<br />
measurement. Finaly pure Nextel 440, Nextel 440 – Basalt in three different ratios and<br />
pure basalt has been measured and so Nextel 550 – Carbon. See table 2.<br />
Table 2. Prepared composites Nextel 440 – Basalt, Nextel 550 – Carbon<br />
Nextel 440 Basalt Nextel 550 Carbon<br />
% Strand % Strand % Strand % Strand<br />
10 66 0 0 10 54 0 0<br />
8 53 2 11 8 43 2 2<br />
5 33 5 28 5 27 5 5<br />
2 13 8 44 2 11 8 8<br />
0 0 10 55 0 0 10 10<br />
RESULTS AND DISCUSSION<br />
DMA-SC-MF analyzis of DFHC<br />
To obtain the data sheet of mechanical properties a three values were gathered. The<br />
storage modulus, loss modulus and tanδ was gathered. For each type of manufactured<br />
DFHC and resin a two figures were constructed.<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 50
Storage modulus<br />
(MPa)<br />
5000<br />
4500<br />
4000<br />
3500<br />
3000<br />
2500<br />
2000<br />
1500<br />
1000<br />
500<br />
0<br />
Storage modulus comparison<br />
40 65 90 115 140 165<br />
Temperature (°C)<br />
N440 N4B1 N1B1 N1B4 Basalt Resin<br />
Figure 1. DMA-SC-MF analyzis of composites Nextel 440 – Basalt<br />
Storage modulus (MPa)<br />
<strong>60</strong>00<br />
5000<br />
4000<br />
3000<br />
2000<br />
1000<br />
0<br />
Storage modulus comparison<br />
40 <strong>60</strong> 80 100 120 140 1<strong>60</strong> 180 200<br />
Temperature (°C)<br />
C C4N1 C1N1 C1N4 N550 Resin<br />
Figure 2. DMA-SC-MF analyzis of composites Nextel 550 – Carbon<br />
Law of mixtures assessement<br />
Composite´s features must obey a law of mixtures (see equation 1), where E<br />
represents an appropriate property of composit, ν volume fraction and subscripts 1, f1,<br />
2 refers to each type of reinforcement (1 for Nextel 440, 550 and 2 for basalt and<br />
carbon respectively) and m to the matrix respectively. η represents a parametr of<br />
ideality of adhesion between matrix and reinforcement. Varying this parameters allowe<br />
us tailoring the material properties.<br />
Equation 1.<br />
E = η1νf1E1 + η2(1-νf1)E2 + νmEm<br />
η1 = η2 = 0,8<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 51
The following figures represents the corelation between theoretical prediction<br />
represented by equation 1 and DMA-SC-MF measurement of each composite. (see<br />
figure 3 and 4). By varying the η1, η2 parameter the equation 1 can be fit to the DMA-<br />
SC-MF (45 °C) dependence. Practicaly it means changing the matrix-reinforcement<br />
interphase features! (see introduction for further informations and conclusion).<br />
E (GPa)<br />
20,00<br />
15,00<br />
10,00<br />
5,00<br />
0,00<br />
Composite Nextel 440/Basalt<br />
0 0,25 0,5 0,75 1<br />
v f1 0,75 0,5 0,25 v f2<br />
Equation 1 DMA-SC-MF (45°C)<br />
Figure 3. Equation 1 vs. DMA-SC-MF (45 °C) Nextel 440 – Basalt composite<br />
E (GPa)<br />
20,00<br />
15,00<br />
10,00<br />
5,00<br />
0,00<br />
Composite Nextel 550/Carbon<br />
0 0,25 0,5 0,75 1<br />
vf2 0,75 0,5 0,25 vf1<br />
Equation 1 DMA-SC-MF (45°C)<br />
Figure 4. Equation 1 vs. DMA-SC-MF (45 °C) Nextel 550 – Carbon composite<br />
Epoxy matrix properties.<br />
The DMA-SC-MF measurement was performed for pure ATLAC 430 resin to<br />
receive the same data sheet as for the DFHC. But another measurement was performed<br />
to gain resin´s curing point – DSC. This was done to get know the safe temperature<br />
interval for heating the resin as a bubbles prevention (see also chapter Preparation of<br />
dual-fibre hybrid composites DFHC specimens).<br />
CONCLUSIONS<br />
DMA-SC-MF analyzis of Nextel 440 – Basalt composites indicates a<br />
correlation between increasing basalt content and decreasing Nextel 440 fibres content.<br />
The prediction of storage modulus was not fulfilled. It should be truly opposite.<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 52
DMA-SC-MF of Nextel 550 – Carbon displays a correlation between increase<br />
in Nextel 550 content and dicrease of carbon fibres. The theoretical prediction prevails<br />
the best mechanical data for carbon reinforced composites.<br />
The explanation of non-corresponding data can be found in the following<br />
sentences. First inhomogenity. It is no easy to ensure an appropriate playcement of<br />
fibres hence they are well sized and <strong>ke</strong>ep their manufactured strand well. Also the<br />
mutual distribution between ceramic basalt or ceramic-carbon fibres wasn´t ideal and it<br />
is one of important assignment that has to be solved in industry too. And third reason<br />
lies with the wettability basalt fibres interact with resin better than the other ones so the<br />
connection between matrix and reinforcement occurs frequently than for carbon or<br />
ceramic fibres where occurs rarely. The non-connected fibres does not sustain any<br />
stress or energy.<br />
The corresponding data between theoretical prediction and practical realization<br />
(figure 3,4) lies with more careful preparation of composites and as well as with<br />
increasing the wettability by varying η. This could be done by a sizing agent, which<br />
permanently binds matrix and the reinforcement, or increasing the time period of<br />
contact between reinforcement and resin befor curing it.<br />
REFERENCES<br />
[1] Composite materials: Engineering and science /F. L. Matthews and R. D.<br />
Rawlings, 1995. p. 1-72, 168-219,251-282.<br />
[2] Introduciton to physical polymer science L. H. Sperling, 2001. <strong>60</strong>4-612.<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 53
ŠTÚDIUM ANTIOXIDANTOV A ICH ÚČINKOV PRI STABILIZÁCII<br />
POLYMÉROV<br />
Bc. Ján Rimarčík, 5. ročník<br />
Vedúci práce: Ing. Erik Klein, PhD<br />
Ústav fyzikálnej chémie a chemic<strong>ke</strong>j fyziky, <strong>Fakulta</strong> chemic<strong>ke</strong>j a potravinárs<strong>ke</strong>j technológie,<br />
Slovenská technická univezita v Bratislave, Radlinského 9, SK-812 37 Bratislava,<br />
Slovenská republika, e-mail: hadica@gmail.com<br />
ÚVOD<br />
Problematika spojená s negatívnym vplyvom prostredia na životnosť polymérov sa<br />
študuje od začiatku ich komerčného využívania. Životnosť polymérov determinuje oxidácia<br />
a mechanická degradácia. Zlúčeniny redukujúce alebo inhibujúce termickú oxidáciu sú<br />
všeobecne nazývané antioxidantmi. Ako špeciálny typ aditív sa pridávajú do polymérov<br />
individuálne alebo ako súčasť kompozitného stabilizačného systému. Cieľom tejto práce bolo<br />
overiť vhodnosť semiempirických kvantovochemických metód AM1 a PM3 na výpočty<br />
disociačných entalpií väzieb N–H, O–H, S–H v molekulách substituovaných anilínov, fenolov<br />
a tiofenolov. Tie slúžia ako modelové štruktúry antioxidantov. Predpoveď vlastností<br />
potenciálnych antioxidantov, ktoré ešte neboli syntetizované, na základe kvantovochemických<br />
výpočtov dokáže ušetriť ekonomické i ľudské zdroje nevyhnutné na syntézu a testovanie<br />
týchto látok. Preto je dôležité zistiť, ako spoľahlivo jednotlivé výpočtové metódy reprodukujú<br />
dostupné experimentálne výsledky.<br />
STABILIZÁCIA – ANTIOXIDANTY<br />
Radikálový mechanizmus navrhnutý Bollandom a Geeom vysvetlil termickú oxidáciu<br />
mnohých polymérov [1]. Rýchlosť oxidácie závisí od najpomalšieho kroku – tvorby voľných<br />
radikálov. Oxidačné reakcie sa vo všeobecnosti uskutočňujú rôznymi spôsobmi. Sú zrýchlené<br />
tepelnou energiou a mechanickým namáhaním pri spracovaní. Často sú katalyzované<br />
prítomnými zvyškami katalyzátora polymerizácie a nečistotami. Využívajú sa 3 hlavné typy<br />
stabilizácie [1, 2]: predstabilizácia, stabilizácia počas procesu spracovania polyméru,<br />
dlhotrvajúca stabilizácia. Antioxidanty môžu ovplyvniť každý krok termic<strong>ke</strong>j oxidácie, čo<br />
záleží od ich zloženia a štruktúry. Pôsobenie stabilizátorov – antioxidantov (InH) počas<br />
radikálovej oxidácie možno opísať rovnicami (P – polymér) [1, 2]:<br />
•<br />
PO 2 + InH ⎯→ ⎯ POOH + In • (1)<br />
PO • + InH ⎯→ ⎯ POH + In • (2)<br />
Radikál na molekule antioxidantu je rezonančne stabilizovaný, čo v konečnom dôsledku<br />
znamená, že tieto radikály prakticky nie sú schopné uskutočniť prenosovú reakciu<br />
na polymér. Preto radikál obyčajne zaniká v terminačnej reakcii s ďalším oxy- alebo<br />
peroxylovým radikálom. Účinnosť antioxidantov sa zvyšuje so znižovaním disociačnej<br />
entalpie (BDE) väzby O–H vo fenolových, resp. N–H amínových antioxidantoch, <strong>ke</strong>ďže prvý<br />
krok terminácie reaktívnych radikálov vznikajúcich počas oxidácie polymérov predstavuje<br />
transfer vodíkového atómu z molekuly antioxidantu na reaktívny radikálový intermediát<br />
[1, 2]. Ďalšou dôležitou charakteristikou antioxidantov je ionizačný potenciál (IP). Níz<strong>ke</strong><br />
hodnoty IP prispievajú k zvýšeniu pravdepodobnosti prenosu elektrónu, čím sa zvyšuje riziko<br />
vzniku superoxidového anión-radikálu prenosom elektrónu priamo na molekulu O2 [1].<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 54
ŠTUDOVANÉ MOLEKULY<br />
Vhodnosť semiempirických kvantovochemických metód AM1 a PM3 na výpočty<br />
disociačných entalpií väzieb (BDE) N–H, O–H, S–H budeme testovať na molekulách<br />
substituovaných anilínov, fenolov a tiofenolov (obr. 1) slúžiacich ako modelové štruktúry<br />
v praxi používaných antioxidantov. Tieto zlúčeniny boli vybrané nielen preto, že existujú<br />
pre ne experimentálne dáta, ale umožňujú nám aj preštudovať efekt substituentov v meta<br />
a para polohách na veľkosť BDE.<br />
X<br />
X<br />
NH 2<br />
NH 2<br />
X<br />
X<br />
SH<br />
SH<br />
X N H COMe X N H NH 2<br />
Obr. 1: Študované molekuly, X = NO2, CF3, CN, Cl, Br, MeCO, MeSO2, PhCO, Me, MeO,<br />
OH, MeO2C, CHO, NMe2, NH2.<br />
VÝSLEDKY A DISKUSIA<br />
Tabuľka 1 uvádza spracované výsledky pre tiofenoly. Hodnota ΔexpBDE je roziel medzi<br />
experimentom a vypočítanou hodnotou pre BDE. Ostatné série zlúčenín boli spracované<br />
analogicky.<br />
Tabuľka 1. Tiofenoly<br />
BDE [kJ/mol] ΔexpBDE [kJ/mol] IP [eV]<br />
názov exp PM3 AM1 PM3 AM1 PM3 AM1 σm, p<br />
p–NO2PhSH 341 394 381 –53 –40 9,53 9,20 0,78<br />
m–CF3PhSH 338 381 367 –43 –29 9,19 8,85 0,43<br />
p–BrPhSH 332 381 369 –49 –37 8,94 8,61 0,23<br />
p–ClPhSH 331 381 366 –50 –35 8,78 8,54 0,23<br />
m–MePhSH 330 378 363 –48 –33 8,75 8,40 –0,07<br />
p–MeO2CPhSH 329 385 375 –56 –46 9,06 8,79 0,45<br />
p–MePhSH 328 377 362 –49 –34 8,66 8,32 –0,17<br />
p–MeOPhSH 322 372 355 –50 –33 8,52 8,18 –0,27<br />
p–NH2PhSH 292 372 348 –80 –56 8,29 7,93 –0,66<br />
m–ClPhSH 335 380 366 –45 –31 8,92 8,63 0,37<br />
PhSH 331 377 363 –46 –32 8,78 8,43<br />
Hodnoty disociačných entalpií väzieb boli vypočítané pomocou programu HYPERCHEM [3].<br />
Experimentálne hodnoty týchto entalpií boli získané metódou termodynamického cyklu.<br />
Autori uvádzajú smerodajnú odchýlku určených hodnôt ±8,5 kJ/mol [4].<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 55<br />
X<br />
X<br />
OH<br />
OH
Na základe experimentálnych údajov pre anilíny sme zistili, že PM3 metóda podhodnocuje<br />
BDE v priemere o 10 kJ/mol. Naopak metóda AM1 entalpie nadhodnocuje v priemere<br />
o 11 kJ/mol. V prípade fenolov obe metódy poskytujú nižšie hodnoty BDE než experiment<br />
pre väčšinu študovaných substituentov. Výnimkou sú meta –NH2, para –OH, para –NH2<br />
pre PM3 a v prípade AM1 aj para –MeO substituent. Priemerná odchýlka vypočítaných BDE<br />
od experimentu je 16 kJ/mol pre PM3 a 9 kJ/mol pre AM1 metódu. Pri substituovaných<br />
tiofenoloch obe metódy významne nadhodnocujú experimentálne entalpie – metóda PM3<br />
v rozmedzí 43–80 kJ/mol, AM1 29–56 kJ/mol. Priemerné odchýlky od experimentálne<br />
určených hodnôt BDE sú 52 a 37 kJ/mol pre PM3 a AM1. Príčinou nepresných výsledkov je<br />
zrejme atóm síry s vyšším počtom elektrónov oproti atómom H, N, alebo O a problémy<br />
súvisiace s výpočtom radikálu, <strong>ke</strong>ďže vypočítaná tvorná entalpia nesubstituovaného tiofenolu<br />
(115 kJ/mol – PM3, resp. 107 kJ/mol – AM1) dobre súhlasí s experimentálnou hodnotou<br />
112 kJ/mol [5].<br />
ZÁVISLOSŤ ΔBDE A ΔIP OD HAMMETTOVÝCH KONŠTÁNT<br />
L. P. Hammett zistil, že pre meta a para substituované deriváty existuje lineárna závislosť<br />
medzi disociačnou konštantou a rýchlosťou hydrolýzy esterov substituovaných benzoových<br />
kyselín [6]. Podobný vzťah platí aj pre rovnovážne konštanty a rýchlostné konštanty. Pratt,<br />
DiLabio a kol. zistili, že ΔBDE (kde ΔBDE = BDEsubstituovaná – BDEzákladná) hodnoty para<br />
substituovaných anilínov a fenolov [7] lineárne závisia od Hammettových konštánt. Všetky<br />
závislosti ΔBDE od σm, σp sú približne lineárne (obr. 2). Najlepšia lineárna korelácia sa<br />
získala pre fenoly a tiofenoly, kde korelačné koeficienty dosiahli hodnoty v rozmedzí 0,916 až<br />
0,954. V prípade substituovaných anilínov sú jednotlivé body rozptýlené okolo regresnej<br />
priamky viac (R=0,813 pre PM3, R=0,873 pre AM1). V prípade substituovaných anilínov<br />
a tiofenolov lepšie korelujú s Hammettovými konštantami výsledky AM1 metódy. Pre fenoly<br />
sú obe metódy rovnocenné – korelačné koeficienty sú zhodné. Podobne sme študovali aj série<br />
para substituovaných acetanilínov a fenylhydrazínov. Aj tu sa potvrdila lineárna závislosť<br />
ΔBDE od Hammettových konštánt. Parasubstituované acetanilíny korelujú lepšie ako<br />
fenylhydrazíny s korelačnými koeficientami podobnými ako pri substituovaných fenoloch<br />
a tiofenoloch. Okrem hodnôt ΔBDE sme korelovali s Hammettovými konštantami aj<br />
vypočítané hodnoty ΔIP. Ionizačné potenciály IP sa rovnajú zápornej hodnote energie<br />
najvyššieho obsadeného orbitálu (HOMO). Hodnoty ΔIP boli určené ako rozdiel IP<br />
substituovanej a nesubstituovanej zlúčeniny. Zistili sme, že tiež lineárne závisia od<br />
Hammettových konštánt. Hodnoty ΔIP korelujú s Hamettovými konštantami lepšie ako<br />
hodnoty ΔBDE (obr. 3). Potenciálnu nepresnosť oboch semiempirických metód možno<br />
pripísať problémom s výpočtom radikálov. Optimálna geometria nemusí zodpovedať<br />
geometrii s minimálnou energiou kvôli metóde polovičného elektrónu, ktorú používajú mnohé<br />
programy [8].<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 56
ΔBDE/kJ mol -1<br />
20<br />
15<br />
10<br />
5<br />
0<br />
-5<br />
-10<br />
-15<br />
-20<br />
-0.8 -0.6 -0.4 -0.2 0.0 0.2 0.4 0.6 0.8<br />
σp, σm<br />
Obr. 2: Závislosť ΔBDE substituovaných<br />
tiofenolov od Hammettových konštánt σm,<br />
σp pre AM1 metódu.<br />
-0.8 -0.6 -0.4 -0.2 0.0 0.2 0.4 0.6 0.8<br />
ZÁVER<br />
Táto práca bola zameraná na posúdenie vhodnosti semiempirických kvantovochemických<br />
metód na predikciu vlastností hlavných skupín antioxidantov. Vypočítané hodnoty<br />
disociačných entalpií väzieb N–H, O–H a S–H sa porovnali s publikovanými<br />
experimentálnymi hodnotami. Odchýlky vypočítaných hodnôt BDE N–H (okrem p –CF3<br />
v prípade PM3) a O–H väzieb od experimentálnych sú blíz<strong>ke</strong> smerodajnej odchýl<strong>ke</strong> BDE<br />
z experimentov. Výpočty BDE S–H väzby v tiofenoloch ukázali, že obe metódy významne<br />
nadhodnocujú experimentálne hodnoty, hoci trendy vyplývajúce z efektu substituentov<br />
na BDE vystihujú uspokojivo. Hodnoty ΔBDE pre všetky série zlúčenín lineárne korelujú<br />
s Hammettovými konštantami σp, σm. Rozdiel medzi IP substituovaných zlúčenín a základnej<br />
tiež lineárne závisí od Hammettových konštánt.<br />
Možno teda konštatovať, že PM3 a AM1 semiempirické kvantovochemické metódy<br />
predstavujú vhodnú alternatívu k DFT (Density Functional Theorem) a ab-initio metódam,<br />
vhodnú predovšetkým pre výpočty molekúl amínových a fenolových antioxidantov s vyšším<br />
počtom atómov. Obe metódy poskytujú výsledky v signifikantne kratšom čase (trvanie<br />
výpočtu niekoľko minút na bežnom PC) ako DFT alebo ab-initio metódy.<br />
LITERATÚRA<br />
[1] Gugumus F.: Oxidation Inhibition in Organic Materials, Vol I, CRC Press, Boca Raton<br />
1990.<br />
[2] Wolf R., Kaul B. L., Plastics Additives, Ullman’s Encyclopedia of Industrial<br />
Chemistry, Vol. A20, VCH, Weinheim 1992.<br />
[3] HYPERCHEM, rel 7.5 for Windows, Hypercube, Inc., 2003.<br />
[4] Zhu Q., Zhang X. M., Fry A. J.: Polym. Degrad. Stab. 57, 43 (1997).<br />
[5] Scott D. W., McCullough J. P., Hubbard W. N., Messerly J. F., Hossenlopp I. A.,<br />
Frow F. R., Waddington G.: J. Am. Chem Soc. 78, 5463 (1956).<br />
[6] Hrnčiar P.: Organická chémia, SPN, Bratislava 1990, str. 110.<br />
[7] Pratt D. A., DiLabio G. A., Mulder P., Ingold K. U.: Acc. Chem. Res. 37, 334 (2004).<br />
ΔIP/eV<br />
0.8<br />
0.6<br />
0.4<br />
0.2<br />
0.0<br />
-0.2<br />
-0.4<br />
-0.6<br />
σp , σm<br />
Obr. 3: Závislosť ΔIP substituovaných<br />
tiofenolov od Hammettových konštánt σm, σp<br />
pre AM1 metódu.<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 57
[8] Stewart J. J. P.: J. Comp-Aided Mol. Design 4, 1 (1990).<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 58
HOŘLAVOST A MECHANICKÉ VLASNOTSTI NANOKOMPOZITŮ<br />
EVA/Mg(OH)2<br />
Sadílek Jiří, 5. ročník<br />
Vedoucí práce: prof. RNDr. Josef Jančář, CSc.<br />
<strong>Vysoké</strong> učení technické v Brně, <strong>Fakulta</strong> <strong>chemická</strong>, Chemie, technologie a vlastnosti<br />
materiálů, Purkyňova 118, 612 00 Brno, e-mail: xcsadilek@fch.vutbr.cz<br />
ÚVOD<br />
Práce se zabývá studiem vlivu velikosti částic hydroxidu hořečnatého (MH) na tepelnou<br />
stabilitu a mechanické vlastnosti poly(ethylen-co-vinyl acetátu), EVAc. Hlavní použití EVAc<br />
je v oblasti kabelové a elektroizolační techniky. Plniva na bázi hydroxidu hořečnatého<br />
představují způsob, jak snížit hořlavost a zároveň modifikovat mechanické vlastnosti EVAc<br />
kopolymeru.<br />
CÍL PRÁCE<br />
• příprava nanočástic Mg(OH)2<br />
• příprava kompozitů<br />
• stanovení tepelné stability, hořlavosti a mechanických vlastností<br />
• interpretace dat<br />
TEORETICKÁ ČÁST<br />
Proces hoření je souborem fyzikálních a chemických procesů, které působí ve třech<br />
rozdílných fázích: plynná (plamen), mezifáze a kondenzovaná (pevná) fáze. Mezifáze je<br />
fázovým rozhraním mezi plynnou a kondenzovanou fází. Plynnou fázi tvoří plynné produkty<br />
degradace polymeru spolu se vzduchem [2]. Kondenzovanou fází je degradující polymer,<br />
případně polymerní kompozit. Vedením tepla z plamene dochází k roztavení pevné fáze,<br />
degradaci kondenzované fáze a k vytvoření hořlavých plynných produktů.<br />
Tepelná degradace EVAc probíhá ve dvou krocích. První krok (300 – 400°C) zahrnuje<br />
odštěpení kyseliny octové a vznik ethylenové struktury na původním řetězci kopolymeru. V<br />
rozmezí teplot 425 – 470 °C nastává rozklad polyethylenového zbytku.<br />
Retardéry hoření lze rozdělit podle mechanismu, kterým působí, na fyzikální inhibitory a<br />
chemické inhibitory. Mg(OH)2 patří mezi fyzikální inhibitory hoření. Fyzikální inhibice<br />
v plynné fázi je založena na působení inertních plynů (H2O), které zřeďují hořlavé plyny a<br />
snižují jejich koncentraci v zóně hoření. Uvolnění H2O je spojeno s výrazným endotermickým<br />
účin<strong>ke</strong>m, který je schopen pohlcovat teplo vznikající v procesu hoření.<br />
Hydroxid hořečnatý se rozkládá na oxid hořečnatý a vodu. Endotermický rozklad<br />
(1450 J/g) začíná při teplotě 300°C a probíhá podle uvedené rovnice:<br />
Mg(OH) 2<br />
2<br />
→ MgO + H O<br />
(1)<br />
Na základě publikovaných výsledků [4] lze usuzovat, že v kompozitech s nanoplnivy<br />
dochází k výrazné imobilizaci polymerních řetězců v důsledku velkého specifického povrchu<br />
plniva. Tato imobilizace má přímý dopad na mechanické vlastnosti nanokompozitů.<br />
V případě tepelné stability a hořlavosti se ovšem mohou imobilizačním efektům konkurovat<br />
katalytické účinky velkého povrchu nanoplniva.<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 59
PRAKTICKÁ ČÁST<br />
Hydroxid hořečnatý byl připraven precipitací síranu hořečnatého a vodného roztoku<br />
amoniaku:<br />
MgSO 4 + 2NH<br />
4OH<br />
→ Mg(OH) 2 + ( NH 4 ) 2SO<br />
4<br />
Kompozitní vzorky byly připraveny míšením kopolymeru EVAc a plniva v roztoku<br />
cykolhexanu. Byly připraveny vzorky s obsahem nanometrického MH připraveného<br />
laboratorně a s obsahem komerčního mikrometrického hydroxidu hořečnatého - FR–20<br />
(DEAD SEA PERICLASE Ltd., Israel). Obsah plniva v kompozitu byl 13obj.% (26hmot.%).<br />
Snímky plniv z transmisního elektronového mikroskopu jsou uvedeny v obr. 1. Snímky<br />
lomových ploch kompozitů připravených v tekutém dusíku jsou na obr. 2.<br />
Obr. 1: TEM použitých MH plniv, nano-MH (vlevo) a mikro-MH FR-20 (vpravo).<br />
Použitá plniva byla charakterizována pomocí následujících metod: (1) BET, (2) rozptyl<br />
světla, (3) transmisní elektronová mikroskopie (TEM), (4) pyknometrické stanovení hustoty,<br />
(5) rentgenová difrakce, (5) termogravimetrická analýza (TGA). Základní charakteristiky<br />
použitých plniv jsou v tab. 1.<br />
Tabulka 1: Charakterizace všech použitých plniv.<br />
Metody stanovení Nanoplnivo FR – 20<br />
TEM (nm) 32 – 34 800 – 900<br />
Spec. povrch (m 2 /g) změřený 43 – 48 5 – 6<br />
Spec. povrch (m 2 /g) počítaný 44 – 47 1 – 2<br />
Teplota rozkladu (°C) 336 – 433 310 - 430<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana <strong>60</strong><br />
(2)
Pro charakterizaci vlastností kopozitních vzorků byly použity následující metody: (1)<br />
termogravimetrická analýza, (2) stanovení kyslíkového čísla, (3) dynamicko-mechanická<br />
analýza, (4) tahové testy.<br />
Obr. 2: SEM snímky lomových ploch kompozitů s nano-MH (vpravo) a s mikro-MH FR-20<br />
(vlevo) .<br />
VÝSLEDKY A DISKUSE<br />
Z průběhu křivek izotermální degradace při 430°C, obr. 3 vlevo, je zřejmé, že vzorky<br />
EVAc plněné nanometrickým hydroxidem hořečnatým vykazovaly rychlejší pokles hmotnosti<br />
v porovnání se vzorky EVAc plněné mikrometrickým MH.<br />
Limitní kyslíkové číslo, kyslíkový index je stanovení relativní hořlavosti plastů podle ČSN<br />
EN ISO 4589-3. LOI je test zpětného hoření tenkých vzorků polymerních proužků v proudu<br />
plynu a vyjadřuje nejnižší koncentraci kyslíku ve směsi s dusí<strong>ke</strong>m (v %), která ještě stačí na<br />
to, aby materiál při podmínkách zkoušky hořel. Nízká hodnota LOI znamená, že materiál hoří<br />
i při malém podílu kyslíku ve směsi.<br />
Obr. 3: TGA s<strong>ke</strong>ny čisté matrice a obou kompozitů při konstantní teplotě 430°C a LOI test.<br />
Uvedené výsledky naznačují, že velký specifický povrch nanometrického MH vede k<br />
výraznému katalytickému působení a snížení retardační schopnosti MH.<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 61
Výsledky tahových zkoušek jsou uvedeny na obr. 4. Z výsledků je patrné, že došlo<br />
k výraznému zvýšení modulu pružnosti (směrnice tahové křivky při hodnotě deformace 0,15<br />
%) u nanokompozitního vzorku ve srovnání s čistou matricí a EVAc plněného<br />
mikrometrickým MH. Na druhé straně, nedošlo k výrazné změně houževnatosti EVAc<br />
kopolymeru s přídav<strong>ke</strong>m MH plniv.<br />
Obr. 4: Mechanická tahová zkouška.<br />
ZÁVĚR<br />
Na základě termogravimetrické analýzy a stanovení limitního kyslíkového čísla bylo<br />
zjištěno, že v případě velký specifický povrch nanometrického hydroxidu hořečnatého může<br />
působit katalyticky na procesy termooxidační degradace. Tento jev je schopen efektivně<br />
snižovat endotermický efekt hydroxidu hořečnatého daný rozkladem na vodu a MgO.<br />
Z mechanických zkoušek vyplynulo, že použití nanometrického MH jako plniva vede<br />
k výraznému zvýšení modulu pružnosti ve srovnání s mikrometrickým hydroxidem<br />
hořečnatým. Tento jev je pravděpodobně dán výraznou imobilizací polymerních řetězců<br />
v přítomnosti velkého specifického povrchu plniva.<br />
LITERATURA<br />
[1] Rychlý, J., Veselý, K., Gál, E., Kummer, M., Jančář, J., Rychlá, L.: Use of Thermal<br />
Methods in the Chracterization of the High-Temperature decomposition and Ignition of<br />
Polyolefins and EVA Copolymers Filled with Mg(OH)2, Al(OH)3 and CaCO3. Polymer<br />
Degradation and Stability, 1990, 30, 57 – 72<br />
[2] Troitzsch, J.: Plastics Flammability Handbook: Principles, Regulations, Testing, and<br />
Approval. 3. vydání. Mnichov: Hanser, 2004. 748 s. ISBN: 1-56990-356-5<br />
[3] Halikia, I., Neou-Syngouna, P., Kolitsa, D.: Isothermal kinetic analysis of the thermal<br />
decomposition of magnesium hydroxide using thermogravimetric data determination of<br />
polymerization shrinkage kinetice in visible-light-cured materials: methods developement.<br />
Thermochimica Acta, 1998, 320, 75 – 88.<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 62
[4] Kalfus, J.: Viscoelastic properties of polyvinylacetate-hydroxyapatite nanocomposites.<br />
Brno : <strong>Vysoké</strong> učení technické, 2005. 106 s.<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 63
STUDIUM MECHANISMU KOORDINAČNÍ POLYMERACE HEXA-<br />
1,5-DIENU KATALYZOVANÉ FENOXYIMINOVÝM KOMPLEXEM<br />
TITANU A METHYLALUMINOXANEM<br />
Jan Ševčík, 4. ročník<br />
Vedoucí práce: Mgr. Soňa Hermanová, Ph.D.<br />
<strong>Vysoké</strong> učení technické v Brně, <strong>Fakulta</strong> <strong>chemická</strong>, ústav chemie a technologie materiálů,<br />
Purkyňova 118, 612 00 Brno, email: xcsevcikj@fch.vutbr.cz<br />
ÚVOD<br />
Koordinační polymerace dienů přitahují pozornost především z důvodu vzniku strukturně<br />
rozmanitých typů monomerních jednotek v polymerních řetězcích. Izolované dieny mohou<br />
podstoupit cyklopolymeraci, ve které je inzerce dvojné vazby monomeru do vazby Mt–C<br />
následována intramolekulární inzercí zbývající dvojné vazby za vzniku methylen-1,3cyklopentanové<br />
jednotky (MCP). 1 Doi a kol. polymerovali hexa-1,5-dien v toluenu pomocí<br />
V(acac)3 a Al(C2H5)2Cl při -78 °C za vzniku homopolymeru obsahujícího 46 mol % 1vinyltetramethylenových<br />
(VTM) jednotek a 54 mol % 1,3-cyklopentylenmethylenových<br />
(MCP) jednotek. 2<br />
Cílem této práce je studium mechanismu polymerace nekonjugovaného dienu<br />
katalyzované fenoxyiminovým komplexem titanu (FITi) a methylaluminoxanem (MAO) jako<br />
kokatalyzátorem. Uvažované způsoby zabudování molekul hexa-1,5-dienu do rostoucího<br />
řetězce jsou znázorněny na obr. 1. Adice dienu může probíhat buď 1,2-(primární) nebo 2,1-<br />
(sekundární) inzercí. Po 1,2-inzerci může následovat inzerce zbývající dvojné vazby v poloze<br />
1,2, čímž dojde k vytvoření cyklopentanového kruhu. Po inzerci dvojné vazby dienu v poloze<br />
2,1 může také následovat 1,2 inzerce zbývající dvojné vazby pří níž vzniká<br />
cyklobutylmethylový meziprodukt, který se β-alkyleliminací přemění na jednotku s boční<br />
vinylovou skupinou.<br />
monomer<br />
FITi<br />
Polymeračně aktivní centrum<br />
FITi<br />
P<br />
(1,2)<br />
MCP<br />
P<br />
P<br />
FITi<br />
+<br />
(2,1)<br />
hexa-1,5-dien<br />
FITi<br />
FITi<br />
cyklobutan<br />
VTM<br />
P<br />
β-alkyl elim.<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 64<br />
P
Obr. 1 Navržené mechanismy inzerce hexa-1,5-dienu<br />
EXPERIMENTÁLNÍ ČÁST<br />
Všechny práce s látkami citlivými na vzduch a vlhkost byly prováděny pod inertní<br />
atmosférou, za použití standardních Schlenkových technik a vakuové linky pracující v režimu<br />
vakuum/inert. Dusík (99,999%, Siad) byl dočištěn pomocí sušících věží naplněných<br />
molekulovými síty (4 Å) a měďným katalyzátorem. Toluen (p.a., Lachema) byl před použitím<br />
sušen pomocí směsi sodík/benzofenon. Hexa-1,5-dien (99 %, Fluka) byl sušen na CaH2<br />
a čerstvě destilován. Kokatalyzátor methylaluminoxan (10 % wt. v toluenu, Crompton<br />
GmbH) byl před použitím zbaven volného trimethylaluminia (TMA) a znova rozpuštěn<br />
v toluenu. Bis[N-(3-terc-butylsalicylidene)-2,3,4,5,6-pentafluoroanilinato]titanium (IV)<br />
dichlorid byl syntetizován dle publikovaného postupu. 3<br />
Polymerace<br />
Polymerace byly prováděny v opláštěném reaktoru (250 ml) s magnetickým míchadlem.<br />
Jednotlivé komponenty byly přidávány v následujícím pořadí: toluen (50 ml), kokatalyzátor<br />
(6 mmol) a monomer (2 mmol). Reaktor byl temperován na příslušnou teplotu po dobu 20<br />
minut a poté byl dávkován roztok katalyzátoru (10 μmol) pro iniciaci polymerace.<br />
Polymerace byla ukončena za <strong>60</strong> min terminací ethanolem (10 ml), polymer byl vysrážen<br />
v roztoku methanolu okyseleném HCl, izolován filtrací a sušen za vakua do konstantní<br />
hmotnosti.<br />
Analýza polymeru<br />
GPC analýza byla provedena na SEC zařízení použitím Polymer Laboratories PL gel<br />
10 μm MIXED-8, 300×7,5 mm koloně, RI detektor RUBY 05. Analýza byla provedena při<br />
25 °C za použití THF jako rozpouštědla. Pro měření 1 H NMR spektra poly(hexa-1,5-dienu)<br />
byl připraven roztok rozpuštěním 20 mg polymeru v 0,5 cm 3 1,1,2,2,-tetrachlorethanu-d2 při<br />
120 °C, který byl následně při této teplotě homogenizován po dobu 8 hodin. Měření NMR<br />
spekter katalyzátorů bylo provedeno na přístroji Bru<strong>ke</strong>r Avance o pracovní frekvenci<br />
300 MHz.<br />
VÝSLEDKY A DISKUZE<br />
Polymerace hexa-1,5-dienu byla provedena v toluenu (50 ml) při teplotě 0 °C za katalýzy<br />
bis[N-(3-terc-butylsalicyliden)-2,3,4,5,6- pentafluoranilinato]titan (IV) dichloridem a MAO<br />
jako kokatalyzátorem. Vzniklý polymer (Mn = 41 kg·mol -1 , Mw/Mn = 1.5) byl po celou dobu<br />
polymerace rozpustný v polymeračním médiu, což indikuje absenci zesítěných struktur.<br />
K zesítění makromolekul by přispívaly nezreagované dvojné vazby monomerních jednotek ve<br />
formě butenylových postranních větví. Během polymerace bylo dosaženo vysokého stupně<br />
konverze (96 %). Počet vzniklých polymerních řetězců však neodpovídal počtu potenciálních<br />
aktivních center. Pouze 39 % aktivních center přispělo k propagaci řetězce, možným<br />
vysvětlením je nedostatečný přebytek kokatalyzátoru, jehož esenciální funkcí je aktivace<br />
katalytického prekurzoru a ochrana aktivního centra. Šířka distribuce molárních hmotností<br />
(Mw/Mn = 1.5) naznačuje, že v systému se uplatňují nežádoucí boční reakce (přenos).<br />
Mikrostruktura řetězce byla analyzována pomocí 1 H NMR spektroskopie. Procentuální<br />
zastoupení VTM jednotek ve vzorku polymeru bylo kvantifikováno na 36,5 % (Obr.2).<br />
Teplota s<strong>ke</strong>lného přechodu polymeru (Tg = -15,3 °C) byla stanovena pomocí DSC.<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 65
Obr. 2 1 H NMR poly(hexa-1,5-dienu), zastoupení jednotek VTM/MCP = 36,5 % / 63,5 %<br />
ZÁVĚR<br />
Prvotní studie prokázala, že systém FITi/MAO je katalyticky aktivní pro polymeraci hexa-<br />
1,5-dienu. Větší podíl MCP jednotek v připraveném poly(hexa-1,5-dienu) při 0 °C naznačuje<br />
upřednostnění adice molekul monomeru na aktivní centrum mechanismem primární 1,2inzerce.<br />
Další výzkum bude zaměřen na studium mechanismu a kinetiky propagace hexa-1,5dienu<br />
za různých polymeračních podmínek (T, vliv koncentrace monomeru, vliv poměru<br />
katalyzátor/kokatalyzátor).<br />
LITERATURA<br />
1. Marvel C. S., Stille J. K.: J Am Chem Soc 80, 1740 (1957).<br />
2. Doi Y., Tokuhiro N., Soga K.: Makromol. Chem. 190, 643 (1989)<br />
3. Fujita T., Mitani M.: J. Am. Chem. Soc. 124, 3327 (2002)<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 66
MOLEKULOVÁ DYNAMIKA C-GLYKOZYLNITROMETÁNOV<br />
Stanislava Šoralová, 5. ročník<br />
Vedúci práce: Mgr. Juraj Kóňa, PhD.*<br />
Ústav fyzikálnej chémie a chemic<strong>ke</strong>j fyziky, FCHPT STU, Radlinského 9, SK-812 37 Bratislava,<br />
Slovenská republika, e-mail: stanislava.soralova@gmail.com<br />
*Chemický ústav SAV, Dúbravská cesta 9, 845 38 Bratislava, SR<br />
ÚVOD<br />
Rezistencia patogénov na existujúce lieky sa stáva čoraz väčším problémom nie len pre<br />
medicínu. To zapríčinilo rozsiahly výskum nových inhibítorov. Pretože výskyt D-rabinofuranózy<br />
a D-galaktofuranózy je v prírode špecifický pre mykobaktérie, protozoa a huby, môžu zlúčeniny<br />
im podobné prekážať enzymaticky katalyzovaným cestám ich spracovania a vytvárať tak<br />
kompetetívnu inhibíciu. Preto sa stále viac stáva žiadanou syntéza C-glykofuranózových<br />
štruktúr.<br />
Monosacharidy v roztoku podliehajú vnútromolekulovej reakcii medzi karbonylovou<br />
skupinou a jednou z hydroxylových skupín. To dáva rôzne možnosti cyklických štruktúr. Ak<br />
daný monosacharid môže poskytovať päť i šesťčlánkové kruhy, sú v rovnovážnom roztoku<br />
zastúpené predovšetkým šesťčlánkové kruhy, relatívne zastúpenie päťčlánkových kruhov je<br />
veľmi níz<strong>ke</strong>, pretože šesťčlánkové kruhy sú oproti päťčlánkovým termodynamicky výhodnejšie<br />
[2].<br />
Napriek tomu bola ako pracovná hypotéza prijatá predstava, podľa ktorej ak má molekula<br />
schopnosť tvoriť oba kruhy, tvorba päťčlánkového cyklu prebieha s väčšou rýchlosťou ako<br />
šesťčlánkového, je kineticky preferovaná. Zrejmým dôvodom je vzdialenosť reakčných<br />
partnerov cyklizačnej reakcie, ktorá je pri tvorbe päťčlánkového kruhu asi o 20% kratšia a z toho<br />
vyplývajúca vyššia vnútromolekulová koncentrácia ako pri šesťčlánkovom cykle.<br />
Pravdepodobnosť cyklizácie klesá s rastúcou dĺžkou reťazca medzi reakčnými partnermi. Táto<br />
hypotéza sa samozrejme nedá aplikovať na menšie kruhy, pretože nárastajúce napätie väzbových<br />
uhlov vo vznikajúcom kruhu by výrazne zvýšilo aktivačnú energiu. [3]<br />
Pri cyklizácii 1,2-dideoxy-1-nitroalk-1-enitolu nie je možné zachytiť primárne vznikajúci<br />
kineticky preferovaný produkt päťčlánkový kruh C-glykozylnitrometánov, pretože sa v dôsledku<br />
rýchlej vratnej konverzie mení na termodynamicky výhodnejší šesťčlánkový kruh. Z tohoto<br />
dôvodu boli rozpracované metódy nerovnovážnej prípravy C-glykozylnitrometánov, ktoré môžu<br />
poskytovať netermodynamické C-glykofuranozyl-nitrometány. [3]<br />
Táto práca naväzuje na výskum prípravy anomérov C-L-arabinofuranozylnitro-metánov<br />
z dostupného 3,4,5,6-tetra-O-acetyl-1,2-dideoxy-1-nitro-L-arabino-1-enitolu 1. V prvom kroku<br />
O-deacetylácia látky 1 v kyslom metanole poskytuje nitroalkén 2, jeho cyklizácia prebieha ako<br />
1,4-adícia poskytujúca C-(L-arabinofuranozyl)-metylnitrónové kyseliny 5 a 6, ktoré po následnej<br />
protonizácii podliehajú novej modifikácii Nefovej reakcie. Táto reakcia zastaví ďalšie zmeny<br />
v cykle, čo umožňuje izolovať kineticky preferované päťčlánkové cykly. Výsledkom sú<br />
dimetylacetálovo chránené C-(L-arabinofuranozyl)metanaly 7, 8 a zmiešaný vnútro-metylový<br />
acetál 9. [3, 4]<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 67
AcO<br />
AcO<br />
Obr. 1<br />
1<br />
NO 2<br />
OAc<br />
OAc<br />
MeOH<br />
+<br />
H<br />
MeOH<br />
H+<br />
HO<br />
O<br />
H<br />
2b<br />
N<br />
OH<br />
OH<br />
HO<br />
O<br />
H<br />
HO OMe<br />
O<br />
OMe<br />
HO OH<br />
7<br />
O<br />
O<br />
2a<br />
NO 2<br />
OH<br />
OH<br />
+<br />
HO NO H 2<br />
O<br />
HO OH<br />
HO OMe<br />
O<br />
OMe<br />
HO OH<br />
HO NO2 O<br />
+<br />
+<br />
HO OH<br />
3<br />
HO NO2H O<br />
HO OH<br />
5 6<br />
H +<br />
MeOH<br />
O OMe<br />
O<br />
HO OH<br />
8 9<br />
+<br />
HO NO2 O<br />
HO OH<br />
METÓDA<br />
Pri voľbe teoretického prístupu bol použitý predpoklad, že aktivačné energie vzniku<br />
anomérov päťčlánkových a šesťčlánkových kruhov sú približne rovnaké a tak nemajú vplyv<br />
na experimentálne získané množstvá produktov. Takisto sa predpokladá, že kineticky<br />
preferované produkty budú mať za rovnaký časový interval menšiu priemernú vzdialenosť<br />
reakčných partnerov, a zároveň bude preferovaná hydroxylová skupina častejšie pod správnym<br />
uhlom k väzbe C=C. U preferovaného anoméru sa bude hydroxylová skupina častejšie<br />
približovať k elektrofilu zo strany typic<strong>ke</strong>j pre tento anomér, prípadne bude mať kratšiu<br />
priemernú vzdialenosť reakčných partnerov ako pri približobaní z opačnej strany.<br />
Vďaka týmto predpokladom bolo možné pre teoretickú analýzu použiť relatívne jednoduché<br />
a rýchle výpočty molekulovej dynamiky pre rôzne C-glykozylnitrometány v rôznych formách<br />
(1,2-dideoxy-1-nitroalk-1-enitolu s protonizovanou nitro skupinou, per-acetylovaný a pod.).<br />
Vzhľadom na nepoužiteľnosť parametrov programu Amber 8 SANDER v tomto prípade,<br />
bolo potrebné ich vyrobiť [1]. Geometrie molekúl boli zoptimalizované v programe<br />
HYPERCHEM, získané geometrie slúžili ako vstup do programu GAUSSIAN G03 verzia D1<br />
Windows. Výsledné geometrie a rozloženia nábojov boli po solvatácii metanolom podrobené<br />
minimalizácií energie počas 300 krokov. Výpočty molekulovej dynamiky boli robené pre čas<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 68<br />
4
10 ns, geometria systému sa zapisovala každé 2 fs. Použitá teplota pri výpočtoch bola 273 K a<br />
243 K.<br />
VÝSLEDKY A DISKUSIA<br />
Tab. 1<br />
Názov a forma<br />
Protonizovaný 1,2-dideoxy-<br />
1-nitro-L-arabino-1-enitol<br />
Protonizovaný 3,4,5,6-tetra-<br />
O-acetyl-1,2-dideoxy-1-nitro-<br />
L-arabino-1-enitol<br />
1,2-dideoxy-1-nitro-<br />
D-galakto-1-enitolu<br />
Protonizovaný 1,2-dideoxy-<br />
1-nitro-D-galakto-1-enitolu<br />
1,2-dideoxy-1-nitro-<br />
D-gluko-1-enitolu<br />
Päťčlánkový<br />
Teplota<br />
273 K 243 K<br />
Priemerná vzdialenosť reakčných<br />
partnerov pre vznik kruhu [Å]<br />
Šesťčlánkový <br />
Päťčlánkový <br />
Šesťčlánkový<br />
4.41 4.99 4.41 4.62<br />
3.76 5.09 4.26 6.34<br />
4.36 4.76 4.17 4.80<br />
4.95 5.05 4.95 5.04<br />
3.95 5.39 4.38 4.62<br />
Ako vidieť z Tab. 1 priemerné vzdialenosti reakčných partnerov sú za rovnakých podmienok<br />
menšie pri vzniku päťčlánkových cyklov. Nejedná sa len o priemerné vzdialenosti ale<br />
i o okamžité vzdialenosti, ktoré sú u päťčlánkových kruhov kratšie (Graf 1).<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 69
vzdialenosť reakčných<br />
partnerov ( )<br />
6.4<br />
5.9<br />
5.4<br />
4.9<br />
4.4<br />
3.9<br />
3.4<br />
2.9<br />
2.4<br />
Protonizovaný 3,4,5,6-tetra-O-acetyl-1,2-dideoxy-1nitro-L-arabino-1-enitol<br />
päťčlánkový<br />
šesťčlánkový<br />
0 0.5 1 1.5 2 2.5 3 3.5 4 4.5 5 5.5 6 6.5 7 7.5 8 8.5 9 9.5 10<br />
čas (ns)<br />
Graf 1<br />
Tento výsledok podporuje pôvodnú predstavu kineticky preferovaných päťčlánkových kruhov,<br />
zároveň i vyhovuje experimentálnym poznatkom získaných z analýzy reakčných produktov<br />
metanolýzy 3,4,5,6-tetra-O-acetyl-1,2-dideoxy-1-nitro-L-arabino-1-enitolu, kde výťažky<br />
päťčlánkových kruhov 7-9 tvoril až 65-78%. Ďalší súhlas výpočtov s experimentom bol<br />
dosiahnutý v určení preferencie jedného anoméru za rôznych teplôt. Z experimentu i výpočtov<br />
vyplýva, že pri níz<strong>ke</strong>j teplote je u rôzne veľkých cyklov tendencia vytvárať len jeden anomér.<br />
Záverom môžeme tvrdiť, že použitá metóda je vhodná na opis kinetického správania<br />
C-glukozylnitrometánov.<br />
LITERATÚRA<br />
[1] T. M. Glennon, K. M. Merz Jr.: J. Mol. Struc. (Theochem), 395-396 (1997) 157-171.<br />
[2] G. Illuminati, L. Mandolini: Acc. Chem. Res. 14 (1981) 95-102.<br />
[3] M. Petrušová, M. Vojtech, Pribulová B., Lattová, E., Matulová M., Poláková M., BeMiller<br />
J. N., Kren V. Petruš L. Carbohydr Res. 341 (2006) 2019-2025.<br />
[4] Ballini R., Petrini M.: Tetraedron <strong>60</strong> (2004) 1017-1047.<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 70
STUDIUM DEGRADACE TISKU NA TENKÝCH POLYMERNÍCH<br />
VRSTVÁCH<br />
Jiří Stančík, 5. ročník<br />
Vedoucí práce: doc. Ing. Michal Veselý, CSc.<br />
<strong>Vysoké</strong> učení technické v Brně, <strong>Fakulta</strong> <strong>chemická</strong>, ústav fyzikální a spotřební chemie,<br />
Purkyňova 118, 612 00 Brno, e-mail: xcstancik@fch.vutbr.cz<br />
ÚVOD<br />
Cílem práce bylo zkoumání vlastností potisků prováděných na polymerních vrstvách<br />
s anorganickými plnivy. Jako médium byl použit filtrační papír, polymerním nosičem byl<br />
modifikovaný polyvinylalkohol (dále jen PVAL). Do roztoku PVAL byl dispergován<br />
modifikovaný oxid titaničitý (TiO2). TiO2 je často používaným polymerním plnivem pro svou<br />
snadnou dostupnost a nízkou cenu. Zlepšuje vlastnosti polymerních vrstev při tisku, ale může<br />
urychlovat negativní vlivy, které způsobují degradaci použitých inkoustů vlivem slunečního<br />
záření.<br />
TEORETICKÁ ČÁST<br />
Na kvalitu výtisku mají vliv tři stěžejní faktory: tisk, skladování a potiskované médium<br />
(papír). Kvalita papírů používaných pro tisk je hodnocena hlavně z hlediska jejich textury,<br />
světlosti a opacity.<br />
• Textura: může být hrubá nebo hladká.<br />
• Světlost: vyjadřuje se v jednotkách od 1 do 100, přičemž hodnota 100 odpovídá<br />
nejsvětlejšímu papíru.<br />
• Opacita: je mírou neprůhlednosti či odrazivosti média. Vypočítá se jako poměr<br />
světelného toku dopadajícího na papír ku světelnému toku, který papírem projde<br />
(odrazí se). Čím je opacita vyšší, tím je papír neprůsvitnější (matnější), má lepší<br />
kvalitu.<br />
Opacitu lze zjistit pomocí veličiny průsvitnost (ν) podle vztahu 100 − ν. Průsvitnost lze<br />
vypočítat ze vztahu<br />
ν = 100<br />
Rb<br />
− Rč<br />
,<br />
R ´ −R<br />
´<br />
(1)<br />
b<br />
kde Rb´ a Rč´ jsou hodnoty reflektance pro referenční bílou a černou podložku a hodnoty Rb a<br />
Rč jsou hodnoty reflektancí vzorků měřených na těchto podložkách [1] . Použitý filtrační papír<br />
nelze nazvat kvalitním médiem pro inkoustový tisk, avšak pro studium procesu je výhodné<br />
použití nosiče složeného z čistých celulózových vlá<strong>ke</strong>n bez jakéhokoli přídavku plniv a<br />
aditiv.<br />
Kvalitu výtisku lze mimo jiné hodnotit podle optické hustoty a ostrosti vytištěných prvků<br />
(sledování nárůstu rastrového bodu). Vysoká optická hustota a ostrost jsou známkami<br />
kvalitního výtisku.<br />
Skladování je také velmi důležité. Blednutí výtisku může být způsobeno účin<strong>ke</strong>m<br />
chemických látek ze vzduchu (ozón), vlhkostí, ale převážně světlem. Největší podíl na<br />
degradaci barviv má přímé sluneční světlo, hlavně jeho UV složka. Podobný účinek, i když<br />
mnohem menších dopadů, má také umělé osvětlení (žárovka). Další negativní faktory<br />
představují teplo a vlhko. Žádné médium by se tedy nemělo vystavovat příliš vysokým<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 71<br />
č
teplotám a nebo nechávat na vlhkých místech Chyba! Nenalezen zdroj odkazů. . Všechny uvedené<br />
faktory způsobují blednutí, či nežádoucí změny odstínů vytištěných barev.<br />
Světlo odrážející se od pozorovaného objektu je modulované vlastnostmi tohoto objektu<br />
(schopnost povrchu pohlcovat či odrážet světlo určitých vlnových délek). Takto modulované<br />
světlo dopadá do oka a ve zrakovém systému vyvolává určitý barevný vjem. Mezinárodní<br />
komise pro osvětlení (Commision Internationale de l’Eclarage – CIE) v roce 1931<br />
standardizovala systém měření barev a definovala standardní zdroje osvětlení (definice<br />
spektrálních charakteristik sady světelných zdrojů, se kterými se nejčastěji pracuje, světelný<br />
zdroj A až F, kdy nejpoužívanější pro grafiku jsou D50 a D65), podmínky osvětlování vzorku<br />
a detekce odraženého světla. Definovala také spektrální citlivost detektorů zavedením funkcí<br />
standardního pozorovatele a doporučila způsob vyhodnocování získaných údajů.<br />
Standardní pozorovatel představuje spektrální citlivost průměrného zdravého lidského oka<br />
na tři základní barvy – červenou, zelenou a modrou. Funkce standardního pozorovatele se<br />
také označují jako CIE trichromatické členitelé x(λ), y(λ), z(λ) nebo 2° standardní<br />
pozorovatel CIE 1931, protože odpovídají pozorování barevného pole v úhlu 2°. V roce 1964<br />
CIE definovala a doporučila tzv. CIE 1964 doplňkové trichromatické členitele pro 10°<br />
standardního pozorovatele.<br />
Ve smyslu barevného vjemu je možné jednoznačně definovat barvu pomocí tří čísel –<br />
trichromatických složek X, Y a Z, vypočítaných z remisních křivek barevného vzorku R(λ),<br />
spektrální distribuce osvětlení Φ 0 (λ) a funkcí trichromatických členitelů x(λ), y(λ) a z(λ). Pro<br />
příklad bude uveden výpočet trichromatické složky X, přičemž dvě ostatní trichromatické<br />
složky se vypočtou podle stejného vzorce, pouze se zaměněním patřičného trichromatického<br />
členitele<br />
730nm<br />
380nm<br />
0<br />
X = K ∫ Φ ( λ) ⋅R( λ) ⋅x(<br />
λ)dλ, (2)<br />
kde konstanta K je dána vztahem<br />
100<br />
K =<br />
. (3)<br />
0<br />
∫Φ<br />
( λ) ⋅ y(<br />
λ)dλ Normováním těchto trichromatických složek lze získat trichromatické souřadnice x, y, z, ze<br />
kterých byl vytvořen barevný model CIE xyY. Mnohem důležitější pro tuto práci je barevný<br />
model, který CIE navrhla v roce 1976 a je označován jako barevný prostor CIE 1976 L*a*b*.<br />
Hodnoty souřadnic udávají polohu barvy v třírozměrném barevném prostoru a získají se<br />
přepočtem z trichromatických složek dle vzorců<br />
*<br />
1 3<br />
L = 116 ⋅ Y Y − , (4)<br />
( n ) 16<br />
( X<br />
1 3<br />
X n ) ( Y<br />
1<br />
n )<br />
1 3 ( Y Y ) ( Z<br />
1 )<br />
3<br />
[ ]<br />
3<br />
[ ]<br />
∗<br />
a = 500 ⋅ − Y<br />
*<br />
b ⋅ n −<br />
n<br />
, (5)<br />
= 200 Z , (6)<br />
kde Xn, Yn, Zn jsou trichromatické složky pro referenční bílou při zvoleném osvětlení a<br />
pozorovateli.<br />
Měrná světlost L* reprezentuje vertikální osu a osy a*, b* tvoří chromatickou rovinu.<br />
Prostor L*a*b* je téměř uniformní, a proto se používá hlavně k vyhodnocování barevných<br />
diferencí a tolerancí (Obr. 1).<br />
Barevnou odchylku ΔE*ab lze vypočítat ze souřadnic L*, a* a b* podle vzorce<br />
*<br />
Eab ( ) ( ) ( ) 2<br />
* 2 * 2 *<br />
ΔL<br />
+ Δa<br />
+ Δb<br />
Δ =<br />
. (7)<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 72
Při měření barev prakticky všechny barevné prostory vycházejí při definici svých<br />
souřadnic z trichromatických složek X, Y a Z. Každý přístroj používaný pro měření barvy by<br />
měl zabezpečit: osvětlení měřeného povrchu zvoleným standardním osvětlením, detekci a<br />
měření změny vlastností odraženého světla od měřeného povrchu a samozřejmě vyhodnocení<br />
a vyčíslení trichromatických složek ve smyslu jejich definice [2] .<br />
−a<br />
zelená<br />
L = 100<br />
bílá<br />
−b<br />
modrá<br />
+b<br />
žlutá<br />
L = 0<br />
černá<br />
Obr. 1 Chromatická rovina barevného prostoru CIE 1976 L*a*b* a osa světlosti L.<br />
POUŽITÉ PŘÍSTROJE<br />
• Barvivová tiskárna Epson R220 obsahující cartridge: C, M, Y, K, LM, LC,<br />
• sonda Spectrolino využívající software Gretag Macbeth Measure Tool 4.1,<br />
• mikroskop Intrecomicro,<br />
• fotoaparát Nikon D70,<br />
• UV metr Lutron 340,<br />
• spektrofotometr Datacolor 3890.<br />
EXPERIMENTÁLNÍ ČÁST<br />
Připravený 5% roztok modifikovaného PVAL byl rozdělen na 5 dílů, do těchto 5 dílů byla<br />
dispergována různá množství modifikovaného TiO2 se sníženou fotokatalytickou účinností.<br />
K nanášení disperzí na filtrační papír byla použita metoda sítotisku. První vzorek byl ovrstven<br />
rozto<strong>ke</strong>m PVAL bez přídavku TiO2, u dalších vzorků byl poměr sušiny PVAL k TiO2 zvolen<br />
následovně: 1:1, 1:2, 1:4, 1:8 a 1:16. Připravené vzorky byly ponechány k volnému vysušení.<br />
U nepotištěných vzorků byla hodnocena světlost a opacita. Poté byla na vzorky vytištěna<br />
sada barevných škál obsahujících azurovou (C), purpurovou (M), žlutou (Y) a kompozitní<br />
černou barvu (K). Škály mají 11 políček na kterých klesá pokrytí inkoustem po 10 % od<br />
100 % po 0 % (Obr. 2). Neexponované vzorky byly vyfotografovány přes mikroskop, na<br />
výsledných snímcích byla hodnocena textura média a ostrost tisku.<br />
Obr. 2 Škála odstínů šedé, tištěná také v barvách C, M, a Y.<br />
+a<br />
červená<br />
Po proměření L*, a*, b* souřadnic a optických hustot (D65, 2° pozorovatel) pro jednotlivé<br />
barvy byly vzorky připevněny na podložku a umístěny na okno nacházející se na slunné<br />
straně budovy. Byly tedy vystaveny extrémním podmínkám pro degradaci potisku. Během 13<br />
dnů po které trvala expozice byla v okně se vzorky prováděna měření intenzity ozáření. Poté<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 73
yly u vzorků opět proměřeny L*, a*, b* souřadnice a optické hustoty. Z výsledků byla<br />
vypočítána barevná odchylka ΔE*ab po první expozici (7). Vzorky byly poté vystaveny druhé<br />
expozici a opět proměřeny.<br />
VÝSLEDKY A DISKUSE<br />
Pro fotografování přes mikroskop byla vybrána políčka s 20% krytím inkoustu, protože<br />
jednotlivé rastrové body jsou na nich snadno rozlišitelné (neslívají se). S rostoucí koncentrací<br />
TiO2 se zlepšuje ostrost tisku. Podrobnějším zkoumáním těchto fotografií bylo zjištěno, že ve<br />
stejném směru se zlepšuje také textura a světlost.<br />
Měření opacity bylo provedeno na přístroji Datacolor 3890 tak, že byla nejprve proměřena<br />
reflektance referenční bílé a černé podložky a poté byly proměřeny jednotlivé vzorky na<br />
těchto podložkách. Ze změřených reflektancí byla vypočítána průsvitnost dle vztahu (1),<br />
z této veličiny pak byla spočtena opacita. Vypočtené hodnoty opacity shrnuje Tabulka 1.<br />
Intenzita ozáření vzorků byla proměřována pravidelně ve stejnou dobu (13:00 SLČ)<br />
v období od 29. 5. do 10. 7. 06. V rámci jednoho dne v daném období byla navíc provedena<br />
měření intenzit ozáření každou hodinu, aby bylo možno udělat si představu o rozložení<br />
intenzit ozáření v průběhu celého dne. Výsledky těchto měření jsou zaznamenány na Obr. 3.<br />
Intenzita ozáření ( μW cm -2 )<br />
300<br />
250<br />
200<br />
150<br />
100<br />
50<br />
0<br />
29.5 12.6 26.6 10.7<br />
Datum<br />
Obr. 3 Výsledky měření intenzit ozáření<br />
Intenzita ozáření ( μW cm -2 )<br />
300<br />
250<br />
200<br />
150<br />
100<br />
a) intenzita ozáření vzorků ve dnech experimentu (13:00 SLČ)<br />
b) průměrná intenzita ozáření vzorku během jednoho dne<br />
50<br />
0<br />
7 9 11 13 15 17 19<br />
Čas (h)<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 74
ΔE * ab<br />
50<br />
40<br />
30<br />
20<br />
10<br />
0<br />
1:0<br />
1. expozice 2. expozice<br />
Azurová<br />
1:1<br />
1:2<br />
1:4<br />
1:8<br />
PVAL:TiO2<br />
1:16<br />
ΔE * ab<br />
50<br />
40<br />
30<br />
20<br />
10<br />
0<br />
1:0<br />
1. expozice 2. expozice<br />
Purpurová<br />
1:1<br />
1:2<br />
1:4<br />
1:8<br />
PVAL:TiO2<br />
Obr. 4 Grafická znázornění barevných odchylek pro azurovou a purpurovou barvu.<br />
Z proměřených barevných škál byla vyhodnocována pouze políčka se 100% krytím<br />
inkoustem. Jejich barevné odchylky ΔE*ab po první i po druhé expozici byly graficky<br />
zaznamenány pro všechny koncentrace TiO2 (Obr. 4, Obr. 5).<br />
Tabulka 1 Nárůst hodnot opacity s koncentrací TiO2.<br />
Poměr PVAL:TiO2 1:0 1:1 1:2 1:4 1:8 1:16<br />
Opacita 57,6 69,4 72,6 74,3 81,8 85,9<br />
ΔE * ab<br />
50<br />
40<br />
30<br />
20<br />
10<br />
0<br />
1:0<br />
1. expozice 2. expozice<br />
Žlutá<br />
1:1<br />
1:2<br />
1:4<br />
1:8<br />
PVAL:TiO2<br />
1:16<br />
ΔE * ab<br />
50<br />
40<br />
30<br />
20<br />
10<br />
0<br />
1:0<br />
1. expozice 2. expozice<br />
Černá<br />
1:1<br />
1:2<br />
1:4<br />
1:8<br />
PVAL:TiO2<br />
Obr. 5 Grafická znázornění barevných odchylek pro žlutou a černou barvu.<br />
Bylo zjištěno, že TiO2 má jako plnivo do polymerních vrstev pozitivní vliv na kvalitu<br />
tisku. Nanášením disperzí na filtrační papíry byla zlepšována jeho textura a světlost, zároveň<br />
docházelo k nárůstu opacity. Všechny tyto faktory se podepsaly na zlepšení kvality výtisku<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 75<br />
1:16<br />
1:16
což bylo prokázáno pozorováním rastrových bodů a textury papíru pod mikroskopem a<br />
následným měřením opacity.<br />
ZÁVĚR<br />
Použitý TiO2 měl mít vlastnosti potlačující jeho fotokatalytické účinky, i přesto však<br />
výrazně přispíval k degradaci potisků což potvrdily vypočtené barevné odchylky. Barevné<br />
odchylky narůstaly s rostoucí koncentrací TiO2. Odchylky také narůstají s počtem expozic,<br />
proto byly po 2. expozici větší než po 1. expozici.<br />
Intenzita ozáření v měřeném období vzrůstala, <strong>ke</strong> konci období její nárůst poklesl.<br />
Průměrná hodnota intenzity ozáření byla 133 μW cm −2 . V průběhu jednoho dne intenzita<br />
ozáření vzrůstala až do 16 hodin, kdy dosáhla svého maxima.<br />
Polymerní vrstvy obsahující TiO2 vykazují zlepšené tiskové vlastnosti, zároveň však<br />
dochází k výrazné degradaci potisků. V další práci tedy bude věnována pozornost použití UV<br />
stabilizátorů za účelem zvýšení archivní stálosti výtisků.<br />
LITERATURA<br />
[1] Nároky na papír při tisku. Univerzita Pardubice [online]. [cit. 6. října 2006]. Dostupné<br />
na www: http://www.upce.cz/priloha/kpf-tiskovepapiry3<br />
[2] Hájek, M., Fade Resistance aneb blednout či neblednout? Fotografování [online]. 2005,<br />
[cit. 6. října 2006]. Dostupné na www:<br />
http://www.fotografovani.cz/art/fotech_fototisk/Fade-resistance-p.html<br />
[3] Veselý, M., Králová, I., Dzik, P., Zita, J., Vnímání barev a jejich měření, <strong>Vysoké</strong> učení<br />
technické v Brně, <strong>Fakulta</strong> <strong>chemická</strong>, Purkyňova 118, 612 00, Brno 2004<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 76
ZMĚNY OBSAHU AMINOKYSELIN V BRA<strong>MB</strong>ORÁCH<br />
V ZÁVISLOSTI NA HNOJENÍ DUSÍKEM<br />
Bc. Petra Valová, 5. ročník<br />
Vedoucí práce: Ing. Otakar Rop, Ph.D.<br />
Univerzita Tomáše Bati ve Zlíně, <strong>Fakulta</strong> technologická, Ústav potravinářského inženýrství<br />
a chemie, Náměstí T.G. Masaryka 275, 762 72 Zlín, e-mail: petra.flajsmanova@email.cz<br />
ÚVOD<br />
Dusík (N) je pro rostliny nejvýznamnější živinou (1). Po přihnojení dusíkatými hnojivy<br />
se zvyšuje intenzita tvorby organických sloučenin v rostlinném organismu. Příliš vysoké<br />
dávky dusíku však mohou inhibovat syntézu zásobních polysacharidů na úkor zvýšené tvorby<br />
některých jiných chemických sloučenin (2). Při hnojení dusí<strong>ke</strong>m se každopádně mění<br />
chemické složení rostlin v důsledku zvýšené schopnosti rostlin přijímat anorganické<br />
i organické složky půdního roztoku (3).<br />
Správně volené hnojení dusí<strong>ke</strong>m má kladný vliv na výnosy a kvalitu brambor. Se zvyšující<br />
se dávkou dusíku však jeho účinnost klesá. To znamená, že v rámci nízkých dávek N na 1<br />
hektar (50 kg) na 1 kg dusíku připadá přírůstek výnosu kolem 100 - 120 kg hlíz, ale u dávek<br />
nad 120 kg N.ha -1 již jenom 20 - 30 kg hlíz (4). Velmi vysoké dávky dusíku snižují obsah<br />
sušiny, škrobu a zhoršují chuť hlíz po uvaření. Existuje i nebezpečí zvýšeného obsahu<br />
dusičnanů v hlízách i když tento jev je více záležitostí průběhu počasí v ročníku a délky<br />
vegetační doby jednotlivých odrůd brambor (5).<br />
Jedním z nejzajímavějších důsledků aplikace dusíku k bramborám může být změna<br />
v obsahu aminokyselin v bramborové hlíze (6). Hnojení dusí<strong>ke</strong>m zvyšuje v bramborových<br />
hlízách obsah celkového dusíku a obsah bílkovinného dusíku i když při velmi vysokých<br />
dávkách N může dojít <strong>ke</strong> stresu rostliny, který má naopak za následek pokles množství<br />
bramborové bílkoviny (7). Přestože se u brambor rostoucích na pozemcích, kde byla<br />
aplikována dusíkatá výživa, většinou setkáváme s vyšším množstvím bílkovin, jejich<br />
výživová hodnota jednoznačně klesá. Tento fakt je zapříčiněn přednostním zabudováváním<br />
dusíku v neesenciálních aminokyselinách (8).<br />
METODIKA<br />
Cílem práce bylo sledovat vliv aplikace dusíku na obsah aminokyselin v dužnině<br />
bramborových hlíz. Pokus byl prováděn v plastových nádobách, do kterých bylo navažováno<br />
po 10 kg stejné zeminy. Nádoby byly umístěny na pokusném pracovišti Ústavu<br />
potravinářského inženýrství a chemie FT UTB ve Zlíně.<br />
Do pokusu byly zařazeny varianty v 8 opakováních, a to bez přídavku dusíku do půdy<br />
a s přihnojením dusí<strong>ke</strong>m na úrovni 20 mg N.kg -1 (odpovídá <strong>60</strong> kg N.ha -1 ) a 40 mg N.kg -1<br />
zeminy (odpovídá 120 kg N.ha -1 ). Dusík byl aplikován ve formě dusičnanu amonného.<br />
K výsadbě byly použity velmi rané brambory odrůdy KORUNA, jejichž hlízy byly pro<br />
analýzu sklízeny v 90 dnech vegetace, kdy jsou v konzumní zralosti.<br />
Pro stanovení celkového dusíku bylo využito metody podle Kjeldahla. Množství dusíku<br />
v hlízách bylo přepočteno na obsah hrubé bílkoviny. Hydrolýza vzorků pro stanovení<br />
aminokyselin byla provedena c (HCl) = 6 mol⋅dm -3 . Aminokyseliny methionin a cystein byly<br />
stanoveny pomocí oxidativně kyselé hydrolýzy ve směsi 85 % kyseliny mravenčí a 30 %<br />
peroxidu vodíku (9). Chromatografická analýza hydrolyzátu byla provedena na přístroji AAA<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 77
400 (INGOS Praha) pomocí sodnocitrátových pufrů a ninhydrinovou detekcí. Obsah<br />
aminokyselin byl vyjádřen a mezi variantami porovnán standardní hodnotou - v g<br />
aminokyseliny na 16g N (10).<br />
Výsledky chemických analýz byly zpracovány metodou analýzy variance.<br />
Pro vyhodnocení průkaznosti rozdílů byl použit Scheffého test při 95 % hladině významnosti<br />
(11).<br />
VÝSLEDKY<br />
Výnosové parametry a výsledky chemických analýz jsou uvedeny v tabulkách I – III.<br />
Se zvyšujícím se přídav<strong>ke</strong>m dusíku do půdy vrůstal výnos bramborových hlíz. U varianty<br />
s 40 mg N.kg -1 zeminy byl ve srovnání s kontrolní variantou výnos větší o 83 %. U této<br />
varianty bylo také množství hlíz získaných z jedné rostliny statisticky průkazně vyšší než<br />
u varianty kontrolní a varianty s 20 mg N.kg -1 zeminy. Naopak průměrná hmotnost 1 hlízy<br />
se snižovala (tab. I).<br />
Stupňované dávky dusíku snižovaly množství sušiny v bramborových hlízách (tab. I).<br />
Statisticky významně se snižovalo také celkové množství aminokyselin a hrubé bílkoviny<br />
(tab. II). Přihnojení dusí<strong>ke</strong>m na úrovni 20 mg N.kg -1 zeminy mělo za následek snížení obsahu<br />
všech aminokyselin v čerstvé hmotě bramborových hlíz ve srovnání s kontrolou (pokles<br />
o 27 %). Další přídavek dusíku (tj. 40 mg N.kg -1 zeminy) opět způsobil nižší obsah většiny<br />
aminokyselin s výjimkou histidinu a tyrosinu jejichž množství se mírně zvýšilo. Ve srovnání<br />
s variantou 20 mg N.kg -1 zeminy byl celkový obsah aminokyselin u varianty s vyšší dávkou<br />
dusíku snížen o 17 % (tab. II).<br />
Při vyjádření obsahu aminokyselin v bramborové hlíze standardní hodnotou v gramech<br />
aminokyseliny na 16 g hlízového dusíku je také patrný trend snižování obsahu všech<br />
aminokyselin v závislosti na stupňovaných dávkách dusíku v půdě (tab. III). Výjimkou je opět<br />
vyšší obsah histidinu a tyrosinu u varianty s vyšší dávkou dusíku v půdě ve srovnání<br />
s variantou s 20 mg N.kg -1 půdy. Zatímco u kontrolní varianty bylo zaznamenáno přibližně<br />
90 % aminokyselin vázaných v bílkovině, u brambor hnojených dusí<strong>ke</strong>m na úrovni<br />
20 mg N.kg -1 zeminy byl jejich obsah jen 67 % a v bramborových hlízách získaných z půdy<br />
s 40 mg N.kg -1 byl obsah aminokyselin v bílkovině pouze 65 % (tab. III).<br />
Tabulka I. Průměrná hmotnost bramborových hlíz v gramech, průměrný počet hlíz v kusech<br />
a průměrná hmotnost 1 hlízy v gramech připadající na 1 nádobu a průměrný obsah sušiny<br />
v bramborových hlízách (v hmotnostních %)<br />
Parametr Kontrolní varianta Varianta s 20 mg<br />
N.kg -1 zeminy<br />
Varianta s 40 mg<br />
N.kg -1 zeminy<br />
Výnos 118,33 g 168,33 g 217,50 g<br />
Počet hlíz 5,5 ks 6,33 ks 10,75 ks<br />
Hmotnost 1 hlízy 21,51 g 26,59 g 20,23 g<br />
Sušina 23,5 % 22,18 % 21,14 %<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 78
Tabulka II. Obsah aminokyselin a hrubé bílkoviny v bramborových hlízách (g.kg -1 čerstvé<br />
hmoty)<br />
Aminokyselina Obsah v kontrolní<br />
variantě<br />
Obsah u varianty s 20<br />
mg N.kg -1 zeminy<br />
Obsah u varianty<br />
s 40 mg N.kg -1<br />
zeminy<br />
Valin 0,84 0,68 0,55<br />
Leucin 1,06 0,88 0,72<br />
Isoleucin 0,63 0,53 0,45<br />
Threonin 0,70 0,57 0,45<br />
Methionin 0,33 0,31 0,26<br />
Lysin 1,01 0,81 0,66<br />
Fenylalanin 0,73 0,61 0,51<br />
Histidin 0,33 0,20 0,25<br />
Arginin 1,06 0,73 0,57<br />
Cystein 0,37 0,33 0,25<br />
Kyselina asparagová 2,23 1,84 1,32<br />
Kyselina glutamová 2,19 1,67 1,39<br />
Serin 0,69 0,<strong>60</strong> 0,48<br />
Prolin 0,96 0,75 0,59<br />
Glycin 0,65 0,52 0,43<br />
Alanin 0,62 0,47 0,43<br />
Tyrosin 2,02 1,41 1,72<br />
Celkové množství<br />
aminokyselin<br />
16,42 12,91 11,03<br />
Hrubá bílkovina 18,20 17,59 16,86<br />
Tabulka III. Obsah aminokyselin v bramborových hlízách (g.16 g -1 dusíku - vyjadřuje<br />
přibližně procentické zastoupení příslušné aminokyseliny v bílkovině)<br />
Aminokyselina Obsah v kontrolní Obsah u varianty s 20<br />
variantě mg N.kg -1 Obsah u varianty s 40<br />
zeminy mg N.kg -1 zeminy<br />
Valin 4,63 3,54 3,28<br />
Leucin 5,80 4,56 4,23<br />
Isoleucin 3,43 2,74 2,64<br />
Threonin 3,81 2,97 2,65<br />
Methionin 1,82 1,61 1,55<br />
Lysin 5,52 4,21 3,90<br />
Fenylalanin 3,99 3,19 3,00<br />
Histidin 1,82 1,06 1,47<br />
Arginin 5,84 3,81 3,37<br />
Cystein 2,06 1,69 1,48<br />
Kyselina asparagová 12,25 9,59 7,83<br />
Kyselina glutamová 12,04 8,69 8,27<br />
Serin 3,75 3,12 2,87<br />
Prolin 5,31 3,9 3,49<br />
Glycin 3,54 2,72 2,56<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 79
Alanin 3,47 2,45 2,54<br />
Tyrosin 11,07 7,36 10,20<br />
Celkové množství<br />
aminokyselin<br />
90,15 67,21 65,33<br />
REFERENCES<br />
1) PURVES W. et al., 2004. Life: The Science of Biology. Sunderland: Sinauer<br />
Associates:1121 p.<br />
2) WESTERMANN D. T. et al., 1994. Nitrogen and potassium fertilization on potatoes –<br />
sugars and starch. American Potato Journal, 71 (7): 433 - 453.<br />
3) LIN S. et al., 2004. Influence of nitrogen nutrition on tuber quality of potato with special<br />
reference to the pathway of nitrate transport into tubers. Journal of Plant Nutrition, 27 (2):<br />
341-350.<br />
4) ROP O., 1999: The toxic elements content in early potato varieties. Thesis. Brno, MZLU:<br />
77 p.<br />
5) VOKÁL B., RADIL B., 1996. Effects of row spacing on tuber yield, dry matter content<br />
and starch in potatoes. Rostlinna vyroba, 42 (1): 5 – 9.<br />
6) FRIEDMAN M., 2000. Nutritional value of proteins from different food sources.<br />
A Review. Journal of Agricultural and Food Chemistry, 70 (1): 6 – 29.<br />
7) TALAAT M., 2003. The effect of mineral and compost fertilization on the nitrogen content<br />
and amino acid composition of potato and oat grains. Thesis. Wien, Universität Wien: 154 p.<br />
8) MITRUS J. et al., 2003. The influence of selected cultivation on the content of total protein<br />
and amino acids in the potato tubers. Plant Soil and Environment, 49 (3): 131 – 134.<br />
9) JANDASEK J., KRÁČMAR S., MILERSKI M et al., 2003. Comparison of the contents<br />
of intramuscular amino acids in different lamb hybrids. Czech Journal of Animal Science, 48<br />
(7): 301 – 306.<br />
10) Official Journal L 206. Eighth Commission Directive 78/633/EEC of June 15, 1978.<br />
Establishing Community Methods of Analysis for the Official Control of Feeding Stuffs, July<br />
29, 1978, 0043 – 0055.<br />
11) UNISTAT : Statistical Package for Windows. London : Unistat House, 2002, s. 406 –<br />
419.<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 80
PRECIPITATION OF DL – VALINE FROM AQUEOUS ISOPROPANOL<br />
SOLUTIONS<br />
Bc. Miroslav Variny, 5. ročník<br />
Vedúci práce: Ing. Ján Šefčík, PhD.<br />
Slovenská Technická Univerzita, <strong>Fakulta</strong> Chemic<strong>ke</strong>j a Potravinárs<strong>ke</strong>j Technológie, oddelenie<br />
chemického a biochemického inžinierstva, Radlinského 9, 812 37 Bratislava, Slovenská<br />
Republika, e-mail: laurelindorenan@zoznam.sk<br />
INTRODUCTION<br />
Protein–coated microcrystals (PCMCs) 1,2 show potential in providing a novel, inexpensive<br />
method of administration of modern drugs. Their formation involves mixing of aqueous<br />
excipient and protein solution with excipient saturated solvent, miscible with water, in which<br />
both protein and excipient are less soluble. However, mechanism and kinetics of PCMC<br />
nucleation and growth are poorly understood. There are some preliminary indications from<br />
previous experimental work that the precipitation process does not proceed through classical<br />
nucleation/growth mechanism, but rather an intermediate liquid-liquid phase separation step<br />
was postulated 3,4 . In order to better design, scale-up and control PCMC formation processes,<br />
better understanding of underlying physico-chemical mechanisms is needed. Therefore, in this<br />
project I focussed on experimental studies of precipitation of a suitable excipient 3,4 , the amino<br />
acid DL–valine, from aqueous isopropanol solutions.<br />
EXPERIMENTAL PART<br />
Materials used in experiments were of laboratory reagent grade: 2-propanol (isopropanol),<br />
DL-valine, deionised water from in–house Millipore Water System and were used without<br />
further purification. Laboratory instruments used for measurements were DU 800<br />
Spectrophotometer (Beckmann Coulter) for turbidimetry and Malvern Mastersizer MS 2000<br />
(Malvern Instruments) for small angle static light scattering measurements. Procedure of<br />
solution preparation can be found elsewhere 3 .<br />
Standard operation procedure of a spectrophotometric measurement included using light<br />
of the wavelength <strong>60</strong>0 nm and manual control of cuvette placement. The experiment itself<br />
consisted of taking blank measurement, sample preparation, injecting a small volume of<br />
prepared sample in cuvette and measuring the sample sufficiently long to achieve a plateau in<br />
absorbance, or for at least <strong>60</strong>0 – 800s. Typical sample preparation included injecting small<br />
volume of valine solution in valine saturated isopropanol and mixing for 20 s on magnetic<br />
stirrer. Isopropanol solution volume ranged from 10 to 20 ml, volume of valine aqueous<br />
solution was between 30 and 100 μl. For one set of experimental conditions, at least five<br />
measurements were performed. In one set of experiments I deliberately changed the initial<br />
mixing time (leaving the mixing intensity unchanged). In dilution experiments, a 1–2 ml<br />
sample was diluted by dilution ratio from 1:1 to 1:4 after 20 s of mixing.<br />
Raw data from Mastersizer measurements were analyzed with standalone to obtain the<br />
mean radius of gyration. Radius of gyration is equal to the geometrical radius divided by 1,41<br />
(=2 1/2 ) for thin disks, which correspond to crystal shape of DL-valine observed in these<br />
experiments.<br />
In batch experiments, samples were prepared identically to those measured by the<br />
spectrophotometer. From samples mixed either for 20 s or all time, at certain times a small<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 81
sample was ta<strong>ke</strong>n. It was immediately injected in a glass bea<strong>ke</strong>r filled with bigger volume of<br />
quenching solution and well stirred for a couple of seconds. Small Volume Dispersion Unit,<br />
attached to measurement cell, had to be wetted with isopropanol, pumped through it for a<br />
couple of minutes, to ensure bubble removal from the cell window. Only after this procedure,<br />
the measurement itself could be started. Each measurement included also background scan.<br />
For the purpose of stopped flow experiments 5 , a complete apparatus has been kindly<br />
provided by XstalBio Inc. This apparatus is typically used to produce PCMCs in amounts of<br />
several hundreds of grams per hour. I managed to obtain a complete data set, having used 50<br />
mg/ml and 70 mg/ml valine aqueous solution, initial isopropanol flow rates 100, 200 and 400<br />
ml/min and resulting water content of 1 or 1,5%. In order to assess the effect of added protein<br />
on kinetics of the process and particle size, I repeated these measurements with addition of 5<br />
% w/w albumine in aqueous valine solution.<br />
RESULTS AND DISCUSSION<br />
Average absorbance<br />
1<br />
0,8<br />
0,6<br />
0,4<br />
0,2<br />
0<br />
0 200 400 <strong>60</strong>0 800 1000<br />
Time (s)<br />
Fig. 1 Spectrometric data for samples with 100 μl of<br />
aqueous valine solution with valine concentration 50<br />
mg/ml added to a certain volume of saturated<br />
isopropanol solution (ml): solid – 10, dash – double<br />
dotted – 12, dotted – 15, dash - dotted – 17, dashed -<br />
20.<br />
Fig. 1 shows sensitivity of<br />
precipitation kinetics to the<br />
volume ratio of aqueous and<br />
isopropanol solutions. In terms<br />
of both initial absorbance<br />
increase as well as the final<br />
absorbance reached, it can be<br />
seen that even a small difference<br />
in the volume ratio has extensive<br />
effects. Absorbances for two of<br />
volume ratios of aqueous and<br />
isopropanol solutions are<br />
depicted separately in Fig. 2,<br />
together with the measured radii<br />
of gyration. In the case of<br />
classical nucleation and growth<br />
mechanism, many small nuclei<br />
would form in first moments of<br />
precipitation and in later stages<br />
only their size increase would<br />
contribute to absorbance increase.<br />
But while absorbance doubles or even triples, particle size grows only by 15 % (diamonds) or<br />
by <strong>60</strong> % (squares). Furthermore, absorbance dependence on particle size is of order 1/3 - 2/3<br />
(A ~ dp 0,33 - 0,67 ) so that it is not particle size growth, but particle number increase that is<br />
responsible for the observed absorbance increase. This confirms that non-trivial precipitation<br />
mechanism is present in this system.<br />
Spectrophotometric data were subjected to supersaturation analysis based on DL – valine<br />
solubility 4 data. In Fig. 3 data from several sets of experiments are shown. (S-1) equals to<br />
supersaturation driving force for crystal growth, where S=C/Cs, C is concentration of valine in<br />
supersaturated solution and Cs is its solubility in corresponding solution. “Seeded” data relate<br />
to solutions with addition of small amount of valine crystals, which resulted in higher<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 82
absorbance growth rate and its lesser sensitivity to amount of valine available in the<br />
supersaturated solution.<br />
Data from dilution experiments are included as well. Generally, tremendous change in<br />
Ln (Absorbance growth rate)<br />
-3<br />
0<br />
-4<br />
0,5 1 1,5 2 2,5<br />
-5<br />
-6<br />
-7<br />
-8<br />
-9<br />
-10<br />
-11<br />
-12<br />
Ln (S - 1)<br />
Fig. 2 Average absorbance and radii of gyration vs.<br />
time for samples prepared batch wise on magnetic<br />
stirrer by injecting 100 μl of 50 mg/ml aqueous valine<br />
solution in: solid line, diamonds - 15 ml; dotted line,<br />
squares – 10 ml of valine saturated isopropanol.<br />
Average absorbance<br />
1<br />
0,8<br />
0,6<br />
0,4<br />
0,2<br />
0<br />
0 100 200 300 400 500 <strong>60</strong>0<br />
Time (s)<br />
Fig. 3 Ln (Absorbance growth rate) vs. Ln (S – 1)<br />
analysis with linear regression of batch wise prepared<br />
samples by using aqueous solution with<br />
concentration: full diamonds – 50 mg/ml, empty<br />
circles – 50 mg/ml, seeded, empty diamonds – 70<br />
mg/ml, triangles – dilution of more supersaturated<br />
solution, squares – dilution of less supersaturated<br />
solution.<br />
5<br />
4<br />
3<br />
2<br />
1<br />
0<br />
Rg (um)<br />
absorbance growth rate<br />
dependence on (S-1) value can be<br />
seen. Slopes of linear regression<br />
fits in this chart give exponents x<br />
in the dependence of the<br />
absorbance growth rate on (S-1).<br />
The values of exponent x range<br />
from 6-7 (full and empty<br />
diamonds) through 4,5 (circles) to<br />
2-3 (triangles and squares). This<br />
observation is again inconsistent<br />
with simple nucleation and growth<br />
mechanism, where nucleation<br />
should cease or at least slow down<br />
after dilution, thus leaving the<br />
already formed nuclei to grow. In<br />
order to obey absorbance<br />
dependence on particle size<br />
(absorbance grew several times),<br />
nuclei would have to grow to tens<br />
or even to hundreds of μm size,<br />
which immediately would cause<br />
sedimentation. However, this was<br />
not observed. One can therefore<br />
conclude that nuclei formed even<br />
after dilution. Moreover, the<br />
absorbance growth rate after<br />
dilution was higher than the one<br />
for indiluted samples at the same<br />
supersaturation. Mechanism<br />
involving liquid-liquid phase<br />
separation of solution prior to<br />
nucleation and growth of solid<br />
particles would explain observed<br />
phenomena.<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 83
Ln (time of clouding)<br />
9<br />
8<br />
7<br />
6<br />
5<br />
4<br />
3<br />
1 2 3 4 5<br />
(Ln(S)) 2<br />
Fig. 4 Ln(Time of clouding) vs. (Ln(S)) 2<br />
analysis without and with linearization.<br />
Samples prepared by using aqueous<br />
solution with concentration (mg/ml):<br />
diamonds – 70, squares – <strong>60</strong>, triangles –<br />
50, full circles – 30, empty circles – 20.<br />
Concentration of valine in<br />
aqueous solution (mg/ml)<br />
A B<br />
70 -1,273 9,896<br />
<strong>60</strong> -1,230 9,630<br />
50 -1,650 10,345<br />
30 -1,957 10,624<br />
20 -2,973 11,643<br />
Tab. 1 Regression coefficients of<br />
linearization<br />
2<br />
Ln ( time of clouding)<br />
= A(<br />
Ln(<br />
S ) )<br />
ding to Fig. 4.<br />
+ B accor<br />
Fig. 4 provides further evidence for the<br />
role of liquid-liquid phase separation and<br />
related mixing effects in the precipitation<br />
mechanism. It shows the induction time<br />
(estimated as time of clouding of<br />
supersaturated solutions) dependence on<br />
squared logarithm of supersaturation. Data<br />
for valine concentration in aqueous solution<br />
<strong>60</strong>, 30 and 20 mg/ml can be found<br />
elsewhere 3 . In classical nucleation and growth theory, such chart should show a single line<br />
regardless what was valine concentration in the original aqueous solution before mixing with<br />
the isopropanol solution. Clearly, it is not the case here. Linearization 8,9 regression<br />
Rg (um)<br />
20<br />
18<br />
16<br />
14<br />
12<br />
10<br />
8<br />
6<br />
4<br />
2<br />
0<br />
0 100 200 300<br />
Time (s)<br />
Fig. 5 Radii of gyration and laser obscuration as a function of elapsed time for samples<br />
prepared by stopped flow experiments, initial isopropanol solution flow rate 400 ml/min,<br />
aqueous solution with 5% w/w albumine First number – volume flow rate ratio (aqueous<br />
solution/isopropanol solution), second number – concentration of valine in aqueous<br />
solution: diamonds – 1 %, 50 mg/ml, big squares – 1,5 %, 50 mg/ml 1. Run, small squares<br />
– 1,5 %, 50 mg/ml 2. Run, triangles – 1 %, 70 mg/ml, circles – 1,5 %, 70 mg/ml.<br />
Laser obscuration (%)<br />
100<br />
80<br />
<strong>60</strong><br />
40<br />
20<br />
0<br />
0 100 200 300<br />
Time (s)<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 84
coefficients, summed up in Tab. 1, show a clear trend of slope and intercept as valine<br />
concentration changes.<br />
In Fig. 5 clear evidence of precipitation rate dependence on solutions flow rate can be<br />
seen, which is caused by imperfect mixing in static mixer. No significant particle size changes<br />
can be observed with changing solutions flow rate or solutions flow rate ratio in this chart, but<br />
generally particles grow quic<strong>ke</strong>r as supersaturation increases. Albumine presence has only<br />
little influence on both particle size and precipitation rate, except an interesting effect that in<br />
some measurement particle size at first decreased. This happened entirely when using aqueous<br />
valine solution with concentration 70 mg/ml and, except one case, always in the presence of<br />
albumine. It remains an open question how did albumine presence modify the precipitation<br />
process. But regardless of albumine addition, valine particle sizes are much bigger than those<br />
seen in batch experiments (Fig. 2), their sizes being similar to those valine particles formed by<br />
mixing in the vortexer unit (without stirring bar present). This indicates existence of<br />
additional shear stress effects 10 on precipitation kinetics.<br />
CONCLUSIONS<br />
During this research project, I obtained a wide range of data characterising precipitation of<br />
DL – valine from aqueous isopropanol solutions under various conditions.<br />
The spectrophotometric data show that the precipitation process is very sensitive both in<br />
respect to supersaturation and to water content in final solution. Seeding the solution with<br />
valine crystals can further speed up the precipitation kinetics. Mixing and dilution<br />
experiments show different absorbance vs. time dependence, compared to standard samples<br />
with the same supersaturation. The conclusion from the spectrophotometric measurements<br />
and supersaturation analysis is that DL-valine precipitation is strongly mixing-dependent 6-8<br />
with indications pointing to intermediate liquid-liquid phase separation, confirming earlier<br />
preliminary experiments.<br />
Small angle static light scattering measurements and the following data analysis show<br />
particle size dependence on the type and length of mixing. Particle sizes from magnetic stirrer<br />
agitated batch experiments are smaller than those from either vortexer mixed batch<br />
experiments or stopped flow experiments. In some cases the visible particles (domains) at first<br />
decrease in size and after passing through a minimum, they start to grow, which is consistent<br />
with existence of liquid particles in precipitation process formed by liquid-liquid phase<br />
separation.<br />
REFERENCES<br />
(1) Kreiner, M.; Moore, B. D.; Par<strong>ke</strong>r, M. C. Chem. Commun. 2001, 1096 – 1097<br />
(2) Kreiner, M.; Moore, B. D.; Par<strong>ke</strong>r, M. C. J. Mol. Cat.B 2005, 65 – 72<br />
(3) De Miguel, S. A. Effects of composition and mixing on formation of valine microcrystals<br />
2006<br />
(4) Vos, J. Understanding the formation mechanism of protein coated microcrystals 2006<br />
(5) Roelands, C. P. M.; Roestenberg, R. R. W. Cryst. Growth Des. 2004, 4, 921 – 928<br />
(6) Vekilov, P. G. Cryst. Growth Des. 2004, 671 – 685<br />
(7) Laferrère, L.; Hoff, C.; Veesler, S. J. Cryst. Growth 2004, 550 – 557<br />
(8) Brick, M. C.; Plamer, H. J; Whitesides, T. H. Langmuir 2003, 19, 6367 – 6380<br />
(9) Mahajan, A. J.; Kirwan, D. J. J. Phys. D.: Appl. Phys. 1993, 26, B176 – B180<br />
(10) Kaneko, S.; Yamagami, Y.; Tochihara, H. J. Chem. Eng. Jpn. 2002, 35, 1219 – 1223<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 85
ÚVOD<br />
VÝVOJ MIKROŠTRUKTÚRY SODNO-BORITO-KREMIČITÝCH<br />
SKIEL S PRÍDAVKOM TIO 2<br />
Oto Vojtechovský, 5. ročník<br />
Vedúci práce: RNDr. Jana Kozánková<br />
FCHPT, Ústav anorganic<strong>ke</strong>j chémie, technológie a materiálov, STU Bratislava,<br />
Radlinského 9, 812 37 Bratislava, Slovenská Republika,<br />
e-mail: OtoVojtechovsky@zoznam.sk<br />
Sklo tvorí celý rad anorganických a organických látok, ktoré ochladzovaním z kvapalného<br />
stavu určitou rýchlosťou nestačia vytvoriť pravidelné štruktúry. Z anorganických látok<br />
môžeme uviesť:<br />
• prvky: S, Se, Te, P<br />
• oxidy: B2O3, SiO2, GeO2, P2O5, As2O3<br />
• boritany a kremičitany: Na2B4O7, Na2Si2O5<br />
• iné zlúčeniny: sulfidy, selenidy, teluridy [1]<br />
Makrolikvácia v magme<br />
Sklá boli považované za homogénne izotropné hmoty. Avšak s objavom sklokryštalických<br />
hmôt sa v súvislosti s cieľavedomým štúdiom nukleácie ukázalo, že mnoho skiel nemožno<br />
zďaleka považovať za skutočne homogénne, ale že sa u nich prejavujú mikrohomogenity,<br />
ktoré sa podarilo dokázať modernými výskumnými metódami. Zistené nehomogenity majú<br />
obdobné charakteristiky, aké sa prejavujú u nemiešateľných kvapalín. Na odmiešanie (tj. na<br />
likváciu) roztavenej lávy upozornili petrológovia Vogt a Levinson Lessing. Odmiešaním sa<br />
snažili vysvetliť mechanizmus magmatic<strong>ke</strong>j diferenciácie a petrogenézy. Okrem<br />
makrolikvácie poznáme aj mikrolikváciu. Obidva tieto pojmy úzko súvisia s teplotou liquidus.<br />
K makrolikvácii dochádza nad teplotou liquidus a k mikrolikvácii pod touto teplotou.<br />
Odmiešanie sa prejavuje aj u skiel typu Vycor.<br />
Teória micelárnej štruktúry skla<br />
Podľa Moriyovej teórie, teórie micelárnej štruktúry skla,sa sklo skladá z dvoch fáz: z<br />
vysoko molekulárnych buniek (mikromiciel) a početných iónov či molekúl, ktoré ich<br />
obklopujú. Táto kombinácia vysokomolekulárnych buniek a ďalších iónov či molekúl<br />
nevykazuje pravidelnosti kryštálov, pretože bunky sú amorfné alebo kvázikryštalické. Medzi<br />
týmito bunkami a obklopujúcimi iónmi existuje rovnováha za každej teploty. Za vysokých<br />
teplôt tavenia sa sklovina skladá z disociovaných iónov a molekúl, ktoré sa vyznačujú veľkou<br />
pohyblivosťou [2]. Pri znížení teploty takéto častice začínajú asociovať a tvoria sa zárodky<br />
mikrofázy. Množstvo a rozmery mikrofázy závisia na tepelnej histórii skla.Pri veľmi rýchlom<br />
ochladzovaní vznikajú pomerne malé zárodky mikrofázy, okolo nej zostáva veľa<br />
disociovaných iónov. Naproti tomu, u pomaly ochladzovaného skla sú rozmery mikrofázy<br />
značne väčšie, pretože je dostatok času k asociácii iónov do väčších útvarov. Za nižších teplôt<br />
sa kvapky navzájom zrážajú (kondenzujú) vo väčšie útvary a sklo tuhne.<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 86
Spinodálny rozklad<br />
• metastabilná oblasť - a<br />
• metastabilná oblasť - b<br />
• nestabilná oblasť – c<br />
Základné charakteristiky Vycor skla<br />
Vycor sklo je zaradené medzi alkalickoboritokremičité sklá. V našom experimente sme<br />
sledovali sklo s prídavkom a bez prídavku TiO2 Toto sklo je zložené z B2O3, SiO2, Na2O.<br />
B2O3<br />
Oxid boritý je bezfarebná sklovitá látka, ktorá len veľmi obtiažne kryštalizuje. Topí sa pri<br />
teplote 450°C. Svoju vysokú sklovitosť a fluiditu si zachováva aj ako zložka viaczložkových<br />
skiel a zlepšuje ich taviteľnosť. Teplota liquidus vstupom oxidu boritého klesá. Skladá<br />
saz nepravidelne usporiadaných skupín BO3 trojuholníkového tvaru, spojených tak, že každý<br />
atóm kyslíka je viazaný s dvoma atómami bóru. V kryštalickom stave sa skladá z dvoch typov<br />
navzájom spojených reťazcov, tvorených tetraedrickými skupinami BO4[4].<br />
SiO2<br />
Oxid kremičitý je veľmi ťažko taviteľná látka. Je to podmienené tým ,že atómy kremíka<br />
a kyslíka tvoria v oxide kremičitom polymérne priestorové útvary. V oxide kremičitom je<br />
každý atóm kyslíka viazaný na dva atómy kremíka. V bežných podmienkach je každý atóm<br />
kremíka viazaný so štyrmi atómami kyslíka, ktoré sú okolo neho usporiadané tetraedricky.<br />
Vzájomné naviazanie tetraédrov umožňuje vznik rôznych modifikácií tuhého oxidu<br />
kremičitého.<br />
Obr. 1. Fázový diagram sústavy Na2O-<br />
B2O3-SiO2 [2].<br />
Obr. 2. Schematické znázornenie<br />
spinodálneho rozkladu pod teplotou<br />
liquidus u modelových sústav SiO2-R2O<br />
(R= Li, Na)[2]<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 87
Pri tepelnom spracovaní odmiešaných skiel dochádza ku zväčšovaniu odmiešaných častíc.<br />
Po počiatočnom stave odmiešania, ktorý môže byť buď nukleácia kvapiek a ich ďalší rast.,<br />
alebo spinodálny rozklad, sa sústava snaží zmenšiť svoje medzipovrchy zväčšovaním častíc.<br />
K tomu môže dôjsť zrážaním (koalescenciou) kvapôčok v dôsledku Brownovho pohybu,<br />
viskóznym tokom a pod. ďlší možný proces vedúci k rastu odmiešaných častíc, ktorý sa<br />
uplatňuje predovšetkým u tuhých skiel je Ostwaldov proces zrenia. Tento proces je založený<br />
na rozdielnej rozpustnosti malých a väčších častíc. Najmenšie častice majú snahu sa<br />
rozpúšťať a rozpustené ióny potom difundujú k väčším stabilnejším časticiam, ktoré rastú.<br />
K zhrubnutiu mechanizmom Ostwaldovho zrenia dochádza u sústavy SiO2 - B2O3<br />
EXPERIMENTÁLNA ČASŤ<br />
V náväznosti na výskum sodno-borito-kremičitého skla (NBS) a sodno-boritokremičitéhoskla<br />
s prídavkom TiO2 (NBST) sme sa podrobnejšie zamerali na sledovanie<br />
vývoja mikroštruktúry NBS a NBST skla v počiatočnom úseku oblasti fázovej separácie pri<br />
tepolte 700°C a teplotnej výdrži v intervale od 0 do 120 minút. Cieľom práce bolo štúdium<br />
zmien v mikroštruktúre NBS a NBST skla temperovaných na teplotu 700°C (pri ktorej<br />
dochádza k fázovej separácii) metódou rastrovacej elektrónovej mikroskopie (REM) .Cieľ<br />
práce bol zvolený na základe pozorovaných zmien optic<strong>ke</strong>j priepustnosti NBS skla v prvých<br />
hodinách výdrže pri 700°C.<br />
Zloženie sledovaných skiel v hmotnostných %:<br />
NBS: 9,09% Na 2O, 26,18% B 2O 3, 64,43 SiO 2 NBST: 9,09% Na 2O, 26,18% B 2O 3, <strong>60</strong>,43 SiO 2,<br />
4,00 TiO 2<br />
Vychladené sklo sme narezali na vzorky veľkosti 5x10x10mm na diamantovej píle. Vzorky<br />
utaveného skla sa fázovo separovali pri teplote 700°C po dobu: 0 min., 30 min., <strong>60</strong> min., 90<br />
min, 120 min. Zmeny v mikroštruktúre vzoriek sme sledovali v rastrovacom elektrónovom<br />
mikroskope REM - TESLA BS – 300. Vzorky skiel boli vodivo upravené naprášením vrstvy<br />
Au v naprašovacej aparatúre BALZERS SCD 050. Sledované boli neleptané a leptané (2,5%<br />
HF) lomové plochy skiel.<br />
Optická priepustnosť (a.u.)<br />
2000<br />
1<strong>60</strong>0<br />
1200<br />
800<br />
400<br />
0<br />
3<br />
2<br />
1<br />
0 5 10 15 20 25 30<br />
Čas (h)<br />
Obr. 3. Zmena optic<strong>ke</strong>j priepustnosti vzoriek NBS skla počas ich temperácie pri<br />
špecifikovaných teplotách: 1 – 700 °C, 2 – 720 °C, 3 – 740 °C [3].<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 88
Obr. 3 poukazuje na pokles priepustnosti vzorky skla v intervale 0-120 min. Tento jav<br />
môžeme vysvetliť tým, že v priebehu spinodálneho odmiešania sa v pôvodnom skle<br />
vytvárajú dve vzájomne penetrujúce fázy s charakteristickou vermikulárnou mikroštruktúrou.<br />
Fázy sa líšia chemickým zložením, a teda aj veľkosťou indexu lomu. Svetelný lúč na každom<br />
fázovom rozhraní mení smer podľa veľkosti indexu lomu prostredia, do ktorého vstupuje.<br />
Opakovanými zmenami smeru lúčov sa časť z nich presmeruje takým spôsobom, že buď<br />
nedochádza k ich vyústeniu na spodnej ploche vzorky, alebo dochádza k ich vyústeniu pod<br />
uhlami divergentnými voči pôvodnému smeru prechádzajúceho žiarenia. Dôsledkom je<br />
zodpovedajúci pokles intenzity registrovaného žiarenia. Veľkosť poklesu žiarenia je úmerná<br />
počtu a veľkosti fázových domén, cez ktoré svetlo prechádza. To súhlasí s<br />
mikroštruktúrou vzoriek (obr. 4, 5) a počtom vytvorených domén (tab.1, 2).<br />
Tabuľka 1<br />
Leptaná vzorka NBS skla<br />
zahrievaná po dobu ( v min.)<br />
Celkový počet domén na ploche<br />
5 μm * 5 μm<br />
Tabuľka 2<br />
Leptaná vzorka NBS skla<br />
zahrievaná po dobu ( v min. )<br />
Celkový počet domén väčších<br />
jako 1 μm *1 μm na ploche<br />
5 μm * 5 μm<br />
Výsledky sledovania mikroštruktúry<br />
0 30 <strong>60</strong> 90 120<br />
140 47 3 ojedinelé Vermikulárna<br />
štruktúra<br />
0 30 <strong>60</strong> 90 120<br />
0 5 3 ojedinelé Vermikulárna<br />
štruktúra<br />
Vybrané obrázky z riadkovacej elektrónovej mikroskopie REM pri zväčšeniach 10000x a<br />
20000x dokumentujú mikroštruktúru skiel tak, ako bola pozorovaná na väčšine pripravených<br />
lomových plôch skiel.<br />
Obr.4. Mikroštruktúra NBS skla po fázovej separácii pri 700°C a) 0 min, b) 30 min, c) <strong>60</strong><br />
min, d) 120 min<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 89
Obr.5. Mikroštruktúra NBST skla po fázovej separácii pri 700°C a) 0 min, b) 30 min, c) <strong>60</strong><br />
min, d) 120 min<br />
ZÁVER<br />
Obrázky 4, 5 vývoja mikroštruktúry v sklách NBS a NBST poukazujú na výrazný vplyv<br />
prídavku TiO2 na rýchlosť fázového odmiešania a tvorby vermikulárnej mikroštruktúry.<br />
Vývoj mikroštruktúry NBS skla (obr. 4) je v súlade s Moriyovou teóriou micelárnej štruktúry<br />
skla. Fázové odmiešanie sledované v sodno-borito-kremičitých sklách umožňuje vylúhovaním<br />
boritanovej fázy pripraviť pórovitý materiál s riadenou veľkosťou pórov a značnou<br />
chemickou a tepelnou odolnosťou kremičitého s<strong>ke</strong>letu. Tento typ pórovitého skla má<br />
významné uplatnenie v oblasti membránových filmov aj pre biologické aplikácie a vzhľadom<br />
na sorpčné vlastnosti aj ako materiál vhodný pre oblasť mikroelektroniky.<br />
LITERATÚRA<br />
1. Hlaváč J., Základy technologie silikátů, str. 135-2712. Voldán J., Odmísení s<strong>ke</strong>l, str. 4-13,<br />
20-253. Mojumdar S. C., Kozankova J., Chocholusek J., Majling J. and Nemecek V., Jour. of<br />
Ther.<br />
Analysis and Calorimetry (2004) 145–152.<br />
4. Volf M. B., Technická skla a jejich vlastnosti, SNTL, Praha 1987, p. 214<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 90
VÝZNAM VOĽNÝCH RADIKÁLOV PRI POŠKODENÍ PAPIERA<br />
(EPR ŠTÚDIUM)<br />
Bc. Zuzana Vrecková, 5. ročník<br />
Vedúca práce: prof. Ing. Vlasta Brezová, DrSc.<br />
Ústav fyzikálnej chémie a chemic<strong>ke</strong>j fyziky, <strong>Fakulta</strong> chemic<strong>ke</strong>j a potravinárs<strong>ke</strong>j<br />
technológie, Slovenská technická univerzita v Bratislave, Radlinského 9, SK-812 37<br />
Bratislava, Slovenská republika, e-mail: zuvrro@gmail.com<br />
ÚVOD<br />
Historické dokumenty na papierových podložkách zahrňujú okrem archívnych a<br />
knižničných materiálov aj ostatné práce, ako sú obrazy, grafiky, kresby, plagáty, plány, mapy<br />
a pod., ktoré predstavujú súčasť kultúrneho dedičstva každého štátu. Veľká časť materiálov,<br />
ktoré boli napísané v posledných 150 rokoch trpí neustálym procesom poškodzovania, ktoré<br />
pri dlhodobom pôsobení môže viesť k úplnej strate tlačených a písaných textov v knižniciach<br />
a archívoch [1, 2].<br />
Pre hodnotenie stability vlastností papierových materiálov je definovaná stálosť<br />
(permanence), ktorá označuje zachovanie pôvodných vlastností papiera s dôrazom na<br />
chemickú stabilitu a jej zmeny sa opisujú chemickými parametrami. Z chemických<br />
parametrov sa sleduje povrchové pH, obsah α-, β-, γ-celulózy, rozpustnosť v zriedených<br />
alkáliách, stupeň polymerizácie, obsah redukujúcich látok (meďné číslo), číslo kappa, obsah<br />
karboxylových skupín, obsah síry resp. rozpustných síranov, obsah dusíkatých látok.<br />
Trvanlivosť (durability) označuje zachovanie mechanických a optických vlastností papiera,<br />
ktoré si uchováva napriek používaniu a pôsobeniu vonkajšieho prostredia. Monitorujú sa<br />
mechanické parametre ako odolnosť v prehýbaní, pevnosť v dotrhnutí, pevnosť v ťahu<br />
a v prietlaku, nulové tržné dĺžky. Z optických parametrov sa hodnotí belosť, špecifický<br />
koeficient svetelnej absorpcie, relatívne dekoloračné číslo, opacita alebo špecifický koeficient<br />
rozptylu svetla [1, 2].<br />
Keďže základný polymérny materiál tvoriaci papier predstavuje celulóza, je proces<br />
degradácie papiera determinovaný poškodením molekulovej a nadmolekulovej štruktúry<br />
celulózy (obr. 1). Poškodenie papiera pri starnutí je<br />
proces, <strong>ke</strong>dy dochádza k značnej zmene<br />
mechanických, optických, chemických a fyzikálnych<br />
vlastností. Proces starnutia papiera pri dlhodobom<br />
skladovaní je determinovaný rôznymi faktormi, ktoré<br />
môžeme rozdeliť na vnútorné (druh, kvalita, chemické<br />
zloženie vlákna, lepidlá, glejidlá, plnidlá, prítomnosť<br />
kyslých skupín a iónov kovov) a vonkajšie<br />
(podmienky uskladnenia, teplota, relatívna vlhkosť<br />
okolitého vzduchu, svetlo, atmosférické znečistenie<br />
Obr. 1.<br />
Štruktúra celulózy a vodíkových väzieb<br />
medzi polymérnymi vláknami [3]<br />
a biologická kontaminácia papiera), pričom počas<br />
dlhodobého skladovania môže dochádzať k vzájomnej<br />
interakcii ako je schematicky znázornené na obr. 2 [4].<br />
Pri laboratórnom hodnotení degradácie papiera je<br />
nevyhnutná simulácia podmienok prirodzeného starnutia metódami urýchleného starnutia, pri<br />
ktorých sa proces degradácie urýchľuje aplikáciou vyššej teploty a rôznej relatívnej vlhkosti<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 91
v klimatizačných komorách [5, 6]. Metódami urýchleného starnutia môžeme zmeny vlastností<br />
papiera zodpovedajúce niekoľkoročnému prirodzenému starnutiu pozorovať už po niekoľkých<br />
dňoch (napr. 3 dni umelého starnutia papiera pri 105 °C zodpovedajú 25 rokom prirodzeného<br />
starnutia [6]).<br />
ENDOGÉNNE FAKTORY<br />
pH<br />
Kovové ióny<br />
Lignín<br />
Produkty degradácie<br />
EMISIA<br />
Prchavé produkty degradácie<br />
ABSORPCIA<br />
Kyslé plyny (SO2, NOx)<br />
EXOGÉNNE FAKTORY<br />
Teplota<br />
Vlhkosť<br />
Kyslík<br />
Svetlo<br />
Znečistenie (prach)<br />
Obr. 2.<br />
Faktory a procesy ovlyvňujúce<br />
degradáciu papiera [4].<br />
Poškodenie polymérneho reťazca celulózy môže prebiehať rôznymi mechanizmami (kyslá<br />
hydrolýza, alkalická degradácia, oxidácia, autooxidácia iniciovaná iónmi kovov,<br />
fotodegradácia, mikrobiologická depolymerizácia), ktoré sa môžu vzájomne ovplyvňovať [4].<br />
Významnú úlohu pri procesoch oxidačného poškodenia papiera zohrávajú kyslíkomcentrované<br />
voľné radikály, ktorých tvorba je iniciovaná prítomnosťou iónov prechodných<br />
kovov a kyslíka (1–4) [7, 8]. Generované reaktívne hydroxylové radikály ( • OH) sú schopné<br />
iniciovať depolymerizáciu celulózy, ako aj neselektívne reakcie s ďalšími zložkami papiera,<br />
napr. s lignínom (5).<br />
Fe(II) + O2 ↔ Fe(III) + O2 •– (1)<br />
Fe(III) + O2 + RH → • R + HOO • + Fe(III) (2)<br />
Fe(II) + HOO • + H + → Fe(III) + H2O2 (3)<br />
Fe(II) + H2O2 → Fe(III) + HO • + OH – (4)<br />
ROH + • OH → RO • + H2O (5)<br />
Výsledkom interakcie reaktívnych radikálov s lignínovými zložkami papiera je vznik<br />
paramagnetických semichinoidných štruktúr, ktoré sa vyznačujú dostatočne dlhým časom<br />
života aj pri laboratórnej teplote a ich prítomnosť v papierovom materiáli je možné priamo<br />
monitorovať pomocou EPR spektroskopie. Predpokladá sa, že koncentrácia semichinoidných<br />
radikálových štruktúr môže reflektovať oxidačné poškodenie papiera [9].<br />
Naša práca je orientovaná na kvantitatívne EPR štúdium semichinoidných radikálových<br />
štruktúr v kyslom papieri, ktorý bol vystavený pôsobeniu urýchleného starnutia v rôznych<br />
experimentálnych podmienkach.<br />
EXPERIMENT<br />
Meranie EPR spektier sme realizovali pri teplote 22 °C na EPR spektrometri EMX<br />
(Bru<strong>ke</strong>r, SRN) s príslušným programovým vybavením. Do okrúhlej kremennej kyvety<br />
(vnútorný priemer 3 mm) na meranie v tuhej fáze sme umiestnili vzorku papiera so známou<br />
hmotnosťou nastrihanú na tenké pásiky (28 mm×1 mm). Kyvetu sme umiestnili do<br />
štandardnej dutiny TE102 (ER 4102 ST) a zmerali sme EPR spektrum. Pri meraniach všetkých<br />
vzoriek sme používali tú istú kyvetu, pričom sme ju vkladali do dutiny spektrometra vždy<br />
rovnakým spôsobom, aby sme zabezpečili reprodukovateľnosť meraní. Pri tejto manipulácii<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 92
s kyvetami je chyba pri určení relatívnej integrálnej intenzity EPR signálu približne 5 %.<br />
Hodnotu g-faktorov radikálov sme určili s presnosťou ±0,0001 simultánnym meraním EPR<br />
spektra semi-stabilného radikálu 1,1-difenyl-2-pikrylhydrazyl (DPPH, Sigma). Na určenie<br />
koncentrácie spinov v jednom grame vzorky sme použili pripravený štandard so známou<br />
koncentráciou DPPH v tuhom NaCl (0,0002 mg DPPH g –1 ).<br />
Na meranie sme použili čiastočne drevitý, kyslý písací papier (80 g m –2 , pH = 4,4);<br />
Slavošovské papierne, Slavošovce, Slovenská<br />
republika, ktorý sme získali z Oddelenia polygrafie<br />
a aplikovanej fotochémie ÚPM FCHPT STU. Papier<br />
bol vystavený pôsobeniu umelého starnutia v rôznych<br />
podmienkach (suché teplo pri teplotách 105 °C<br />
a 120 °C s; vlhké teplo pri teplote 80°C a relatívnej<br />
vlhkosti 25%, 45% a 65%) počas rôznych časových<br />
intervalov (0, 8, 24, 72, 168, 336 a 672 hodín). Reálny<br />
objekt poškodeného papierového materiálu<br />
predstavovala kniha (rok vydania 1977), ktorá bola<br />
v roku 1980 poškodená kyselinou sírovou a 26 rokov<br />
bola uložená v knižnici pri laboratórnej teplote<br />
Obr. 3.<br />
Kniha poškodená v roku 1980 kyselinou<br />
sírovou.<br />
(obr. 3). Z knihy sme odobrali vzorky s rôznym<br />
stupňom poškodenia a hore uvedeným spôsobom sme<br />
v nich určili koncentráciu spinov g –1 .<br />
VÝSLEDKY A DISKUSIA<br />
Obrázok 4 zobrazuje EPR spektrum namerané vo vzorkách papiera pri laboratórnej teplote,<br />
ktoré predstavuje singlet charakterizovaný parametrami gef = 2,0054; polšírkou ΔHpp = 0,85<br />
mT, ktorý je typický pre semichinoidné štruktúry<br />
nachádzajúce sa často v materiáloch rastlinného<br />
pôvodu [10]. Počas starnutia papiera sme<br />
Relatívna intenzita EPR signálu<br />
g ef =2,0054<br />
326 330 334 338 342<br />
Magnetické pole, mT<br />
Obr. 4.<br />
EPR spektrum vzorky papiera merané<br />
pri teplote 22 C.<br />
v podmienkach EPR experimentov nepozorovali vznik<br />
iných paramagnetických signálov. Problémom<br />
stanovenia koncentrácie semichinoidných<br />
paramagnetických štruktúr je stabilita EPR<br />
spektrometra a subjektívne chyby a nepresnosti pri<br />
nastavení EPR spektrometra, ktoré sme minimalizovali<br />
použitím identic<strong>ke</strong>j kyvety a jej rovnakého<br />
umiestnenia v dutine a EPR merania sme realizovali<br />
v čo najkratšom časovom úseku.<br />
Obrázok 5 vyjadruje závislosť koncentrácie spinov<br />
v jednom grame vzorky papiera od času urýchleného<br />
suchého starnutia pri teplote 120 °C () a pri teplote<br />
80°C s relatívnou vlhkosťou (RH) 45% (tzv. mokré<br />
starnutie) (). Už po 8 hodinách pozorujeme nárast<br />
koncentrácie spinov u oboch metód starnutia, ktorá<br />
dosahuje maximum po 336 hodinách (14 dní). Rozdiel v absolútnej koncentrácii spinov pri<br />
suchom (120°C) a mokrom (80°C/45% RH) starnutí dobre korešponduje s degradačným<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 93
efektom metódy starnutia. Pokles koncentrácie spinov pozorovaný po 672 hodinách (28 dní)<br />
starnutia je pravdepodobne spôsobený premenami paramagnetických semichinoidných<br />
štruktúr na diamagnetické produkty v súlade s predpokladaným 2-elektrónovým<br />
mechanizmom oxidácie fenolických zlúčenín [9].<br />
Spiny, g -1<br />
1.6x10 16<br />
1.4x10 16<br />
1.2x10 16<br />
1.0x10 16<br />
8.0x10 15<br />
6.0x10 15<br />
0 48 96 144 192 240 288 336 384 432 480 528 576 624 672<br />
Urýchlené starnutie, hodiny<br />
Obr. 5.<br />
Závislosť koncentrácie spinov v jednom<br />
grame vzorky papiera od času urýchleného<br />
suchého starnutia pri teplote 120 °C ()<br />
a pri teplote 80 °C s relatívnou vlhkosťou<br />
45% ().<br />
Spiny, g –1<br />
1 2 2 6 7 8 10<br />
Stupeň poškodenia papiera<br />
EPR merania realizované na papieri získanom z knihy poškodenej kyselinou sírovou ukázali,<br />
že koncentrácia paramagnetických semichinoidných zlúčenín je približne 10-násobne vyššia<br />
(obr. 6), pričom najvyššiu koncentráciu vykazovali silne poškodené časti papiera, ktoré sa pri<br />
manipulácii rozpadali na prach. Stupeň poškodenia chápeme ako rozsah poliatia jednotlivej<br />
vzorky kyselinou. Je pravdepodobné, že koncentrácia semichinoidných štruktúr demonštruje<br />
komplexný mechanizmus poškodenia papiera kyslou hydrolýzou a radikálovými oxidačnými<br />
procesmi.<br />
ZÁVER<br />
EPR spektroskopiou sme monitorovali zmeny koncentrácie paramagnetických<br />
semichinoidných zlúčenín v drevitom papieri obsahujúcom lignín, ktorý bol vystavený<br />
pôsobeniu suchého a vlhkého tepla (120 °C a 80 °C/45% RH) alebo poškodený kyselinou<br />
sírovou. Predpokladáme, že plánovaná kombinácia EPR spektroskopie s inými technikami<br />
sledovania poškodenia papiera (meranie mechanických, optických a chemických vlastností)<br />
môže priniesť zaujímavé informácie o degradačných mechanizmoch a tým aj ich<br />
predchádzaniu.<br />
POĎAKOVANIE<br />
Predovšetkým chcem poďakovať prof. Ing. Vlaste Brezovej, DrSc. za odbornú<br />
a pedagogickú pomoc. Zároveň ďakujeme Slovens<strong>ke</strong>j grantovej agentúre za finančnú podporu<br />
EPR výskumu (projekt VEGA/1/3579/06) a RNDr. Bohuslave Havlínovej, CSc. za<br />
poskytnutie vzoriek papiera.<br />
1.2x10 17<br />
1.0x10 17<br />
8.0x10 16<br />
6.0x10 16<br />
4.0x10 16<br />
2.0x10 16<br />
Obr. 6.<br />
Závislosť koncentrácie spinov v jednom<br />
grame vzorky papiera od stupňa poškodenia<br />
papiera H2SO4.<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 94
LITERATÚRA<br />
[1] B. Havlínová, V. Brezová, F. Belányi, G. Szeiffová, Papír a Celulóza 57 , 338-342<br />
(2002).<br />
[2] B. Havlínová, V. Brezová, Ľ. Horňáková, J. Mináriková, M. Čeppan, Journal of<br />
Materials Science 37, 303-308 (2002).<br />
[3] http://www.steve.gb.com/science/molecules.html<br />
[4] http://www.heritage.xtd.pl/pdf/full_strlic.pdf<br />
[5] ISO 5630. Paper and board – accelerated ageing. – Part 1: Dry heat treatment at 105° C.<br />
Last revision 1991 – Part 2: Moist heat treatment at 90° C and 25% RH. Last revision<br />
1985 – Part 3: Moist heat treatment at 80° C and 65% RH. Last revision 1986 – Part 4:<br />
Dry heat treatment at 120° or 150° C. Last revision 1986. Part 1 is equivalent to the US<br />
American Standard ASTM (1987). Standard Test Method for Determination of Effect of<br />
Dry Heat on Properties of Paper and Board. American Society for Testing and Materials<br />
(ASTM-D776-87; 72 hours at 105±2 °C).<br />
[6] http://www.uni-muenster.de/Forum-Bestandserhaltung/kons-restaurierung/agebansa.html<br />
[7] L. Botti, O. Mantovani, D. Ruggiero, Restaurator 26, 44-62 (2005).<br />
[8] D. F. Guay, B. J. W. Cole, R. C. Fort Jr., M. C. Hausman, J. M. Genco, T. J. Elder,<br />
Tappi Fall Conference & Trade Fair, 2002), http://www.umche.maine.edu/pilot/docs/<br />
2002%20TPC%20O2%20Delig%20Reactions.pdf.<br />
[9] C. Felby, B. R. Nielsen. P. O. Olesen, L. H. Skibsted, Appl. Microbiol. Biotechnol. 48,<br />
459-464 (1997).<br />
[10] M. Polovka, V. Brezová, A. Staško, Biophys. Chem. 106, 39-56 (2003).<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce STČ 2006, strana 95
Příspěvky soutěže<br />
doktorských studijních programů<br />
„O cenu děkana 2005”<br />
(Sekce DSP 2005)
CHITOSAN - A NEW TYPE OF POLYMER COAGULANT<br />
Ing. Marcela Borovičková 1 , 3. DSP<br />
Supervisor: doc. Ing. Petr Dolejš, CSc. 1,2<br />
1 Institute of Chemistry and Technology of Environmental Protection, Faculty of Chemistry,<br />
Brno University of Technology, Purkyňova 118, 612 00 Brno, Czech Republic,<br />
e-mail: borovickova@fch.vutbr.cz<br />
2 W&ET Team, box 27, Písecká 2, 370 11 České Budějovice, Czech Republic<br />
ABSTRACT<br />
Results of laboratory experiments into the removal of humic substances by cationic<br />
biopolymer chitosan are presented. Chitosan is a natural product derived from chitin, a<br />
polysaccharide found in the exos<strong>ke</strong>leton of shellfish li<strong>ke</strong> shrimp or crabs. The high content of<br />
amino groups provide to chitosan very interesting heavy metals chelating properties. Chitosan<br />
is partially soluble in dilute mineral acids such as HNO3, HCl, H3PO4. We have used 1%<br />
solutions of chitosan diluted in 0,05M; 0,1M and 0,15M HCl. Aggregates of humic<br />
substances after inorganic coagulant or chitosan addition were separated by centrifugation.<br />
Tests were made with two model humic waters having different concentration of humic<br />
substances. Residual concentration of coagulant (Fe and Al) and absorbance at 387 nm and<br />
254 nm were evaluated.<br />
INTRODUCTION<br />
Impurities present in raw water are in suspended, colloidal, and dissolved forms. These<br />
impurities are dissolved organic and inorganic substances, microscopic organisms, and<br />
various suspended inorganic materials. It is necessary to destabilize and bring together<br />
(coagulate) the suspended and colloidal material to form particles. Afterwards these particles<br />
are removed by filtration.<br />
Coagulation is accomplished by the addition of ions having the opposite charge to that of<br />
the colloidal particles. Since the colloidal particles are almost always negatively charged, the<br />
ions which are added should be cations or positively charged. Typically, two major types of<br />
coagulants are added to water. These are aluminium salts and iron salts. The most common<br />
aluminium salt is aluminium sulphate, the most comon iron salt is ferric sulphate.<br />
Iron and aluminium salts are used as primary coagulants and the reactions that occur after<br />
addition of these coagulants are fairly well elucidated. More recently, organic polyelectrolyte<br />
coagulants have become also used. Organic coagulants are sometimes used in combination<br />
with inorganic coagulants. Depending on the specific chemistry of the target water, polymer<br />
use can vary from as little as 5 % of the total coagulant dosage to as much as 100 % [1].<br />
Chitosan is a derivative of chitin, a polysaccharide that is the major component of the<br />
shells of crustaceans and insects. Chitin consists of long chains of acetylated D-glucosamine,<br />
that is, glucosamine with acetyl groups on the amino groups (N-acetylglucosamine). Chitosan<br />
is N-deacetylated chitin, although the deacetylation in most chitosan preparations is not<br />
complete (see Fig. 1). Chitin itself is usually prepared from crab or shrimp shells or fungal<br />
mycelia. Treatment with an alkali then produces chitosan with about 70% deacetylation.<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 97
Fig. 1 Sructures of chitin and chitosan<br />
Chitosan has been widely used in vastly diverse fields, ranging from waste management to<br />
food processing, medicine and biotechnology. It becomes an interesting material in<br />
pharmaceutical applications due to its biodegradability and biocompatibility, and low toxicity.<br />
The protonization of amino groups in solution ma<strong>ke</strong>s chitosan positively charged, and<br />
thereby very attractive for flocculation and different kinds of binding applications. Since most<br />
natural colloidal particles, including bacteria and macromolecules, are negatively charged,<br />
attractive electrostatic interactions may lead to flocculation [2-5].<br />
EXPERIMENTAL<br />
Chitosan solutions<br />
Chitosan is partially soluble in dilute mineral acids such as HNO3, HCl, H3PO4. We have<br />
used 0,05M; 0,1M and 0,15M HCl solutions. Five hundred milligrams of chitosan powder<br />
was dissolved in a glass bea<strong>ke</strong>r and mixed with 30 mL of HCl solution. The dissolution of<br />
chitosan was slow. Afterwards, it was further diluted to 50 mL with HCl solution to obtain a<br />
solution containing 10,0 mg chitosan per mL of solution. The solutions were prepared fresh<br />
before each set of experiments for consistency.<br />
Metal-based coagulants<br />
Ferric sulphate [Fe2(SO4)3] and aluminium sulphate [Al2(SO4)3.18 H2O], supplied by<br />
Kemifloc a.s. (Přerov, Czech Republic) or Kemwater ProChemie s.r.o., (Bakov nad Jizerou,<br />
Czech Republic) respectively were used for the experiments.<br />
Model humic waters<br />
Tests were made with two model humic waters (A, B) having different concentration of<br />
humic substances. Model waters were mixtures of distilled water, tap water and natural<br />
concentrate of humic substances sampled from a peatbog near Radostín. Selected model<br />
humic water parameters are given in Table 1. Absorbance at 254 nm was measured in 1 cm<br />
cell and absorbance at 387 nm was measured in 5 cm cell. Absorbance at 387 nm is<br />
proportional to colour by multiplying by factor 255 [6].<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 98
Table 1 Parameters of model humic waters<br />
Model<br />
water<br />
pH χ [mS/m] ANC4,5 A254 A387<br />
A 6,5 18,2 0,4 0,163 0,148<br />
B 6,5 13,8 0,4 0,307 0,295<br />
χ - conductivity; ANC4,5 – acidic neutralising capacity to pH 4,5; A254 – absorbance at 254<br />
nm; A387 - absorbance at 387 nm<br />
Coagulation tests<br />
Coagulation test using centrifugation as the separation method was employed. This test did<br />
allow us to study formation of NOM particles by Brownian motion (perikinetic coagulation)<br />
and has the highest possible degree of reproducibility (in any place in the world) as<br />
temperature is the only parameter influencing the kinetics of particles formation. Aggregation<br />
time was 10 and 40 minutes. Residual concentration of coagulant (Fe and Al) and absorbance<br />
at 387 nm and 254 nm were evaluated.<br />
RESULTS<br />
Fig. 2 shows comparison of removal of UV absorbance between ferric sulphate; aluminium<br />
sulphate and chitosan, which was dissolved in different concetrations of HCl. Optimum<br />
coagulant dose ranged in the order of several units of mg/l for chitosan, and tens of mg/l<br />
respectively for aluminium and ferric sulphate.<br />
Model water A: optimum dose was between 21 and 28 mg/l of Fe2(SO4)2, 25 - 35 mg/l<br />
of Al2(SO4)3 and about 5 mg/l of chitosan.<br />
Model water B: optimum dose ranged in interval 30 - 39 mg/l of Fe2(SO4)2,<br />
41 - 48 mg/l of Al2(SO4)3 and about 10 mg/l of chitosan.<br />
Generally, the results show relatively good removal of humic substances by chitosan,<br />
which is comparable with Al and Fe coagulants. Main disadvantage of chitosan is its price,<br />
which should be compensated by low dosage. The optimum pH value for chitosan coagulation<br />
was between 5 – 7.<br />
The results show that chitosan is a promising substitute of metal-based coagulants, which<br />
are traditionally applied in treatment of humic waters..<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 99
A254<br />
A254<br />
A254<br />
0,25<br />
0,20<br />
0,15<br />
0,10<br />
0,05<br />
Ferric sulphate versus Chitosan in 0,05 M HCl<br />
Fe(A)-10´ Fe(A)-40´ Fe(B)-10´ Fe(B)-40´<br />
Chit(A)-10´ Chit(A)-40´ Chit(B)-10´ Chit(B)-40´<br />
0,00<br />
0 10 20 30 40 50 <strong>60</strong><br />
0,25<br />
0,20<br />
0,15<br />
0,10<br />
0,05<br />
Dose [mg/l]<br />
Ferric sulphate versus Chitosan in 0,1 M HCl<br />
0,00<br />
0 10 20 30 40 50 <strong>60</strong><br />
0,25<br />
0,20<br />
0,15<br />
0,10<br />
0,05<br />
Fe(A)-10´ Fe(A)-40´ Fe(B)-10´ Fe(B)-40´<br />
Chit(A)-10´ Chit(A)-40´ Chit(B)-10´ Chit(B)-40´<br />
Dose [mg/l]<br />
Ferric sulphate versus Chitosan in 0,15 M HCl<br />
Fe(A)-10´ Fe(A)-40´ Fe(B)-10´ Fe(B)-40´<br />
Chit(A)-10´ Chit(A)-40´ Chit(B)-10´ Chit(B)-40´<br />
0,00<br />
0 10 20 30 40 50 <strong>60</strong><br />
Dose [mg/l]<br />
A254<br />
A254<br />
A254<br />
0,25<br />
0,20<br />
0,15<br />
0,10<br />
0,05<br />
Aluminium sulphate versus Chitosan in 0,05 M HCl<br />
Al(A)-10´ Al(A)-40´ Al(B)-10´ Al(B)-40´<br />
Chit(A)-10´ Chit(A)-40´ Chit(B)-10´ Chit(B)-40´<br />
0,00<br />
0 10 20 30 40 50 <strong>60</strong><br />
0,25<br />
0,20<br />
0,15<br />
0,10<br />
0,05<br />
Dose [mg/l]<br />
Aluminium sulphate versus Chitosan in 0,1 M HCl<br />
Al(A)-10´ Al(A)-40´ Al(B)-10´ Al(B)-40´<br />
Chit(A)-10´ Chit(A)-40´ Chit(B)-10´ Chit(B)-40´<br />
0,00<br />
0 10 20 30 40 50 <strong>60</strong><br />
0,25<br />
0,20<br />
0,15<br />
0,10<br />
0,05<br />
Dose [mg/l]<br />
Aluminium sulphate versus Chitosan in 0,15 M HCl<br />
Al(A)-10´ Al(A)-40´ Al(B)-10´ Al(B)-40´<br />
Chit(A)-10´ Chit(A)-40´ Chit(B)-10´ Chit(B)-40´<br />
0,00<br />
0 10 20 30 40 50 <strong>60</strong><br />
Dose [mg/l]<br />
Fig. 2 UV254 removal comparison between metal-based coagulants and chitosan (black<br />
curves for model water A, grey curves for model water B)<br />
REFERENCES<br />
1. Gulbrandsen: Organic polymers [online]. 2002 [cit. 2005-08-23].<br />
.<br />
2. Dalwoo Corporation: Chitin, chitosan and chitosan oligomer from Crab Shells. [online].<br />
Last updated April 12, 1999 [cit. 2005-07-31]. http://members.tripod.com/~dalwoo/.<br />
3. Beaulieu E.B.: Marinard Biotech [online]. [cit. 2005-07-31]. http://www.marinard.com/.<br />
4. Li J. et al.: Polymer Degradation and Stability, 87, 441, 2005.<br />
5. Vander P. et al: Plant Physiol., 118, 1353, 1998.<br />
6. Dolejš P.: Spektrofotometrické stanovení barvy huminových vod. In Sborník konference<br />
"Hydrochémia ‘83". Bratislava: ČSVTS VÚVH, 1983, s. 361-370.<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 100
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 101
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 102
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 103
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 104
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 105
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 106
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 107
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 108
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 109
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 110
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 111
STUDY OF PHYSICOCHEMICAL AND ANTIOXIDATIVE<br />
PROPERTIES OF YEAST ME<strong>MB</strong>RANE COMPONENTS<br />
Michaela Drabkova, 3 rd year of PGS<br />
Supervisor: Prof. Ing. L. Omelka, DrSc.<br />
Doc. RNDr. I. Márová, Ph.D.<br />
Department of Food Chemistry and Biotechnology, Faculty of Chemistry, Brno University of<br />
Technology, Purkynova 118, 612 00, Czech Republic, email: drabkova@fch.vutbr.cz<br />
INTRODUCTION<br />
Carotenoids are isoprenoid membrane-protective antioxidant pigments produced by plants, algae,<br />
bacteria and fungi. They belong to the most widespread natural pigments with many important<br />
biological activities. Production of carotenes, the great variety of natural carotenoids, is more than<br />
100 million tons per year. They are produced by specific branch of common isoprenoid biosynthetic<br />
pathway occuring in all types of organisms. With regard to different applications in food and feed<br />
industry most attention is now being focused on the natural production of carotenoids by microbial<br />
technology using yeast and/or bacteria.<br />
There are two major ways to influence the microbial production of carotenoids: i) by<br />
modification of cultivation conditions and ii) by construction of genetically modified<br />
overproducers. Qualitative and quantitative changes in a cell metabolite complement can be induced<br />
by environment, stress and other factors. Thus, identification of metabolic mar<strong>ke</strong>r characteristics for<br />
certain events provides important insight into the mechanisms of pathways occurring in the organism<br />
and can also lead to the regulation of production of industrially significant metabolites. Especially in<br />
microorganisms, production of metabolites is strongly influenced by various external factors.<br />
Environmental stress surrounding of yeast cells evo<strong>ke</strong>s various changes in their behaviour in order to<br />
survive under unfavourable conditions. Under stress, various specific compounds including lipidic<br />
substances are overproduced (e. g. glycerol, phospholipids, carotenoids, ergosterol etc.). However,<br />
more information is needed about regulation of production of these substances. Factors that influence<br />
efficiency of natural carotenoid biosynthesis are important for commercial applications.<br />
Carotenoids are membrane-bound lipid-soluble pigments, which can act as effective<br />
antioxidants and scavenge singlet oxygen. In red yeast they probably act as adaptive and/or<br />
protective mechanism against exogenous oxidative stress and UV-irradiation. Those compounds<br />
are accumulated in particular cell organelles. It is not clear so far, whether carotenoids are present<br />
in plasma membrane only or in other inner membrane systems as well as in cell wall. Also distribution<br />
of individual carotenoid derivatives in individual sub-cellular fractions has not been studied<br />
yet. Moreover, significant changes of these parameters under exogenous stress could occur, which<br />
could influence potential biotechnological use of red yeasts to industrial production of carotenoids.<br />
In this work some techniques for isolation and separation of sub-cellular fractions (cell wall,<br />
membrane fraction, cytosol) of red yeast cells grown in optimal conditions and under osmotic and<br />
oxidative stress were tested. Further, analysis of carotenoids in these fractions as well as in<br />
whole cells was done. Results of antioxidant properties of sub-cellular fractions were compared<br />
with carotenoid composition and antioxidant activity of some standard carotenoids. The aim of this<br />
work is to study, what is the distribution and trafficking of carotenoids in the cell and which<br />
carotenoids are the main contributors of antioxidant activity in individual cell compartments.<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 112
MICROORGANISMS<br />
Yeast strains Rhodotorula glutinis CCY 20-2-26, Sporidiobolus salmonicolor CCY 19-4-8,<br />
Phaffia rhodozyma CCY 77-1-1 and Saccharomyces cerevisiae CCY 21-4-88 were used.<br />
Saccharomyces cerevisiae was used as comparative strain because of very high importance in<br />
genetic engineering.<br />
CULTIVATION CONDITIONS<br />
Yeasts were cultivated on glucose medium aerobically with light at 28 °C to the exponential phase<br />
of growth. Exogenous stress was induced by 2-5 % NaCl added into inoculation media and 2-5 mM<br />
H2O2 added into production media.<br />
Saccharomyces cerevisiae was cultivated on YPD medium anaerobicaly at 28 °C for 24 hours .<br />
PREPARATION OF SPHEROPLASTS<br />
Spheroplasts were prepared by incubation of yeast cells with some enzymes to softly disrupt<br />
plasmatic membrane. Different concentrations of lyticase and glucuronidase, other detergents (e.g.<br />
SDS, beta-merkaptoethanol, etc.) and osmotic lysis were used. Membrane fraction and cytosol were<br />
separated by ultracentrifugation. Spheroplasts were detected by light-microscopy and with addition of<br />
fluorescein dye by UV-microscopy. Further, spheroplasts were used for isolation of chromosomal<br />
DNA from red yeasts. Isolation was performed in 1% low melting agarose plugs and analyzed then<br />
by PFGE.<br />
a) Spheroplasts of R. glutinis – Light microscopy b) Bursting cells in isotonic environment<br />
ISOLATION OF SUB-CELLULAR FRACTION<br />
Sub-cellular fractions of cells were obtained by gradually separation using combination of<br />
enzymes and detergents. Cell wall fraction (surface layer) was obtained using sonification followed<br />
by ethanol precipitation. In selected fraction lipid profiles were analyzed using TLC. Levels of<br />
carotenoids - lycopene, alpha-carotene, beta-carotene, torulen and phytoene were obtained from yeast<br />
cells using acetone extraction and saponification by ethanolic KOH solution. The sample was<br />
repeatedly extracted by diethylether, evaporated and dissolved into ethanol for HPLC. We analyzed<br />
those samples using HPLC/MS. Antioxidant activity of individual sub-cellular fractions was tested<br />
using ABTS Metod (Randox kit). Protein profiles under stress conditions were compared too. Those<br />
were analyzed by PAGE-SDS.<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 113
RESULTS AND DISCUSSION<br />
The most important industrial red yeast Rhodotorula glutinis was used as tested strain for<br />
identification of carotenoids and other membrane compounds. Dry biomass of these yeasts is used as<br />
feed complement for improvement of appearance and nutritional properties of animal products (e. g.<br />
the color of eggs and milk, the color of salmon meat).<br />
This microorganism was chosen based on results of previous screening study on selected yeast<br />
strains which produce carotenoids as natural pigments. Carotenogenic cultures were influenced by<br />
several exogenic chemical and physical stress factors added into cultivation media to clear the role of<br />
carotenoids in general stress response. We found that all types of stress conditions led to increased<br />
production of beta-carotene according to phase of application or concentration of stress factor. Crossprotection<br />
and production of similar metabolites in various types of stress conditions suggests the<br />
existence of an integrating mechanism that senses and responds to different forms of stress.<br />
Preparation of individual sub-cellular fractions from red yeast cells was strongly complicated by<br />
lipotrophic character of this strain. Especially preparation of spheroplasts was very difficult because of<br />
rigid character of cell wall membrane and yields of membrane fractions were therefore very low.<br />
Under stress conditions, about hundred proteins were overproduced by this strain. Expecting shock<br />
proteins, it could be some enzymes that catalyze overproduction of stress metabolites including<br />
carotenoids. Under both oxidative and osmotic stress pigments were overproduced, higher amount<br />
was detected in surface cell structures (cell wall and plasma membrane). Ergosterol was found both<br />
in cytosol and membrane lotions and its production under stress changed simultaneously with<br />
carotenoid formation. Glycerol was detected above all in cytosol fraction and its production under<br />
stress was inversely to carotenoid and ergosterol production.<br />
As surprising finding can be noted high content of carotenoids found in upper cell wall fraction.<br />
Presence of carotenoids, mainly beta-carotene, was detected in plasma membrane as well as in inner<br />
membrane fraction. The highest antioxidant activity was found in surface structures.<br />
This work was supported by project MSM 0021630501 H/'Czech Ministry of Education and by<br />
project IAA400310506 f Grant Agency of the Academy of Sciences of the Czech Republic.<br />
REFERENCES<br />
.l. Marova I., Breierova E., Koci R., Friedl Z., Slovak B., Pokorna J.: Annals Microbiol. 54, 73<br />
(2004).<br />
2. Farina J. I., Molina O. E., Figueroa L. I.. C.: Journal of Applied Microbiology 96, 254-262 (2004)<br />
3.Pardo M. Monteoliva L., Pla J. Sanchez M., Gil C. Nombela C.: Yeast 15, 459-472 (1999)<br />
4. Misawa N. Shimada H.: J. Biotechnol. 59, 169 (1997).<br />
5. Armstrong G. A., Hearst J. E.: FASEB J. 10, 228 (1996).<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 114
DETERMINATION OF YEAST VIABILITY BY MEANS OF<br />
FLUORESCENCE MICROSCOPY AND IMAGE ANALYSIS<br />
Ing. Petra Jeřábková<br />
Supervisor: doc. Ing. Oldřich Zmeškal, CSc.<br />
Institute of Physical and Applied Chemistry, Faculty of Chemistry, Brno University of<br />
Technology, Purkyňova 118, 612 00 Brno, e-mail: jerabkova@fch.vutbr.cz<br />
1. INTRODUCTION<br />
In a condition assessment of microbiological populations, the important factor is a<br />
representation of live and dead individuals that it is possible to differentiate by means of<br />
fluorescent labelling. A direct microscopic count is usually used for direct determination of<br />
the cell number in counting chambers (by Thom, Bür<strong>ke</strong>r). The number of cells of microorganisms<br />
can frequently be determined indirectly with the assistance of cultivation when it is<br />
assumed that one colony will grow up from a viable cell and these colonies will be counted<br />
after that. It is possible to use an image analysis for the detection of defined objects<br />
(e.g. yeasts) without the necessity to count them.<br />
In this work, the wavelet transformation at a fractal analysis of an image, the so-called box<br />
counting method, was utilised. The wavelet transformation (or the Haar one) ma<strong>ke</strong>s the<br />
calculation of squares of different sizes of a laid mesh more effective with the box counting<br />
method provided that a square area is being analysed. An analysed image with a size that is<br />
given by the power of a definite number (2 i ) is transformed into the space of spectra. On this<br />
spectra, the filter of a low pass type with the size of square 2 i , where i = 1, 2, 4… n – 1 is<br />
gradually applied and the number of black, white and black and white squares is determined<br />
in images that arise from the inverse transformation in the same way as with a box counting<br />
method. It is possible to substitute the process of transformation and inverse transformations<br />
of an image by the addition of neighbouring foursome pixels and to analyse a relevant<br />
sequence of these pictures. Three fractal dimensions of the black and white area and their<br />
interface (DBBW, DBWB, DBW) and fractal measures (KBBW, KBWB, KBW) will again be the result.<br />
When analysing a black and white picture that may be obtained from a coloured picture by the<br />
process called thresholding, a method identical with a box counting method is described [1].<br />
With the assistance of the fractal analysis, characteristic data about an analysed structure,<br />
namely the fractal dimension D and the fractal measure K are determined. These parameters<br />
can be used for picture ordering evaluation as well as e.g. for specifying the number of<br />
defined objects or for specifying their radius [2].<br />
Pictures for the analysis were obtained by means of fluorescence microscopy because this<br />
method is quick and simple. Results are obtained within minutes, whereas classic cultivation<br />
methods require the minimum of 24 hours.<br />
2. EXPERIMENTAL PART<br />
The fractal analysis was used for specifying the number and dimension of images of live<br />
and heat-killed yeast cells. The fluorescent dye of acridine orange (AO) was used for the<br />
simultaneous distinction of live and dead cells (dead cells give out red or orange fluorescence<br />
and live cells yellow-green; 180 µM AO, pH 6). Fluorescein diacetate (FDA) was used for the<br />
distinction of live cells (greenish fluorescence; 6 mM FDA, pH 7.2). With the assistance of<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 115
fluorescent labelling it is possible to threshold either dead or live cells on a black colour in<br />
one picture. Images of cells were thresholded by the intensity or coloured components of the<br />
RGB space. To detect the convenient thresholding, it is possible to use the fractal spectrum,<br />
i.e. fractal dimension dependence on the intensity or selected RGB component, which is<br />
accessible in the HarFA software as a tool referred to as Fractal Analysis – Range [3]. The<br />
number of cells x and their radius r were determined provided that images of cells were of a<br />
circular shape, similar in size and distinguishable on the background according to relations<br />
x =<br />
4π<br />
N<br />
2<br />
( ) ,<br />
BW<br />
N + N<br />
B<br />
BW<br />
r =<br />
( N + N )<br />
B<br />
where NB is the number of black boxes, NBW is the number of black and white boxes with the<br />
size of ε × ε pixels. The resulting cell number x is derived from the value x, which is the<br />
maximum from the calculated values for different sizes of the mesh. The maximum value is<br />
selected because the fractal structure is bordered most conveniently for the given mesh size.<br />
Yeasts from Culture Collection of Yeasts – CCY, the Institute of Chemistry, Slovak<br />
Academy of Sciences in Bratislava and from the Faculty of Chemical and Food Technology<br />
of the Slovak University of Technology in Bratislava were used for the image analysis. Yeast<br />
Saccharomyces cerevisiae FV1 and yeast Saccharomyces cerevisiae subsp. cerevisiae CCY<br />
21-4-102 have ellipsoidal and spherical cells with width about 2.6–6.4 µm and with length of<br />
3.7–9.7 µm. Yeast Kloec<strong>ke</strong>ra apiculata CCY 25-6-22 has the shape of a lemon and size of<br />
(1.5–5) µm × (2.5–11) µm. Vegetative cells of yeast Hansenula anomala CCY 38-1-30 are<br />
spherical or ellipsoidal with size of 2–4 and 2–6 µm [4].<br />
Yeasts that were cultivated on an inclined agar by the room temperature were, then,<br />
inoculated into a liquid culture medium (glucose peptone yeast extract medium) where they<br />
were cultivated for a minimum of 24 hours again by the room temperature under aerobic<br />
conditions. After that, a half of the volume of the culture medium with cells was exposed to<br />
95°C for 3–6 hours (according to the yeast species). A drop of live and dead cells mixture was<br />
put on a slide and a drop of fluorescent dye was added. Microscopical preparation was<br />
observed immediately at staining by fluorescein diacetate and after 30 minutes at staining by<br />
acridine orange with 40× objective.<br />
The number of cells was evaluated in 35 visual fields. A suitable neutral density filter<br />
(ND4, ND8, ND16), which decreases the intensity of the excitation light, was inserted<br />
between the excitation light (mercury lamp) and excitation filter, when required. For the<br />
recording of pictures, the combination of the epifluorescence microscope Nikon Eclipse E200<br />
with the filter cube and a digital camera Nikon Coolpix5400 with the resolution of 2592 ×<br />
1944 was used. For the storage of pictures, the software Lucia Net 1.16.6. was used. The<br />
obtained pictures with the size of 1024 × 1024 were analysed by means of the software<br />
HarFA [3].<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 116<br />
πx<br />
BW<br />
ε 2
Table 1<br />
Average numbers of cells from 35 pictures of sample of different yeast species that were<br />
obtained by fractal analysis<br />
AO FDA<br />
Yeast<br />
species<br />
Saccharomyces<br />
cerevisiae<br />
Saccharomyces<br />
cerevisiae FV1<br />
Kloec<strong>ke</strong>ra<br />
apiculata<br />
Hansenula<br />
anomala<br />
Dead<br />
cells<br />
number<br />
Average<br />
error<br />
(%)<br />
Live<br />
cells<br />
number<br />
Average<br />
error<br />
(%)<br />
Total<br />
number<br />
of cells<br />
Average<br />
error<br />
(%)<br />
Live cells<br />
number<br />
Average<br />
error<br />
5.42 8.05 6.<strong>60</strong> 8.66 11.97 6.10 11.40 4.02<br />
4.19 7.23 7.00 4.85 11.19 4.61 8.96 3.74<br />
4.94 6.61 10.34 6.94 14.90 5.66 10.53 5.93<br />
10.17 8.44 10.38 6.92 20.57 5.22 8.05 8.11<br />
Table 2<br />
Average radii of cells from 35 pictures of sample of different yeast species that were obtained<br />
by fractal analysis<br />
AO FDA<br />
Yeast<br />
Dead cells<br />
Live cells Live and dead cells Live cells<br />
species<br />
radius<br />
radius<br />
radius<br />
radius<br />
Saccharomyces<br />
cerevisiae<br />
Saccharomyces<br />
cerevisiae FV1<br />
Kloec<strong>ke</strong>ra<br />
apiculata<br />
Hansenula<br />
anomala<br />
3.25 4.12 3.74 3.78<br />
3.47 3.70 3.55 3.75<br />
2.88 2.72 2.96 2.62<br />
2.61 2.65 2.72 3.17<br />
3. CONCLUSION<br />
The fluorescence microscopy in the connection with the image analysis seems to be a<br />
suitable method for the counting of yeast cells. For the staining of dead and live cells<br />
simultaneously, the acridine orange dye is suitable, for the staining of live cells it is the<br />
fluorescein diacetate. When counting live and dead cells of different yeast species by means<br />
of the fractal analysis it was determined that the average error of determination of the yeast<br />
number from 35 pictures is less than 10 % with the number of cells up to 20 in a picture<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 117<br />
(%)
(Table 1). This error can also be caused by an unequal size of cells and shapes, differences in<br />
colouring of the individual cells and quality of the recorded picture. If cells are killed by heat,<br />
the cytoplasm is sometimes stained greenly as well, which complicates the thresholding<br />
process with cells that were stained by the acridine orange. Budding cells were counted as one<br />
cell identically as in the direct determination of the cell number whereas if a daughter cell<br />
carried another bud still not separated from a mother cell, it was counted as two.<br />
Obtained radiuses of cells image provided that they have a circular shape are listed in<br />
Table 2 and are within the range of 2.61–4.12 µm. The sizes of the radii fall within values that<br />
are listed in literature or, in some case, are slightly greater [4].<br />
4. REFERENCES<br />
[1] Tománková, K. Studium vlastností hrubě disperzních soustav pomocí metod obrazové<br />
analýzy. Brno, 2004, 42 s. Diploma thesis, Faculty of Chemistry, Brno University of<br />
Technology. Supervisor: Oldřich Zmeškal.<br />
[2] Zmeškal O., Sedlák O., Nežádal M.: Metody obrazové analýzy dat, Digital Imaging in<br />
Biology and Medicine, Czech Academy of Science České Budějovice: May 13., 2002, pp.<br />
34 - 43, ISBN 80-901250-8-5.<br />
[3] Zmeškal O., Nežádal M., Buchníček M., Bžatek T.: HarFA 5.0, Harmonic and Fractal<br />
Image Analyzer, http://www.fch.vutbr.cz/lectures/imagesci, Brno, 2003.<br />
[4] Kocková-Kratochvílová, A.: Taxonómia kvasinek a kvasinkovitých mikroorganizmov.<br />
1. vyd. Bratislava: Alfa, 1990. ISBN 80-05-00644-6.<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 118
FLOW BEHAVIOUR OF DILUTED SUSPENSIONS IN<br />
CARBOXYMETHYLCELLULOSE-WATER SOLUTION<br />
MICHAL KLIMOVI<br />
Brno University of Technology, Faculty of Chemistry, Institute of Physical and Applied<br />
Chemistry, Purkyova 118, 612 00 Brno, Czech Republic, e-mail:<br />
klimovic@fch.vutbr.cz<br />
Introduction<br />
Recently we have reported on rheological problems in processing lignite pastes to form<br />
elements for lignite alternative applications outside the power generation industry [1, 2].<br />
We have also shown how these problems can be overcome using cellulosic derivative as<br />
a rheological aid [3]. Further our work was concerned with study of much more diluted<br />
suspensions of liquid-li<strong>ke</strong> appearance. We have found that small amount of lignite<br />
surprisingly lowered apparent viscosity of the solution comparing with the pure solvent.<br />
This work reports on flow behaviour of inorganic particle dispersions in the same<br />
medium. The aim was to find whether this thinning effect is specific to lignite or not.<br />
Experimental<br />
Carboxymethylcellulose (in the form of sodium salt; CMC) was supplied by Aldrich.<br />
Two preparations were used with following producer’s specifications: high molecular<br />
weight, Mw = 700 000 g mol -3 , degree of substitution 0.9; low molecular weight, Mw =<br />
90 000 g mol -3 , degree of substitution 0.7. Moisture content was determined as 8.2 and<br />
9.4 %.<br />
Two types of filler were used for preparation of dispersions, the foundry sand from<br />
laboratory of FSI VUT Brno and microsil from Silchem. The fraction of foundry sand<br />
captured between 0.15 and 0.30 mm sieves was used for subsequent experiments.<br />
Microsil is Silchem trademark of glass microspheres; type G080 was used.<br />
The peparation of suspensions consisted in suspending weighted amounts of filler and<br />
CMC in corresponding amount of warm (70 °C) deionized water. Mixture was stirred<br />
for one hour, temperature was maintained for the first 30 min only.<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 119
Measurements of flow properties were carried out at 25 °C on Haa<strong>ke</strong> RS100 rheometer<br />
equipped with the double cylinder sensor Z20 DIN both in constant rate and in constant<br />
stress mode. All reported data points are averages from three replicates.<br />
Results and discussion<br />
Figure 1 shows flow curves for increasing sand contents measured immediately after<br />
preparation with suspensions based on the high molecular weight cellulose derivative.<br />
Higher sand loadings (7 % and more) shift flow curves up, to higher shear stress values,<br />
on increasing sand contents and also apparent viscosity rises. On contrary, flow curves<br />
for low sand loadings (5 %) lie bellow the curve of blank CMC solution giving also<br />
lower apparent viscosities. Thus, adding sand to the carboxymethylcellulose solution<br />
does not increase its (apparent) viscosity as expected, unless the sand concentration is<br />
sufficiently high. The same effect was observed for lignite as a filler.<br />
Suspensions prepared from the low molecular weight cellulose derivative did not show<br />
this untypical behaviour.<br />
The same behaviour of suspensions was observed at carboxymethylcellulose-microsilwater<br />
dispersions (Fig 2).<br />
Fig 1 The dependance of shear stress on shear rate for dispersions of pure CMC<br />
solution and different concentrations of sand in CMC solution (2 %, 5 %, 7 %, 10 %).<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 120
Fig 2 The dependance of shear stress on shear rate for dispersions of pure CMC<br />
solution and different concentrations of microsil in CMC solution (0.5 %, 1 %, 3 %,<br />
5 %).<br />
Measured flow curves can be adequately fitted by the Ostwald-de Waele model<br />
n<br />
= K<br />
Model parameters reflected the influence of lignite on suspension flow behaviour.<br />
Results of this study indicate that lowering apparent viscosity of suspensions prepared<br />
from water solution of high molecular weight carboxymethylcellulose may be quite<br />
common phenomenon when concentration of dispersed particles is low (approximately<br />
in orders of tenths up to ten weight percent). The cause is probably in easier movement<br />
of particles entrapped within long biopolymer chains li<strong>ke</strong> balls in ball-bearing.<br />
Conclusion<br />
Flow curves determined in this study showed that lowering suspension apparent<br />
viscosity at moderate amount of particles suspended in high molecular weight<br />
carboxymethylcellulose solution is not specific effect of lignite particles. The same<br />
effect was observed also for sand and even for glass microspheres, which are usually<br />
used as thic<strong>ke</strong>ner. Results thus confirm our hypothesis on some “ball-bearing” effect,<br />
which enables easier flow of long macromolecular chains then when they are in closer<br />
contact in pure solution.<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 121
References<br />
1. Lapík ., Lapíková B., Filgasová G.: Colloid Polym. Sci. 278, 65 (2000)<br />
2. Peka M.: Flow behaviour of concentrated lignite dispersions. In Proceedings of the<br />
XIIIth International Congress on Rheology, Cambridge, vol.4. British Society of<br />
Rheology, Glasgow, p.4, 105, 2000.<br />
3. Peka M., Diviová P., Klimovi M.: submitted to Colloid. Polym. Sci. (2005)<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 122
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 123
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 124
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 125
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 126
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 127
SPECTROPHOTOMETRIC STUDY OF SOME BIOLOGICAL<br />
BUFFERS SOLUTIONS<br />
Ing. Miroslava Krčmová a , 3 rd year of DSP<br />
Supervisor: doc. Ing. Radim Vespalec, DrSc. b<br />
a Brno University of Technology, Faculty of Chemistry, Institute of Physical and Applied<br />
Chemistry, Purkyňova 118, CZ 612 00 Brno, Czech Republic,<br />
e-mail: xckrcmova@fch.vutbr.cz<br />
b Institute of Analytical Chemistry, Czech Academy of Science, Veveří 97, CZ 611 42 Brno,<br />
Czech Republic, e-mail: vespalec@iach.cz<br />
INTRODUCTION<br />
Biological buffers are predominantly synthetic compounds serving widely to the pH<br />
stabilization and for the pH control in the pH range of 5.5–11.4 1 . They are predominantly<br />
white crystallized powders that are good soluble in water and they have a high chemical<br />
stability and compatibility in various biological systems. Today, the biological buffers are<br />
predominantly utilized as chemical agents for analytical chemistry. They are used in buffer<br />
exchange processes and as mobile phases during some chromatography steps, too 2 . These<br />
buffer compounds provide alternate buffer compounds with pKa values near physiological<br />
conditions for biological applications 2, 3 . The biological buffers are free of chromophores<br />
absorbing light above 200 nm. In spite of this, it was reported recently that some of the<br />
buffers wea<strong>ke</strong>n mar<strong>ke</strong>dly the passing light at some experimental condition 4 .<br />
GOOD BUFFERS<br />
In the year 1966 discovered, developed and introduced Goods research team the special<br />
group of zwitterionic biological buffers, called Good buffers 2 . Later, in 1980, was patented<br />
and introduced a new generation of biological buffer compounds based on the zwitterionic<br />
character of Good buffers 5 . Good buffers stabilise pH of solution and they are used as protein<br />
solubilization agents in aqueous solutions 6 , too. The aforesaid buffers are non-toxic to cells,<br />
they are not interferenced through biological membranes (for example: penetration,<br />
adsorption surface, solubilization, etc.). The pKa values of these biological buffers lie at or<br />
near physiological pH area 2, 5 .<br />
Popularity of using of biological buffers in analytical chemistry follows from two<br />
phenomenons. At first it is a photometric detection that is accounted as a very frequently and<br />
popular in contemporary modern instrumental analytical methods. The second phenomenon is<br />
the fact that biological buffers are free of chromophores absorbing ultraviolet light above<br />
200 nm. Regardless it was detected modify of intensity of the passing UV-light beam that<br />
passed through aqueous solutions of same biological buffers in the short wavelength area 4, 7 .<br />
The strain of our study is the verification of this finding for three most frequently and popular<br />
used zwitterionic Good buffers, 3-(N-morpholino)propanesulfonic acid (MOPS), 2-(Nmorpholino)ethanesulfonic<br />
acid (MES), 3-(cyclohexylamino)-2-hydroxy-1-propanesulfonic<br />
acid (CAPSO), see Fig. 1 1, 7 and Table 1 1 .<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 128
A B C<br />
Fig. 1: The structure of research biological buffers: A) MOPS, B) MES and C) CAPSO 1 .<br />
Table 1: Inquired zwitterionic GOOD buffers 1 .<br />
Buffer Formula M W<br />
(g.mol -1 )<br />
pKa<br />
(25 °C)<br />
pH range<br />
MOPS C7H15NO4S 209.26 7.2 6.5 – 7.9<br />
MES C6H13NO4S 195.24 6.1 5.5 – 6.7<br />
CAPSO C9H19NO4S 237.32 9.6 8.9 – 10.3<br />
EXPERIMENTAL PART<br />
The UV-Vis spectrophotometer VARIAN – CARY 50 Probe (Varian BV, The<br />
Netherlands) equipped with the quartz cell of 1 cm optical path length served for photometric<br />
measurements in the range of 190-300 nm. The acquired experimental data were processed by<br />
the program Cary WinUV, the program equipment to the spectrometer Varian (Varian BV,<br />
The Netherlands). The examined buffers (MES, MOPS and CAPSO) are from Sigma-Aldrich<br />
(Prague, Czech Republic). The buffer solutions were prepared from redistilled water and<br />
theirs concentration was between the value 4 and 200 mmol.l -1 . The pH of researched<br />
solutions was adjusted with sodium hydroxide (Lachema, Brno, Czech Republic). The<br />
experimental temperature has the value 25 °C (common laboratory temperature).<br />
RESULTS<br />
Investigated zwitterionic biological Good buffers, MES, MOPS and CAPSO cover almost<br />
completely pH range of the Good buffers applicability. The investigated buffers are free of<br />
chromophores absorbing light above 190 nm disregarding their chemical identity and pH.<br />
Wea<strong>ke</strong>ning of a light beam passing through their aqueous solutions cannot be therefore<br />
classified the light absorption even if it is displayed in absorbance units by the used<br />
instrument. Three pH units below their pKa values, the buffers are only in the zwitterionic<br />
form considering the measurement precision. At pH = pKa, the concentration of the<br />
zwitterionic form and of the anionic form is identical. At pH = pKa + 3, practically only<br />
anions exist in solution.<br />
Experiments with purely zwitterionic solutions of MES, MOPS (Fig. 2) and CAPSO<br />
revealed that their formal absorbance increases with both increasing concentration and with<br />
the decreasing wavelength of the passing light. The maximum measured absorbance depended<br />
on the dissolved compound. In the range of usually utilized concentrations of Good buffers,<br />
ranging from 20 to <strong>60</strong> mmol.l -1 , the light absorption was acceptably low above 200 nm. Zero<br />
absorbance was obtained at the wavelength of 250 nm and higher disregarding the compound<br />
and its concentration.<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 129
Fig. 2: Typical course of the dependence of the formal absorbance of dissolved<br />
zwitterionic buffers on the passing light wavelength documented on the example of MOPS.<br />
The MOPS concentration is given as a parameter: 1) 4.1 mmol.l -1 , 2) 50 mmol.l -1 , 3)<br />
100 mmol.l - 1 and 4) 200 mmol.l -1 .<br />
The (pH-pKa) difference affects the light beam wea<strong>ke</strong>ning dramaticly at concentrations up<br />
to pH = pKa (Fig. 3). At pH > pKa, the absorbance rises up much less. The dependences of<br />
formal absorbance at pH = pKa and pH = pKa + 3 on the light beam wavelength differ<br />
therefore only slightly (Fig. 4). The steepness of the concentration dependence of the apparent<br />
absorbance rises up with the decreasing wavelength (Fig. 5) as may be expected from<br />
previous experiments.<br />
Fig. 3: Typical course of the dependence of the formal absorbance of dissolved<br />
zwitterionic buffers on the buffer pH documented on the example of MOPS. The wavelength of<br />
the passing light is given as a parameter: (A) 1) 200 nm, 2) 220 nm and 3) 250 nm,<br />
(B) 1) 210 nm and 2) 230 nm.<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 130
Fig. 4: Typical course of the dependence of the formal absorbance of dissolved<br />
zwitterionic buffers on the buffer pH at 1) pH = pKa and 2) pH = pKa + 3 documented on the<br />
example of MOPS.<br />
Fig. 5: Typical course of the dependence of the formal absorbance of dissolved<br />
zwitterionic buffers on their concentration documented on the example of MOPS. The<br />
wavelength of the passing light is given as a parameter: (A) 1) 200 nm, 2) 220 nm and 3)<br />
250 nm, (B) 1) 210 nm and 2) 230 nm.<br />
CONCLUSIONS<br />
Photometric experiments evidenced general aggregation capability of investigated<br />
zwitterionic Good buffers MES, MOPS and CAPSO and, in agreement with the previous<br />
communication 4 , dramatic increase of this capability in the pH range in which the<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 131
concentration of the anionic form of the buffer approaches 50 % of its total concentration.<br />
Formation of aggregates from dissolved MES, MOPS and CAPSO, which disperse the<br />
passing UV-light beam, is a possible interpretation of presented experiments. Conductivity<br />
measurements not presented here evidence that the aggregates are not micelles. This finding<br />
controverts the explanation proposed in the article 4 . Presented results of photometric<br />
experiments evidence correctness of experiments given in Ref. 4. The results also substantiate<br />
continuation of the presented research.<br />
REFERENCES<br />
1. Sigma-Aldrich catalogue 2000-2001.<br />
2. Accessible in the Internet: http://www.jtba<strong>ke</strong>r.com/biopharm/biopharm_buffer_bio.html<br />
[citation 22 February 2005].<br />
3. Ferguson, W.J., Braunschweiger, K.I., Braunschweiger, W.R., Smith, J.R., McCormick,<br />
J.J., Wasman, C.C., Jarvis, N.P., Bell, D.H. and Good, N.E.: Anal. Biochem. 104 (2), 300<br />
(1980).<br />
4. Vespalec, R., Vlčková, M., Horáková, H.: Aggregation and intermolecular interactions of<br />
biological buffers observed by capillary electrophoresis and UV photometry. Journal of<br />
Chromatography A, 2004, vol. 1051, p. 75 - 84.<br />
5. Good, N.E., Winget, G.D., Winter, W., Connolly, T.N., Izana, K., Singh, R.M.M.:<br />
Biochemistry 5, 467 (1966).<br />
6. Hames, B.D., Rickwood, D.: Gel Electrophoresis of Proteins. 2-nd edition, p. 153, OIRL<br />
Press, Oxford, 1990.<br />
7. Beynon, R.J., Easterby, J.S.: Buffer Solutions: The basics. p. 48, 69 – 70, 74, 78, OIRL<br />
Press, Oxford, 1996.<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 132
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 133
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 134
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 135
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 136
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 137
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 138
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 139
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 140
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 141
Investigation of environmental and varietal influences on distribution of<br />
starch granules in barley <strong>ke</strong>rnels by GFFF and laser particle size analysis<br />
Ing. Karel Mazanec<br />
Supervisor: RNDr. Josef Chmelík, CSc.<br />
Institute of Material Chemistry, Faculty of Chemistry, Brno University of Technology,<br />
Purkyňova 118, 612 00 Brno, Czech Republic, e-mail: mazanec@iach.cz<br />
INTRODUCTION<br />
The beer production has great tradition and is much extended in this country. Because the<br />
production of beer is very energy-consuming, any decrease of input and process loads <strong>ke</strong>eping<br />
taste and qualities of resulting product may play a significant role at volumes of production.<br />
Therefore understanding of processes joined with brewing and malting may give promising<br />
results.<br />
Starch is formed in green plants as the final product of photosynthesis. Starch is one of the<br />
barley components important for its malting quality. The starch present in barley <strong>ke</strong>rnels is in<br />
form of large (type A) and small (type B) granules [1]. Distribution of starch granules in<br />
barley <strong>ke</strong>rnels plays important role in technological processes of beer production. Because the<br />
small granules are less susceptible to enzymatic digestion and causing technological problems<br />
during brewing (they can form starch haze and can block filtration beds in lauter tuns) for a<br />
long time it was presumed that the malting varieties experimentally selected as<br />
technologically suitable for brewing should have only minimal content of small starch<br />
granules. The so-called malting varieties of spring barley have different distribution of size of<br />
starch granules in comparison to other varieties. Measurement of starch particles size<br />
distribution gives good idea of technological quality of individual malting varieties. Several<br />
techniques have been generally employed for this purpose. Nevertheless implementation of<br />
field-flow fractionation (FFF) fitted the needs of low cost and short separation time.<br />
MATERIALS AND METHODS<br />
Plant material and sample preparation<br />
Twelve spring and winter barley varieties (obtained from the testing stations of the Central<br />
Institute for Supervising and Testing in Agriculture (CISTA) of the Czech Republic) were<br />
used for isolation and size measurements. Each year the above given variety assortment was<br />
grown in three testing stations. External conditions influenced all varieties equally.<br />
Monitoring was conducted for three years (2001-2003) and thus 108 samples were processed<br />
during whole project. This work shows data from the last harvest of this three years project.<br />
Starch granules were isolated by way described in [2]. Barley grains were ground on the<br />
adjusted milling equipment, stirred with 0.02M HCl and refrigerated for 15 hours (4 °C).<br />
When ta<strong>ke</strong>n out pH was adjusted with 0.2M NaOH to 5.0 ± 0.2 and cellulase and β-glucanase<br />
were added, content was sha<strong>ke</strong>n in bath (30 °C for 3 hours), then pH was adjusted to<br />
11.0 ± 0.2 (50 °C for 90 minutes). After this pH was adjusted to 7.0 ± 0.2 and the content was<br />
filtered. Filtrate containing starch was poured into a storage pot. Obtained starch was<br />
repurified with an aqueous solution of CsCl (80%). Isolated starch was purged with acetone<br />
and dried out. Suspension of starch granules in solvent (10 -3 % SDS – sodium dodecyl sulfate)<br />
were measured on GFFF and with commercial particle size and shape analyzer.<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 142
Gravitational Field-Flow Fractionation<br />
Gravitational FFF is the experimentally simplest and cheapest among the family of FFF<br />
techniques. Separation in GFFF is based on combination of gravitational field and a nonuniform<br />
flow velocity profile of carrier liquid in channel (fig 1). Gentle experimental<br />
conditions (low pressure and weak force field) and the possibility to use isotonic buffer<br />
solutions as carrier liquids ma<strong>ke</strong> GFFF unique possible also for separations and purifications<br />
of biological samples [3].<br />
Plexiglass<br />
Glass<br />
Detector<br />
Spectra 100<br />
(λ = 470 nm)<br />
Sample injection<br />
High-pressure<br />
pump<br />
HPP 4001<br />
Fig.1: Principe of Gravitational FFF Fig.2: Arrangement of GFFF channel<br />
The arrangement of GFFF channel is shown in the fig. 2. The separation channel was cut in<br />
0.150 mm thick foil and inserted between two glass plates. The channel dimensions were 3<strong>60</strong><br />
mm x 20 mm x 0.150 mm. The carrier liquid and the sample were introduced into the channel<br />
via an inlet capillary situated at the channel head, and ta<strong>ke</strong>n out via an outlet capillary located<br />
at the end of the channel. The high-pressure pump HPP 4001 (Laboratory Instruments,<br />
Prague, Czech Republic) was used to introduce the carrier liquid into the channel. As the<br />
detector the UV/VIS Spectra 100 (Development Workshop AS CR, Prague, Czech Republic)<br />
operating at 470 nm was used. Data were collected on 386’s (Intel, Santa Clara, CA, USA)<br />
PC equipped with digitalization card.<br />
Starch samples were suspended (the concentration of 10 mg/ml) in 10 -3 % SDS and soa<strong>ke</strong>d<br />
for at least 24 h. Then the sample was sonicated for 1h prior to the FFF experiment. Samples<br />
were injected during the stopped flow. Just after injection, a loading flow at rate 0.2 ml/min<br />
was applied for 10 s. Relaxation time period (stopped-flow period) was for 1.5 min. After this<br />
a linear flow rate of 0.8 ml/min was applied.<br />
CIS-100 Particle Size and Shape Analyzer<br />
The CIS-100 (An<strong>ke</strong>rsmid Ltd., Yokneam, Israel) is a Computerized Inspection System for<br />
Particle Size Analysis (PSA) in the range of 0.5-3<strong>60</strong>0 micron employing laser and video<br />
measurement channels. Together, both channels provide all necessary information to analyze<br />
and characterize spherical, non spherical and elongated particles of large variety of materials.<br />
A He-Ne laser beam in the first channel is scanned circularly by a rotating wedge prism and<br />
focused down to a 1.2 μm spot, which scans the sample measurement volume. As the particles<br />
(moving or stationery) within the sample volume are individually bisected by the laser spot,<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 143
interaction signals are generated. These signals are then detected by a PIN photodiode. Since<br />
the beam rotates at a constant speed, the duration of interaction provide a direct measurement<br />
of each particle's size. The interaction signals are collected by a data acquisition card and<br />
analyzed in <strong>60</strong>0 discrete size intervals. Pulse analysis algorithms are employed to reject outof-focus<br />
and off-center interactions.<br />
The shape analysis channel uses a CCD video camera microscope to provide an optimal<br />
image for processing. Illumination is provided by a synchronized strobe light. Acquired<br />
images are passed to a frame grabber card for analysis, and then displayed on a monitor for<br />
viewing.<br />
Samples were prepared at the concentration of 50 mg/ml in 10 -3 % SDS. In storing reservoir<br />
of instrument they were diluted to 0.5 l and were sonicated with nominal frequency 65 KHz<br />
during whole analysis. The stirring was set up to 226 rpm. The pump drove the suspension to<br />
the channel at flow rate 190 ml/min.<br />
RESULTS<br />
The results of both methods presented in this work give principally the same interval<br />
ranges of starch granule size in barley <strong>ke</strong>rnels as in the preceding works [4]. The main size<br />
fractions of starch granules were detected in the areas of 0-8 μm with peak around 3 μm (9<br />
min in case of GFFF) and 8-30 μm with maximum around 20 μm (4 min). This classification<br />
resulted from the natural bimodal distribution of starch granule size. The smallest amount of<br />
starch particles is among 7.5 and 8 μm, so 8 μm was ta<strong>ke</strong>n as a boundary between small (A)<br />
and large (B) particles. Due to the different principle of measurement of both methods, the<br />
results are not comparable in absolute values. Nevertheless the starch distributions of<br />
individual barley varieties obtained by both methods correspond well.<br />
Cultivar Winter/Spring Row Quality GFFF PSA<br />
CHT LIB STA CHT LIB STA<br />
Tolar Spring 2 VG 0,612 0,584 0,753 3,921 3,228 4,453<br />
Orthega Spring 2 Feed 0,503 0,5<strong>60</strong> 0,970 6,358 6,123 5,988<br />
Heris Spring 2 Feed 1,009 0,709 0,992 7,496 4,015 9,799<br />
Jersey Spring 2 VG 1,388 0,913 1,252 7,091 5,317 5,083<br />
Prestige Spring 2 VG 0,748 0,739 1,205 4,534 4,086 5,631<br />
Scarlett Spring 2 G 0,891 1,042 0,720 6,199 7,834 7,711<br />
Luran Winter 6 Feed 0,961 0,684 1,215 5,4<strong>60</strong> 5,277 5,911<br />
Luxor Winter 6 Feed 1,021 0,903 1,543 7,842 7,123 9,020<br />
Nelly Winter 6 Feed 1,089 1,098 1,328 8,328 7,354 7,150<br />
Vilna Winter 2 Feed 1,589 1,536 1,467 6,402 6,117 7,039<br />
Camera Winter 2 Feed 2,052 2,844 1,250 8,921 8,662 6,764<br />
Tiffany Winter 2 G 2,974 2,541 2,745 9,163 10,521 6,918<br />
Tab.: shows results of individual barley varieties. Quality means: Feed, good malting quality<br />
(G) and very good malting quality (VG). Values are A/B peaks areas from individual test<br />
stations.<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 144
The set of the studied varieties was split into three groups according to the results obtained<br />
by the GFFF and PSA methods. Winter, two-row varieties Tiffany, Camera and Vilna<br />
belonged to a group of varieties with the smallest ratio of small starch granules. Conversely,<br />
the two-row varieties of spring barley Heris, Jersey, Tolar and Orthega formed a group with<br />
the largest ratio of small starch granules. This study did not confirm the assumption that the<br />
malting barley varieties in the studied set compared to the non-malting varieties had a lower<br />
number of small starch granules.<br />
The variety of barley affects the starch granule size distribution most significantly. Effect<br />
of locality and local environmental conditions in the testing stations during growing of the<br />
seeds and year is significantly lower.<br />
CONCLUSIONS<br />
The isolated starch granules from <strong>ke</strong>rnels of twelve barley varieties cultivated in three<br />
different regions were analyzed using gravitational FFF instrument and particle size and shape<br />
analyzer. Significant intervarietal and also some interregional and year differences were<br />
detected in starch granule size distribution. Because of different principle of both methods the<br />
results are not the same in absolute values, but show the same tendencies. This study did not<br />
confirm the assumption that the malting barley varieties in the set observed compared to the<br />
non-malting varieties had a smaller number of small starch granules.<br />
FUTURE WORK<br />
Because malting varieties of barley have other enzymatic apparatus quality, future work will<br />
be focused on characterization of enzymatic activity and starch degradation by defined<br />
enzymes by combination of separation techniques (IEF, GPC) with MALDI-TOF/TOF MS.<br />
REFERENCES<br />
[1] May L.H., Buttrose M.S.: Physiology of cereal grain. II. Starch formation in the<br />
developing barley <strong>ke</strong>rnel. Aust. J. Biol. Sci. 12, 146 - 159, 1959.<br />
[2] Psota V., Bohačenko I., Pytela J., Vydrová H., Chmelík J.: Determination of size<br />
distribution of size distribution of barley starch granules using low angle laser light scattering.<br />
Plant production 46 (10), 433-436, 2000.<br />
[3] Giddings J.C.:Field-Flow Fractionation – Analysis of macromolecular colloidal, and<br />
particulate materials. Science 2<strong>60</strong> (5113), 1456 – 1465, 1993.<br />
[4] MacGregor A.W., Fincher G.B.: Carbohydrates of the barley grain. MacGregor, A.W.,<br />
Bhatty, S.R. (eds.) Barley: Chemistry and Technology. AACC St.Paul, MN, USA. 1993.<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 145
A granules<br />
(8 – 30 μm)<br />
0 5 10<br />
Fig.3: GFFF curves of a) Tiffany, b) Luxor and c) Tolar varieties<br />
a)<br />
b)<br />
0 10 20 30<br />
Size [μm]<br />
B granules<br />
(0 – 8 μm)<br />
Time [min]<br />
0 10 20 30<br />
Size [μm]<br />
Fig.4: Differential and integral PSA volume distribution curves of a) Tiffany, and b) Tolar<br />
varieties<br />
a)<br />
b)<br />
c)<br />
100%<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 146
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 147
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 148
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 149
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 150
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 151
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 152
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 153
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 154
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 155
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 156
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 157
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 158
INFLUENCE OF TRIETHYLALUMINIUM COCATALYST ON<br />
INITIAL KINETIC OF PROPENE POLYMERIZATION<br />
Ing. Miroslav Skoumal 1 , 3 th year Ph.D. student<br />
Supervisor: Ing. Jan Kratochvíla, CSc. 2<br />
1 Institute of Materials Chemistry, Faculty of Chemistry, Brno University of Technology,<br />
Purkyňova 118, 612 00 Brno, Czech Republic, e-mail: skoumal@fch.vutbr.cz<br />
2 Polymer Institute Brno, Tkalcovská 2, 656 49 Brno, Czech Republic<br />
INTRODUCTION<br />
The triethylaluminium (TEA) influence on the initial stage of propene polymerization with<br />
heterogeneous TiCl4/MgCl2-supported Ziegler-Natta catalysts is presented in this study. It is<br />
generally known that alkylaluminium compound (TEA is the most common) plays the<br />
essential role in active center formation which is able to produce poly-α-olefins under the low<br />
temperature and pressure. A wide range of experimental studies, summarized in several<br />
reviews [1,2], was performed for the evaluation of the alkylaluminium role in Ziegler-Natta<br />
catalysts. It was defined that the activation of the catalyst proceeds in two steps. First,<br />
alkylaluminium reduces Ti (IV) to Ti (III). Then it alkylates the Ti (III) forming the first<br />
metal-polymer bond accessible for monomer insertion. Due to high reactivity of TEA, it was<br />
also found that a large number of side reactions with TEA proceeds during the polymerization<br />
such as transfer (broadening of MWD) or termination (further reduction to non-propagative<br />
Ti (II) species).<br />
The heterogeneous nature of MgCl2-supported catalyst, the presence of different types of<br />
active centers on its surface, the activity decay during the polymerization, influence of<br />
catalyst compounds and experimental conditions on the rate of polymerization ma<strong>ke</strong> difficult<br />
to study the polymerization kinetics with this catalysts. These problems are significant<br />
especially during the first seconds of polymerization, where the MgCl2-supported catalyst<br />
exhibits high activity. The investigations of the initial kinetics recently became a subject of<br />
intensive research, assuming that the catalyst behavior during the first seconds of<br />
polymerization determines its efficiency and consequent polymer particle morphology.<br />
For the better understanding of the reactions involved in the initial period, the novel<br />
method for the assessment of initial polymerization kinetic and determination of the number<br />
of active sites was developed [3]. This method allows us to evaluate the kinetic profiles from<br />
1 to 300 seconds with high accuracy and reproducibility.<br />
EXPERIMENTAL PART<br />
The presented study was performed with commercial high-activity TiCl4/MgCl2-supported<br />
Ziegler-Natta catalysts commonly used for polypropene production in industry. Catalyst was<br />
slurried in mineral oil and stored in glass vessels covered by a nitrogen flow. Polymerization<br />
grade propene originated from Chemopetrol Litvínov. Contents of critical impurities CO and<br />
COS were less than 10 ppb, water and oxygen levels were under 0.1 ppm. Triethylaluminum<br />
(TEA) originating from Witco, GmbH (Germany) was dissolved in heptane and <strong>ke</strong>pt in glass<br />
vessels similarly as the catalyst. n-Heptane used as the polymerization medium was purchased<br />
from Aldrich (99 % spectrophotometric grade). Prior to polymerization, removal of water and<br />
oxygen was conducted inside the reactor by stripping. About 10 % of heptane was stripped-<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 159
off by high-purity nitrogen (70°C, 40 min) to remove water and oxygen. The contents of O2<br />
and H2O in the nitrogen were under 0.5 ppm.<br />
The slurry polymerizations were carried out in a water-jac<strong>ke</strong>d glass reactor (240 ml). The<br />
reactor was equipped with magnetic stirrer and connected to a low-pressure polymerization<br />
apparatus. The apparatus was under the PC control, operating the thermostatic circuit,<br />
switching-on the magnetic stirrer, injection of a quenching agent and measuring the<br />
polymerization time.<br />
The PC control operates the automatic injection of a quenching agent into the reactor at a<br />
pre-set time. The main part of the injector was a syringe connected by tube to a solenoidoperated<br />
valve. Before the polymerization, the syringe was loaded with ca. 0.5 ml of a<br />
quenching agent (methanol, 2-propanol, and concentrated hydrochloric acid, volume ratio<br />
4:4:1) and connected to the neck of the glass reactor. After the preset polymerization time had<br />
elapsed, the solenoid valve was automatically opened. Water pressure pushed down the<br />
syringe piston and injected the quenching agent into the reactor. This ensured a fast and<br />
complete termination of the polymerization reaction.<br />
Polymerizations were carried out in ca. 180 ml of previously stripped and cooled heptane<br />
saturated with propene at 30°C (0.61 mol C3H6/l) under a propene flow blan<strong>ke</strong>t at<br />
atmospheric pressure. The defined amount of TEA was introduced via a syringe and the<br />
content of the reactor was homogenized by short stirring. Next, the stirrer was turned off and<br />
the appropriate amount of catalyst oil slurry was slowly dosed into the reactor by means of a<br />
syringe with a long metal injector leading close to the bottom of the reactor. The application<br />
of viscous mineral oil prevents sedimentation of homogenized catalyst slurry during the<br />
charging procedure. Thanks to a nearly zero mass transfer between the oil and the heptane<br />
phases, the catalyst remained separated from cocatalyst (TEA) dissolved in the heptane phase<br />
until the stirrer was turned on (Figure 1).<br />
Figure 1: Schematic drawing of the oil/heptane diffusion interface before the start of stirring.<br />
Typically, 10-15 s after the catalyst was dosed the magnetic stirrer was turned on. The<br />
stirrer speed was high enough to homogenize the oil/heptane mixture within ca. 0.2 s. The<br />
stirrer was operated at <strong>60</strong>0 rpm during polymerization and polymerization conversion<br />
corresponded to a max. 5 % of the monomer dissolved in the polymerization slurry; therefore<br />
the effect of mass transfer through the gas-liquid interface was negligible. It was proven that<br />
the reproducibility, in the range of polymer yields 10 – 300 mg and polymerization times<br />
1 − 300 s, was always within about 7 %. For more information see [3].<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 1<strong>60</strong>
RESULTS AND DISCUSSION<br />
The presented study was focused on influence of starting TEA concentration on the<br />
catalyst behavior during first seconds of propene polymerization. Using the recently<br />
developed method for the initial kinetic determination, it was observed that the applied<br />
MgCl2-supported catalyst exhibits the decay kinetic profile with high initial activity followed<br />
by rapid deceleration to the level of 15% of starting value during 10 seconds<br />
([TEA] = 0.0092 mmol/ml). The high level of initial activity is a negative phenomenon,<br />
accompanied with intensive releasing of heat, which could cause an overheating of polymer<br />
particles and consequent adverse changes in particle morphology. Typical example is<br />
formation of solid polymer particle agglomerates dangerous mainly in industrial reactors.<br />
Yield [g-PP/mmol-Ti]<br />
Activity [g-PP/(mmol-Ti*h)]<br />
30<br />
25<br />
20<br />
15<br />
10<br />
5<br />
0<br />
[TEA] = 0.0092 mmol/ml<br />
[TEA] = 0.0044 mmol/ml<br />
[TEA] = 0.0007 mmol/ml<br />
0 20 40 <strong>60</strong> 80 100 120<br />
Time [s]<br />
7000<br />
<strong>60</strong>00<br />
5000<br />
4000<br />
3000<br />
2000<br />
1000<br />
0<br />
[TEA] = 0.0092 mmol/ml<br />
[TEA] = 0.0044 mmol/ml<br />
[TEA] = 0.0007 mmol/ml<br />
0 20 40 <strong>60</strong> 80 100 120<br />
Time [s]<br />
Yield [g-PP/mmol-Ti]<br />
Activity [g-PP/(mmol-Ti*h)]<br />
10<br />
8<br />
6<br />
4<br />
2<br />
0<br />
7000<br />
<strong>60</strong>00<br />
5000<br />
4000<br />
3000<br />
2000<br />
1000<br />
[TEA] = 0.0092 mmol/ml<br />
[TEA] = 0.0044 mmol/ml<br />
[TEA] = 0.0007 mmol/ml<br />
0 2 4 6 8 10 12 14 16 18 20<br />
Time [s]<br />
0<br />
[TEA] = 0.0092 mmol/ml<br />
[TEA] = 0.0044 mmol/ml<br />
[TEA] = 0.0007 mmol/ml<br />
0 2 4 6 8 10 12 14 16 18 20<br />
Time [s]<br />
Figure 2: Kinetic profiles for different starting TEA concentrations presented as dependence<br />
of the polymer yield and its derivative (activity) on polymerization time. Polymerization<br />
conditions: Atmospheric pressure, temperature 30°C, heptane 180 ml, propene: 0.61 mol/l.<br />
The kinetic profiles from the first 120 s of polymerization indicate that the initial activity<br />
directly depends on the starting TEA concentration in the solution. Changes in TEA<br />
concentration cause significant effect on the initial activity and the following deceleration<br />
period. The Figure 2 shows that the 50% decrease in TEA concentration reduces the initial<br />
activity to one-half while the activity after 120 s is comparable. It suggests that the<br />
unfavorable high initial activities of MgCl2-supported catalyst can be controlled by<br />
application of a suitable starting TEA concentration or by catalyst prepolymerization under<br />
the mild conditions. Further lowering of the cocatalyst concentration reduces the initial<br />
activity to 15%. However, such TEA lowering also causes more significant decrease in the<br />
catalyst productivity.<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 161
Obtained polymer samples were utilized for the determination of molecular weight<br />
distribution by GPC (gel permeation chromatography) analyses. Then the number of active<br />
sites was evaluated from the number-average molecular mass values by a method based on<br />
the number of macromolecules [3-5]. The active sites are determined from the dependence of<br />
the number of macromolecules versus polymer yield (Figure 3), where the intercept of the<br />
linear extrapolation to zero corresponds to the number of propagative centers formed at the<br />
beginning of polymerization.<br />
N [mol/mol-Ti]<br />
0.25<br />
0.20<br />
0.15<br />
0.10<br />
0.05<br />
0.00<br />
y = 0.0105x + 0.0259<br />
R 2 = 0.9977<br />
y = 0.0107x + 0.012<br />
R 2 = 0.9955<br />
y = 0.0102x + 0.0045<br />
R 2 = 0.9965<br />
[TEA] = 0.0092 mmol/ml<br />
[TEA] = 0.0044 mmol/ml<br />
[TEA] = 0.0007 mmol/ml<br />
0 5 10 15 20<br />
Yield [g-PP/mmol-Ti]<br />
Figure 3: Dependence of the number of macromolecules N versus polymer yield extrapolated<br />
to zero.<br />
It was found that the number of active sites decreases linearly in dependence on the<br />
starting TEA concentration. From the determined values of initial catalyst activity A(0) and<br />
number of active sites [C*] the propagation rate constant kp can be calculated for each TEA<br />
concentration. The equation for the kp evaluation follows from the expression [4]:<br />
( 0)<br />
= k ⋅[<br />
M]<br />
[ C*<br />
]<br />
A p ⋅<br />
where [M] is the monomer concentration.<br />
Table 1: Calculated values of propagation rate constant kp for each TEA concentration.<br />
[TEA]<br />
A(0)<br />
[C*]<br />
kp<br />
[mmol/ml] [mol-C3/(mol-Ti*s)] [mol-%]<br />
[l/(mol*s)]<br />
0.0092 47.8 2.6 3000<br />
0.0044 22.7 1.2 3100<br />
0.0007 7.2 0.5 2700<br />
The Table 1 shows that contrary to the number of active sites the kp value remains almost<br />
unchanged under the different cocatalyst concentrations. From the above-presented results are<br />
obvious that the TEA effect on the catalyst activity is more affected by the change of the<br />
number of active sites than by propagation rate constant. This finding is in a good agreement<br />
with results obtained by other researchers using different procedures [6].<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 162
The slope of linear equation in Figure 3 depends on the intensity of transfer reactions<br />
during polymerization. The higher extent of the transfer reactions causes the increase of the<br />
slope value, at negligible level of the transfer reactions the linear dependency (N vs. yield)<br />
would be parallel with x-axis. The results in Figure 3 show that the TEA concentration did not<br />
affect the slope of the fitted lines. This observation suggests that TEA does not act as an<br />
active transfer agent during the polymerization under the mild conditions (atm. pressure,<br />
30°C). So, it could be assumed that the polymer chain transfer reaction proceeds mainly with<br />
monomer or solvent.<br />
LITERATURE<br />
[1] Dusseault J. J. A., Hsu Ch. C., J. Macromol. Sci., Rev. Macromol. Chem. Phys. C33, 103<br />
(1993)<br />
[2] Albizzati E., Giannini U., Collina G., Noristi L., Resconi L., Polypropylene Handbook<br />
Part I., ed. Moore E. P., p. 11, Hanser Publishers, Munich Vienna New York 1996<br />
[3] Skoumal M., Cejpek I., Cheng C. P., Macromol. Rapid Commun. 26, 357 (2005)<br />
[4] Kashiwa N., Yoshita<strong>ke</strong> J., Makromol. Chem., Rapid Commun. 3, 211 (1982)<br />
[5] Chu K.-J., Eur. Polym. J. 34, 577 (1998)<br />
[6] Liu B., Matsuoka H., Terano M., Macromol. Rapid Commun. 22, 1 (2001)<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 163
PHYSICO-CHEMICAL PROPERTIES OF PLASMA-POLYMERIZED<br />
TETRAVINYLSILANE<br />
Mgr. Jan Studýnka<br />
Supervisor: doc.RNDr. Vladimír Čech, Ph.D.<br />
<strong>Vysoké</strong> učení technické v Brně, <strong>Fakulta</strong> <strong>chemická</strong>, Ústav chemie materiálů, Purkyňova<br />
118, 612 00 Brno, email: xcstudynka@fch.vutbr.cz<br />
INTRODUCTION<br />
Plasma-Enhanced Chemical Vapor Deposition (PE CVD) is a suitable technique for<br />
preparation of thin films on a basis of organosilicones [1,2]. The technique enables<br />
reproducible deposition of plasma polymer films with desired physico-chemical properties [3]<br />
by changing the deposition conditions (power, monomer flow rate, pressure).<br />
Tetravinylsilane (TVS) was used as a monomer for deposition of pp-TVS films for the<br />
first time.The helical coupling system [4] was applied to deposit single films using an RF<br />
glow discharge operated in pulsed mode. Various spectroscopic methods were used to<br />
evaluate atomic composition and content of the chemical bonds. Ellipometric measurements<br />
were employed to determine thicknesses of the films and optical constants of the layers.<br />
Selected mechanical properties of single films were determined from nanoindentation<br />
measurements.<br />
EXPERIMENTAL DETAILS<br />
Tetravinylsilane, Si–(CH=CH2)4<br />
(TVS), was used as the monomer. The<br />
silicon wafer was pretreated by O2<br />
plasma (5 sccm, 4 Pa, 25 W) for 10 min<br />
in order to improve the film adhesion.<br />
Pulsed plasma was operated at<br />
conditions given in Table 1.<br />
The elemental composition of thin<br />
films was studied by conventional and<br />
Table 1. Deposition conditions tested for preparation of<br />
pp-TVS films using a stable plasma.<br />
Frequency 13.56 MHz<br />
ton, toff<br />
1 ms, 1 – 999 ms<br />
Effective power 0.05 – 10 W<br />
Effective power density 8 ×10 -4 – 1 ×10 -1 W cm -3<br />
Basic pressure 2 × 10 -3 Pa<br />
Process gas pressure 0.1 – 4.4 Pa<br />
Monomer vapor flow rate 0.45 sccm<br />
resonant Rutherford Backscattering Spectrometry (RBS) and Elastic Recoil Detection<br />
Analysis (ERDA) methods. Chemical structure of plasma polymer was investigated using a<br />
NICOLET IMPACT 400 Fourier transform infrared (FTIR) spectrophotometer. Measuring<br />
range was from 400 to 4000 cm -1 . We employed a Nano Hardness Tester (NHT) by CMS<br />
Instruments in order to carry out nanoindentation tests. The Young’s modulus and hardness of<br />
films were determined from unload-displacement curves measured at 12% of the film<br />
thickness.<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 164
RESULTS AND DISCUSSION<br />
A) Ellipsometry<br />
Ellipsometric measurements were performed in order to determine the film thickness and<br />
optical constants, i.e. refractive index and extinction coefficient, for pp-TVS films.<br />
Dispersions of refractive index and extinction coefficient for pp-TVS films deposited at<br />
different effective power are given in Fig. 1. The results correspond to pp-TVS films with a<br />
thickness about 400 nm deposited at the same deposition conditions but different power. The<br />
value of refractive index at the wavelength of 633 nm increased with power from 1.63<br />
(0.05 W) to 1.75 (10 W). There is a growth of the refractive index and extinction coefficient<br />
in ultraviolet region. A<br />
different shape of<br />
dispersion in ultraviolet<br />
region for the refractive<br />
index corresponding to a<br />
film deposited at higher<br />
power (10 W) may be<br />
caused by higher<br />
absorption in UV region.<br />
The extinction coefficient<br />
rose sharply with the<br />
power from 0.09<br />
(0.05 W) to 0.37 (10 W)<br />
at the wavelength of 240<br />
nm.<br />
300 400 500 <strong>60</strong>0 700 800<br />
B) Elemental composition and chemical structure of films<br />
Atomic concentration [at%]<br />
Refractive index<br />
1.95<br />
1.90<br />
1.85<br />
1.80<br />
1.75<br />
1.70<br />
1.65<br />
1.<strong>60</strong><br />
0.40 Film thickness: 0.4 μm<br />
0.35<br />
0.05 W<br />
0.5W<br />
0.30<br />
5 W<br />
0.25<br />
10 W<br />
300 400 500 <strong>60</strong>0 700 800<br />
Wavelenght<br />
Wavelenght<br />
Figure 1. Dispersions of refractive index and extinction coefficient<br />
for pp-TVS films deposited at different power.<br />
The pp-TVS films of a film thickness about 1 µm were used to characterize chemical<br />
70<br />
<strong>60</strong><br />
50<br />
40<br />
30<br />
20<br />
10<br />
0<br />
C<br />
O<br />
Si<br />
H<br />
C/Si<br />
O/Si<br />
0.1 1 10<br />
Effective power [W]<br />
Figure 2. Elemental composition of pp-TVS films deposited<br />
at different power.<br />
14<br />
12<br />
10<br />
8<br />
6<br />
4<br />
2<br />
0<br />
Extinction coefficient<br />
0.20<br />
0.15<br />
0.10<br />
0.05<br />
0.00<br />
-0.05<br />
properties of the plasma polymer.<br />
Elemental composition of films was<br />
determined from RBS and ERDA<br />
spectra. The RBS spectra were<br />
evaluated by computer code GISA 3<br />
[5] and the ERDA ones by<br />
SIMNRA code [6] both using crosssection<br />
values from SigmaBase. An<br />
estimated accuracy of evaluated<br />
concentration was up to 5 at%. A<br />
dependence of atomic<br />
concentrations on the effective<br />
power is given in Fig. 2. The<br />
concentration of hydrogen atoms<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 165
was about 56 at% and did not vary with the effective power. The same trend or a slight<br />
decrease was observed for the concentration of silicon atoms in plasma polymer deposited at<br />
different power. A mean concentration of silicon atoms was about 5 at%. However, the<br />
oxygen concentration decreased rapidly at the expense of carbon atoms with increasing<br />
power. The concentration of oxygen atoms descended from 13 at% (0.05 W) to 1 at% (10 W),<br />
while carbon concentration rose from 27 at% (0.05 W) to 42 at% (10 W). The C/Si ratio<br />
indicated great changes in organic/inorganic character of plasma polymer with enhanced<br />
power. The film deposited at a power of 10 W was hydrogenated amorphous carbon<br />
containing a small amount of silicon-carbide phase.<br />
Table 2. Assignment of IR absorption bands.<br />
Absorbance [a.u.]<br />
Adsorption<br />
band<br />
Wavenumber<br />
[cm -1 ]<br />
Assignment<br />
A 3650 – 3200 O–H stretching<br />
B 3000 – 2800 CH2, CH3 stretching<br />
C 2122 Si–H stretching<br />
D 1714 C=O stretching<br />
E 1591 C=C stretching in vinyl<br />
F 1461 CH2 scissoring<br />
G 1412 CH2 deformation in vinyl<br />
H 1255 CH2 waging in Si–CH2–R<br />
I 1100 – 1000 Si–O–C stretching<br />
J 1015 =CH wagging in vinyl<br />
K 959 =CH2 wagging in vinyl<br />
L 845 Si–H bending<br />
M 732 Si–C stretching<br />
A<br />
B<br />
0.1 W<br />
0.5 W<br />
2.5 W<br />
10 W<br />
C<br />
D<br />
G<br />
E<br />
F<br />
J<br />
L<br />
M<br />
I K<br />
3500 3000 2500 2000 1500 1000 500<br />
Wavenumber [cm -1 ]<br />
H<br />
TVS molecule does not contain<br />
any oxygen and thus, the oxygen<br />
atoms incorporated in plasma<br />
polymer could have their origin in<br />
pretreatment procedure using O2<br />
plasma. Most li<strong>ke</strong>ly, the residual<br />
oxygen was desorbed from the<br />
wall of plasma chamber during<br />
the deposition process and was<br />
incorporated in plasma polymer.<br />
We estimated the flow rate of<br />
desorbed oxygen at < 0.05 sccm<br />
from the pressure increase in<br />
separate deposition chamber. The<br />
results on elemental composition<br />
revealed that the residual oxygen<br />
was effectively built in plasma<br />
polymer only if a lower power (<<br />
5 W) was used. The lower power<br />
the higher oxygen concentration<br />
in pp-TVS film.<br />
Typical infrared spectra of pp-<br />
TVS films deposited at different<br />
power are given in Fig. 3.<br />
The assignment of IR<br />
absorption bands using Ref. 7 and<br />
8 was summarized in Table 2. One<br />
of the dominated peaks, the<br />
absorption band B, was assigned<br />
to CH2, CH3 stretching vibrations.<br />
However, both the species cannot<br />
be distinguished with respect to<br />
symmetric character of the band<br />
Figure 3. Infrared spectra of pp-TVS films prepared at<br />
Sborník different soutěže effective Studentské power. tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 166
Young's modulus [GPa]<br />
B. The intensity and area of absorption bands A, D, and I, corresponding to species such as<br />
OH, C=O, and Si–O–C, respectively, descended with increased power and the trend was in<br />
good agreement with a descent of oxygen concentration in plasma polymer revealed by RBS<br />
measurements. A decrease of bands C, H, L, and M assigned to species containing silicon<br />
atoms was observed as well and the trend corresponded to an increase of C/Si ratio with<br />
enhanced power. An occurrence of vinyl groups in plasma polymer was evident from<br />
IR spectra corresponding to pp-TVS films deposited at lower power (≤ 2.5 W). Quantity of<br />
vinyl groups decreased with enhanced power as a descent of bands E, J, and especially G, K<br />
indicated.<br />
C) Nanoindentation<br />
Load-displacement curves were measured at a depth of 50 nm for all the films with the<br />
film thickness about 400 nm. Five indents were produced on each sample with spacing<br />
between the indentations of 5 µm. Nanoindentation measurements were evaluated by Oliver<br />
& Pharr method [9] in order to determine the Young’s modulus and hardness of pp-TVS<br />
40<br />
30<br />
20<br />
10<br />
0<br />
Film thickness: 0.4 μm<br />
0.1 1 10<br />
Effective power [W]<br />
films. The Poisson’s ratio used for<br />
plasma polymer was 0.3. The mean<br />
modulus and hardness was<br />
calculated from five values for each<br />
film. The results on mechanical<br />
properties of pp-TVS films are<br />
summarized as power dependence in<br />
Fig. 4. The modulus increased<br />
almost 3 times and the hardness<br />
increased almost 4 times with<br />
enhanced power. The increase of<br />
modulus could be related to a higher<br />
crosslinking and/or an alteration of<br />
microstructure with increasing<br />
organic character of plasma polymer.<br />
CONCLUSIONS<br />
Plasma polymer films of tetravinylsilane were deposited on silicon wafers by plasmaenhanced<br />
chemical vapor deposition. Pp-TVS films deposited at a flow rate of 0.45 sccm<br />
(process pressure 1.5 Pa), and an effective power of 0.05 – 10 W were analyzed by<br />
spectroscopic techniques extensively to elemental composition and chemical structure (RBS,<br />
ERDA, FTIR). A phase-modulated spectroscopic ellipsometer was employed to determine the<br />
film thickness and optical constants (refractive index, extinction coefficient) of deposited<br />
films. The refractive index increased by 7.4% at the wavelength of 633 nm when the power<br />
was enhanced from 0.05 to 10 W. The extinction coefficient in ultraviolet region (240 nm)<br />
rose sharply by 4 times with enhanced power (0.05 – 10 W). The results revealed that an<br />
14<br />
12<br />
10<br />
8<br />
6<br />
4<br />
2<br />
0<br />
Hardness [GPa]<br />
Figure 4. Power dependence of mechanical properties for pp-<br />
TVS films.<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 167
organic/inorganic character (C/Si ratio) of films and a content of vinyl groups could be<br />
controlled by deposition conditions precisely. Nanoindentation measurements enabled us to<br />
evaluate the elastic modulus and hardness of films prepared at different power. The elastic<br />
modulus and hardness could be varied from 11 GPa (0.05 W) to 30 GPa (10 W) and from 1.4<br />
to 5.9 GPa for an increased power, respectively.<br />
Acknowledgements: This work was supported in part by the Czech Ministry of Education<br />
(contracts COST 527.110 and P12.001) and the Czech Science Foundation (contract<br />
104/03/0236).<br />
References<br />
1. A.M. Wrobel and M.R. Wertheimer, in Plasma Deposition, Treatment, and Etching of<br />
Polymers, (R. d’Agostino, Editor), Academic Press, New York (1990) p.163.<br />
2. Y. Segui, in Proceedings of NATO ASI Plasma Processing of Polymers (R. d’Agostino, P.<br />
Favia, F. Fracassi, Eds.). Acquafredda di Maratea, Kluwer Academic Publ. (1997) p.305.<br />
3. V. Cech, J. Vanek, J. Zemek, L. Zajickova, and R. Prikryl, Variety of functional<br />
interlayers utilizable for polymer composites, Proc. 11 th Int. Conf. on<br />
Composites/NanoEngineering, August 8-14, 2004, Marriott Hilton-Head Beach & Golf<br />
Resort, South Carolina, USA, pp. 2.<br />
4. V. Cech, R. Prikryl, R. Balkova, J. Vanek, and A.Grycova, J. Adhesion Sci. Technol. 17<br />
(2003) 1299.<br />
5. J. Saarilahti and E. Rauhala: Interactive personal-computer data analysis of ion<br />
backscattering spectra, Nucl. Instrum. and Methods in Physics Research B64, 734 (1992).<br />
6. M. Mayer, SIMNRA User`s Guide, Forschungscentrum Julich, Inst. fur Plasmaphysik,<br />
1998.<br />
7. D. Lin-Vein, N.B. Colthup, W.G. Fateley, J.G. Grasselli, The Handbook of Infrared and<br />
Raman Characteristic Frequencies of Organic Molecules, Academic Press, San Diego, 1991.<br />
8. V.P. Tolstoy, I.V. Chernyshova, V.A. Skryshevsky, Handbook of Infrared Spectroscopy<br />
of Ultrathin Films, John Wiley & Sons, New Jersey, 2003.<br />
9. W. C. Oliver, G.M. Pharr, J. Mater. Res. 7 (1992) 1564.<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 168
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 169
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 170
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 171
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 172
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 173
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 174
SPECIATION OF FIVE SELENIUM COMPOUNDS BY<br />
HPLC-HEATING-UV-HG-AFS<br />
Ing. Eva Vitoulová<br />
Supervisor: doc. Ing. Miroslav Fišera, CSc.<br />
Brno University of Technology, Fakulty of Chemistry, Department of Food Chemistry and<br />
Biotechnology, Purkyňova 118, 612 00 Brno, e-mail: xcvitoulova@fch.vutbr.cz<br />
INTRODUCTION<br />
Selenium is an essential nutrient at low concentrations, but is toxic for humans and animals at<br />
high doses. It is a component of the enzyme glutathione peroxidase, which is one of the antioxidant<br />
defence systems of the body, catalyses intermediate metabolic reactions, and inhibits the toxicity of<br />
some heavy metals. 1<br />
The effect of the element on human health is highly dependent on the chemical species under<br />
which it is consumed. Selenium exists in different chemical forms, as inorganic (selenite, selenate)<br />
and as organic species (selenoaminoacids, selenoproteins), in environmental and biological<br />
matrices. The nutritional bioavailability and cancer chemoprotective activity of selenium depend on<br />
the concentration and the chemical form in which it is present. 2, 3<br />
The absorption of selenium is greater in the case of organic species and the possibility that the<br />
selenium supplementation of food might protect against the development of cancer in humans has<br />
generated great interest in the speciation of the various chemical forms in foodstuffs and in dietary<br />
supplements. 1 Each species adsorbed differently by the human body and has a different tendency to<br />
bioaccumulate in organisms. 4<br />
The availability of analytical techniques for the separation and determination of the compounds<br />
of an element at trace level has gained considerable importance. In this context, hyphenated<br />
techniques are those most frequently used. For selenium, speciation is necessary because of the<br />
differing mobilities, toxicities and bioavailabilities of its compounds. 5<br />
Analytical systems developed for the speciation of selenium species employ a powerful highperformance<br />
liquid chromatography (HPLC) coupled to a specific atomic detector with a high<br />
efficiency sample introduction system. 6 Spectrometry methods are those most widely used as a<br />
detection system. Atomic fluorescence spectrometry (AFS) and inductively coupled plasma - mass<br />
spectrometry (ICP-MS) have been incorporated with very good results. Atomic spectrometry<br />
methods are the most widely used because of their high selectivity and sensitivity. 5<br />
EXPERIMENTAL<br />
Instrumentation<br />
The HPLC system consisted of an HPLC pump Gynkotech P580 (Gynkotech, Softron, Germany)<br />
equipped with a six-port sample injection valve (C&D, Ecom, Prague, Czech Republic) and a 20 μl<br />
loop for sample introduction. The separation of the selenium species occurred in column Hamilton<br />
PRP X-100 (250 x 4.1 mm, 10 μm) strong anion exchange column.<br />
Hydride generation atomic fluorescence spectrometry (AFS) was performed using PSA<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 175
Millenium Excalibur continuous system (PS Analytical, Orpington, Kent, UK) with a PSA 10.570<br />
UV crac<strong>ke</strong>r and heating system (Chromspec, Czech Republic) (Fig. 1.). Measurements were carried<br />
out using a boosted discharge hollow cathode lamp for selenium (Photron, Pty. Ltd., Australia) at<br />
196,0 nm line, with a 20,0 mA primary current and 25,0 mA boost current.<br />
A Heidolph Instruments – Promax 1020 and a Heidolph Instruments – Incubator 1000<br />
(Germany) were used for extracting the Se-compounds from the samples. The samples were<br />
centrifuged in Boeco U 32 – R (Hettich zentrifugen, Germany).<br />
The supernatants were filtred through a 0,45 μm PTFE membrane Minisart SRP 15 (Sartorius,<br />
UK).<br />
Fig. 1 The scheme of interconection between HPLC-Heating-UV-HG-AFS<br />
Reagents and materials<br />
All the solutions were prepared with deionised water (18,2 MΩ.cm) purified through a Purelab<br />
Classic purification system (Elga LabWater, UK).<br />
Stock solutions of 100 μg.ml -1 were prepared by dissolving sodium selenite (Sigma-Aldrich,<br />
Germany), sodium selenate (Sigma-Aldrich, Germany), Seleno-DL-ethionine (Sigma-Aldrich,<br />
Germany), L-selenomethionine (Merck, KGaA, Germany) and Se-(methyl)-selenocystein (Sigma-<br />
Aldrich, Germany). Diluted solutions of concentration 500 ng.ml -1 for analysis of standard mixtures<br />
were prepared immediately before using.<br />
Protease from Bacillus licheniformis (13,1 units/mg) (Fluka, Denmark), Protease XIV from<br />
Streptomyces griseus (4,4 units/mg) (Sigma-Aldrich, Germany) and Subtilisin A, type VIII, from<br />
Bacillus licheniformis (8 units/mg) (Sigma-Aldrich, Germany) were used for enzymatic hydrolysis<br />
of the samples.<br />
Sodium tetrahydroborate solution (1%) was prepared by dissolving the solid product (Sigma-<br />
Aldrich, Germany) in 0,1M NaOH (Riedel de Haën, Germany). This reductive solution was<br />
prepared daily.<br />
The mobile phase was a 40 mmol.l -1 ammonium dihydrogenphosphate solution of pH 6.2<br />
prepared daily from the commercial product (Sigma-Aldrich, Germany) and pH was adjusted by<br />
adding dropwise 25 % ammonium hydroxide solution (Sigma-Aldrich, Germany).<br />
Hydrochloric acid 32% solution was prepared from 37% hydrochloric acid p.a. (Analytika,<br />
Prague, Czech Republic).<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 176
Samples<br />
The samples were different nutritional selenium supplements, two of them were based on<br />
selenized yeast and one on the sodium selenite, all of which were commercially obtained. All the<br />
supplements were in tablets and labeled with the quantity of selenium in the each tablet.<br />
1. Sample was a nutritional suplement based on selenized yeast<br />
2. Sample was a nutritional suplement based on selenized yeast<br />
3. Sample was a nutritional suplement based on sodium selenite<br />
PROCEDURES<br />
Enzymatic hydrolysis<br />
Tablets of nutritional supplements were properly ground in a agate mortar. Approximately 0,5 g of<br />
the powdered sample and 20 mg of enzyme:<br />
1. Protease XIV from Streptomyces griseus (4,4 units/mg)<br />
2. Subtilisin A, type VIII, from Bacillus licheniformis (8 units/mg)<br />
3. Protease from Bacillus licheniformis (13,1 units/mg)<br />
4. Without enzyme<br />
were placed in a 25 ml polypropylene bottle. After addition of 5 ml of deionised water, the samples<br />
were sha<strong>ke</strong>n at 170 rpm for 24 hours using mechanical sha<strong>ke</strong>r at 37 °C.<br />
After extraction, the extracts were separated from the samples by centrifugation for 15 min at<br />
3000 rpm. The supernatants were filtered through 0,45 μm membrane to eliminate suspended<br />
solids. The clear filtrate was injected into the chromatograph.<br />
RESULTS AND DISCUSSION<br />
Optimisation of chromatographic separation of Se species<br />
For the separation of the five selenium species we used a polystyrene-divinylbenzene-based<br />
anion exchange column Hamilton PRP X-100.<br />
Firstly we studied the dependence of the retention time of the five species on the pH of the<br />
phosphate solution used as mobile phase. The flow of the mobile phase was 1 ml.min -1 . Inorganic<br />
species selenite and selenate required the use of an alkaline solution as mobile phase to obtain low<br />
retention times. Selenoaminoacids showed high retention times at alkaline pH, whereas at lower pH<br />
values retention times were shortened. We tried to change pH values between 5.5 to 7.5 and the best<br />
results were obtain by working at pH 6,2. The species were eluted in the following order: SeEt,<br />
SeCys, selenite, SeMet and selenate (Fig. 2.).<br />
Secondly we varied the concentration of the mobile phase. Several concentrations of the mobile<br />
phase were tested, ranging from the 20 mmol.l -1 to the 80 mmol.l -1 . The concentration 40 mmol.l -1<br />
was chosen as the best, because this concentration gave sufficient resolution.<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 177
Thirdly we tried to change the composition of the mobile phase, by adding of the methanol to the<br />
mixture. This addition of methanol had no significant influence on the separation.<br />
Extraction of selenium species from nutritional supplements<br />
The common procedures used for the speciation of selenium in yeast, plants and vegetables have<br />
been hot water extraction, enzymatic hydrolysis, buffers, water-methanol and HCl extraction. 7 In<br />
this study, protease XIV, Subtilisin A and Protease from Bacillus licheniformis (13,1 units/mg) were<br />
used for enzymatic extraction to cleave peptide bonds in proteins. The examples of chromatograms<br />
obtained for the samples are given in Fig. 3 -5<br />
Fig. 2 The HPLC-Heating-UV-HG-AFS chromatogram of mixed selenium standard solution: (A)<br />
SelenoEthionine, (B) selenocysteine, (C) selenite, (D) selenomethionine, (E) selenate<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 178
Fig. 3 The HPLC-Heating-UV-HG-AFS chromatogram of selenium species after enzymatic<br />
extraction (Protease XIV) - Sample 1 : (A) selenocysteine, (B) selenite, (C) selenomethionine, (D)<br />
selenate<br />
Fig. 4 The HPLC-Heating-UV-HG-AFS chromatogram of selenium species after enzymatic<br />
extraction (Protease XIV) - Sample 2 : selenate<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 179
Fig. 5 The HPLC-Heating-UV-HG-AFS chromatogram of selenium species after non enzymatic<br />
extraction - Sample 3 : selenite.<br />
CONCLUSION<br />
The speciation of selenite, selenate, selenoethionine, selenocysteine and selenomethionine was<br />
satisfactory when using the combination HPLC-Heating-UV-HG-AFS. This method is relatively<br />
free of interferences, involving low cost and maintenance. The procedure was applied to the<br />
speciation of Se-species in nutritional supplements. In the sample number 1 (suplement contained<br />
selenised yeast) selenoethionine, selenocysteine, selenite, selenomethionine and selenate were<br />
identified. In the sample number 2 (suplement contained selenised yeast) only selenate was<br />
identified. In the sample number 3 (suplement contained sodium selenite) selenite was identified.<br />
Three different enzymes for enzymatic extraction were tested. The best results were obtained with<br />
Protease XIV from Streptomyces griseus (4,4 units/mg). Forthcoming work will be directed to the<br />
evaluation of selenium recoveries.<br />
ACKNOWLEDGEMENTS<br />
This work was supported by Ministry of Education (MŠMT) of the Czech Republic (Project<br />
G4/1259/2005 FRVŠ).<br />
REFERENCES<br />
[1] Vińas P., López-García I., Merino-Merońo B., Campillo N., Hernández-Córdoba M.: Anal.<br />
Chim. Acta 535, 49 (2005).<br />
[2] Dumont E., De Cremer K., Van Hulle M., Chéry C., Vanhaec<strong>ke</strong> F., Cornelis R.: Journal of<br />
Chrom. A 1071, 191, (2005).<br />
[3] Gómez-Ariza J.L., Caro de la Torre M., Giraldez I., Sánchez-Rodaz D., Velasco A.,<br />
Morales E.: App. Organometallic Chem. 16, 265, (2002).<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 180
[4] Tsopelas F. N., Ochsenkühn-Petropoulou M. T., Mergias I. G., Tsakanika L. V.: Analytica<br />
Chimica Acta 539, 327 (2005)<br />
[5] Vilanó M., Padró A., Rubio R., Rauret G.: Journal of Chrom. A 819, 211, (1998).<br />
[6] Ipolyi I., Stefánka Z., Forot P.: Anal. Chim. Acta 435, 367, (2001).<br />
[7] Kahakachchi Ch., Boakye H. T., Uden P.C., Tyson J. F.: Journal of Chrom. A 1054, 303,<br />
(2004)<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 181
EPITAXIAL GROWTH OF Ni AND Co ON (111) METALLIC<br />
SUBSTRATES<br />
Ing. Martin Zelený, 2 nd year<br />
Supervisor: Prof. RNDr. Mojmír Šob, DrSc.<br />
Faculty of Chemistry, Brno University of Technology, Purkyňova 118, 612 00 Brno,<br />
Czech Republic, zeleny@fch.vutbr.cz<br />
INTRODUCTION<br />
Transition-metal thin films and multilayers have recently attracted a lot of interest because of<br />
their technological importance in data storage and sensor applications as well as in fundamental<br />
research of magnetism.<br />
Bulk Co exists in two ferromagnetic thermodynamically stable phases at ambient pressure:<br />
hcp α-Co below 388 °C and fcc β-Co above 450 °C. In case of Ni, only the fcc modification is<br />
known. It turns out that Ni and Co thin films exhibit similar structures as in the bulk. For<br />
example, only distorted fcc films of Ni have been observed [1, 2]. On the other hand, cobalt thin<br />
films with the hcp structure [3] as well as with a trigonally distorted fcc structure [4] were found<br />
on fcc (111) substrates. In some cases, both structures were reported [5].<br />
The goal of this contribution is to advance our fundamental understanding of epitaxial growth<br />
of Ni and Co thin films on various fcc(111) substrates. For this purpose, we employ ab initio<br />
electronic structure calculations based on fundamental quantum mechanics. Pseudomorphic<br />
overlayers adopt the lattice dimensions of the fcc substrate in the (111) plane and relax the<br />
interlayer distance. There is a stress in the (111) plane <strong>ke</strong>eping the structure of the film and of the<br />
substrate coherent, and the stress perpendicular to this plane vanishes due to relaxation (Fig. 1).<br />
On the (111) surface, there are two possible stackings of the layers in the film: ABABAB...,<br />
giving the hcp structure, and ABCABC..., providing the trigonally distorted fcc structure. The<br />
nearest-neighbor distance DNN in the plane of the substrate is equal to asub 2 /2, where asub is the<br />
lattice constant of the substrate. If there is a large difference between the lattice dimensions of the<br />
substrate and material of the film (lattice mismatch), the stress in the (111) plane is too large. As<br />
a consequence, the film becomes incoherent and has a structure similar to the ground state of the<br />
bulk of the film material.<br />
Our investigations allow us to understand, from the first principles, the structure of Ni and Co<br />
thin films on various fcc(111) substrates. In particular, we explain why some of those films<br />
exhibit the hcp structure and some of them the trigonally distorted fcc structure.<br />
METHOD<br />
To simulate the epitaxial growth, we first calculate the total energy of the film material (i. e.<br />
Ni or Co) in equilibrium hcp and fcc structures. Here a transformation of fcc cell to hexagonal<br />
coordinates is useful (Fig. 1). Then, in the second step, we apply some elongation of the bulk<br />
crystal along both crystallographic axes by a fixed amount ε that provides the same atomic<br />
spacing as in the (111) plane of substrate, DNN. For each ε, we minimize the total energy, relaxing<br />
the stress in the direction perpendicular to the loading axes. This is a very simple model, but it<br />
turns out that it is quite realistic.<br />
For the total-energy calculations, we employ the full-potential linearized augmented plane<br />
waves method incorporated in the WIEN2k code [6]. The calculations are performed using the<br />
generalized gradient approximation (GGA). The muffin-tin radius of atoms of 2.0 a.u. is <strong>ke</strong>pt<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 182
Figure 1. Epitaxial growth of thin films on the fcc(111) substrate. The fcc cell is characterized<br />
also by hexagonal coordinates (ahex, chex).<br />
constant for all calculations, the number of k-points in the whole Brillouin zone is equal to 8000,<br />
and the product of the muffin-tin radius and the maximum reciprocal space vector, RMTkmax, is<br />
equal to 9. For Ni, the maximum value of l for the waves inside the atomic spheres, lmax, and the<br />
size of the largest reciprocal vector G in the charge Fourier expansion, Gmax, is set to 9 and 16,<br />
respectively. For cobalt, lmax is set to 11 and Gmax is set to 16.<br />
RESULTS AND DISCUSSION<br />
The total energy of bulk Ni as a function of DNN is shown in Fig. 2(a). Triangles correspond<br />
to trigonally distorted fcc structures and circles to hcp structures. There are several interesting<br />
points on these dependences. Each curve has the global minimum, which corresponds to the<br />
equilibrium state. The equilibrium state with the lowest energy is the fcc ground state of Ni. The<br />
structural energy difference between the fcc and hcp structures at this point is 2.0 mRy/atom.<br />
Second interesting point on each curve is the inflexion point. The inflexion point with the lowest<br />
energy corresponds to the maximum strain in the (111) plane and to maximum DNN (5.31 a.u.),<br />
where the Ni film may be expected to be coherent. Next interesting point is a crossing point<br />
between the curves at 5.25 a.u. It represents the position where a phase transition between<br />
trigonally distorted fcc structures and hcp structures may ta<strong>ke</strong> place.<br />
It follows from the analysis of Fig. 2(a) that the fcc layer stacking (ABCABC...) is stable in<br />
the neighborhood of the ground state up to DNN equal to 5.25 a.u. This is in a good agreement<br />
with the experimental data for Ni film on Cu(111) [5] and on Pt(111) [7] substrates. If the<br />
substrates have the DNN larger than 5.25 a.u., the film should exhibit the hcp layer stacking<br />
(ABAB…). However, a maximum value of DNN, where the film is coherent, is equal to 5.31 a.u.<br />
(the inflexion point). The value of DNN for Pt of 5.24 a.u. is nearly equal to 5.25 a.u., so we can<br />
expect hcp islands in Ni films on Pt(111). Because Au and Ag substrates have larger DNN than the<br />
value corresponding to the inflexion point, the Ni thin films on these substrates have not the hcp<br />
structure, but are incoherent and have a structure similar to fcc Ni in the ground state [8].<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 183
Figure 2. Total energy of Ni (a) and Co (b) as a function of nearest-neighbor distance DNN in the<br />
fcc(111) substrate. Full symbols denote our predictions of structures and coherence of Ni and Co<br />
thin films on different fcc(111) substrates. The inflexion point is shown only for the stable<br />
structures.<br />
Cobalt films behave differently (Fig. 2(b)). Total energies of hcp structures are lower than<br />
energies of trigonally distorted fcc structures, which means that hcp phases are energetically<br />
more favorable along the whole deformation curve. The structural energy difference between the<br />
fcc and hcp Co at the energy minimum is only 1.1 mRy/atom, which is half the value for Ni. Hcp<br />
structure is experimentally found in Co films on Cu(111) substrate [9], but islands with trigonally<br />
distorted fcc structures have also been reported [5]. This is due to a small structural energy<br />
difference between the hcp and fcc phases of Co, so that a transition between these phases is very<br />
easy. To the best of our knowledge, the only coherent films of cobalt were found on Cu(111)<br />
substrate. Our calculations predict also coherent Co layers on Pt(111) and Pd(111) substrates,<br />
because the DNN for these substrates is smaller than the value corresponding to the inflexion point<br />
(5.30 a.u.) – see Fig. 2(b). However, experimentally prepared films on these substrates are<br />
incoherent. Co thin films on Pd(111) substrate have the hcp structure [10], and the structure of Co<br />
thin films on Pt(111) strongly depends on the way of preparation [4, 11]. The difference between<br />
our prediction and experiment is probably due to the diffusion of Pt into the Co film and<br />
formation of alloys based on CoPt [12]. For Co on Au(111) substrate we predict incoherent film<br />
with the hcp structure, which is in agreement with available experimental data [3].<br />
The interlayer distance d in the film as a function of the nearest-neighbor distance DNN in the<br />
substrate is shown in Fig. 3 for Ni (a) and Co (b). Comparison of numerical values is given in<br />
Table 1. Our predictions are in a good agreement with the experimental data.<br />
CONCLUSIONS<br />
We have calculated the total energies of Ni and Co as a function of nearest-neighbor distance<br />
DNN in the fcc(111) substrate for two different structures – trigonally distorted fcc structure and<br />
hcp structure, and applied these results to understand the atomic configuration of Ni and Co thin<br />
films. We have found an inflexion point on the dependence of total energy on DNN. It corresponds<br />
to the maximum value of DNN, where the film <strong>ke</strong>eps to be coherent with the substrates.<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 184
Figure 3. Interlayer distance d of Ni (a) and Co (b) as a function of nearest-neighbor distance<br />
DNN in the fcc(111) substrate. Full symbols represent the values from experimentally prepared<br />
thin films. The inflexion point is shown only for the stable structures.<br />
Our predictions for Ni films are in a good agreement with available experimental data. For<br />
Co films the agreement is not so good, because the structural energy difference between the hcp<br />
and fcc structures is too small and the structure of films strongly depends on the way of<br />
preparation. Some deviations may be also due to the simplicity of our model of epitaxial growth.<br />
Our investigations show that the results of ab initio calculations may be used to understand<br />
and predict the structure of nic<strong>ke</strong>l and cobalt overlayers on various fcc(111) substrates.<br />
ACKNOWLEDGEMENTS<br />
This research was supported by the Grant Agency of the Czech Republic (Projects No.<br />
202/03/1351 and 106/05/H008), by the Grant Agency of the Academy of Sciences of the Czech<br />
Republic (Projects No. IAA1041302 and S2041105), and by the Research Projects<br />
AV0Z20410507 and MSM0021622410. The use of the computer facilities at the MetaCenter of<br />
the Masaryk University, Brno, is acknowledged.<br />
Table 1. Comparison between theoretical results and experimental data. Predictions of interlayer<br />
distance d are shown only for coherent films.<br />
film/sub.<br />
Ni/Cu(111)<br />
asub<br />
[a.u.]<br />
DNN<br />
[a.u.] d [a.u.]<br />
theory<br />
struc. film<br />
experiment<br />
d [a.u.] struct. film<br />
a<br />
6.82 4.82 3.78 fcc coh. 3.82 fcc coh.<br />
Ni/Pt(111) b<br />
7.40 5.24 3.57 fcc/hcp coh. 3.61 fcc coh.<br />
Ni/Au(111) c<br />
7.69 5.44 - fcc incoh. 3.78 fcc incoh.<br />
Ni/Ag(111) c<br />
7.71 5.45 - fcc incoh. 3.78 fcc incoh.<br />
Co/Cu(111) d<br />
6.82 4.82 3.76 hcp coh. 3.82 hcp coh.<br />
Co/Pd(111) e<br />
7.34 5.19 3.57 hcp coh. 3.72 hcp incoh.<br />
Co/Pt(111) f<br />
7.40 5.24 3.52 hcp coh. 3.68 hcp/fcc incoh.<br />
Co/Au(111) g<br />
7.69 5.44 - hcp incoh. 3.84 hcp incoh.<br />
a b c d e f g<br />
Ref. [5], Ref. [7], Ref. [8], Ref. [9], Ref. [10], Ref. [4, 11], Ref. [3].<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 185
REFERENCES<br />
1. D. W. Gidley, Phys. Rev. Lett. 62, 811, (1989).<br />
2. M. Sambi, E. Pin, G. Granozii, Surf. Sci. 340, 215 (1995).<br />
3. N. Marsot, R. Belkhou, H. Magnan, P. Le Fevre, C. Guillot, and D. Chandesris, Phys. Rev. B<br />
59, 3135 (1999).<br />
4. M. Galeotti, A. Atrei, U. Bardi, B. Cortigian, G. Rovida, and M. Torrini, Surf. Sci. 297, 202<br />
(1993).<br />
5. F. Huang, M. T. Kief, G. J. Man<strong>ke</strong>y, R. F. Willis, Phys. Rev. B 49, 3962 (1994).<br />
6. P. Blaha, K. Schwarz, G. K. H. Madsen, D. Kvasnicka, and J. Luitz: WIEN2k, An<br />
Augmented Plane Wave + Local Orbitals Program for Calculating Crystal Properties<br />
(Karlheinz Schwarz, Techn. Universität Wien, Austria), 2001.<br />
7. M. Sambi, G. Granozii, Surf. Sci. 400, 239 (1998).<br />
8. S. Morin, A. Lachenwitzer, F. A. Möller, O. M. Magnussen, and R. J. Behm, J. Electrochem.<br />
Soc. 146, 1013 (1999).<br />
9. P. Le Fevre, H. Magnam, O. Heckmann, V. Briois, and D. Chandesris, Phys. Rev. B 52,<br />
11 462 (1995).<br />
10. S. K. Kim, Y. M. Koo, V. A. Chernov, and H. Padmore, Phys. Rev. B. 55, 114 (1996).<br />
11. J. Thiele, R. Belkhou, H. Bulou, O. Heckmann, H. Magnam, P. Le Fevre, D. Chandesris,<br />
C. Guillot, Surf. Sci. 384, 120 (1997).<br />
12. S. Ferrer, J. Alvarez, E. Lundgren, X. Torrelles, P. Fajardo, and F. Boscherini, Phys. Rev. B<br />
56, 9848 (1997).<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 186
ACETYLCHOLINESTERASE INHIBITOR FROM NOSTOC SLIZ. KOL.<br />
Ing. Petr Zelík<br />
Supervisor: Ing. Jiří Kopecký, CSc.<br />
Institute of Physical and Applied Chemistry, Faculty of Chemistry, Brno University of<br />
Technology, Purkyňova 118, 612 00 Brno, e-mail: zelik@fch.vutbr.cz<br />
INTRODUCTION<br />
Alzheimer´s disease is the most frequent dementia with progressive degeneration of central<br />
nervous system 1 . Selective decrease of nerve cells in the brain is associated with cognitive<br />
disorders and memory lost. There have been proposed several ways how to improve cognitive<br />
functions. Current symptomatic treatment use especially acetylcholinesterase (AChE)<br />
inhibitors, eg. donepezil, galanthamine and rivastigmine. Requirement for a new AChE<br />
inhibitors leads either to screening of natural sources, mainly higher plants, or modification of<br />
chemical structures of known AChE inhibitors via organic synthesis. Only a few works have<br />
been focused on screening of microorganisms (actinomycetes and fungi). Surprisingly, there<br />
isn´t any publication dealing with screening of autotrophic microorganisms – algae and<br />
cyanobacteria.<br />
About 250 species of algae and cyanobacteria have been tested for AChE inhibitory<br />
activity in attempt to find a new AChE inhibitors of nature origin. The most active strain,<br />
Nostoc sliz. kol., has been used for the next study. Fraction with AChE inhibitory activity was<br />
determined in the crude biomass extract of Nostoc sliz. kol. and subsequently isolated.<br />
Purified AChE inhibitor has been used in preliminary determination of the inhibition nature of<br />
AChE and for structure elucidation by IR, MS and NMR techniques.<br />
METHODS<br />
Assay of AChE activity<br />
Measuring of AChE activity was modified from the assay described by Ellman et. al. 2<br />
Method was optimized for microplate assay. Reaction mixutres containing 25 μl of 0,76 mM<br />
DNTB, 50 μl of 0,04 U/ml AChE, 20 μl of sample, and 125 μl of buffer were incubated for<br />
30 min at 30°C. After incubation, 30 μl of 6,22 mM ATCI (substrate) was added and the<br />
increase of absorbance was read at 412 nm every 18 s for twenty times. The results were<br />
corrected for spontaneous hydrolysis of the substrate. Enzyme activity were calculated as a<br />
percentege compared to an assay without any sample. Every experiment was done in<br />
triplicate.<br />
HPLC- MS analysis<br />
The active extracts were analysed by HPLC/ESI-MS/MS on Agilent 1100 MSD SL-Ion<br />
Trap mass spectrometer. The ZORBAX Eclipse XDB-C8 column (150x4,6 mm, 5 μm) was<br />
used for HPLC analyses. Elution was realized in gradient mode of methanol – water with<br />
addition of formic acid and UV detection was performed at 280 nm.<br />
Nostoc sliz. kol. – determination of active fraction (IN6)<br />
The crude methanolic extract was fractionated using HPLC and every fraction was tested<br />
for AChE inhibitory activity.<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 187
Nostoc sliz. kol. – isolation of active fraction (IN6)<br />
The scheme of the isolation procedure is shown in Fig. 2. Freeze-dried biomass was<br />
desintegrated with sea sand and extracted with methanol. The crude extract was evaporated<br />
under reduced pressure to the dryness and dissolved in the mixture of acetone-hexane (3:7,<br />
V/V). This solution was chromatographed in the system of acetone-hexane (gradient mode;<br />
3:7→7:3, V/V) on silicagel. All fractions were analyzed by HPLC/MS. Middle fraction<br />
containing mainly AChE inhibitor was evaporated to the dryness at low pressure. Residue of<br />
evaporation was dissolved in the minimum volume of methanol. Final separation of the active<br />
compound was realised by preparative HPLC. Watrex Reprosil C8 column (250x10 mm,<br />
5 μm) and mobile phase of methanol-water in gradient mode were used. UV detection was<br />
performed at 280 nm. Peak belongs to active compound was collected manually.<br />
Determination of AChE inhibition nature by IN6<br />
Initial reaction velocities were measured at two fixed concentrations of ATCI (0,175 mM<br />
and 0,75 mM) and different IN6 concentrations (final range in reaction mixure was 0-30 μM).<br />
Data were graphically processed according to Dixon.<br />
RESULTS AND DISCUSSION<br />
Approximately 250 species of algae and cyanobacteria were screened in quest to find<br />
potential producers of new AChE inhibitors among autotrophic microorganisms. The<br />
dichloromethane extracts of cultural mediums, methanolic and methanolic-tetrahydrofuran<br />
biomass extracts were tested for AChE inhibitory activity. There was found out AChE<br />
inhibitory activity higher than 90 % in biomass extracts of Monodus subteratus, Nostoc sliz.<br />
kol. (SV-mol, ISB 93, tok Soj, 5/97, DE) and Nostoc ellipsosporum (2, GM, Štěbal). Extract<br />
from the biomass of Nostoc clipsespor proved 85 % AChE inhibitory activity. Geminella<br />
terricola and Monodopsis subterranea biomass extracts proved inhibitory activity of 50 –<br />
<strong>60</strong> %. The rest of biomass extracts and dichloromethane medium extracts showed AChE<br />
inhibitory activity either no or lower than 50 %.<br />
The crude extract of the most active strain Nostoc sliz. kol. (SV-mol, ISB 93, tok Soj, 5/97,<br />
DE) was analyzed by HPLC-MS and fraction proved anti-AChE activity was determined. The<br />
only fraction responsible for AChE inhibitory activity of the extract was the peak with<br />
retention time of 23,6 min (Fig. 1).<br />
Isolation of IN6 was developed (Fig. 2). At first, methanol extract of the freeze-dried<br />
biomass is prepared. Concentrate of IN6 is obtained in second isolation step, through the<br />
column chromatography of the crude extract in the system of acetone-hexane on silicagel.<br />
Thereafter is IN6 purified using preparative HPLC. Sufficient amount of IN6 (in 96% and<br />
higher purity) was isolated for structural analysis and determination of the inhibition nature in<br />
this way.<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 188
mAU<br />
<strong>60</strong>0<br />
300<br />
0<br />
0 20 23,6<br />
min<br />
40<br />
Fig. 1: HPLC chromatogram of the crude methanolic extract from biomas of Nostoc sliz. kol.;<br />
I – fraction with AChE inhibitory activity<br />
Freeze-dried biomass of Nostoc sliz. kol.<br />
Crude methanolic extract<br />
Concentrate of IN6<br />
IN6 of > 96 purity<br />
Fig. 2: Scheme of IN6 isolation procedure.<br />
Isolated AChE inhibitor was analyzed by MS, IR and NMR but structure is still unknown.<br />
Nature of AChE inhibition by IN6 was also studied. Initial reaction velocities were<br />
measured at two fixed concentrations of substrate and different concentrations of IN6 (0-<br />
30μM). Data were graphically processed according to Dixon (Fig. 3). Two possible types of<br />
inhibition resulting from shapes of curves in Fig. 3 - either reaction of inhibitor with substrate<br />
or non-competitive inhibition with strongly bounded inhibitor to enzyme.<br />
Current interest is focused on more detailed kinetic study of IN6 and structure elucidation<br />
of IN6.<br />
I<br />
extraction with methanol<br />
column chromatography<br />
preparative HPLC<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 189
[v (μmol.l -1 .s -1 )] -1<br />
120<br />
100<br />
80<br />
<strong>60</strong><br />
40<br />
20<br />
0<br />
ATCI - 0,75 mM<br />
ATCI - 0,175 mM<br />
0,0E+00 5,0E-06 1,0E-05 1,5E-05 2,0E-05 2,5E-05 3,0E-05 3,5E-05<br />
IN6 [mol.l -1 ]<br />
Fig. 3: Dixon plot for AChE inhibition by IN6 at two substrate concentrations.<br />
REFERENCES<br />
1. Jirák, R., Obenberger, J., Preiss, M.: Alzheimerova choroba. 1998. 15. ISBN 80-85800-<br />
88-8.<br />
2. Ellman, G.L., Courtney, K.D., Andres, V., Featherstone: A new and rapid colorimetric<br />
determination of acetylcholinesterase activity. Biochem. Pharm., Vol. 7, 1961, 88-95.<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 190
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 191
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 192
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 193
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 194
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2005, strana 195
Příspěvky soutěže<br />
doktorských studijních programů<br />
„O cenu děkana 2006”<br />
(Sekce DSP 2006)
PRODUCTION OF SELECTED SECONDARY METABOLITES IN<br />
TRANSFORMED BACTERIAL CELLS<br />
Ing. Jana Hrdličková, 3. ročník DSP<br />
Vedoucí práce: Doc. RNDr. Ivana Márová, Csc.<br />
<strong>Vysoké</strong> učení technické v Brně, fakulta <strong>chemická</strong>, ústav chemie potravin a biotechnologií,<br />
Purkyňova 118, 612 00 Brno, e-mail: hrdlickova@fch.vutbr.cz<br />
INTRODUCTION<br />
Poly<strong>ke</strong>tides are a large group of secondary metabolites of varied structure. They exhibit<br />
many physiological actions on organisms. Some OF them are useful drugs for humans,<br />
including antibacterials (e.g. erythromycin, tetracycline), anticancer agents (e.g. daunomycin),<br />
antifungal agents (e.g. amphotericin), cholesterol-lowering agents (e.g. lovastatin),<br />
immunosuppressants (e.g. rapamycin) and veterinary products (e.g. the antiparasitic,<br />
avermectin; the feed additive, monensin), others can be allergenic, toxic, and in some cases<br />
carcinogenic. Genetic engineering may lead to new poly<strong>ke</strong>tide drugs.<br />
The tetracyclines were the first major group of antimicrobial agents for which the term<br />
broad-spectrum was used, they exhibit activity against both gram-positive and gram-negative<br />
bacteria. Most natural tetracyclines have a common structure with the β-di<strong>ke</strong>tone system in<br />
rings B and C. Streptomyces rimosus is used for the production of natural tetracyclines by<br />
commercial fermentation. In S. rimosus a mixture of tetracycline and oxytetracycline is<br />
produced, but the 5-hydroxylase enzyme is extremely active, thus, the equilibrum is far in<br />
favor (>95%) of oxytetracycline production. Oxytetracycline (OTC) is a broad-spectrum<br />
antibiotic produced by S. rimosus. OTC is a member of the "poly<strong>ke</strong>tide" class of secondary<br />
metabolites biosynthesized by condensation of coenzyme A derivatives of metabolic<br />
precursors. The backbone of the antibiotic, consisting of 19 carbon atoms, is thought to be<br />
derived from an aminated starter unit (most li<strong>ke</strong>ly malonamyl-CoA), to which eight acetyl<br />
(malonyl-CoA) extender units are added sequentially.<br />
Streptomyces rimosus has a linear chromosome of about 8 Mb. The chromosome has<br />
inverted repeats of 550 kb, which are the longest yet reported for a Streptomyces species.The<br />
otc biosynthetic gene cluster is located about <strong>60</strong>0 kb from one of the chromosome ends, just<br />
outside the inverted repeat structure.<br />
Carotenoids are naturally occuring membrane-protective antioxidant pigments, that<br />
efficiently scavenge singlet oxygen and peroxyl radicals. Carotenoids are produced in higher<br />
plants, algae and phototrophic bacteria as well as in non-phototrophic bacteria, yeasts and<br />
fungi. In recent years, there is evidence that accumulating of carotenoids plays an important<br />
role in human health by preventing degenerative diseases. From a commercial point of view,<br />
there is an increasing demand of special carotenoids in nutrient supplementation, for<br />
pharmaceutical purposes, as food colorants and in animal feeds. Biotechnological production<br />
of carotenoids by exploiting the carotenoid genes cloned from different species is of<br />
increasing interest.<br />
Lutein, lycopene and beta-carotene belong to industrially important carotenoids, widely<br />
used in food and feed industry as natural pigments, provitamins and food/feed supplements.<br />
Carotenoids act as antioxidants and protect organism from photooxidative damage.<br />
Availability of carotenoids for industrial usage is limited by partial problems associated with<br />
their chemical synthesis as well as with isolation from natural sources. According to this fact,<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2006, strana 197
in last years some ways for overproduction of carotenoids including modern methods of<br />
molecular cloning and genetic engineering are studied.<br />
Erwinia carotovora is nonphotosynthetic bacterium pathogenic for higher plants. Beside<br />
its agricultural significance, Erwinia strains are of increasing interest as industrial microbes<br />
producing pectinolytic, cellulolytic and proteolytic enzymes as well as antileukaemic<br />
asparaginase. Erwinia strains form carotenoids which cause yellow-orange coloured<br />
phenotype.<br />
METHODS<br />
Bacterial strains. For production of oxytetracyclines bacterium Streptomyces rimosus 4018<br />
was used. For production of carotenoids bacterium Erwinia carotovora CCM 1008 was used.<br />
For transformation DH5α, DH10β and ET 12567 Escherichia coli competent cells were<br />
prepared.<br />
Cultivation. Escherichia coli was cultivated in 2TY medium at 37ºC. 2TY medium<br />
contained, per litre: tryptone, 16 g; yeast extract, 10 g; NaCl, 5 g .<br />
Plasmids. pHSG298, pGEM-T (for PCR product), pSGset2 (containing Erm, OriC, Apr,<br />
OriT, attP, phi-C31 int), pTS55 (containing Amp, tsr, att, int, repSA) and pIJ 4026<br />
(containing Erm, bla) were used as transformation vectors.<br />
Isolation. Plasmid DNA isolation was performed using commercial kits (Gen Elute<br />
Plasmid Miniprep Kit).<br />
Transformation. Transformation of E. coli cells was done using electroporation by BioRad<br />
GENEPULSER apparatus at following conditions: voltage 2500 V, resistance 200 Ω,<br />
capacitance 25 μF.<br />
Electrophoresis. DNA was analysed using agarose electrophoresis and pulsed field gel<br />
electropohoresis (PFGE).<br />
Analysis of carotenoids. Production of carotenoids by transformants was analysed<br />
chromatographically. Carotenoids were extracted from E. coli transormant cells by ethanol.<br />
Individua pigments were separated and quantified by RP-HPLC using a Nucleosil 100 C18<br />
column and methanol (analysis of lycopene, lutein and carotenes) or mixture<br />
acetonitril:methanol 95:5 (phytoene analysis) as eluent.<br />
RESULTS<br />
Presented work was focused on production of selected secondary metabolites in<br />
transformed bacterial cells. First, regulation of poly<strong>ke</strong>tide antibiotik production in Escherichia<br />
coli cells transformed by otc genes from Streptomyces rimosus was studied. Further, isolation<br />
and cloning of crt gene cluster from bacteria Erwinia carotovora in E.coli DH5α cells was<br />
tested. Most OF experiments were performed in co-operation with Biotechnical Faculty,<br />
University of Ljubljana (Socrates/Erasmus exchange).<br />
The DNA sequence of Streptomyces rimosus was analysed by FramePlot (FramePlot is a<br />
web-based tool for predicting protein-coding regions in bacterial DNA with a high G+C<br />
content, such as Streptomyces). Primer structure was derived from sequence analysis results.<br />
The genes were amplified by PCR. Recommended sizes of PCR products were 714 bp and<br />
470 bp. The sequence of 714 bp was named CEL and the sequence of 470 bp was named<br />
MUT. After purification by Gen Elute PCR Clean-Up Kit the PCR products were ready to be<br />
cloned. CEL and MUT PCR products were ligated into the pGEM-T and introduced into<br />
E. coli competent cells. The cells with CEL or MUT were selected on LB agar plates<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2006, strana 198
containing ampicillin, IPTG and x-gal (blue-white test). Plasmids incorporated into<br />
transformants were isolated by Gen Elute Plazmid Miniprep Kit. To confirm that genes CEL<br />
and MUT were successfully cloned into pGEM, CEL+pGEM and MUT+pGEM were<br />
digested with EcoRI. Electrophoretic separation showed that inserts CEL and MUT were<br />
present in transformed cells. Verification of CEL and MUT sequences was done in cooperation<br />
with Macrogen Inc. Company (Soul, Korea).<br />
The CEL gene was extracted from pGEM-T using Nde I. After electrophoretic separation<br />
and extraction from the gel genes were ligated into the pSGset2/Nde I-deP and introduced<br />
into Escherichia coli competent cells. The cells containing CEL+pSGset2/NdeI were selected<br />
on LB agar plates with apramycin.<br />
The MUT gene was extracted from pGEM-T using Eco RI and after electrophoretic<br />
separation and extraction from the gel. After ligation into the pIJ 4026/Eco RI-deP<br />
transformation vestors were introduced into ET 12567 Escherichia coli competent cells. The<br />
cells containing MUT+pIJ 4026/Eco RI were selected on LB agar plates with<br />
ampicillin+chloramphenicol.<br />
We can summarize, that in this part of work PCR amplification of obtainable sequence and<br />
cloning and incorporation to expression vector was performed. Further, target sequences were<br />
incorporated into transformation vectors and recombinant plasmids were then incorporated<br />
into Escherichia coli recipient cells. The presence of recombinant plasmids was verified at the<br />
level of genotype and phenotype. Transformation of Streptomyces rimosus recipient cells by<br />
recombinant plasmids will be conduct in the future.<br />
In second part of this work, regulation of carotenoid production using genetic<br />
engineering was tested. Several methods of isolation and transfer of crt genes from bacteria<br />
Erwinia carotovora to recipient strain Escherichia coli DH5α cells were tested and<br />
optimized. Identification of carotenoids produced by recombinant cells was verified by HPLC<br />
analysis.<br />
In individual Escherichia coli transformants production of lutein, lycopene and βcarotene<br />
was demonstrated. Production of carotenoids in Escherichia coli cells transformed<br />
by several recombinant vectors pHSG298/crt was substantially higher then those found in<br />
Erwinia carotovora cells. Further, the posibility of regulated high-yield carotenoid production<br />
in laboratory fermentor was tested. Production of lutein in Escherichia coli transformants was<br />
about 8x higher then amount of lutein found in Erwinia carotovora cells, which were<br />
cultivated in the same conditions.<br />
ACKNOWLEDGEMENTS<br />
I would li<strong>ke</strong> to thank Dr. Hrvoje Petkovič and Ms. Urška Lešnik for help and technical<br />
assistance. Both from Biotechnical Faculty, University of Ljubljana, Slovenia.<br />
REFERENCES<br />
1. Petkovič H., Thamchaipenet A., Zhou L-H., Hranueli D., Raspor P., Waterman P.G.,<br />
and Hunter I. S.: Disruption of an Aromatase/Cyclase from the Oxytetracycline<br />
Gene Cluster of Streptomyces rimosus Results in Production of Novel Poly<strong>ke</strong>tides<br />
with Shorter Chain Lengths. The Journal of Biological Chemistry 46, p.32829<br />
-32834, 1999.<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2006, strana 199
2. Pandza K., Pfalzer G., Cullum J. and Hranueli D.: Physical mapping shows that the<br />
unstable oxytetracycline gene cluster of Streptomyces rimosus lies close to one end<br />
of the linear chromosome. Microbiology, p.1493-1501, 1997.<br />
3. Bentley R.: Poly<strong>ke</strong>tides. Encyclopedia of life sciences, 2001.<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2006, strana 200
THE SIMPLE METHOD FOR THE RECOGNITION OF REDUCING AND<br />
NONREDUCING NEUTRAL CARBOHYDRATES BY MALDI-TOF MS<br />
Ing. Markéta Laštovičková a,b , 5. DSP<br />
Supervisor: RNDr. Josef Chmelík b<br />
a Faculty of Chemistry, Brno University of Technology, Purkyňova 118, 612 00 Brno, Czech<br />
Republic; e-mail: lastovickova@iach.cz<br />
b Institute of Analytical Chemistry, Academy of Sciences of the Czech Republic, 611 42 Brno,<br />
Czech Republic<br />
1. INTRODUCTION<br />
The Matrix-Assisted Laser Desorption/Ionization Time-of-Flight mass spectrometry<br />
(MALDI-TOF MS) can provide valuable information on several aspects of carbohydrate<br />
structural analysis, such as the determination of sequence, branching, and linkage. For the<br />
analyses of neutral oligosaccharides, more frequently the positive-ion MALDI-TOF mode has<br />
been performed. The negative-ion mode has been applied mainly in the analysis of charged<br />
carbohydrates that more simply form the deprotonated molecules ([M–H] – ) [1–4].<br />
Inulin and maltooligosaccharides (MOSs) are the representatives of neutral carbohydrates.<br />
Inulin belongs to the fructan group. It is a nonreducing polysaccharide containing Dfructofuranosyl<br />
units lin<strong>ke</strong>d by α–1–2 glycosidic bonds and ended with one glucose unit.<br />
MOSs are the linear glucose oligomers containing only 1–4 linkages. Glucose syrups are<br />
concentrated, aqueous solutions of reducing, low molecular mass oligosaccharides (containing<br />
1–4 and 1–6 glycosidic bonds) obtained by hydrolysis of starch. These starch hydrolysates<br />
can be trespassed as the cheap sweeteners at the adulteration of food, e.g., fruit juices,<br />
therefore they are important for food industry [5].<br />
This study demonstrates a great potential of the negative-ion mode MALDI-TOF MS for<br />
the characterization of underivatized neutral oligosaccharides.<br />
2. EXPERIMENTAL<br />
PREPARATION OF NEUTRAL CARBOHYDRATES STANDARD SAMPLES: Inulin from<br />
Dahlia tubers Mw 5000 (Fluka, Buchs, Switzerland) and MOSs G4-G10 (Sigma, St. Louis,<br />
MO) were prepared at concentrations of 1 mg/mL in deionized water.<br />
EXTRACTION OF NEUTRAL CARBOHYDRATES FROM REAL SAMPLES: Red onion and<br />
Jerusalem articho<strong>ke</strong>s were acquired from a private producer from the Czech Republic. A<br />
procedure for the carbohydrate extraction from fresh samples was described previously [4, 6]<br />
Low glucose syrup (LGS) from 80% wheat starch (w/v), obtained from Amylon Co.<br />
(Havlickuv Brod, Czech Republic), was used at a concentration of 4 mg/mL in deionized<br />
water.<br />
MALDI-TOF MS: The reflectron negative-ion mode experiments were performed with an<br />
Applied Biosystems 4700 Proteomics Analyzer (Applied Biosystems, Framingham, MA)<br />
utilizing a Nd:YAG laser (355 nm). The optimal laser power was selected from the relative<br />
scale 0-8800. 2,4,6-trihydroxyacetophenone (THAP; 100 mg/ml acetone) was used as a<br />
matrix.<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2006, strana 201
3. RESULTS AND DISCUSSION<br />
The main task of this study was the application of the negative-ion mode MALDI-TOF MS<br />
to the determination of a structure of storage oligosaccharides isolated from plants.<br />
The proper experimental conditions (the most convenient matrix, optimal concentrations of<br />
samples and matrix, optimal laser power etc.) were determined for inulin and MOSs standard<br />
samples (the details are not shown). Moreover, these initial experiments showed a potential of<br />
negative-ion mode MALDI-TOF MS for the differentiation of reducing (MOSs) and<br />
nonreducing (inulin) oligosaccharides, because of easy fragmentation of reducing end ring<br />
(the production of in-source fragment ions [M – H – 120] – ; see Figure 1). This in-source<br />
fragmentation has already been described for negative-ion mode MALDI-TOF mass spectra<br />
of dextrans (the polymer containing the main chain with 1–6 glycosidic bonds and different<br />
degree of branching) [7]. This information is very useful for the identification of storage<br />
carbohydrates isolated from plants as is shown below.<br />
All real samples (LGS and oligosaccharides isolated from Jerusalem articho<strong>ke</strong> and red<br />
onion) were analyzed with THAP that was selected as the most convenient matrix. While<br />
[M – H] – ions formed the dominant distribution for oligosaccharides from both vegetables, the<br />
main distribution of LGS was formed by the in-source fragment ions [M – H – 120] – (see<br />
Figure 2). LGS showed this fragmentation because of its structure that contains the main<br />
chain formed by 1–4 glycosidic bonds and branches with 1–6 glycosidic bonds and a reducing<br />
end group. The mass spectra differed in the quantity of the adducts which was dependent on<br />
the amount of ions in the samples.<br />
Thus there are some important conclusions on the mass spectrometric behavior of the<br />
neutral oligosaccharides. Although the negative-ion mode MALDI-TOF MS is ignored in<br />
connection with neutral carbohydrates it is possible to determine the main characteristics of<br />
their distribution without any carbohydrate derivatization (see Table 1). In addition, the<br />
negative-ion mode MALDI-TOF mass spectra is able to differentiate reducing<br />
maltooligosaccharides and nonreducing fructooligosaccharides extracted from real samples,<br />
because of easy fragmentation of reducing end ring, which is not evident in the positive-ion<br />
mode MALDI-TOF MS where both types of oligosaccharides form the alkali-ion adducts.<br />
REFERENCES<br />
[1] Harvey, D. J. Matrix-assisted laser desorption/ionization mass spectrometry of<br />
carbohydrates and glycoconjugates. Int. J. Mass Specrom. 2003, 226, 1-35.<br />
[2] Zaia, J. Mass spectrometry of oligosaccharides. Mass Spectrom. ReV. 2004, 23, 161-227.<br />
[3] Harvey, D. J. Mass Specrom. Reviews. 2006, 25, 595-662<br />
[4] Štikarovská, M.; Chmelík, J. Analytica Chimica Acta 2004, 520, 47–55<br />
[5] Robyt, J.D. Essentials of carbohydrate chemistry, Springer, New York, 1998<br />
[6] Laštovičkova,M; Chmelik, J. J. Agric. Food Chem. 2006, 54,5092-5097<br />
[7] Čmelík, R.; Štikarovská, M.; Chmelík, J. J. Mass Spectrom. 2004, 39, 1467-1473.<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2006, strana 202
Figure 1 Negative-ion mode MALDI-TOF mass spectra of standard oligosaccharides: inulin<br />
(A) and MOSs (B) where<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2006, strana 203
Figure 2 Negative-ion mode MALDI-TOF mass spectra of oligosaccharides from the real<br />
samples: Jerusalem articho<strong>ke</strong> (A); red onion (B); and LGS (C).<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2006, strana 204
Table 1 Evaluation of negative-ion MALDI-TOF mass spectra of LGS and oligosaccharides<br />
isolated from red onion and Jerusalem articho<strong>ke</strong>; where the range between the shortest and<br />
highest detected oligomers = the range of the degree of polymerization (DP), the total<br />
number of detected oligomers = np, number-average molecular mass = Mn, weight-average<br />
molecular mass = Mw and polydispersity = δ.<br />
Jerusalem<br />
articho<strong>ke</strong><br />
Red onion LGS<br />
np 17 6 19<br />
DP 6–22 6–11 7–25<br />
Mn 1805 1392 1763<br />
Mw 1962 1426 1966<br />
δ 1.09 1.02 1.11<br />
Peak types<br />
[M–H] –<br />
[M+K–2H] –<br />
[M–H] –<br />
[M+K-2H] –<br />
[M–H–120] –<br />
[M+Na–2H–120] –<br />
[M+K–2H–120] –<br />
[M+HSO4] –<br />
The most abundant peak<br />
Mr 1313.06 1313.06 1193.31<br />
Intensity 471.2 mV 371.8 mV 534.5 mV<br />
DP 8 8 8<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2006, strana 205
MINIATURE METALLIC DEVICE FOR COLLECTION OF HYDRIDE<br />
FORMING ELEMENTS<br />
Pavel Krejčí 1,2<br />
Bohumil Dočekal 2<br />
1 Department of Environmental Chemistry and Technology, Faculty of Chemistry,<br />
Brno University of Technology, Purkyňova 118, CZ-61200 Brno, Czech Republic,<br />
e-mail:krejci-p@fch.vutbr.cz<br />
2 Institute of Analytical Chemistry, Czech Academy of Sciences, Veveří 97, CZ-<strong>60</strong>200 Brno,<br />
Czech Republic, e-mail: docekal@iach.cz<br />
INTRODUCTION<br />
Collection of hydride forming elements (As, Bi, Ge, In, Pb, Sb, Se, Sn and Te) has become<br />
a simple and useful tool for pre-concentration and separation purposes in the trace and<br />
ultratrace analysis by atomic spectrometric methods [1,2]. It is typically performed "in situ"<br />
using conventional graphite (GF) or tungsten (WETA) heated atomizers with subsequent<br />
analyte detection by electrothermal atomic absorption spectrometry (ETAAS) method [1].<br />
Nevertheless, the trapping technique can also be applied in other spectrometry methods<br />
(atomic emission spectrometry, atomic fluorescence spectrometry and mass spectrometry).<br />
For this purpose, conventional electrothermal atomizers are modified and/or special, mostly<br />
laboratory made devices are designed [2-5].<br />
Capability of trapping of hydrides on a prototype of a miniature electrothermal<br />
vaporization (ETV) device was studied employing antimony, arsenic, bismuth and selenium<br />
hydrides as volatile species of analytes. This device is based on a strip of the molybdenum<br />
foil (which is typically used in production of "halogen" bulbs) and combined with miniature<br />
hydrogen diffusion flame for specific analyte detection in atomic absorption spectrometry.<br />
Influence of trapping temperature, modification of the molybdenum surface with noble metals<br />
– Ir, Pt and Rh, distance between the orifice of the injection capillary and the strip and<br />
composition of the gaseous phase (argon-hydrogen-oxygen) was studied in order to clarify the<br />
general hydride trapping mechanism.<br />
EXPERIMENTAL<br />
A Perkin–Elmer (Norwalk, USA) model 3110 atomic absorption spectrometer equipped<br />
with a deuterium background correction system was employed in this study. The Photron<br />
Super Lamps® (Photron, Victoria, Australia) of antimony, bismuth, arsenic and selenium<br />
were used as a specific radiation sources.<br />
The device for electrothermal induced collection of hydrides and their subsequent<br />
electrothermal release was based on a piece of the molybdenum foil (Metallwerk Plansee,<br />
Reutte, Austria), 85 µm thick, 2.15 mm wide and 56 mm long. It was bent to form a U–<br />
profile. Both ends of the strip were pressed between boron nitride cylindrical body (6 mm in<br />
diameter) and two brass contacts. A part of the bent strip (9 mm) remained free. Boron nitride<br />
electric insulation and the brass contacts maintained also an efficient cooling of the strip<br />
providing reproducible temperature setting in series of experiments. In this arrangement,<br />
maximum power of about 130 W was supplied at the highest temperature applicable<br />
(2<strong>60</strong>0°C).<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2006, strana 206
Fig. 1: Scheme of the hydride generation aparature coupled with the heated molybdenum-foil<br />
trap system and simple hydrogen diffusion flame atomic absorption system.<br />
The implementation of the electrothermal trapping/vaporisation (ETV) device in the<br />
instrumental arrangement is shown in Fig. 1. Only a very simple atomiser based on<br />
a miniature hydrogen diffusion flame was used for atomic absorption spectrometry detection<br />
in trapping experiments. The flame was supported by the gas mixture of hydrogen and argon<br />
at a flow rate of 215 ml min -1 and 800 ml min -1 , respectively. The injection capillary, made of<br />
a wide bore quartz GC–capillary (8 cm × 0.53 mm id), was inserted through the narrow hole,<br />
drilled in the axis of the boron nitride body. The tip of the capillary was precisely positioned<br />
by means of an adjusting screw made of PEEK. A laboratory made pulse-width modulation<br />
power supply was used for heating the molybdenum strip. It was based on a high power<br />
MOS-FET transistor and a car battery (12 V, <strong>60</strong> Ah). The power supply was controlled by<br />
a PC by using software created in Visual Basic ver.3. In trapping experiments, the actual<br />
temperature of the heated central part of the molybdenum strip was simultaneously measured<br />
by pyrometers.<br />
A laboratory made flow injection hydride generation system is depicted also in Fig. 1. The<br />
peristaltic pump (model 72624–71, Ismatec, Switzerland) was fitted with Tygon tubes. The<br />
generation system was based on 3-channel peristaltic pump, PTFE-reaction loop and gasliquid<br />
separator with a forced outlet for liquid phase and with a PTFE-filter in an outlet for<br />
gaseous phase. The flow rates were 1.1, 3.6 and 5.0 ml min -1 for 0.5% m/v NaBH4 solution,<br />
sample solution in 1 mol l -1 HCl and waste solution, respectively. The sample channel was<br />
equipped with a Knauer (Berlin, Germany) 6–port injection valve made of PEEK with<br />
a 100 μl sampling loop for performing flow injection of the sample solution. Argon was<br />
introduced in two channels, upstream of the reaction loop as reaction gas and into the gas–<br />
liquid separator as stripping gas at a flow rate of 55 ml min -1 and 10 ml min -1 , respectively.<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2006, strana 207
The gaseous hydrides were directed towards the center of the bent part of the molybdenum<br />
foil via a wide bore quartz GC–capillary. See Ref. [5] for more details.<br />
RESULTS<br />
The influence of the molybdenum foil temperature and concentration of hydrogen in the<br />
gaseous phase on trapping behavior of bismuth and antimony were investigated for a bare<br />
molybdenum surface and argon-hydrogen atmosphere of the flame supporting gas mixture.<br />
Maximum trapping efficiencies were found for antimony and bismuth in the temperature<br />
ranges of 650-750 °C and 500-<strong>60</strong>0 °C, respectively. These optimum temperatures are<br />
approximately 300-500 °C lower than those found for arsenic and selenium (1100-1200°C).<br />
Capability of trapping antimony and bismuth hydrides on a modified surface of the<br />
molybdenum trap was also investigated. Rhodium, platinum and iridium were chosen as<br />
permanent modifiers and were introduced stepwise on the surface in amounts of 10, 30, 100<br />
and 200 μg. Significant depletion of signals of collected antimony and bismuth was observed<br />
when the amount of any modifier used exceeded 30 μg. Evidently, all modifiers inhibit the<br />
interaction of both analytes with active sites on the molybdenum surface. In contrary, these<br />
modifiers do not significantly affect trapping of arsenic and selenium on the molybdenum<br />
surface. The maximum trapping efficiency of arsenic and selenium was independent of the<br />
modifier amount applied in the range from 0 to 200 μg. Their signal profiles were higher,<br />
more reproducible and symmetrical when increasing modifier amount.<br />
The overall efficiency of generation of hydrides and their transport into the trapping<br />
chamber is independent on the injection gas flow rate between the minimum and the<br />
maximum achievable rates of 40 ml min -1 and 2<strong>60</strong> ml min -1 , respectively. Maximum trapping<br />
efficiency was reached at a flow rate close to 70 ml min -1 , and at a distance of 2 mm between<br />
the tip of the introduction capillary and the foil surface. Obviously, aerodynamic conditions<br />
prevailing near the capillary orifice and the molybdenum foil during the trapping step play the<br />
same role in trapping of all analytes studied.<br />
Vaporization experiments showed that antimony, arsenic and selenium are strongly bonded<br />
to the molybdenum surface. Collected antimony is completely released at temperatures above<br />
2200 °C and arsenic and selenium at temperatures above 2400 °C. To the contrary, bismuth<br />
exhibits a different behavior. A relative low temperature of 1200 °C is sufficient for complete<br />
vaporization of trapped Bi. The heating vaporization pulse should be very short to prevent<br />
losses of analyte on the inner quartz wall of the trap chamber and to perform an efficient<br />
transport of the analyte into the diffusion flame. In the present experimental arrangement, the<br />
optimum heating pulse in duration of 0.4 s was found.<br />
ACKNOWLEDGEMENT<br />
This work was supported by The Grant Agency of the Czech Republic (Project<br />
No. 203/06/1441) and by Ministry of Education, Youth and Sports of the CZ<br />
(FRVS 1054/2006).<br />
REFERENCES<br />
[1] J. Dedina, D. L. Tsalev: Hydride Generation Atomic Absorption Spectrometry,<br />
Wiley & Sons, Inc., Chichester (1995).<br />
[2] H. Matusiewicz and R. E. Sturgeon, Spectrochim. Acta, Part B, 51 (1996) 377-397.<br />
[3] F. Barbosa Jr., S. Simiao de Souza, F.J. Krug, J. Anal. At. Spectrom., 17 (2002) 382-388.<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2006, strana 208
[4] H. Matusiewicz, M. Kopras, J. Anal. At. Spectrom., 18 (2003) 1415-1425.<br />
[5] P. Krejci, B. Docekal, Z. Hrusovska, Spectrochim. Acta, Part B, 61 (2006) 444-449.<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2006, strana 209
INFLUENCE OF POLYUNSATURATED FATTY ACIDS INTAKE<br />
ON LIPID METABOLISM IN PATIENTS WITH HYPERLIPIDAEMIA<br />
Simona Macuchová<br />
Mentor: Doc. RNDr. Ivana Márová, CSc.<br />
Brno University of Technology, Faculty of Chemistry, Department of Food Technology<br />
and Biotechnology, Purkyňova 118, 612 00, Brno, email: macuchova@fch.vutbr.cz<br />
INTRODUCION<br />
Cardiovascular diseases are the main cause of mortality in most of industrialized countries.<br />
Risk factors include smoking, diabetes, obesity, high level of blood cholesterol, a diet high in<br />
fats, and having a personal or family history of heart disease. Cerebrovascular disease,<br />
peripheral vascular disease, high blood pressure, and kidney disease involving dialysis are<br />
disorders that may also be associated with atherosclerosis (1).<br />
Atherosclerosis is characterized by deposition of cholesterol rich plaques in the<br />
endothelium. This observation stimulated research on the metabolism of cholesterol and<br />
revealed that cholesterol is transported in esterified form to cells by the low density<br />
lipoprotein (LDL). LDL is recognized by an endothelial cell receptor and introduced into the<br />
cell by endocytosis. There the esters are cleaved. The resulting free cholesterol is transferred<br />
to the cell walls. The process is strictly regulated. In atherosclerotic patients LDL is altered by<br />
oxidation. This altered LDL is ta<strong>ke</strong>n up in unlimited amounts by macrophages. Dead<br />
macrophages filled with cholesterol esters are finally deposited in arteries (1).<br />
The fact that LDL is rendered toxic by oxidation raises the question which constituents of<br />
LDL are prone to undergo oxidation. LDL consists of a core of cholesterol esters which is<br />
surrounded by a phospholipid membrane in which the protein is inbedded. The latter is<br />
required to recognize the LDL cell receptor.<br />
Polyunsaturated fatty acids (PUFAs) esterified to cholesterol or present as phospholipids<br />
represent the most oxygen sensitive compounds of all these LDL constituents.<br />
Dietary polyunsaturated fatty acids (PUFA) have effects on diverse physiological<br />
processes impacting normal health and chronic diseases, such as the regulation of plasma lipid<br />
levels, cardiovascular and immune function, insulin action, and neural development and<br />
visual function (1).<br />
Ingestion of PUFA would lead to their distribution to virtually every cell in the body with<br />
effects on membrane composition and function, eicosanoid synthesis, and signaling as well as<br />
the regulation of gene expression.<br />
Cell specific lipid metabolism, as well as the expression of fatty acid-regulated<br />
transcription factors li<strong>ke</strong>ly play an important role in determining how cells respond to changes<br />
in PUFA composition.<br />
Chemically, PUFA belong to the class of simple lipids, as are fatty acids with two or more<br />
double bonds in cis position. There are two main families of PUFA: n-3 and n-6. These fatty<br />
acids family are not convertible and have very different biochemical roles.<br />
Dietary n-3 PUFA have several beneficial properties:<br />
- act favorably on blood characteristics by reducing platelet aggregation and blood<br />
viscosity;<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2006, strana 210
- are hypotriglyceridemic;<br />
- exhibit antithrombotic and fibrinolytic activities;<br />
- exhibit antiinflammatory action;<br />
- reduce ischemia/reperfusion-induced cellular damage. This effect is apparently due to the<br />
incorporation of eicosapentaenoic acid in membrane phospholipids.<br />
Linoleic acid (n-6) (LA) and alfa-linolenic acid (n-3) (LNA) are two of the main<br />
representative compounds, known as dietary essential fatty acids (EFA) because they prevent<br />
deficiency symptoms and cannot be synthesized by humans (1).<br />
Reduction of blood lipids and inhibition of LDL oxidation are the main therapeutic<br />
approaches to the treatment of atherosclerosis. Antioxidant agents, alone or in combination<br />
with hypolipidaemic drugs are considered useful for this treatment. Food supplements<br />
containing such substances can serve as additional therapeutical agents (1).<br />
CLINICAL EXPERIMENT<br />
The aim of this work was to contribute to current knowledge of the influence of an<br />
antioxidant supplement type on metabolic and antioxidant status in a group of hyperlipidemic<br />
patients. Influence of complex food supplement containing tocopherol as antioxidant<br />
component and polyunsaturatd fatty acids as hypolipidaemic component on antioxidant status<br />
and parameters of lipid metabolism in 30 patients with hyperlipidaemia was studied. Food<br />
supplement (180 mg of eicosapentaneic acid EPA, 120 mg of docosahexaneic acid DHA, 1.12<br />
mg of vitamin E in 1 tbl.) was ta<strong>ke</strong>n for 3 months, two tbl. daily; blood samples of each<br />
subject were ta<strong>ke</strong>n in regular intervals.<br />
METHODS<br />
Determination of antioxidant activity<br />
Total antioxidant status was determined using ABTS method (Randox Laboratories, USA).<br />
Serum AGE (Advanced Glycation End Products) were analysed fluorimetrically at<br />
350 nm/440 nm. Total amount of serum oxidation products „AOPP“ (Advanced Oxidation<br />
Protein Products) was analysed spectrophotometrically according to Witko-Sarsat et al. 1998<br />
(2), in Kalousová et al. 2001 modification (3).<br />
Biochemical parameters<br />
A set of biochemical parameters characterizing lipid metabolism was measured<br />
at Department of Clinical Biochemistry in the Kyjov Regional Hospital. Levels of total<br />
cholesterol, triacylglycerols, HDL and LDL – cholesterol, apolipoproteine A and B, urea,<br />
creatinine, uric acid, alaninaminotransferase, aspartataminotransferase, albumin and glycated<br />
haemoglobin were determined using automatically system HITACHI 717.<br />
HPLC analysis<br />
As parameters of antioxidant status levels of serum carotenoids, α-tocopherol and retinol<br />
were measured using HPLC method. Separation of carotenoids, retinol and α-tocopherol was<br />
carried out using the Biospher column C18 (4,6 mm × 150 mm, particulation size 7 μm),<br />
methanol as the mobile phase and flow rate 1.1 ml.min -1 . Content of trans-all-retinol was<br />
detected at 325 nm, α-tocopherol at 289 nm and carotenoids at 450 nm.<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2006, strana 211
Determination of fatty acids<br />
A simplified method for analysis of fatty acids in human serum was used according to<br />
Kang at al. 2005 (4). Fatty acids were methylated using BF3/methanol reagent and extracted<br />
to hexane phase. Fatty acids methyl esters were measured using GC-FID method. In each<br />
sample 30 different fatty acids were detected using external standards.<br />
RESULTS<br />
After 3-month inta<strong>ke</strong> OF PUFA/tocopherol supplemet a significant decrease of serum<br />
cholesterol, LDL-cholesterol (10-15%) and mainly triglycerides (about 35%) in<br />
hyperlipidaemic patients was observed. No similar changes in controls was shown. While the<br />
decrease of cholesterol as well as LDL-cholesterol levels was caused predominantly by<br />
tocopherol effect, TAG levels could be influenced by combined effect of PUFA and<br />
tocopherol. Further, PUFA/tocopherol inta<strong>ke</strong> led to a significant increase of tocopherol levels<br />
and TAS and, thus, to corresponding decrease of serum AGEs and AOPPs.<br />
The profiles of serum fatty acids (FA) shown also some changes after supplementation. A<br />
significant decrease of saturated FA (myristic, palmitic, stearic acid) was observed in all<br />
groups. Different changes of unsaturated FA were observed in hyperlipidaemics when<br />
compared with controls. A significant increase of PUFA mixture, EPA and DHA was found<br />
in controls, while no changes of PUFA, EPA and moderate changes of DHA were observed<br />
in hyperlipidaemics.<br />
The main problem with any epidemiological study is that correlation does not imply<br />
causation. There are many other factors, that could be responsible for biological effect.<br />
Additional problems are connected with group composition as well as with biomar<strong>ke</strong>r<br />
selection (1). Moreover, in Czech population basal levels of antioxidants were changing in<br />
the course of time.<br />
Despite these problems, our results indicated, that inta<strong>ke</strong> of food supplement containing<br />
PUFA and tocopherol can positively influence lipid metabolism and antioxidant status in<br />
patients with hyperlipidaemia. Very important is composition of vitamin preparative; many<br />
commercial PUFA are extensively oxidized and, thus, isoprostan formation can occur.<br />
Acknowledgements: This work was supported by project FRVS 3150/G1/2006 the Czech<br />
Ministry of Education, Youth and Sport.<br />
References:<br />
1. Halliwell B., Gutterdige J.M.C.: Free Radicals in Biology and Medicine. 3rd Edition,<br />
Oxford University Press, 1999<br />
2. Witko-Sarsat et al.: Advanced Oxidation Protein Products as Novel Mediators of<br />
Inflammation and Monocyte Activation in Chronic Renal Failure. The Journal of<br />
Immunology 2524-2532, 1998<br />
3. Kalousová M. et al.: Advanced Glycation End-Products and Advanced Oxidation<br />
Protein Products in Patients with Diabetes Mellitus. Physiol. Res. 51: 597-<strong>60</strong>4, 2002<br />
4. Kang J.X., Wang J.: A simplified method for analysis of polyunsaturated fatty acids.<br />
BMC Biochemistry 6:5, 2005<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2006, strana 212
HYDROPHOBIZED SODIUM HYALURONATE IN AQUEOUS<br />
SOLUTION - A FLUORESCENCE STUDY<br />
Ing. Filip Mravec, 3 rd year PGS<br />
Supervisor: doc. Ing. Miloslav Pekař, CSc.<br />
Brno University of Technology, Faculty of Chemistry, Institute of Physical and Applied<br />
Chemistry, Purkyňova 118, 612 00 Brno, e-mail: mravec@fch.vutbr.cz<br />
INTRODUCTION<br />
Polysaccharides and their derivatives have become as major components for the<br />
development of biocompatible and biodegradable materials with many areas of interests (e.g.<br />
tissue engineering, drug delivery). Chemical modification, which no affected<br />
biodegradability, can lead to the expansions of medicine and engineering applications.<br />
Hyaluronan is major component of pericellular and extracellurar matrices. It is a linear<br />
polymer of the disaccharide D-glucuronic acid-1-β-3-N-acetylglukosamine (Figure 1a). It<br />
plays important role in stabilizing the extracellular matrix in many tissues by binding to<br />
specific proteins called hyaladherines. The main hyaluronan fraction is localized in skin<br />
tissue.<br />
The preparing of the hyaluronan derivatives are generally based on the esterifcation on the<br />
D-glucuronic subunit. Our derivatives were modified on the second carbon on the glucuronic<br />
subunit (Figure 1b). Because carboxylic groups are still free, we obtained the amphiphilic<br />
polyelectrolyte - hydrophobized hyaluronan (hHA). From its structure we predict in aqueous<br />
solution modified hyaluronan will aggregate to form micelle-li<strong>ke</strong> structures with non-polar<br />
core. This aggregation behavior can be study by non-polar fluorescence probes solubilized to<br />
H<br />
H<br />
O<br />
O<br />
HO<br />
a<br />
O<br />
O<br />
HO<br />
b<br />
O<br />
O -<br />
O -<br />
O<br />
OH<br />
O<br />
Na +<br />
Na +<br />
O<br />
NH<br />
R<br />
HO<br />
O<br />
HO<br />
O<br />
C<br />
H 3<br />
C<br />
H 3<br />
Figure 1 Structure of the native hyaluronan (a) and its hydrophobized derivative (b),<br />
R = C10.<br />
this core.<br />
Pyrene, benzo[d,e,f]fenanthrene is the even and alternating hydrocarbon (Figure 2a). The<br />
“Pyrene I1:I3 ratio method” is widely used method to determine the critical aggregation<br />
concentration (cac) for a lot of surfactant-based systems. Its unique response to the<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2006, strana 213<br />
OH<br />
OH<br />
O<br />
NH<br />
O<br />
O<br />
NH<br />
O<br />
n<br />
n<br />
OH<br />
OH
microenvironment polarity is well known and described (Aguiar 2003). We evaluated<br />
experimental data using non-linear fitting with Boltzman’s curve with four parameters –<br />
maximum (a), minimum (b), inflex point (x0), and width of the gradient (Δx) (Equation 1).<br />
We voted and subsequently confirmed the “x-coordinate” of the inflex point as the cac value.<br />
For the confirmation we used the perylene’s fluorescence measurements.<br />
a − b<br />
y = + b<br />
1)<br />
( 0<br />
x − x )<br />
Δx<br />
1+<br />
e<br />
Perylene, dibenz[de,kl]anthracene, is also the even and alternating hydrocarbon (Figure<br />
2b). The perylene’s measurements are quite simple for evaluation. The fluorescence intensity<br />
of the perylene rise with the number of non-polar domains in the hHA’s solution. Earlier than<br />
domains are presented in solution no fluorescence is observed. When domains are formed we<br />
observe sharp increasing of the fluorescence. These two trends can be fitted by the linear<br />
curves and the x-coordinate from their point of intersection defines the cac value directly.<br />
a) b)<br />
Figure 2 Fluorescence probes - a) pyrene b) perylene<br />
MATERIALS AND METHOD<br />
The hyaluronan and its derivatives were obtained from CPN Ltd. (Dolní Dobrouč, Czech<br />
Republic). Hyaluronans were in these molecular weights: 97, 5<strong>60</strong>, and 1630 kg·mol -1 .<br />
Derivatives were in molecular weights 134, 183, 3<strong>60</strong>, and 1470 kg·mol -1 , respectively, and<br />
theirs substitution degrees were in range from 10 to 70 %. The substitution degree is defined<br />
as the ratio of the number of the monomer with and without the alkyl chain per polymer<br />
chain, and it was determined from 1 H NMR spectra. All molecular weights were determined<br />
by SEC-MALLS (Mlčochová, 2006). Pyrene and perylene were obtained both from Fluka<br />
GmbH. Acetone p.a. was obtained from Lachema Ltd.<br />
The samples were dissolved in doubly distilled water to the concentration 2 g l -1 . This<br />
stock solution was stabilized by addition sodium azide (NaN3) in final concentration 10 -3 M.<br />
Sample nomenclature. The samples are named in correspondence to their characteristics. The<br />
first come alkyl-type abbreviation, next are basic molecular weight (before derivatization),<br />
and after the solidus the substitution degree. For example D 134/10 means C10-derivate with<br />
the molecular weight 134 kg·mol -1 and the substitution degree 10 %.<br />
Fluorescence Method. The acetone stock solutions of the pyrene and perylene were prepared.<br />
Probes stock solution was introduced into a flask and acetone was evaporated. The stock<br />
solution of hHA was introduced into a flask with evaporating probe, it was diluted to the<br />
desirable concentration, and the resulting solution was sonificated during 4 hours and stored<br />
during next 20 hours. The fluorescence emission spectra were monitored with a luminiscence<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2006, strana 214
spectrophotometer (AMINCO-Bowman, Series 2) at 293.15 ± 0.1 K. The excitation and<br />
emission slit widths were set to 4 nm, where for pyrene and perylene the excitation<br />
wavelength was 335 nm and 408 nm, respectively.<br />
By the pyrene way, the ratio of the fluorescence intensity at 373 nm (I1) and at 383 nm (I3)<br />
was plotted against the logarithm of the concentration. These data was fitted by sigmoid curve<br />
with the nonlinear curve fitting with Origin 75. From non-linear fitting we obtain two possible<br />
cac points - directly cac1-point as the inflex point. Second one, cac2-point, is defined as<br />
cac2 = x0 + 2Δx<br />
. 2)<br />
Perylene’s data evaluation was based on fit of two linear trends. From equations related to<br />
these linear curves was evaluated “x-coordinate”, cacPe, of the point of intersection.<br />
Fluorescence (a.u.)<br />
1<br />
0.8<br />
0.6<br />
0.4<br />
0.2<br />
0<br />
Fluorescence (a.u.)<br />
RESULTS AND DISCUSSION<br />
It is useful to use only one probe for the CAC determination. Pyrene’s data contain not<br />
only information about aggregation but even polarity information. Because pyrene is partially<br />
water soluble, it is necessary to know exactly which cac-point is the right.<br />
F int. norm.<br />
250 300 350 400 450 500<br />
1.0<br />
0.8<br />
0.6<br />
0.4<br />
0.2<br />
I3<br />
a) Emission 0.8 b)<br />
Excitation<br />
0.6<br />
I1<br />
wavelength (nm)<br />
1<br />
0.4<br />
0.2<br />
0<br />
Excitation<br />
Emission<br />
320 370 420 470 520 570<br />
wavelength (nm)<br />
Figure 3 Spectral properties of the pyrene (a) and the perylene (b)<br />
Perylene<br />
Pyrene<br />
R 2 = 0.9702<br />
R 2 = 0.9998<br />
0.0<br />
0.8<br />
-3 -2 -1 0 1<br />
Log C<br />
1.6<br />
1.4<br />
1.2<br />
1.0<br />
I 1 /I 3<br />
Figure 4 The plot of the normalized integral fluorescence and the I1/I3 vs. the Log C. The<br />
perylenes data are separate and fit with the linear curves. The pyrenes data are fit by sigmoid<br />
curve with mar<strong>ke</strong>d cac1 (×). The point of intersection (↑) from perylenes dependence(x-coordinate<br />
- 0.747) is identical with the pyrenes cac1 point (x-coordinate - 0.750).<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2006, strana 215
Table 1 Sum of the cac values for all samples<br />
sample SD cac<br />
- % x10 6 mol·l -1<br />
D 44<br />
D 134<br />
D 183<br />
D 3<strong>60</strong><br />
D 1470<br />
In Figure 4 there are typical<br />
dependencies of the pyrene and perylene<br />
in hydrophobized hyaluronan solution.<br />
Pyrene ratio shows typical decreasing Stype<br />
curve. Perylene, on the other hand,<br />
shows two line system. For pyrene it was<br />
used equations 1) and 2) to solve<br />
parameter cac1 and cac2, respectively.<br />
Perylene’s data, cacPe was solved by<br />
combination of two linear equations as<br />
point of intersection. Final values (in<br />
g·l -1 ) are: cac1 = 0.179, cacPe = 0.178,<br />
cac2 = 0.758. So it was established the<br />
best value for the cac determination is<br />
cac1 point, realized as the inflex point x0.<br />
The cac values are showing two<br />
trends. First, the cac values are<br />
decreasing with increasing SD (except for<br />
samples D 1470). Second, the cac values<br />
are decreasing with increasing M. These trends are obvious in Figure 5 and summarized in<br />
Table 1. The greatest decreasing of the cac values show D 44 samples. It is possible to relate<br />
this behavior to the fact that D 44 samples have the shortest chains and new types of<br />
interactions can be founding. On the other hand, heavy weighted chains show stable behavior<br />
against the SD changes. These results can be explained if we accept that next addition of the<br />
alkyls to the chain only fortify chain-chain interaction. And in fact, it does not lead to the<br />
formation of new hydrophobic cores.<br />
cac (10 -6 mol l -1 )<br />
100.00<br />
10.00<br />
1.00<br />
0.10<br />
0.01<br />
10<br />
30<br />
50<br />
10<br />
30<br />
50<br />
70<br />
10<br />
30<br />
50<br />
10<br />
30<br />
50<br />
30<br />
50<br />
70<br />
12.27<br />
1.02<br />
0.06<br />
4.35<br />
1.21<br />
0.76<br />
0.20<br />
0.99<br />
0.73<br />
0.47<br />
0.51<br />
0.19<br />
0.18<br />
0.15<br />
0.06<br />
0.10<br />
D 44 D 134<br />
D 183 D 3<strong>60</strong><br />
D 1,470<br />
0 20 40 <strong>60</strong> 80<br />
SD (%)<br />
Figure 5 The dependences of the cac values on the SD. Except for D 1470 all samples<br />
show the decreasing tendencies with increasing M and SD.<br />
CONCLUSION<br />
Fluorescence determination of cac for the novel hyaluronate derivatives was presented. It<br />
was showed cac1 as the best point for determination of the critical aggregation concentration.<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2006, strana 216
Hyaluronate derivatives showed with increasing SD decreasing tendency of the cac through<br />
the light weighted chains, and stable value for heavy weighted ones.<br />
REFERENCES<br />
Aguiar J. et al.: J. Colloid Interface Sci 2003, 258, 116-122<br />
Angelescu D., Vasilescu M.: J. Colloid Interface Sci 2001, 244, 139-144<br />
Dong, D. C.; Winnik, F.: Can. J. Chem. 1984, 62, 25<strong>60</strong>-2564<br />
Mlčochová P. et al.: Biopolymers 2006, 82, 74-79<br />
Molina-Bolívar J.A. et al.: J. Phys. Chem. B 2004, 108, 12813-12820<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2006, strana 217
CHARACTERIZATION AND DEGRADATION BEHAVIOUR OF<br />
TRIBLOCK COPOLYMER<br />
Ing. Ludmila Nová<br />
Supervisor: Prof. RNDr. Milada Vávrová, CSc.<br />
Consultant: Ing. Lucy Vojtová, PhD.<br />
Institute of Chemistry and Technology of Environmental Protection, Faculty of Chemistry,<br />
Brno University of Technology, Purkyňova 118, 612 00 Brno<br />
E-mail:nova@fch.vutbr.cz<br />
ABSTRACT<br />
This work is focused on the studying of the thermoreversible behaviors of two copolymers,<br />
PLGA-PEG-PLGA and the same but modified with itaconic acid (ITA-PLGA-PEG-<br />
PLGA-ITA). The critical gel concentrations (CGC) and the critical gel temperatures (CGT)<br />
were determined. As for PLGA-PEG-PLGA the CGC and CGT equal to 19,2 w% and 34,5<br />
°C, respectively, was observed. Second polymer of ITA-PLGA-PEG-PLGA indicated the<br />
shift of the sol-gel transition curve down to the lower values of both CGC (15,3 w%) and<br />
CGT (25 °C). The degradation behaviors of PLGA-PEG-PLGA in a phosphate buffer (pH<br />
7.4) at 37 °C were investigated. A significant decrease in the molecular weight and increase in<br />
the polydispersity within 10 days (until the samples have dissolved) was observed.<br />
INTRODUCTION<br />
Poly(lactic acid) and poly(glycolic acid) have been under extensive study since they were<br />
introduced as biodegradable polymers having hydrolytically unstable backbones. Therefore,<br />
these biopolymers can be used for a certain type of biomedical application such as injectable<br />
polymer drug delivery systems, tissue implants or resorbable bone adhesives. The prevailing<br />
mechanism for the biopolymer degradation is a simple random chemical hydrolysis [1]. The<br />
most common explanation for this heterogeneous degradation process comes from the<br />
absorption of water, followed by a hydrolytic cleavage of ester bonds, which generates chain<br />
fragments with the acidic groups (Fig. 1). This process is characterized by a decrease in<br />
molecular weight, an increase in polydispersity (PD = Mw/Mn) and polymer mass loss<br />
accompanied by an increase in low molecular chain compound concentration in the<br />
surrounding medium [2 – 7].<br />
H<br />
CH 3<br />
O<br />
O<br />
CH 3<br />
O<br />
O<br />
x<br />
O<br />
poly(lactic-co-glycolic acid)<br />
O<br />
O<br />
y<br />
OH<br />
CH<br />
H 3<br />
OH<br />
HO<br />
O<br />
lactic acid<br />
Fig. 1: Scheme of the PLGA degradation.<br />
+<br />
2x 2y<br />
H<br />
HO<br />
H<br />
O<br />
OH<br />
glycolic acid<br />
The injectable biodegradable thermosensitive ABA triblock copolymers consisting of<br />
hydrophobic biodegradable copolymer of poly(lactic acid-co-glycolic acid) (PLGA) and<br />
hydrophilic poly(ethylene glycol) (PEG) acting as A and B block, respectively, were<br />
synthesized via ring-opening polymerization method in a bulk at 155 °C. The ABA<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2006, strana 218
copolymers were additionally functionalized by itaconic acid (ITA), which can be gained<br />
from renewable resources by pyrolysis of citric acid or by fermentation of polysaccharides.<br />
ITA brings reactive double bonds and functional carboxylic acid groups to the end of<br />
copolymer resulting in preparation of biodegradable ITA/PLGA-PEG-PLGA/ITA<br />
macromonomer. Successful end-capping of ITA to PLGA-PEG-PLGA copolymer was proved<br />
by 1H NMR and FT-IR analysis followed by characterization with GPC method. The above<br />
mentioned thermosensitive polymers are soluble in water forming free-flowing solution that<br />
spontaneously gels as the temperature increases generating a water-insoluble physical<br />
hydroge. The sol-gel transition behaviors were studied by the test tube inverting method. The<br />
tests of degradation behaviors of PLGA-PEG-PLGA copolymer were carried out in vitro in<br />
the phosphate buffer medium at 37 °C.<br />
EXPERIMENTAL WORK<br />
Materials<br />
The triblock copolymers of PLGA-PEG-PLGA and ITA- PLGA-PEG-PLGA-ITA were<br />
synthesized at our faculty by Dr. Lucy Vojtová.<br />
Tetrahydrofurane for GPC analyses (THF for HPLC, gradient grade, Merck, Germany),<br />
was used as received. Polystyrene standards (Mp= 316 500 – 162) were purchased from<br />
Polymer Laboratories, Germany. Milli-Q water, phosphoric acid (p.a., 85 %, Czech Republic<br />
and potassium phosphate dibasic (p.a., for HPLC, Fluka, USA) were used for the polymer<br />
degradation studies.<br />
Method<br />
The molecular weight and the molecular weight distribution of the copolymers were<br />
determined by GPC method using Agilent Technologies 1100 Series instrument equipped<br />
with a refractive index detector, PLgel Mixed C column of 300 x 7.5 mm with particle size 5<br />
μm, degasser, pump, auto sampler and fraction collector. Tetrahydrofurane was used as the<br />
mobile phase at a flow rate equal to 1 ml.min -1 . The average molecular weight was calculated<br />
using a series of polystyrene standards (Mp = 316 500 – 162). Samples of triblock copolymers<br />
were prepared in the concentration of 0.5 mg/ml in tetrahydrofurane for HPLC.<br />
The sol-gel transition was determined by the test tube inverting method. 4 ml vials<br />
containing 1 ml of the triblock copolymer were heated from 10 to <strong>60</strong> °C in a water bath. The<br />
transition temperatures were determined by a flow (sol) – no flow (gel) criterion when the vial<br />
was inverted with a temperature increment of 1 °C per step.<br />
The degradation behavior study was performed using 0.3 ml of 23 w% polymer aqueous<br />
solutions. The 1.8 ml vials with polymer solutions were put into an incubator at 37 °C (the<br />
temperature of a human body) to formed the hydrogels. Consequently, 0,5 ml of the<br />
phosphate buffer (pH 7.4, 37 °C) was added to the vials and the samples were placed into the<br />
incubator for 10 days with a view to degrade during this time. At regular intervals, the<br />
samples were withdrawn from of the incubator, lyophilized and analyzed.<br />
RESULTS AND DISCUSSION<br />
The sol-gel transition diagrams of the triblock copolymers (PLGA-PEG-PLGA, ITA-<br />
PLGA-PEG-PLGA-ITA) were created on the base of the test tube inverting method (Fig. 2).<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2006, strana 219
Temperature (°C)<br />
45<br />
40<br />
35<br />
30<br />
25<br />
20<br />
PLGA/PEG/PLGA<br />
ITA-PLGA/PEG/PLGA-ITA<br />
white viscous<br />
suspension<br />
suspension<br />
white gel<br />
white viscous cloudy gel<br />
cloudy viscous<br />
cloudy viscous<br />
amber sol<br />
amber sol<br />
15<br />
0 5 10 15 20 25<br />
Concentration (w% )<br />
white gel<br />
amber viscous<br />
cloudy gel<br />
amber viscous<br />
amber gel<br />
amber gel<br />
Fig. 2: The sol-gel transition phase diagram of the triblock copolymers PLGA-PEG-PLGA<br />
and ITA-PLGA-PEG-PLGA-ITA<br />
The diagram demonstrates three basic areas - sol, gel and suspension (precipitate). The<br />
phase diagram of PLGA-PEG-PLGA shows the critical gel concentration (CGC) of 19.2 w%<br />
and the critical gelation temperature (CGT) of 34.5 °C. For the next study, the solution of 23<br />
%wt was used because of forming the amber hydrogel at around 37°C. As for the copolymer<br />
modified by ITA, CGC and CGT were determined to be 15.3 w% and 25.0 °C, respectively.<br />
The values were shifted below the curves of the polymer without the itaconic acid which<br />
pointed that the presence of hydrophilic –COOH groups causes better interaction between the<br />
polymer and water molecules. Therefore, CGC and CGT were observed to be lower than<br />
those of the copolymer without ITA.<br />
The change from sol to gel, which occurred by increasing the temperature, was not sharp.<br />
At the temperature much lower than the critical gel temperature; unimers, individual micelles,<br />
and grouped micelles coexisted in the sol state (Fig. 2 amber sol). The unimer fraction<br />
decreased with the temperature increasing (Fig. 2 amber viscous). At the same time, the<br />
grouped micelle size grew rapidly resulting in sol-gel transition (Fig. 2 amber gel). The<br />
aggregation and packing interactions between micelles increased to form denser gel with the<br />
raising temperature (Fig. 2 cloudy viscous state, cloudy gel). When the temperature was<br />
further raised, the hydrophobic chains in the micelle core shrank tightly. Also, the hydrophilic<br />
PEG block underwent dehydration and the second gel-sol transition arisen (Fig. 2 white<br />
viscous state, white gel, suspension). The over shrunk micelle groups precipitated in water<br />
and the solution separated into two phases of water and precipitated polymer (Fig. 2<br />
precipitation) [8].<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2006, strana 220
Each micelle has a hydrophobic core and a hydrophilic shell, and can move relatively<br />
freely without any bridging connection between the micelles. Sol-gel transition occurs when<br />
the total volume of micelles fraction is larger than the maximum packing fraction volume [8].<br />
The degradation behavior of PLGA-PEG-PLGA was investigated. Four samples of the<br />
copolymer in 0.7 ml of the phosphate buffer (pH 7.4) were placed into the incubator at 37 °C<br />
(the normal temperature of a human body). The samples were ta<strong>ke</strong>n out and lyophilized after<br />
3, 5, 7 and 10 days. The rest of the copolymer was analyzed by GPC. The decrease in the<br />
molecular weight during these periods can be seen in Fig. 2. After 3, 5 and 7 days two phases<br />
in the vials can be observed, the copolymer gel and the buffer. The total dissolving of the<br />
copolymer occurred after 10 days when the measured molecular weight (Mn) was found to be<br />
4170 (Tab. 1).<br />
Tab. 1: The change of the molecular weight and the polydispersity during the degradation<br />
in the phosphatic buffer (pH 7.4, 37 °C)<br />
Time (day) Molecular weight Polydispersity<br />
0 <strong>60</strong>10 1.21<br />
3 5840 1.23<br />
5 5220 1.31<br />
7 4770 1.33<br />
10 4170 1.35<br />
The change of polydispersity (D) was related to the change of Mn. Mn decreased while D<br />
increased with the growing number of the shorter copolymer chains.<br />
Molecular weight<br />
6500<br />
<strong>60</strong>00<br />
5500<br />
5000<br />
4500<br />
4000<br />
3500<br />
0 2 4 6 8 10 12<br />
Time (day)<br />
Fig. 3: The change of the molecular weight (Mn)● and the polydispersity (D)∆ after 0, 3,<br />
5, 7, and 10 days.<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2006, strana 221<br />
1,36<br />
1,34<br />
1,32<br />
1,30<br />
1,28<br />
1,26<br />
1,24<br />
1,22<br />
1,20<br />
Polydispersity
CONCLUSION<br />
The copolymers of PLGA-PEG-PLGA and ITA-PLGA-PEG-PLGA-ITA were synthesized<br />
and characterized by GPC and the test tube inverting method. The presence of the itaconic<br />
acid in the polymer chain caused the decrease in both CGC and CGT.<br />
The degradation of PLGA-PEG-PLGA was attended by the decreasing of molecular<br />
weight during ten days until the gel has dissolved. The number of the low molecular weight<br />
chains increased with time causing the increasing in polydispersity of the polymer.<br />
Acknowledgement<br />
I would li<strong>ke</strong> to thank Dr. Lucy Vojtová for the copolymers preparation and expert advice.<br />
This work was supported by the Ministry of Education of the Czech Republic under the<br />
research project MSM 0021630501.<br />
REFERENCES<br />
[1] A. Göpferich, Biomaterials 1996, 17, 103 – 114.<br />
[2] P. Giunchedi, B. Conti, S. Scalia, U. Conte: J. Contr. Rel. 1998, 56, 53 – 62.<br />
[3] S. Li,: Biomed. Mater. Res. 1999, 48, 342 – 353.<br />
[4] I. Grizzi, H. Garreau, S. Li, M. Vert: Biomaterials 1995, 16, 305 – 311.<br />
[5] B. Marcato, G. Paganetto, G. Ferrara, G. Cecchin: J. Chromatogr. B. 1996, 682, 147 –<br />
156.<br />
[6] T. G. Park: J. Contr. Rel. 1994, 30, 161 – 173.<br />
[7] G. Schliec<strong>ke</strong>r, C. Schmidth, S. Fuchs, T. Kissel: Biomaterials 2003, 24, 3835 – 3844.<br />
[8] M. S. Shim, H. T. Lee, W. S. Shim, I. Park, H. Lee, T. Chang, S. W. Kim, D. S. Lee:<br />
J. Biom .Mat. Res. 2002, 61, 188 - 196<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2006, strana 222
STUDY OF THE COPPER(II) IONS NON–STATIONARY DIFFUSION<br />
IN HUMIC GEL<br />
Ing. Petr Sedláček, 1 st year of study<br />
Supervisor: doc. Ing. Martina Klučáková, PhD.<br />
Brno University of Technology, Faculty of Chemistry, Institute of Physical and Applied<br />
Chemistry, Purkyňova 118, 612 00 Brno, e–mail: sedlacek@fch.vutbr.cz<br />
INTRODUCTION<br />
Severe reduction of use of solid fossil fuels in the power–producing industry has mar<strong>ke</strong>dly<br />
encouraged the investment to alternative applications of these materials. Humic acids (HA),<br />
which are one of their <strong>ke</strong>y components, are because of their rich natural sources, simple<br />
isolation methods and profitable chemical ad physical behavior predetermined for the wide–<br />
spread use in different industrial and agricultural branches. Although the intensive study of<br />
this material lasts for several decades the knowledge level (mainly concerning the structure of<br />
HA) is still quite low (it is often compared with the knowledge of proteins fifty years ago).<br />
Application of HA in a form of humic gel (see [1]) is quite new and unexplored field of<br />
their study. The gel form of HA is easy to prepare, suitable for the exploration of transport<br />
phenomena and mainly simulates the natural conditions; HA are usually found in the highly<br />
humid environment (water sediments, peat etc.) and thus in the swollen form.<br />
One of most famous and promising HA properties is the ability to bind metal ions. They<br />
can form stable complexes among others with heavy metals, which influences their toxicity in<br />
environment and this fact encourages potential applications of humic substances mainly in<br />
environmental industry, in the production of fertilizers and in pharmacy. Most important<br />
binding sites in HA molecule are carboxylic and phenolic groups and aromatic cycles.<br />
Besides the lower mobility of metal ions in humic gels comparing water solution, the<br />
diffusion in gels are influenced also by the retention or immobilization of the ions, both<br />
caused by chemical reaction between metals and HA. The result is that mathematical<br />
apparatus used in the description of diffusion phenomena is very complicated.<br />
Copper(II) ion is well–known for its high affinity to humic substances [2]. Besides this the<br />
HA–Cu(II) binding is among the highest strengths. Therefore and also because of easy<br />
quantification of copper content by means of<br />
spectroscopy, the copper(II) ions has been chosen<br />
as model metal ions for this work.<br />
The main aim of this research was the study of<br />
copper(II) ions diffusion from solutions with<br />
different Cu 2+ concentration into humic gel across<br />
the phase interface and the diffusion in the gel<br />
itself. Other experiment interested in the influence<br />
of other properties of the copper(II) source<br />
solution, namely the type of anion of copper salt,<br />
solution pH and ionic strength.<br />
EXPERIMENTAL PART<br />
HA were obtained from South-Moravia lignite by means of the alkaline extraction (see<br />
[2]). Humic gel was prepared by the technique optimized in [3]: HA were diluted in 0.5 M<br />
NaOH in the 8 g HA in 1 dm 3 Fig. 1 Humic gel sample<br />
NaOH ratio. This sodium humate solution was acidified by<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2006, strana 223
concentrated HCl to pH ~ 1 and leaved in<br />
refrigerator overnight. After the centrifugation (10<br />
min., 4000 rpm, cooling for 15 °C) the<br />
supernatant was poured out and the gel was<br />
washed three times by deionized water. Each<br />
washing was followed by centrifugation, the last<br />
one took 30 minutes. Finally the gel was<br />
deposited in dessicator with water to stabilize its<br />
humidity. The picture of prepared gel sample is<br />
shown on the Fig. 1.<br />
The HA sample was characterized by means of<br />
table 1 Elementary analysis results<br />
content in dried<br />
element ash-free HA<br />
[atomic %]<br />
H 42,12<br />
C 41,16<br />
O 15,64<br />
N 0,91<br />
S 0,17<br />
the elementary analysis (Microanalyser Flash 1112, Carlo Erba), ash content analysis (sample<br />
contained 6.8 weight % of water and 28.5 weight % of ash), and UV–VIS and FT–IR spectral<br />
analysis (Hitachi U 3300 and Nicolet impact 400 respectively). From the exact results of these<br />
analyses, which are listed elsewhere ([4]), it is clear, that used HA were of high degree of<br />
humification. The humic gel sample was analyzed for its solid matter content<br />
(14.5 weight %), total gel acidity was determined by potentiometric titration (gel contained<br />
8.12 mmol acid equivalents per 1 g of HA). The FT–IR spectroscopy analysis of dried gel<br />
sample results in fact that no chemical structure changes occur while gelation of HA.<br />
Diffusion experiment was divided into several parts; in the first one the pre-determined<br />
(see [3]) value of the diffusion coefficient of copper(II) ions in humic gel was verified by the<br />
stationary diffusion from the saturated copper(II) chloride solution. In detail, experimental<br />
procedure is listed in [4].<br />
In all remaining parts the non-stationary diffusion was studied. All experiments took place<br />
in apparatus presented on Fig. 2. Humic gel was pac<strong>ke</strong>d into plastic tube as 5 cm long<br />
cylinder and 2 ml of both copper ions solution and deionized water were filled into side<br />
containers. First of all diffusion dependency on Cu(II) initial concentration in solution was<br />
monitored by changing the initial concentration of CuCl2 solution placed in the apparatus<br />
container, while the duration of experiment was maintained at 24 hours. After the end of the<br />
experiment, each gel sample was sliced and each slice was separately extracted by 1 mol.dm –3<br />
HCl. By the UV–VIS quantification of Cu(II) content in each extract, the concentration<br />
profile of the gel cylinder and the total diffusion flux across the solution–gel interface were<br />
determined.<br />
Next part deals with the influence of time duration of diffusion experiment. The 1 day,<br />
3 days and 5 days periods have been chosen. The experiment was repeated for three Cu(II)<br />
initial concentration values: 0.1 mol.dm –3 , 0.3 mol.dm –3 and 0.6 mol.dm –3 . Following<br />
experimental technique was adopted from previous experiment without changes.<br />
In the last part, the influence of other copper(II) solution properties has been investigated.<br />
The influence of the type of copper(II) salt anion was studied by using CuCl2, Cu(NO3)2,<br />
Cu(II)<br />
plugged solution<br />
containers<br />
HUMIC GEL<br />
plastic tube<br />
H2O<br />
Fig. 2 Scheme of the aparatus used in all<br />
diffusion eperiments<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2006, strana 224
CuSO4, Cu(ClO4)2 and Cu2P2O7 of the same Cu 2+ concentration (0.1 mol.dm –3 ) as a stock<br />
solution for diffusion experiments of the same time duration (24 hours). Finally, the solution<br />
pH – dependency and solution ionic strength dependency of the diffusion process was<br />
chec<strong>ke</strong>d by diluting CuCl2 salt using the differently concentrated hydrochloric acid and<br />
sodium chloride, respectively. For all of these experiments the same Cu 2+ concentration<br />
(0.1 mol.dm –3 ) as well as the time duration (24 hours) was used and the same experimental<br />
technique (see above) was adopted.<br />
RESULTS AND DISCUSSION<br />
The value of calculated effective diffusion coefficient of copper(II) ions in humic gel was<br />
7.96×10 -10 m 2 .s –1 . This value is in good agreement with the one pre–determined by another<br />
experimental method (see [3]) and was used for all following calculations. As can be seen on<br />
Fig. 3 and Fig. 4, total diffusion flux of Cu 2+ through solution–gel phase interface linearly<br />
increases with the initial concentration of copper(II) in solution and with the square root of<br />
time duration of diffusion experiment. The later dependency is however much more<br />
complicated; the nonzero intercept of the regression line indicates that the linearity is just<br />
illusory and in fact the dependency is curved. Klučáková et al. in [5] stated the equation for<br />
the total diffusion flux across the solution–gel interface:<br />
ε c D τ<br />
0<br />
g<br />
m = (1)<br />
1+<br />
ε D / π<br />
g D<br />
in which m stands for the total diffusion flux, c0 is initial concentration of the ion in solution,<br />
τ is the time duration of the diffusion, ε is ratio of ion concentration in the gel and in the<br />
solution in final equilibrium (at the “end of diffusion”), Dg and D is diffusion coefficient of<br />
cupric ions in humic gel and in solution, respectively. Following this equation the value of ε<br />
was calculated from total diffusion flux corresponding to c0 and τ values. It was found that for<br />
the same duration of diffusion experiment this value is constant for different initial<br />
concentrations of the solution but it varies for different duration of experiment (Fig. 5). This<br />
fact could be explained by the apparatus construction (small solution volumes – gel weight<br />
ratio) or by the time–consuming formation of some stable structural or chemical complexes<br />
between gel and ions. This affects the mobility of ions (retention of ions can lead up to their<br />
immobilization in gel) and their equilibrium in gel and solution. This fact could explain<br />
mentioned deformation of total flux time dependency as well.<br />
The knowledge of the ε value allows<br />
the calculation of theoretical<br />
y = 6.454E-03x<br />
concentration profiles of the copper (II)<br />
R<br />
ions in the humic gel (used<br />
mathematical apparatus is presented in<br />
detail in reference [5]) for individual<br />
experiment conditions (initial<br />
concentration, time duration). The<br />
example on Fig. 6 shows very good<br />
agreement between calculated and<br />
measured concentration profiles.<br />
2 50<br />
40<br />
= 1.000E+00<br />
30<br />
20<br />
10<br />
0<br />
0 2000 4000 <strong>60</strong>00 8000<br />
total flux [mol.m -2 ]<br />
c0 [mol.m -3 ]<br />
Fig. 3 The total diffusion flux dependency on<br />
the initial concentration of Cu 2+ in the<br />
solution (diffusion duration 24 hours)<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2006, strana 225
ε<br />
total flux [mol.m -2 ]<br />
7<br />
6<br />
5<br />
4<br />
3<br />
2<br />
1<br />
y = 0.0099x<br />
R 2 = 0.9997<br />
y = 0.0066x<br />
R 2 = 0.9983<br />
y = 0.0084x<br />
R 2 = 0.9985<br />
0<br />
0 200 400 <strong>60</strong>0 800<br />
c0 [mol.m -3 ]<br />
1 day<br />
3 days<br />
5 days<br />
The results of the part which deals with the influence of another copper(II) solution<br />
properties shows dramatically smaller effect of the type of anion in the copper(II) salt and<br />
solution ionic strength and pH compared with initial copper(II) concentration and time<br />
duration of diffusion. On Fig. 7, it can be seen that for all anions with unit valence (CuCl2,<br />
Cu(NO3)2 a Cu(ClO4)2) the total diffusion flux is similar even if for example anion size differs<br />
mar<strong>ke</strong>dly. On the other hand, anions with higher valence (CuSO4 and Cu2P2O7) show<br />
inconsiderable decrease of total diffusion flux. The pH and ionic strength measurement<br />
excluded these properties as causer of this<br />
decrease, so it can be supposed that this shift<br />
can be explained as a consequence of higher<br />
charge of multivalent anions.<br />
Total diffusion flux is also affected by the<br />
copper solution pH. Fig. 8 shows that with<br />
higher acidity of the solution total diffusion<br />
flux decreases. This could be explained for<br />
example by well–known decrease of HA<br />
sorption ability in acid media (see [2]) or by<br />
the change of solution–gel equilibrium.<br />
Higher acidity affects salt hydrolysis and<br />
thus actual ion concentration in solution.<br />
total flux [mol.m -2 ]<br />
7<br />
6<br />
5<br />
4<br />
3<br />
2<br />
1<br />
y = 5.517E-03x + 2.250E+00<br />
R 2 = 9.950E-01<br />
y = 2.449E-03x + 1.343E+00<br />
R 2 = 9.994E-01<br />
y = 1.095E-03x + 3.265E-01<br />
R 2 = 9.999E-01<br />
0<br />
200 300 400 500 <strong>60</strong>0 700<br />
t 1/2 [s 1/2 ]<br />
Fig. 4 The total diffusion flux dependencies on the initial concentration of Cu 2+ and on the<br />
duration of the experiment<br />
2<br />
1.5<br />
1<br />
0.5<br />
0<br />
0 1 2 3 4 5 6<br />
τ [days]<br />
Fig. 5 The time shift of ε<br />
copper(II) ions concentration<br />
[mol/dm 3 gel]<br />
0.15<br />
0.1<br />
0.05<br />
0<br />
experimental results<br />
(0.3 M; 3 days)<br />
calculated<br />
concentration profile<br />
(0.3 M; 3 days)<br />
0.1M<br />
0.3M<br />
0.6M<br />
0 10 20 30 40 50<br />
distance from the interface [mm]<br />
Fig. 6 Concentration profiles of Cu 2+<br />
in humic gel<br />
total flux [mol.m –2 ]<br />
0.7<br />
0.6<br />
0.5<br />
0.4<br />
0.3<br />
0.2<br />
0.1<br />
0<br />
CuCl2<br />
0.614 0.620 0.<strong>60</strong>8<br />
Cu(NO3)2<br />
Cu(ClO4)2<br />
CuSO4<br />
0.476<br />
Cu2P2O7<br />
Fig. 7 Total diffusion flux for different<br />
copper(II) salts<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2006, strana 226<br />
0.355
total flux [mol.m –2 ]<br />
0.7<br />
0.65<br />
0.6<br />
0.55<br />
0.5<br />
0.45<br />
0.4<br />
0 1 2 3 4 5<br />
pH<br />
total flux [mol.m –2 ]<br />
0.4<br />
0 1 2 3 4 5 6<br />
Finally the total diffusion flux showed complex dependency on the ionic strength of<br />
copper(II) solution. For small NaCl additions, the decrease of total flux comes with the<br />
increase of ionic strength, on the contrary substantial increase appeares in higher NaCl<br />
concentration range. Former decrease can be caused by decrease of HA sorption ability,<br />
competitive diffusion of Na + ions or by affecting Cu 2+ solvation. The later increase is not very<br />
clear. It can be supposed that in the solution with high ionic strength bigger Cu 2+ clusters are<br />
solvated and easily took out from the solution.<br />
CONCLUSIONS<br />
This paper deals with the study of copper(II) ions non–stationary diffusion in humic gel.<br />
Main aim was to determinate the influence of individual copper(II) solution properties on the<br />
total diffusion flux across the solution–gel interface.<br />
It was proved that copper(II) ions diffusion across the phase interface is affected mainly by<br />
the initial concentration of copper in the solution and by the time duration of the diffusion.<br />
The influence of other solution properties, such as its pH or ionic strength as well as the type<br />
of copper(II) salt anion is far less important however show interesting dependencies.<br />
Although the applied apparatus was very simple and could be improved for following<br />
experiments (the higher solution volumes should be used) experimental data are fitted well by<br />
the theoretical calculations, so it can be said that this method presented itself suitable for the<br />
wide spectrum of diffusion experiments using humic gel as a very usefull natural–li<strong>ke</strong> model.<br />
REFERENCES<br />
[1] Klučáková, M.: Huminový gel jako model pro studium transportu těžkých kovů<br />
v přírodních systémech. CHEMagazín 2004, Vol. 14, No. 3, p. 8–9. ISSN 1210–7409<br />
[2] Klučáková, M., Kaláb, M., Pekař, M., Lapčík, L.: Study of Structure and properties of<br />
Humic and Fulvic Acids. II. Study of Adsorption of Cu + ions to Humic Acids Extracted<br />
from Lignite, J.Polym.Mater 19 (3) 2002<br />
[3] Malenovská, M.: Studium difúzních procesů v huminových gelech. 55 stran. Diplomová<br />
práce na VUT, FCH Brno 2005. Vedoucí diplomové práce Ing. Martina Klučáková<br />
PhD.<br />
[4] Sedláček, P..: Difúze kovových iontů v huminových gelech. 70 stran. Diplomová práce<br />
na VUT, FCH Brno 2006. Vedoucí diplomové práce Doc. Ing. Martina Klučáková PhD.<br />
[5] Klučáková, M., Pekař, M.: Diffusion of Metal Cations in Humic Gels. In Humic<br />
Substances: Nature´s most Versatile Materials (E. Ghabbour, G. Davies Eds.), Francis<br />
& Taylor, New York, 2004, p. 263–74<br />
0.7<br />
0.65<br />
0.6<br />
0.55<br />
0.5<br />
0.45<br />
ionic strength [mol.dm –3 ]<br />
Fig. 8 Total diffusion flux dependency on solution pH (left) and ionic strength (right)<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2006, strana 227
LIVING RADICAL POLYMERIZATIONS INITIATED WITH<br />
SILSESQUIOXANES<br />
Author: Ing. Ondřej Smrtka – 3 rd year of DSP<br />
Supervisor: Prof. RNDr. Josef Jančář, CSc.<br />
Brno University of Technology, Faculty of Chemistry, Institute of Material Chemistry<br />
Purkyňova 118, 612 00 Brno, Czech Republic; email: smrtka@fch.vutbr.cz<br />
INTRODUCTION<br />
It has been discovered that small variations in molecular structure at the nano-meter scale<br />
level allow substantial changes in macroscopic behavior of nanostructured systems.<br />
Nanocomposites with polymeric matrix belong among the most intensively investigated<br />
materials since they promise extensive industrial and biomedical applications.<br />
The polyhedral oligomeric silsesquixanes (POSS) can be included into the large group of<br />
nano-particles used for nanocomposite preparation. POSS, due to their well-defined structure,<br />
are considered to simulate the surface of silica nano-particles as the smallest possible silica<br />
particle. Nanocomposites containing POSS nanoparticles are considered extremely interesting<br />
class of nanostructure organic-inorganic materials.<br />
Polyhedral oligomeric silsesquioxans are compounds of general formula (RSiO1.5)x, where<br />
x is an even number higher than 4, mostly being 8. R is any of large number of organic<br />
groups, or e.g. hydrogen or halogen atom.<br />
R<br />
R<br />
O<br />
Si<br />
O<br />
R<br />
Si<br />
O<br />
O<br />
Si<br />
O<br />
R<br />
Si<br />
O<br />
Fig. 1: Polyhedral oligomeric silsesquioxan (POSS)<br />
POSS are prepared preferentially by hydrolytic condensation of trifunctional silanes [1].<br />
Organic groups are carried into the molecule either directly during the synthesis (as the fourth<br />
non-reactive group of the silane precursor), or by a transformation of existing groups [2].<br />
Monofunctional POSS are synthesized from incompletely condensed molecules of<br />
silsesquioxane by condensation of another silane with appropriate functional group [3].<br />
POSS particles can be incorporated to the polymeric materials directly by blending with<br />
polymer as the additive, or as the component of polymeric chain. The POSS containing<br />
macromolecules can be synthesized using monomers with POSS pendant groups, or<br />
afterwards, by grafting to the polymer using various condensation or addition reactions (e.g.<br />
hydrosilylation). Another way how to connect POSS with the polymer chain is to use it as the<br />
initiator for ATRP.<br />
ATRP (Atom Transfer Radical Polymerization) has been developed in 1995 by<br />
Matyjaszewski [4]. It is based on rapid attainment of dynamic equilibrium between low<br />
amount of growing free radicals and much higher amount of dormant species. Because the<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2006, strana 228<br />
O<br />
Si<br />
O<br />
O<br />
Si<br />
R<br />
R<br />
O<br />
O<br />
Si<br />
Si<br />
O<br />
R<br />
R
number of free radicals is very low, the termination is reduced and the polymerization proves<br />
living character.<br />
P –X + Cu –Y/2L ⎯⎯→ P + M + X–Cu –Y/2L<br />
I kakt<br />
•<br />
II<br />
n ←⎯ ⎯ k<br />
n<br />
deakt<br />
Pn + Pm<br />
Fig. 2: Scheme of ATRP<br />
The dormant species are mostly alkylhalides, from which the halogen atom is transferred to<br />
catalyst (copper halide + organic ligand); the specie then becomes a radical, efficient to<br />
propagate radical polymerization. The oxidation state of copper increases by one with<br />
admitting the halogen; with the transfer back to the polymer the original oxidation state is<br />
retrieved. Other transition-metal salts can be used as the ATRP catalyst, with the requirement<br />
to the metal center to have at least two readily accessible oxidation states separated by one<br />
electron.<br />
A variety of monomers have been successfully polymerized using ATRP, typical<br />
monomers include styrenes, methacrylates, acrylamides and acrylonitrile.<br />
In ATRP, alkylhalides are typically used as the initiator. A variety of transition-metal<br />
complexes have been studied as ATRP catalyst, but copper catalysts are superior in ATRP in<br />
terms of versatility and cost. Presence of organic ligand is necessary for dissolution of copper<br />
salt in reaction mixture. Above all, the nitrogen-based ligands are used (e.g. bipyridine,<br />
PMDETA, HMTETA).<br />
Initiation of ATRP with the POSS-based initiator has not been intimately described so far,<br />
only some notes were published [5, 6]; however initiation of ATRP with polysiloxane-based<br />
initiators has been described [7, 8] much more and due to the silimilar structures the depicted<br />
techniques might be used also for POSS-based initiators.<br />
EXPERIMENTAL<br />
Materials used<br />
Styrene was distilled from calcium hydride prior to use. Tetrahydrofurane was distilled<br />
from purple sodium/benzophenon solution. Copper (I) bromide was stirred in glacial acetic<br />
acid overnight, decanted, then washed with absolute ethanol and diethyl ether and vacuum<br />
dried at <strong>60</strong>°C. Ethyl-α-bromo-isobutyrate (EBIB), p-dimethoxybenzene (D<strong>MB</strong>), POSShydride,<br />
allyl-bromoisobutyrate, pentamethyldiethylene triamine (PMDETA), and Karstedt<br />
catalyst were used as received.<br />
Synthesis of POSS-Br<br />
POSS-Br was synthesized using hydrosilylation reaction. 1.37 g (1.44 mmol) of POSS-H,<br />
16 ml dried THF, 0,24 ml (1.44 mmol) allyl-α-bromoisobutyrate and 55μL of Karstedt<br />
catalyst solution in xylene (3 wt.%, 3,64 μmol) were placed into a 50 mL two-neck roundbottom<br />
flask under nitrogen blan<strong>ke</strong>t and stirred. The flask was fit with rubber septum and a<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2006, strana 229<br />
kp<br />
k´t<br />
P • m<br />
kt<br />
Pn+m
eflux condenser equipped with bubble flask filled with silicon oil. Then 55μL of Karstedt<br />
catalyst solution in xylene (3 wt.%, 3,64 μmol) was injected to the reaction flask and the<br />
mixture was heated in 80 °C oil bath under reflux for 24 hours. THF was evaporated and the<br />
product POSS-Br was vacuum dried.<br />
R<br />
R<br />
R<br />
Si<br />
O<br />
O<br />
Si O<br />
O Si<br />
O<br />
R<br />
Si<br />
O<br />
O<br />
R<br />
O<br />
Si<br />
Si O<br />
O Si<br />
O<br />
O<br />
Si<br />
R<br />
H<br />
C<br />
H 3<br />
R<br />
+<br />
C<br />
H 2<br />
RH3<br />
= C<br />
O<br />
O<br />
CH 3<br />
Br<br />
CH 3<br />
Karstedtův Karstedt's katalyzátor catalysator catalyst<br />
THF, reflux<br />
24 hours<br />
Fig. 3: Synthesis of POSS-Br initiator<br />
Polymerizations<br />
Polymerization 1:<br />
A typical polymerization is as follows: 31 mg CuBr was placed into a 50 mL double-neck<br />
flask; the flask was evacuated and filled with nitrogen. Under a nitrogen blan<strong>ke</strong>t 5 mL of<br />
styrene, 40 μL of PMDETA and 4.6 g of p-dimethoxybenzene was added. The mixture was<br />
stirred until a homogenous solution formed and was placed into a 120°C oil bath under<br />
nitrogen. After heating to the reaction temperature 0,48 g of POSS-Br (initiator) was added.<br />
The solution turned dark green as the reaction began. Periodically, small amounts of reaction<br />
mixture were removed for kinetic and molecular weight analysis (conversion was<br />
determinated by weight analysis, molecular weight of polymers was determined using gel<br />
permeation chromatography). Molar ratios: Styrene:POSS-Br:CuBr:PMDETA -<br />
100:1:0.5:0.5.<br />
Polymerization 2:<br />
This polymerization is similar to the polymerization 1, except the POSS-Br initiator is<br />
replaced with 65 μL of ethyl-α-bromo-isobutyrate, which was injected to the hot mixture via<br />
rubber septum. Molar ratios: Styrene:EBIB:CuBr:PMDETA - 100:1:0.5:0.5.<br />
RESULTS<br />
Reason for using POSS as an initiator of ATRP is to get a polystyrene macromolecule with<br />
one bulky POSS group at one of the chain ends; the initiator of ATRP remains the inseparable<br />
part of the macromolecule.<br />
Molecular weight plots prove living character of polymerizations; molecular weight grows<br />
linearly with conversion, with conversion growing to almost 100%. Linear dependence of<br />
conversion on time in semi logarithmic coordinates (Fig. 5) shows the first-order kinetic with<br />
respect to monomer.<br />
From the molecular weight plot of polymerization 1 (Fig. 6) it is obvious, that initiation<br />
efficiency is low and only about one half of the POSS initiator molecules is used for<br />
initiation; this causes the molecular weight to be twice as high as is should be according to the<br />
theoretical value determined from molar ratio monomer:initiator. In contrast, polymerization<br />
2 initiated with low molecular weight initiator EBIB behaves in accordance with theory<br />
(Fig. 7).<br />
R<br />
R<br />
O<br />
Si<br />
O<br />
R<br />
Si<br />
O<br />
O<br />
Si<br />
O<br />
R<br />
Si<br />
O<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2006, strana 230<br />
O<br />
Si<br />
O<br />
O<br />
Si<br />
R<br />
R<br />
O<br />
O<br />
Si<br />
Si<br />
O<br />
R<br />
O<br />
O<br />
CH 3<br />
Br<br />
CH 3
Differences can be caused by several reasons; no one of these reasons was studied in this<br />
work in detail, so the following ideas must be ta<strong>ke</strong>n as a speculations.<br />
One factor could be different solubility of catalyst system due to the presence of different<br />
initiator in reaction mixture; it is also important to rule out the possibility of conventional free<br />
radical polymerization taking place to form polystyrene homopolymer, although the catalyst<br />
system should inhibit this reaction; part of POSS based initiator molecules could be<br />
deactivated with some side reaction, or there is just a steric block for approach of molecules<br />
of catalyst or monomer to the bulky POSS molecule; these ideas must be ta<strong>ke</strong>n into<br />
consideration in subsequent work on this project.<br />
Convers ion [%]<br />
ln([M]0/[M]t)<br />
M<br />
100<br />
2<br />
1<br />
80<br />
<strong>60</strong><br />
40<br />
20<br />
0<br />
0 200 400 <strong>60</strong>0 800 1000<br />
t [min]<br />
Fig. 4: Kinetic plots<br />
0<br />
0 100 200 300 400 500<br />
25000<br />
20000<br />
15000<br />
10000<br />
5000<br />
t [min]<br />
Fig. 5: Semilogarithmic kinetic plots<br />
0<br />
1.00<br />
0 20 40 <strong>60</strong> 80 100<br />
Co nve rs io n [%]<br />
1.70<br />
1.<strong>60</strong><br />
1.50<br />
1.40<br />
1.30<br />
1.20<br />
1.10<br />
D<br />
Polymerization 1<br />
Polymerization 2<br />
Polymerization 1<br />
Polymerization 2<br />
M(th e o r.)<br />
Fig. 6: Molecular weight plot for polymerization 1<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2006, strana 231<br />
M<br />
D
CONCLUSION<br />
M<br />
12000<br />
10000<br />
8000<br />
<strong>60</strong>00<br />
4000<br />
2000<br />
0<br />
0 20 40 <strong>60</strong> 80 100<br />
Conve rs ion [%]<br />
1.3<br />
1.25<br />
1.2<br />
1.15<br />
1.1<br />
1.05<br />
1<br />
D<br />
M(theor.)<br />
Fig. 7: Molecular weight plot for polymerization 2<br />
ATRP has been employed to produce polystyrene macromolecules containing polyhedral<br />
oligomeric silsesquioxanes (POSS). Polymerizations initiated with POSS prove slower<br />
growth of polymers than polymerizations initiated with low-molecular initiators, and also<br />
lower initiation efficiency, which causes the molecular weight to be much higher than<br />
expected. Further work is underway to optimize the conditions of the polymerizations with<br />
respect to the efficiency of initiation, polydispersity of polymer and polymerization rate.<br />
REFERENCE<br />
[1] Agaskar P.A., Inorg. Chem., 30 (1991), p. 2707-2708<br />
[2] Zhang C., Laine R.M., J. Organomet. Chem., 521 (1996), p. 199-201<br />
[3] Lichtenhan J.D. et al., Mat. Res. Soc.Symp. Proc., 435 (1996), p. 3-11<br />
[4] Wang J.S., Matyjaszewski K., J. Am. Chem. Soc., 117 (1995), p. 5614-5615<br />
[5] Pyun J. et al., J. Am. Chem. Soc. 123 (2001), p. 9445-9446<br />
[6] Matyjaszewski K. et al., ACS Symp. Ser. 729 (2000), p. 270-283<br />
[7] Brown D.A., Price G.J., Polymer 42 (2001), p. 4767-4771<br />
[8] Miller P.J., Matyjaszewski K., Macromolecules 32 (1999), p. 87<strong>60</strong>-8767<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2006, strana 232<br />
M<br />
D
MEASUREMENT OF THERMOPHYSICAL PROPERTIES OF PMMA<br />
BY PULSE TRANSIENT METHOD<br />
Ing. Pavla Štefková<br />
Supervisor: Prof. Ing. Oldřich Zmeškal, CSc.<br />
Institute of Physical and Applied Chemistry, Faculty of Chemistry, Brno University of<br />
Technology, Purkynova 118, 612 00 Brno, Czech Republic, email: stefkova@fch.vutbr.cz<br />
INTRODUCTION<br />
Polymethyl methacrylate (PMMA) is the synthetic polymer of methyl methacrylate. This<br />
thermoplastic and transparent plastic was developed in 1928 in various laboratories and was<br />
brought to mar<strong>ke</strong>t in 1933 by the German Company Rohm and Haas. This material is used as<br />
the standard reference material for thermal conductivity measurements in metrology.<br />
Chemical analysis and materials testing are becoming ever more important as science,<br />
trade and society are getting more complex worldwide. The number and significance of<br />
decisions based on the results of chemical analysis and materials’ testing is ever increasing in<br />
all spheres of life. For this purpose results of analysis and testing have to be reliable and<br />
comparable as well as acceptable worldwide. The use of certified reference materials is an<br />
efficient and proper tool to achieve these goals [1].<br />
Measurement of the thermophysical properties of PMMA shows that some effects<br />
influencing the measurement process have to be known when one want to use it as laboratory<br />
reference or standard reference material (SRM). This material should be used for validation of<br />
apparatuses upon well-known experimental conditions, to obtain reliable data [2].<br />
The pulse transient method allows investigating the thermal diffusivity, specific heat and<br />
thermal conductivity within single measurement. The principle of this method and the<br />
arrangement of the measured sample are shown in Figure 1. The heat pulse is generated by<br />
the passing of the electrical current through the plane electrical resistor made of metallic foil.<br />
A sensor measures the time development of the temperature field (temperature response) in a<br />
point of the tested body. Then the temperature is characterized by a function [3]<br />
⎟ 2<br />
Q S ⎛ h ⎞<br />
ΔT =<br />
⋅ exp ⎜<br />
⎜−<br />
. (1)<br />
( E−<br />
D)<br />
/ 2<br />
cp<br />
ρ ( 4π<br />
at<br />
) ⎝ 4a<br />
t ⎠<br />
The thermophysical parameters are calculated from the characteristic parameters of the<br />
temperature response (time and the maximum of temperature response to the heat pulse).<br />
The thermal diffusivity is given by<br />
2<br />
h<br />
a =<br />
2tmax f a<br />
the specific heat by<br />
2<br />
h<br />
=<br />
2(<br />
E − D)<br />
tmax<br />
, (2)<br />
Q<br />
cp = ⋅<br />
ρ ΔTmaxh<br />
and thermal conductivity by<br />
f c<br />
=<br />
2π exp( 1)<br />
Q<br />
E−<br />
D<br />
ρ ΔTmaxh<br />
( E −D<br />
) / 2<br />
⎛ E − D ⎞<br />
⋅ ⎜<br />
2 exp( 1)<br />
⎟<br />
⎝ π ⎠<br />
(3)<br />
Q<br />
λ = cp ρ a =<br />
E−<br />
D−2<br />
2(<br />
E − D)<br />
ΔTmaxt<br />
maxh<br />
( E −D<br />
) / 2<br />
⎛ E − D ⎞<br />
⎜<br />
2 exp( 1)<br />
⎟ .<br />
⎝ π ⎠<br />
(4)<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2006, strana 233
It is possible to definite the coefficient fa (fractal dimension D respectively) for every point of<br />
the experimental dependence<br />
2ln(<br />
ΔTmax<br />
ΔT<br />
)<br />
f a = E − D =<br />
. (5)<br />
ln( t t ) + ( t t −1)<br />
t t<br />
specimen<br />
tm t m<br />
Figure 1 The principle of measurement of thermophysical parameters by the pulse transient<br />
method.<br />
max<br />
planar source thermocouple<br />
current current pulse pulseplanar<br />
source<br />
h<br />
thermocouple<br />
I I<br />
t t0 o<br />
I II III<br />
max<br />
T<br />
temperature response response<br />
EXPERIMENT AND RESULTS<br />
For measuring of the responses to the pulse heat the Thermophysical Transient Tester 1.02<br />
was used. It was developed at the Institute of Physics, Slovak Academy of Science. The<br />
specimen of 30 mm in diameter and 6 mm thick was used for the pulse transient method. Its<br />
density is ρ = 1184 kg m –3 . Thermophysical properties of material were measured in air.<br />
1. Comparison between experimental and recommended data of the thermophysical<br />
parameters of PMMA measured at 25 °C<br />
The pulse width of 4 – 40 s, the heat power of 0.18 up to 3.03 W was used and adequate<br />
the pulse heat energy of 3000 – 42000 J m –2 was obtained. The typical heat energy of pulse<br />
was about 13000 J m –2 that is low enough to avoid temperature damage of this material. The<br />
temperature response ΔTmax in the range of 0.1 up to 1.4 °C was obtained. Analysis of these<br />
sets of data was carried out to find optimal experimental conditions.<br />
ΔT (°C)<br />
1.0<br />
0.8<br />
0.6<br />
0.4<br />
0.2<br />
0.0<br />
T m<br />
0 150 300 450 <strong>60</strong>0 750<br />
t (s)<br />
15065 J m<br />
20197<br />
23753<br />
28706<br />
40081<br />
-2<br />
J m -2<br />
J m -2<br />
J m -2<br />
J m -2<br />
Figure 2 Temperature responses of PMMA measured by the PTM for different heat powers<br />
and various pulse widths; see Table 1.<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2006, strana 234<br />
ΔTm
The typical time responses of temperature for the rectangle (Dirac) pulse of different input<br />
power of heat with various pulse widths are presented in Figure 2. The heat power and width<br />
of pulse were changed for find the optimal measurement conditions and subsequently<br />
determine the reliable data of thermal diffusivity, specific heat and thermal conductivity of<br />
studied material. These results are summarized in Table 1.<br />
Table 1 Thermophysical parameters of PMMA measured in optimum exp. conditions.<br />
Q/S (J m –2 ) P (W) ΔTmax (°C) tmax (s) a (m 2 s –1 ) cp (J kg –1 K –1 ) λ (W m –1 K –1 )<br />
13 069 0.58 0.307 144 0.113 1425.0 0.190<br />
17 325 0.67 0.398 146 0.110 1457.1 0.190<br />
17 267 0.67 0.397 148 0.109 1456.9 0.188<br />
19 789 0.76 0.4<strong>60</strong> 144 0.112 1440.5 0.190<br />
17 990 0.92 0.424 148 0.113 1420.0 0.190<br />
20 197 1.21 0.477 141 0.120 1419.1 0.202<br />
23 753 1.75 0.563 139 0.124 1414.1 0.207<br />
28 706 2.88 0.680 141 0.123 1415.2 0.207<br />
40 081 2.88 0.955 148 0.115 1405.6 0.192<br />
(0.115 ± 0.005) (1428.2 ± 17.8) (0.195 ± 0.007)<br />
The thermophysical parameters were calculated from the parameters of the temperature<br />
response to the heat pulse. Typical temperature increases for reliable data were between<br />
0.3 °C and 1.0 °C that were equivalent to the heat powers of 0.58 W up to 2.88 W and to the<br />
heat energies of 13000 J m –2 up to 40000 J m –2 .<br />
The pulse transient method gives data within the experimental error less than 4.35 % for<br />
thermal diffusivity, less than 1.25 % for specific heat and 3.59 % for thermal conductivity.<br />
Average values a = 0.115 m 2 s –1 , cp = 1428.2 J kg –1 K –1 and λ = 0.195 W m –1 K –1 are in<br />
reasonable coincidence with the recommended values, see Table 2.<br />
Table 2 Recommended values of thermophysical parameters of PMMA at 25 °C [4].<br />
D (–)<br />
ρ (kg m –3 ) a (m 2 s –1 ) cp (J kg –1 K –1 ) λ (W m –1 K –1 )<br />
3.0<br />
2.5<br />
2.0<br />
1.5<br />
1.0<br />
0.5<br />
0.0<br />
1188 0.112 1450.0 0.193<br />
0 50 100 150 200 250<br />
t (s)<br />
15065 J m<br />
20197<br />
23753<br />
28706<br />
40081<br />
-2<br />
J m -2<br />
J m -2<br />
J m -2<br />
J m -2<br />
Figure 3 Fractal dimension of the heat distribution in the specimen determined from<br />
increased part of characteristics.<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2006, strana 235
The coefficient fa (fractal dimension D respectively) of the fractal heat source for every<br />
point of the experimental dependence was calculated using the Eq. (5).<br />
The fractal heat source characterizes the distribution of the temperature in the specimen in<br />
specific time. The character of these dependences differs for different thicknesses of measured<br />
sample. Dependencies of the fractal dimension on the time are plotted in Figure 3.<br />
Generally we can say that all measured results were started from the value of the fractal<br />
dimension D ≈ 3 and then they were saturated at the some constant value of the fractal<br />
dimension. This value of the fractal dimension depends on the losses during the transport of<br />
heat throw the sample.<br />
2. Study of thermophysical parameters of PMMA at 25 °C and 30 °C<br />
The measurements were carried out with the sample temperature stabilized at 25 °C and<br />
30 °C in atmosphere of air and in the same experimental conditions. The temperature<br />
response occurred in the range of 0.1 up to 1.4 °C. Experimental data was obtained for the<br />
pulse width of 4 – 40 s and the pulse heat energy of <strong>60</strong>00 up to 3<strong>60</strong>00 J m –2 .<br />
a (mm 2 s -1 )<br />
c p (J kg -1 K -1 )<br />
λ (W m -1 K -1 )<br />
0.17<br />
0.16<br />
0.14<br />
0.13<br />
0.11<br />
15705000<br />
11000 17000 23000 29000<br />
1510<br />
1450<br />
1390<br />
1330<br />
0.28<br />
0.26<br />
0.24<br />
0.22<br />
0.20<br />
5000 11000 17000 23000 29000<br />
5000 11000 17000 23000 29000<br />
Q /S (J m -2 )<br />
25 °C<br />
30 °C<br />
Figure 4 Thermophysical parameters of PMMA measured at 25 °C and 30 °C.<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2006, strana 236
Figure 4 illustrates the typical dependencies of the thermophysical parameters of PMMA<br />
on the heat energy for the heat power 1.75 W. This figure shows that thermal diffusivity of<br />
PMMA slightly decreases with the increasing temperature as well as thermal conductivity. On<br />
the contrary, the specific heat increases with the increasing temperature of measured sample.<br />
CONCLUSIONS<br />
This paper presents the results of the measurements in optimal experimental conditions and<br />
of the study of the dependency of the thermophysical parameters on the temperature of<br />
studied specimen that were obtained on PMMA. The measurements were made in air. To<br />
interpret the outcomes, the simplified heat conductivity model is used [3]. Results show the<br />
image of heat distribution in the specimen, in various time intervals after the heat supply from<br />
the source.<br />
The analysis of experimental data measured by the pulse transient method for various heat<br />
power and pulse width of measurement was performed on polymethyl methacrylate (perspex).<br />
The optimization of the procedure of conditions metering was used to find the optimal range<br />
of measuring process at 25 °C where data stability interval exists, e.g. the values of<br />
thermophysical parameters are reliable. The value of thermal diffusivity calculated from the<br />
data stability interval was determined as 0.115 m 2 s –1 , 1428.2 J kg –1 K –1 for the specific heat<br />
and the value of thermal conductivity was calculated as 0.195 W m –1 K –1 and they are close to<br />
the recommended values; see Table 2. The pulse transient method gives data within the<br />
experimental error less than 4.35 % for thermal diffusivity, less than 1.20 % for specific heat<br />
and 3.59 % for thermal conductivity. These evaluations could be used for more accurate<br />
determination of the thermal parameters of studied (homogeneous and heterogeneous)<br />
matters.<br />
Data measured on PMMA clearly show difference of thermophysical parameters measured<br />
in different temperatures for the same specimen and experimental conditions. Thermal<br />
diffusivity of PMMA slightly decreases with the increasing temperature as well as thermal<br />
conductivity. On the contrary, the specific heat increases with the increasing temperature.<br />
ACKNOWLEDGEMENTS<br />
This work was supported by the Grant Agency of the Czech Republic, contract No.<br />
2239/2006/G1.<br />
REFERENCES<br />
[1] Steiger T., Pradel R.: Update on COMAR – the Internet Database for Certified<br />
Reference Materials. Journal of Metrology Society of India. Vol. 19. No. 4. 2004.<br />
p. 203-207.<br />
[2] Boháč V., Kubičár L.: Investigation of Surface Effects on PMMA by Pulse<br />
Transient Method. In Thermophysics 2001, Meeting of the Thermophysical<br />
Society, Working Group of the Slovak Physical Society. Račkova dolina. October<br />
23, 2001. p. 21-27. ISBN 80-8050-491-1.<br />
[3] Stefkova, P., Zmeskal, O., Capousek, R. Study of Thermal Field in Composite<br />
Materials. In Complexus Mundi. WS. London, World Scientific. 2006. p. 217 –<br />
224. ISBN 981-256-666-X.<br />
[4] Vohlídal J., Julák A., Štulík K.: Chemické a analytické tabulky. Grada Publishing<br />
1999; 1. vydání; Praha 1999; 652 s. ISBN 80-7169-855-5.<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2006, strana 237
THE ANALYSIS OF GAMMA LINOLENIC ACID IN EVENING<br />
PRIMROSE OIL<br />
Hana Štoudková, 2.ročník DSP<br />
Školitel: doc. Ing. Miroslav Fišera, CSc.<br />
<strong>Vysoké</strong> učení technické v Brně, <strong>Fakulta</strong> <strong>chemická</strong>, Ústav potravinářské chemie<br />
a biotechnologie, Purkyňova 118, 61200 Brno, email: stoudkova@fch.vutbr.cz<br />
INTRODUCTION<br />
Evening primrose oil (Oenothera biennis L.) is rich source of ω-6 series of polyunsaturated<br />
fatty acids. One of these fatty acids is gamma linolenic acid – GLA (cis-6,cis-9,cis-12octadecatrienoic<br />
acid). The content of GLA is the single most important parameter to be<br />
determined [1,2].<br />
The oil content of evening primrose seeds varies with such factors as the age of the seed,<br />
cultivar and growth conditions, and typically varies between 18 and 25 %. The oil consists of<br />
about 98 % triacylglycerols, with small amounts of other lipids (free acids, diacylglycerols,<br />
phospholipides) and about 1–2% unsaponifiable matter, of which sterols and tocopherols are<br />
of some importace [1].<br />
Evening primrose oil is used in increasing amount in nutritional and pharmaceutical<br />
preparations. Essential fatty acids are vital components of all membrane structures in the body<br />
and there are also involved in the production of prostaglandins, which regulate the immune<br />
system. A prostaglandin deficiency can cause skin rashes, hair loss, lowered immunity [3,4].<br />
One of the most important unsaturated fatty acids involved in beneficial prostaglandin<br />
production is gamma linolenic acid.<br />
Gamma linolenic acid and evening primrose oil have been the focus of many medical<br />
studies. Some of these studies claim that evening primrose oil supplementation boost natural<br />
immunity, help lower cholesterol levels, reduce blood pressure, can help ease premenstrual<br />
syndrome, eczema, diabetes, osteoporosis [3,4].<br />
MATERIALS AND METHODS<br />
Samples<br />
A total of eight evening primrose oils were tested. Samples were obtained from Aromatica,<br />
v.o.s. and the other producers. Common lipophilic compounds with stabilized properties were<br />
chosen as antioxidants: coenzyme Q10, β-carotene, vitamin E and Origanox - extract of<br />
Origanum Vulgare containing flavonoids. Various ways of storage were chosen for<br />
assessment of stability of oils. Maximum time of storage was 183 days from opening the vial<br />
with sample.<br />
Methanol esterification method<br />
The fat sample (1,0 g) was saponified with 15 ml methanolic solution of potassium<br />
hydroxide (c = 0,5 mol⋅dm -3 ) for 30 minutes in distilling flask with condenser and was<br />
esterified after neutralization by sulphuric acid on methyl orange for 30 minutes again.<br />
After cooling methyl esters were sha<strong>ke</strong>n with 10 ml of heptane three times. The extract<br />
was dried by anhydrous sodium sulphate and filtered to a 50 ml volumetric flask again. Both<br />
heptane portions were rinsed with 20 ml of water twice. The extract was dried by anhydrous<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2006, strana 238
sodium sulphate and filtered to a 50 ml volumetric flask and filled up to the mark with<br />
heptane [5].<br />
GC analysis<br />
The GC method – gas chromatography was used for identification of the fatty acids. So<br />
prepared heptane methyl esters solutions were injected to gas chromatograph using<br />
autosampler. The compounds were identified according to available standards.<br />
GC conditions: gas chromatograph TRACE GC (ThermoQuest Italia S. p. A., I) equipped<br />
with flame ionization detector, split/splitless injector and capillary column SPTM 25<strong>60</strong><br />
(100 m × 0,25 mm × 0,2 μm) with the temperature programme <strong>60</strong> °C held for 2 min, ramp<br />
10 °C⋅min -1 up to 220 °C, held for 20 min. The injector temperature was 250 °C and the<br />
detector temperature was 220 °C. The flow rate of the carrier gas N2 was 1,2 ml⋅min -1 .<br />
RESULTS AND DISCUSSION<br />
Gamma linolenic acid (GLA) is found naturally to varying extents in the fatty acid fraction<br />
of some plant seed oils. The content of GLA is the one of the most important parameter for<br />
tested evening primrose oils. It is comprised of 18 carbon atoms and three double bonds.<br />
Double bonds can be cause of the reduce GLA and it leads to changes GLA during time of<br />
storage and used antioxidants.<br />
Virgin evening primrose oils<br />
Virgin evening primrose oils samples (Arnaud, Gustav Heess) were stored at 4 °C during<br />
time of storage. Evening primrose oil Arnaud was also stored under laboratory conditions.<br />
The percentage numbers GLA are shown in producer´s certificates (Arnaud – minimal 9 %,<br />
Gustav Heess - in range from 8 to 12 % GLA). The GLA amount changed significantly<br />
during storage. In the case of virgin evening primrose oil Arnaud the measure values GLA<br />
were lower for up to 1 %. In the case evening primrose oil sample Gustav Heess the content<br />
of GLA was about 8 % and it´s in the certificate range.<br />
No-stabilized sample of evening primrose oil which was held on laboratory conditions<br />
recorded a considerable loss GLA (decrease of the GLA content – 2,9 %).<br />
Commercially produced oil samples<br />
Samples of evening primrose oils with antioxidants (vitamin E, vitamin E and coenzyme<br />
Q10, vitamin E and β-carotene) obtained from Aromatica, v.o.s. The GLA content decreased<br />
during time of storage slightly. The loss GLA was observed independently of added kind,<br />
combination or amount antioxidants.<br />
Dependent the content of GLA on time of storage three samples commercially produced<br />
evening primrose oils with the addition of antioxidants is shown in Fig. 1. In the case evening<br />
primrose oil with coenzyme Q10 a vitamin E a decrease was 0,6 % GLA, the sample with<br />
β-carotene and vitamin E 1,3 % GLA and in the case evening primrose oil with vitamin E a<br />
loss was 1,0 % GLA. The decrease GLA related to the total content of fatty acids.<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2006, strana 239
GLA (%)<br />
10<br />
9<br />
8<br />
7<br />
6<br />
5<br />
4<br />
3<br />
2<br />
1<br />
0<br />
coenzyme Q10 + vitamin E β-carotene + vitamin E vitamin E<br />
1 23 52 91 106 147 183<br />
time (days)<br />
Fig. 1: Variation over time of storage in gamma linolenic acid content of commercially<br />
produced evening primrose oils with antioxidants<br />
Laboratory prepared samples<br />
Two samples (evening primrose oil with Origanox TM and evening primrose with mixture<br />
antioxidants coenzyme Q10, β-carotene and vitamin E) were laboratory prepared. In the case<br />
both of samples the GLA content changed slightly over time of storage (Fig. 2).<br />
GLA (%)<br />
10<br />
9<br />
8<br />
7<br />
6<br />
5<br />
4<br />
3<br />
2<br />
1<br />
0<br />
coenzyme Q10 + β-carotene + vitamin E Origanox<br />
1 30 51 84 125 161 183<br />
time (days)<br />
Fig. 2: Variation over time of storage in gamma linolenic acid content of laboratory<br />
prepared evening primrose oils with antioxidants<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2006, strana 240
Compare all tested evening primrose oils<br />
The GLA content was similar for the all tested evening primrose oils and range of GLA<br />
was from 5 to 11 % of total fatty acids. The content of GLA decreased gradually during time<br />
of storage. Compare of the GLA content in various tested evening primrose oils at first and<br />
last (183) day of storage is shown in Table 1.<br />
Maximum level GLA recorded virgin evening primrose oil Arnaud in 147 day of storage<br />
(10,40 ± 0,17 %). Minimal amount GLA was found in the sample evening primrose oil at 183<br />
day of storage, which was stored under laboratory conditions (5,65 ± 0,06 %). Evening<br />
primrose oil with antioxidant Origanox changed minimally. Whereas, most losses recorded<br />
no-stabilized evening primrose oil which was hold on laboratory conditions.<br />
Table 1: The content of GLA in evening primrose oils (first and last day of storage)<br />
Time of storage [days]<br />
Evening primrose oils<br />
1 183<br />
GLA [%] sr [%] GLA [%] sr [%]<br />
+ Q10 + vitamin E 8,00 ± 0,10 1,27 7,39 ± 0,03 0,38<br />
+ β-carotene + vitamin E 8,47 ± 0,09 1,07 7,24 ± 0,10 1,39<br />
+ vitamin E 8,77 ± 0,00 0,01 7,75 ± 0,04 0,53<br />
+ Q10 + β-carotene + vitamin E 7,26 ± 0,18 2,44 7,00 ± 0,06 0,86<br />
+ Origanox 8,84 ± 0,04 0,43 8,72 ± 0,18 2,07<br />
virgin, Arnaud 8,50 ± 0,14 1,63 8,01 ± 0,13 1,66<br />
virgin, Gustav Heess 8,06 ± 0,05 0,68 7,63 ± 0,02 0,26<br />
virgin, laboratory conditions 8,55 ± 0,00 0,04 5,65 ± 0,06 1,01<br />
CONCLUSION<br />
Primrose is a pure natural vegetable oil processed from the seeds of the evening primrose<br />
plant. Evening primrose oil is higher in total essential fatty acids than any other vegetable oil.<br />
The oil contains 74 % linolenic acid (LA) and 8-10 % gamma linolenic acid (GLA).<br />
The purpose of this work was to find out and compare the influences of used antioxidants<br />
and time of storage in respect of the content of fatty acids in evening primrose oil. Results of<br />
gas chromatography were statistically compared to determine the relationship between the<br />
stability of oils and the type of the antioxidant or the way of storage.<br />
A total of eight evening primrose oils (virgin or with addition of antioxidants - coenzyme<br />
Q10, β-carotene, vitamin E and Origanox) were considered.<br />
Various ways of storage were chosen for assessment of stability of oils. Methanol<br />
esterification method using potassium hydroxide catalysis was applied to oil for preparing<br />
fatty acids methyl esters. Gas chromatography (GC) was applied for the determination of fatty<br />
acids content (especially gamma linolenic acid - GLA). The compounds were identified by<br />
their retention times relative to authentic standards<br />
Gamma linolenic acid was present most often in concentration 7 – 9 % of total fatty acids<br />
in the samples of evening primrose oils. The content of GLA decreased gradually depending<br />
on increasing time of storage. Evening primrose oil, which was stored under laboratory<br />
conditions, showed the greatest losses GLA. Whereas, sample evening primrose oil<br />
containing Origanox had minimal changes. It was observed great dissimilarity GLA during<br />
another way of storage of oils with addition antioxidants. Virgin evening primrose oils have<br />
comparable levels of GLA.<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2006, strana 241
Addition of antioxidants, their amounts or combination have no influence on changes in<br />
oils during the storage. However, the way of storage can influence on the content of GLA.<br />
REFERENCES<br />
[1] Christie, W.W.: The analysis of evening primrose oil. Industrial Crops and Products,<br />
1999, vol. 10, pp. 73-83. ISSN 0926-6690.<br />
[2] Court, W. A., Hendel, J.G., Pocs, R.: Determination of fatty acids and oil content of<br />
evening primrose (Oenothera biennis L.). Food Reearch International, 1993, vol. 26,<br />
pp. 181-186. ISSN 0963-9969.<br />
[3] Fan,Y.-Y., Chapkin, R.S.: Importance of dietary γ-linolenic acid in human health and<br />
nutrition. Recent Advantages in Nutritional Science, 1998, vol. 128, pp.1411-1414.<br />
ISSN 0022-3166.<br />
[4] Horrobin, D. F.: Nutritional and medical importance of gamma-linolenic acid. Progress In<br />
Lipid Research, 1992, vol. 31, pp. 163-194. ISSN 0163-7827.<br />
[5] ČSN EN ISO 5509: Animal and vegetable fats and oils – preparing of methyl esters of<br />
fatty acids, 2000.<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2006, strana 242
IMPROVED FLUORIMETRIC DETERMINATION OF Al, Ga AND In<br />
BY MICELLE ENHANCED 8-HYDROXYQUINOLINE -5-SULPHONIC<br />
ACID COMPLEX<br />
Šimon Vojta, 3. ročník DSP<br />
Prof.RNDr.Lumír Sommer, DrSc.<br />
Faculty of Chemistry, Brno University of Technology, Institute of Chemistry and Technology<br />
of Environmental Protection, Purkyňova 118, 61200 Brno, e-mail: vojta@fch.vutbr.cz<br />
INTRODUCTION<br />
The enhancement (sensitization) of fluorescent reactions of metal ions with chelating dyes<br />
by the presence of surfactants provides an inexpensive alternative to fluorimetric methods,<br />
when determination of lower concentrations of elements is required.<br />
The complexes formed in micellar media are often characterized by high molar<br />
absorptivities, high stability over a wide pH range, and usually by large bathochromic shift<br />
caused by addition of surfactants to the binary complex formed in water. Moreover, some<br />
fluorescent binary chelates may dramatically increase their quantum yield in micellar media,<br />
providing exceptionally sensitive fluorimetric methods.<br />
The 8-hydroxyquinoline-5-sulphonic acid (QSA) should provide surfactant-sensitized<br />
reactions with Al(III), Ga(III) and In(III), because it is strong acid readily yielding<br />
a negatively charged group (sulphonate) to interact with the positive head of cationic<br />
surfactants.<br />
The importance of monitoring aluminium concentrations in the blood of patients<br />
undergoing haemodialysis has been realised because of the suspicion that aluminium<br />
intoxication is responsible for encephalopathy, oesteopathy and Alzheimer's disease.<br />
In this work, the influence of various surfactants on the reaction of QSA with Al, Ga and<br />
In is studied in details. Improved method of determination of Al, Ga, In by QSA in presence<br />
of cationic surfactant Zephyramine is brought and optimized.<br />
EXPERIMENTAL<br />
REAGENTS<br />
Aluminium standard, containing 1,000±0,002 g l -1 of Aluminium in 5% HCl, purchased<br />
from Analytica, LTD. Prague<br />
Gallium standard, containing 1,000±0,002 g l -1 of Gallium in 10% HCl, purchased from<br />
Analytica, LTD. Prague<br />
Indium standard, containing 1,000±0,002 g l -1 of Indium in 10% HCl, purchased from<br />
Analytica, LTD. Prague<br />
8-hydroxyquinoline-5-sulphonic acid – Sigma-Aldrich Co.<br />
Benzyldimethyltetradecylammonium chloride (Zephyramine ® ) – Sigma-Aldrich Co.<br />
1-ethoxykarbonylpentadecyltrimethylammonium bromide (Septonex ® )–Sigma-Aldrich Co.<br />
Dodecylbenzyldimethylammonium bromide (Ajatin ® ) – Sigma-Aldrich Co.<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2006, strana 243
Didodecyldimethylammonium bromide – Sigma-Aldrich Co.<br />
Polyoxyethylene(23) lauryl ether (Brij 35) – Sigma-Aldrich Co.<br />
Hexadecyltrimethylammonium chloride – Sigma-Aldrich Co.<br />
4-(1,1,3,3-Tetramethylbutyl)phenyl-polyethylene glycol (Triton X-100) – Calbiochem Co.<br />
APPARATUS<br />
A single beam spectrofluorimeter Aminco Bowman ® , series 2 with a xenon lamp working<br />
in a continuous regime, 4 nm slits and constant photomultiplier voltage was used for the<br />
measurements.<br />
RESULTS<br />
The 8-hydroxyquinoline-5-sulphonic acid forms with Al, Ga and In fluorescent complex with<br />
emission maxima at 495 nm (Al), 503 nm (Ga) and 505 nm (In). Excitation maxima for Al is<br />
3<strong>60</strong> nm, 365 nm for Ga and 367 nm for In. There is no fluorescence observed for isolated<br />
QSA. Excitation and emission spectra of Al-QSA in dependence on concentration of Al and<br />
reaction blank without Al are shown in Fig.1.<br />
The formation of fluorescent complex is instantaneous and fluorescence is stable at least for<br />
twelve hours (Fig.3). Fluorescence intensity grows from In to Al and strongly depends on pH<br />
(Fig.2).<br />
F i<br />
80<br />
70<br />
<strong>60</strong><br />
50<br />
40<br />
30<br />
20<br />
10<br />
1a<br />
2a<br />
3a<br />
4a<br />
5a<br />
6a<br />
6b<br />
0<br />
270 320 370 420 470 520 570 620<br />
Fig.1.: Excitation(a) and Emission(b) spectra of Aluminium(III) in the presence of 7,4.10 -5<br />
mol.dm -3 QSA in dependence on concentration of Al(III).<br />
1-1,6μg.cm -3 ,2-0,8μg.cm -3 , 3-0,4μg.cm -3 ,4- 0,2 μg.cm -3 ,5-0,05 μg.cm -3 ,6–0μg.cm -3<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2006, strana 244<br />
λ (nm)<br />
1b<br />
2b<br />
3b<br />
4b<br />
5b
Al<br />
F i<br />
80<br />
70<br />
<strong>60</strong><br />
50<br />
40<br />
30<br />
20<br />
10<br />
0<br />
0<br />
1 2 3 4 5 6 7 8 9 10<br />
Fig.2.: Fluorescence intensity dependence on pH for each complex<br />
pH<br />
The fluorescence intensity can be significantly increased in the presence of cationic<br />
surfactant and small bathochromic shift in excitation spectra is also observed. The<br />
fluorescence is increased in one hour (Fig.3) and after it reaches maximum, it’s stable at least<br />
10 hours. The best positive effect was found for Zephyramine (Fig.4.). No positive effect was<br />
observed for anionic and nonionic surfactants.<br />
20<br />
Ga<br />
In<br />
18<br />
a b<br />
Fig.3.: Comparison of emission spectra time trace for Al-QSA complex without surfactant (a)<br />
and in presence of cationic surfactant Zephyramine (b)<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2006, strana 245<br />
Al<br />
In<br />
Ga<br />
16<br />
14<br />
12<br />
10<br />
8<br />
6<br />
4<br />
2
F i<br />
90<br />
70<br />
50<br />
30<br />
10<br />
-10<br />
-30<br />
Zephyramin<br />
Septonex Ajatin<br />
DDAB<br />
Triton<br />
SDS<br />
-50<br />
0 0,002 0,004 0,006 0,008 0,01 0,012 0,014 0,016<br />
Fig.4.: Surfactants influence on Ga-HQS complex at pH 3. Septonex, Zephyramine, Ajatin,<br />
SDS – mol.dm -3 , CTAC (Hexadecyl trimethyl ammonium chloride)– c/4 (mol.dm -3 ),<br />
DDAB (Didodecyldimethyl ammonium bromide) – c/5 (mol.dm -3 ), Triton X-100 –<br />
c/4.10 -5 (ppm), Brij – c/15 (mol.dm -3 ).<br />
Critical micellar concentrations are highlighted by the fulfilled marks.<br />
The nature of the complex strongly depends on the pH. The molar ratio of Me to QSA in<br />
the binary complex determined by the continuum variations method is 1:1 in acidic range and<br />
1:3 in alkalic range (Fig.5). In the presence of cationic surfactant Zephyramine, the molar<br />
ratio of ternary complex was found to be always 1:3 (Fig.5).<br />
F i<br />
100<br />
80<br />
<strong>60</strong><br />
40<br />
20<br />
0<br />
0 0,1 0,2 0,3 0,4 0,5 0,6 0,7 0,8 0,9 1<br />
x L<br />
1<br />
2<br />
3<br />
c<br />
80<br />
<strong>60</strong><br />
40<br />
20<br />
CTAC<br />
Brij<br />
a 100<br />
b<br />
F i<br />
0<br />
0 0,1 0,2 0,3 0,4 0,5 0,6 0,7 0,8 0,9 1<br />
Fig.5.: (a) Continuum variations of Al-HQS complex at pH 4. 1-c0= 1,1.10 -4 M, 2- c0= 5,6.10 -5 M,<br />
3-5,6.10 -5 M, 0,0012M Zephyramine<br />
(b) Continuum variations of In-HQS complex. 1-c0= 3,5.10 -5 M, pH8, 2- c0= 1,8.10 -5 M,<br />
pH8, 3-1,8.10 -5 M, 0,0012M Zephyramine, pH8, 4- c0= 1,8.10 -5 M, pH4<br />
Finally, the improved method with enhanced sensitivity by the presence of cationic<br />
surfactant Zephyramine was optimized with concentration of Zephyramine 0,0012M and five<br />
times excess of QSA to highest molar concentration of Me. Optimal pH is 4 for Al, 3 for Ga<br />
and 8 for In.<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2006, strana 246<br />
x L<br />
1<br />
4<br />
2<br />
3
F i<br />
<strong>60</strong><br />
50<br />
40<br />
30<br />
20<br />
10<br />
Data<br />
UCL<br />
LCL<br />
A<br />
B<br />
Calibration Plot<br />
y = 51,982x + 0,9186<br />
R 2 = 0,9991<br />
0<br />
0 0,1 0,2 0,3 0,4 0,5 0,6 0,7 0,8 0,9 1<br />
c Al (μg.cm -3 )<br />
Fig.6.: Calibration plot for Al in the presence of 1,4.10 -4 M QSA and 0,012M Zephyramine,<br />
pH4<br />
F i<br />
80<br />
70<br />
<strong>60</strong><br />
50<br />
40<br />
30<br />
20<br />
10<br />
Data<br />
UCL<br />
LCL<br />
A<br />
B<br />
Calibration Plot<br />
y = 78,46x + 1,1235<br />
R 2 = 0,9992<br />
0<br />
0 0,1 0,2 0,3 0,4 0,5 0,6 0,7 0,8 0,9 1<br />
c Ga (μg.cm -3 )<br />
F i<br />
200<br />
180<br />
1<strong>60</strong><br />
140<br />
120<br />
100<br />
80<br />
<strong>60</strong><br />
40<br />
20<br />
Data<br />
UCL<br />
LCL<br />
A<br />
B<br />
Calibration Plot<br />
y = 185,06x + 10,75<br />
R 2 = 0,9995<br />
0<br />
0 0,1 0,2 0,3 0,4 0,5 0,6 0,7 0,8 0,9 1<br />
Fig.7.: Calibration plots for Ga (In) in the presence of 5,75.10 -4 M (resp.3,5.10 -4 ) QSA and<br />
0,012M Zephyramine, pH3 (resp. 8)<br />
Tab.1.: Comparison of calibration parameters for each complex. U is photomultiplyer voltage<br />
and detection limits were calculated according to Graham 4<br />
a<br />
c In (μg.cm -3 )<br />
Al-HQS Ga-HQS In-HQS<br />
Concentration range (μg.cm -3 ) 0,004-1<br />
U(V) 610 700 680<br />
α<br />
X D (μg.cm -3 ) 7,476E-05 0,0149 6,720E-05<br />
blank<br />
X (μg.cm -3 ) 0,0304 0,0349 0,0333<br />
β<br />
D<br />
−3<br />
( g ⋅ cm )<br />
X b μ<br />
x + 3s<br />
blank<br />
6, a 0,00015 0,00022 0,0042<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2006, strana 247
CONCLUSIONS<br />
Cationic surfactants sensitize the reaction of Al, Ga and In with 8-hydroxyquinoline-5sulphonic<br />
acid. The effect of various surfactants was studied in details. The best results were<br />
obtained for Zephyramine and Septonex. The nature of the complex was investigated by the<br />
continuum variations method. Improved method for determination of Al, Ga and In in<br />
aqueous solutions was optimized and calibration plots were constructed.<br />
REFERENCES<br />
1. Vercruysse A.: Hazardous Metals in Human Toxicology, Part B, Elsevier Amsterdam<br />
1984<br />
2. Garcia Alonzo J. I., Diaz Garcia M. E., Sanz Medel A.: Talanta. 31, 361 (1984)<br />
3. Hinze W. L., Singh H. N.: Trends in Analytical Chemistry. 3, 193 (1984)<br />
4. Graham R. C.: Data Analysis for the Chemical Sciences. A Guide to Statistical<br />
Techniques. VCH Publishers, New York 1993<br />
5. ČSN ISO 8466-1: 1993, 15. Praha, 1993.<br />
6. Anonyme: Anal.Chem. 52, 2245 (1980)<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2006, strana 248
PHYSICAL-CHEMICAL ASPECTS OF PAINT FILMS DEGRADATION<br />
Ing. Lucie Wolfová<br />
Supervisor: prof. RNDr. Zdeněk Friedl, CSc.<br />
Brno University of Technology, Purkyňova 118, Brno 612 00, e-mail:wolfova@fch.vutbr.cz<br />
INTRODUCTION:<br />
My dissertation thesis deals with the physical-chemical aspects of paint film degradation.<br />
This work is focused on the study and examination of physical-chemical aspects of paint film<br />
degradation by selected solvents. Mainly it is considered to the determination of solubility<br />
parameters of used polymers δ2 and the examination of the influence of especially polymer<br />
structure to the value of this δ2. The main goal of this study is to verify the coating solubility<br />
theory based on the knowledge of their solubility parameters in practise and its application for<br />
coatings. The other goals include the projection and formulation some new solvent’s mixture<br />
and additives intended for removing paint films and graffiti. Such formulation need to be well<br />
performing and ecological acceptable at the same time, which is the precondition for their<br />
wide application in practise.<br />
THEORETICAL BACKGROUND:<br />
Chemical degradation of paint films could be defined as the physical and chemical<br />
interactions between film-forming components of the paint and chemical surroundings.<br />
During the chemical degradation of polymer a disruption of chains and reticulation, a change<br />
of chemical structure of chains, a change of side groups or combination of all these<br />
phenomena ta<strong>ke</strong> place. This leads to changes in paint qualities and coats lose their visual<br />
appearance and original qualities upon the exposure to organic solvents.<br />
From the thermodynamic point of view, a solubility of polymer and a degree of solvent<br />
affinity to polymer is given by the Gibb´s energy change, which is determined by the<br />
interaction between solvent molecules and macromolecular chain elements. ΔGmix = ΔHmix -<br />
TΔSmix < 0.<br />
The more negative is value of ΔGmix the better is solubility of polymer in solvent. Then the<br />
dissolving of polymers is mainly a question of value ΔHmix because during swelling or<br />
dissolving always increase system’s disorderliness. According to thermodynamic conditions<br />
than is necessary that ΔHmix
Then, the solubility parameter can be estimated from general equations:<br />
ΔHmix = Vs(δ1 – δ2) * φ1 φ2 .<br />
- This is one of the simple expressions of solubility parameter deduced for nonpolar<br />
systems. In practise, we have solvents and solutions with different polar forces such as<br />
dispersion, polar and hydrogen ones. Therefore the cohesive energy Ecoh is divided into three<br />
parts, corresponding to three types of interaction forces Ecoh = Ed + Ep + Eh (contribution of<br />
dispersion forces Ed, polar forces Ep and hydrogen-bonding Eh). Then every substance could<br />
be characterized according to free contributions of total solubility parameter<br />
2 2 2<br />
δtotal.= σ + σ + σ .<br />
disp.<br />
polar.<br />
hydrogen.<br />
By this is predicted that if δ1 = δ2 the ethalpy of mixing is ΔHmix = 0 and ΔGmix = ΔHmix -<br />
TΔSmix < 0 and this is in accorrdance with the general rule for thermodynamic view of<br />
solubility and mixing. Then, the smaller is the difference of value δ of solvent and polymer,<br />
the better is possibility of its dissolving. The polymers should be mostly soluble in that<br />
solvents, which their < δ > correspond to the middle of the interval of polymer solubility or<br />
approximately ± 2 units.<br />
AIM OF STUDY:<br />
The aim of conducted research is the evaluation of influence of several aspects – especially<br />
structure of samples – on the chemical resistance of PUR coatings and the value of their<br />
solubility parametr δ2. As far as these aspects are concerned, these include especially the type<br />
of used monomers (in case of PUR polymers for example li<strong>ke</strong> type and funcionality of used<br />
alcohols, acids and isocyanates, type of bonds, etc.), the weight, size and type of film forming<br />
polymer and the type of interactions between individual chain parts.<br />
METHOLOGY:<br />
The solubility parameter δ2 of polymer could be determined according to its interaction<br />
with solvents of known solubility parameter δ1. For example, the solubility parameter of a<br />
linear polymer can be determined from its limiting viscosity number and of a cross-lin<strong>ke</strong>d<br />
one according to the degree of swelling, both of these values being the function of solubility<br />
parameter of used solvent δ1. In case of best solvent, is the value of [η] or Q of polymer the<br />
heights and solubility parameter of this solvent δ1 correspondents with solubility parameter of<br />
polymer δ2<br />
As far as measuring of viscosity is concenred, the samples were dissolved in selected<br />
group of solvents whose solubility parameters δ1 varied from 18 to 24 (30). Then their<br />
dynamic viscosity η were masured. It was done using capillar or Ubelohde viscosimeter.<br />
η sp<br />
Subsequently, specific viscosity ηspec and the limiting viscosity number [η] = lim were<br />
c→0<br />
c<br />
calculated.<br />
The degree of sample’s swelling Q = (m-m0/m0)*1/ςsolv was calculated by weighting<br />
samples after soaking in chosen solvents for 5 days.<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2006, strana 250
Finally, the solubility parameter of sample δ2 was determined as a function of [η] or Q and<br />
compared with values of these δ2 obtained by calculation from sample’s structures. The<br />
calculation of δ2 from sample’s structures is possible from the contibutions of dispersion Ed,<br />
polar Ep and hydrogen-bonding Eh forces of the polymer.<br />
RESULTS AND DISCUSSION:<br />
The exact sample composition can not be published at the moment because of their<br />
intended patent protection. Therefore, the samples are named by the manufacturer code.<br />
The result of influence of different structure of PUR samples to the value of their<br />
solubility parameter δ2 (10 -3 J 1/2 m -3/2 ):<br />
- Differences in sample structures were especially polymer’s molecular weight and size of<br />
chain, type and functionality of used alcohols, acids and isocyanates.<br />
Limiting viscosity number of<br />
samples [η] (m 3 /kg)<br />
Solubility parametrs of PUR samples δ2 determined according to<br />
0,09<br />
limiting viscosity number [η]<br />
U14EG2000<br />
0,08<br />
Diexter G214<br />
C36EG34<br />
0,07<br />
Priplast 3192<br />
0,06<br />
0,05<br />
0,04<br />
0,03<br />
0,02<br />
0,01<br />
0,00<br />
19,0 19,5 20,0 20,5 21,0 21,5 22,0 22,5 23,0 23,5 24,0 24,5 25,0 25,5 26,0<br />
Solubility parametr of solvent δ 1(10 -3 J 1/2 m -3/2 )<br />
U14EG56<br />
U14BD2000<br />
According to the theory, the solubility parameter δ2 value corresponds to the point where<br />
the plot of [η] as a function of δ1 has its maximum.<br />
Determination of solubility parameter δ2 as the function of limiting viscosity number<br />
and by calculation from structure:<br />
Solvents:<br />
Solubility<br />
parameter<br />
δ1<br />
Limiting viscosity number of tested samples [η] (m 3 /kg)<br />
U14EG2000 Diexter C36EG34 Priplast U14EG56 U14BD2000<br />
Dichloromethane 19,8 0,0076 0,0173 0,0094 0,0071 - 0,0266<br />
1,4-dioxane 20,5 0,0135 0,0117 0,0093 0,0048 0,0515 0,0540<br />
Acetyl acetone 22,1 0,0180 0,0087 0,0199 0,0068 0,0856 0,0570<br />
N,Ndimethylacetamide 22,7 0,0618 0,0337 0,0677 0,0243 0,0638 0,0580<br />
N methylpyrrolidone 22,9 0,0150 0,0425 0,0193 0,0163 0,0686 0,0514<br />
N,Ndimethylformamide 24,7 0,0400 0,0144 0,0000 0,0185 - 0,0413<br />
Solubility parameter δ2(10 -3 J 1/2 m -3/2 )<br />
Determined from measuring 22,7 22,9 22,7 22,7 22,1 22,7<br />
Determined from structure 21,37 23,87 21,31 21,78 20,92 21,25<br />
According to the obtained results and the theory, we can predict that the total solubility<br />
parameters δ2 of our tested PUR samples are about 22,7×10 -3 J 1/2 m -3/2 and in case of U14EG56<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2006, strana 251
22,1×10 -3 J 1/2 m -3/2 . However, if we calculate the value δ2 from the structure, we obtain<br />
approximately similar values of δ2 in a range of 20,9 to 23,9×10 -3 J 1/2 m -3/2 . The biggest<br />
differences are in case of Diexter G214 and U14EG56 where the biggest differences between<br />
their structures are also.<br />
According to these results, it should be possible to dissolve these samples in solvents with<br />
total solubility parameter value of δ1 about 20 to 24 units.- And in most cases, experimental<br />
results comply with theory and samples could be dissolved in solvents with value of δ1≈δ2 ± 2.<br />
Solubility of PUR sample U14EG56 in various type of solvent:<br />
solubility U14EG56<br />
solubility U14EG56<br />
Solvent: p. δ1 δ2 ≈ 22,1 Solvent:<br />
p. δ1 δ2 ≈ 22,1<br />
Toluene 18,2 * N,N dimethylacetamide 22,7 +<br />
Ethylmethyl<strong>ke</strong>tone 19 + N methylpyrrolidone 22,9 +<br />
Dichloromethane 19,8 + Isopropanol 23,6 -<br />
Acetone 20 * N,N dimethylformamide 24,7 +<br />
1,4-Dioxane 20,5 + Ethanol 26 -<br />
Acetyl acetone 22,1 + Butyrolactone 27 -<br />
Ethylene glycol 30 -<br />
In the case, when sample is not soluble in right one solvent according to the theory, it can<br />
be interpreted by different chemical nature of used solvents and it relates to different value of<br />
individual components of their total solubility parameter.<br />
The result of study of influence of different values of rigid segment fraction and crosslin<strong>ke</strong>d<br />
fraction content of sample U14BD2000 to its solubility parameter, according its<br />
parameter of swelling:<br />
During the study of the influence of rigid segment content and cross-lin<strong>ke</strong>d fraction<br />
content to the solubility parameter, it was observed that the more rigit segment fraction and/or<br />
cross-lin<strong>ke</strong>d fraction is present, the more resistent the sample was. All these changes should<br />
be connected with a changes of sample solubility parametr and should be observable<br />
experimentally.<br />
Study of the influence of different percentage distribution of rigid segment fraction in<br />
case of sample U14BD2000:<br />
Parameter of swelling Q<br />
(Type A with the less quantity, type D with the most quantity of rigid segment content)<br />
16<br />
14<br />
12<br />
10<br />
8<br />
6<br />
4<br />
2<br />
Influence of different quantity of rigid segment to the<br />
solubility parametr of U14BD2000 (according its<br />
parameter of swelling)<br />
type A<br />
type B<br />
type C<br />
type D<br />
0<br />
19,0 20,0 21,0 22,0 23,0 24,0 25,0<br />
Solubility parameter of solvent δ1 (10-3J1/2m -3/2 )<br />
Type of<br />
sample:<br />
δ2 - from<br />
measuring<br />
δ2 - from the<br />
structure<br />
A 19,8 20,11<br />
B 20,5 20,72<br />
C 22,9 21,34<br />
D 22,9 21,96<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2006, strana 252
Study of the influence of different percentage distribution of cross-lin<strong>ke</strong>d fraction in<br />
sample U14BD2000:<br />
(Type AA with the less quantity, type DD with the most quantity of cross-lin<strong>ke</strong>d fraction<br />
content)<br />
Parameter of swelling Q<br />
14,00<br />
12,00<br />
10,00<br />
8,00<br />
6,00<br />
4,00<br />
2,00<br />
Influence of different quantity of cross-lin<strong>ke</strong>d to the<br />
solubility parametr of U14BD2000 (according its<br />
parameter of swelling)<br />
type AA<br />
type BB<br />
type CC<br />
type DD<br />
0,00<br />
18,0 19,0 20,0 21,0 22,0 23,0 24,0 25,0<br />
Solubility parameter of sample δ1 (10-3J1/2m -3/2 )<br />
Obtained results show that in the case of the increasing quantity of rigid segment the<br />
solubility parameter δ2 is changing and moving to higher values. In the case of increasing of<br />
cross-lin<strong>ke</strong>d fraction, the value of δ2 is more or less stable and similar and changes only<br />
parameter of swelling. – Both of these agree with the theory.<br />
CONCLUSION:<br />
Type of<br />
sample:<br />
δ2 - from measuring<br />
AA 19,8<br />
BB 20,5<br />
CC 20,5<br />
DD 20,5<br />
According to the obtained data, experimental results comply with the theory about possible<br />
evaluation of coating’s solubility on the base of knowledge their solubility parameters and<br />
also structures in most cases.<br />
In next experimental part of my dissertation work I would li<strong>ke</strong> to improved, extend and<br />
apply this theory and information to the area of dissolving and removing especially water<br />
based coats and modify all of this for use in practice.<br />
Acknoledgments:<br />
I would li<strong>ke</strong> to thank to C. B. Coreia and Prof. J. C. Bordado, Instituto Superior Technico,<br />
Lisboa for their help and support during this research.<br />
LINKS:<br />
1. Barton A. F. M., Handbook of solubility parameters and other cohesion parameters,<br />
London 1991<br />
2. Van Krevelen D. W., Hoftyzer P. J., Properties of polymers – their estimation and<br />
correlation with chemical structure, Amsterdam 1976<br />
3. Bicerano J., Prediction of polymer properties (second edition), New York, 1996<br />
4. Munk P., Aminabhavi T.M., Macromolecular science (second edition), New York, 2002<br />
5. Mleziva J., Šňupárek J., Polymery-výroba, struktura, vlastnosti a použití, Praha, 2002<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2006, strana 253
Using low-molecular mass pI mar<strong>ke</strong>rs in proteomic staining-free method<br />
for study of posttranslationally modified proteins<br />
Karel Mazanec 1,2 , Karel Šlais 2 and Josef Chmelík 2<br />
1 Institute of Material Chemistry, Faculty of Chemistry, Brno University of Technology,<br />
Purkyňova 118, 612 00 Brno, Czech Republic, e-mail: mazanec@iach.cz<br />
2 Institute of Analytical Chemistry, Academy of Sciences of the Czech Republic, Veveří 97,<br />
<strong>60</strong>2 00 Brno, Czech Republic<br />
INTRODUCTION<br />
At present the knowledge of proteome plays very important role in the study of living<br />
processes. Special attention in our lab is paid to the investigation of the proteome of barely<br />
grains. The aim is to contribute to understanding of malting processes and to selection of<br />
malting barley cultivars. Proteins are the major functional molecules of life made of amino<br />
acids arranged in a linear chain lin<strong>ke</strong>d together by peptide bonds. The residues in a protein are<br />
often chemically altered in a process known as post-transcriptional modification (PTM):<br />
either before the protein can function in the cell, or as part of control mechanisms. The<br />
identification of the PTM is experimentally difficult.<br />
Due to the fact that many samples, mainly of biological origin, are complex mixtures of<br />
proteins with a wide range of molecular masses, salts and other compounds, proteins should<br />
be separated and purified prior to their identification by mass spectrometry. Traditionally,<br />
most proteomics researches are based upon two-dimensional gel electrophoresis involving<br />
isoelectric focusing (IEF) as one dimension. Separated compounds are focused into very<br />
sharp bends according to their pI values during IEF. They are twice or more concentrated in<br />
this zones. However, ampholytes present in gel and creating pH gradient during IEF<br />
complicate the visualizing the separated proteins by staining and ma<strong>ke</strong> this method<br />
impracticable for common proteomic utilization.<br />
The aim of this work was to develop and optimize the novel staining-free proteomic<br />
procedure for separation of intact proteins by gel IEF in presence of low-molecular mass pI<br />
mar<strong>ke</strong>rs and subsequent determination of molecular masses of separated compounds in the<br />
gels by MALDI-TOF/TOF MS. The selected set of pI mar<strong>ke</strong>rs includes both colored and<br />
colorless compounds with known low-molecular mass. Thus, the identification of both pI<br />
mar<strong>ke</strong>r and proteins in the same excised piece of gel by MS technique is expected to give<br />
reliable information about the correct pI values of analyzed proteins even in complex samples.<br />
MATERIALS AND METHODS<br />
pI mar<strong>ke</strong>rs – The substituted phenols (I - Mw 314.08, pI 3.9, yellow color; II - Mw 359.10,<br />
pI 4.3, orange; III - Mw 272.06, pI 5.3, yellow; IV - Mw 252.12, pI 5.7, colorless; V - Mw<br />
285.09, pI 6.4, yellow; VI - Mw 315.10, pI 7.0, yellow; VII - Mw 337.17, pI 7.5, yellow; VIII<br />
– Mw 265.14, pI 7.9, yellow; IX - Mw 363.23, pI 8.4, yellow; X - Mw 267.16, pI 8.9, yellow;<br />
XI - Mw 352.20, pI 9.0, colorless; XII - Mw 333.21, pI 10.1, yellow) used as pI mar<strong>ke</strong>rs were<br />
prepared by the procedure described by Šlais and Friedl 1 at the Institute of Analytical<br />
Chemistry (Brno, Czech Republic). The pI values were determined by potentiometric<br />
titration. The chemical structures of pI mar<strong>ke</strong>rs are shown in Fig. 1.<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2006, strana 254
Fig.1: Chemical structures of pI mar<strong>ke</strong>rs used in this work.<br />
Polyacrylamide gels and IEF - The protein samples (2 mg/ml) were mixed with pI mar<strong>ke</strong>rs<br />
(10 mg/ml each) and the 4 μl of the mixtures were loaded onto the polyacrylamide gel (16%).<br />
The carrier BioLyte 3/10 ampholyte (final concentration 2%) (Bio-Rad, CA, USA) were<br />
added prior to gel polymerization. Separation was performed with constant electric power of<br />
0.6 W for 2 h supplied by VNZ 22 power supply (CSAV Development Workshop, Czech<br />
Republic) using a model 111 Mini IEF Cell (Bio-Rad, CA, USA). After focusing completion<br />
the gels were gently removed from electrodes and scanned.<br />
Ladders of color pI mar<strong>ke</strong>rs simplify the orientation in the gel even when the separated<br />
proteins cannot be seen. Thus, the bands with proteins were excised according to positions of<br />
pI mar<strong>ke</strong>rs. The excised gel pieces were then transferred to 0.5 ml tubes. The pI mar<strong>ke</strong>rs were<br />
then eluted from gel by 30 μl water/ethanol 1:1 (v/v) for 15 min.<br />
Protein digestion and identification - Excised gel pieces after the extraction of the pI mar<strong>ke</strong>rs<br />
were washed twice with water/acetonitrile 1:1 (v/v) for 15 min. Then proteins were identified<br />
according to standard proteomic protocol utilizing in-gel digestion of reduced and alkylated<br />
proteins with trypsin. 2<br />
For comparison, the other IEF focused gels were stained with Coomassie Brilliant Blue R<br />
250. Due to the carrier 3/10 ampholytes used for creating of pH gradient in gels precipitated<br />
the Coomassie dye during staining causing strong blue haze on the background, the washing<br />
step (10% TCA in water/methanol (10/3 v/v); 12-14 h) had to be inserted prior to own<br />
staining procedure.<br />
Mass spectrometry - MS measurements were carried with 4700 Proteomics Analyzer<br />
(Applied Biosystems, USA) MALDI TOF/TOF mass spectrometer (equipped with Nd/YAG<br />
laser; 355 nm). Argon was used as a collision gas. A solution of sinapinic acid (SA) (20<br />
mg/ml in acetonitrile/water (3/2 v/v)) was used as matrix for mar<strong>ke</strong>rs. Alpha-cyano-4hydroxycinnamic<br />
acid (CHCA) (both Sigma-Aldrich, Germany) was used as matrix for<br />
peptides in concentration of 10 mg/ml acetonitrile/water (3/2 v/v).<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2006, strana 255
RESULTS<br />
More than forty pI mar<strong>ke</strong>rs of different types of structures were studied by MALDI(LDI)-<br />
TOF MS. Twelve selected suitable pI mar<strong>ke</strong>rs are used and more closely characterized in this<br />
work.<br />
At analysis of low-molecular mass pI mar<strong>ke</strong>rs, several features were observed that help to<br />
their identification. In the positive ion mode mass spectra of nitro-substituted pI mar<strong>ke</strong>rs the<br />
characteristic peaks at -16 and -32 Da caused by loss of oxygen atoms during the process of<br />
ionization (also reminded by Strohalm et al. 3 ) can be seen (Fig. 2, spectrum a, c). A<br />
characteristic double-peak pattern is obtained for mar<strong>ke</strong>rs containing a chlorine atom (Fig. 2,<br />
spectrum b, c). These groups of peaks are very helpful for identification of pI mar<strong>ke</strong>rs<br />
especially in complex mixtures where one peak may be hidden with a cluster or by another<br />
well-ionizable substance.<br />
Fig.2: The positive ion mass spectra of the pI mar<strong>ke</strong>rs XII (a); II (b) and I (c) obtained after<br />
extraction from the IEF gel. A characteristic peak patterns are clearly seen.<br />
Isoelectric focusing and gel treatment - The 3/10 carrier ampholytes were used to create pH<br />
gradient in this work. At 50-mm distance of electrodes it means that the slope of the gradient<br />
is 0.14 pH/mm. The color pI mar<strong>ke</strong>rs enable the orientation in gels and supervision of the IEF<br />
process. Each abnormality in pH gradient can be immediately and very easily recognized.<br />
Moreover, the gels can be cut up according to the position of the visible mar<strong>ke</strong>rs. The IEF<br />
gels were immediately after focusing scanned (Fig. 3A) and the bands with focused pI<br />
mar<strong>ke</strong>rs were excised. For comparison, another IEF gel was stained with Coomassie Brilliant<br />
Blue R 250. The focused proteins visualized by Coomassie staining are shown in Fig. 3B,C.<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2006, strana 256
Identification of pI mar<strong>ke</strong>rs - First, the pI mar<strong>ke</strong>rs were extracted from each excised gel<br />
piece and identified from extracts by MALDI-TOF/TOF MS. The experiments confirmed that<br />
pI mar<strong>ke</strong>rs are easily and unambiguously identifiable in extracts from excised gel pieces after<br />
IEF.<br />
Fig.3: IEF gel of pI mar<strong>ke</strong>rs and proteins scanned immediately after focusing (A); standards<br />
(B) and beta-amylase extract (C) after Coomassie staining. The positions of electrodes are<br />
mar<strong>ke</strong>d with E. Letters at right show positions where the proteins were identified (see Tab. 1).<br />
Identification of proteins - First experiment was provided with standard proteins (cytochrome<br />
c, myoglobin and albumin). After extraction of pI mar<strong>ke</strong>rs from particular gel pieces, the<br />
proteins in the same gel pieces were treated by enzyme digestion and identified by MS. These<br />
proteins were identified in positions according to their real pI values and validated our<br />
approach.<br />
Then, the extract from barley malt was used as a model mixture of glycated proteins. The<br />
separated protein mixture after the referential Coomassie staining is shown in Fig. 3C. Table 1<br />
shows the proteins identified in the gel pieces from which the pI mar<strong>ke</strong>rs were extracted and<br />
identified. Nine proteins were identified in eight excised gel pieces of twelve ones excised<br />
according to the locations of the pI mar<strong>ke</strong>rs used. Some proteins were found in two different<br />
gel pieces. During malting process the barley proteins are gradually cleaved and modified.<br />
This is the reason why some proteins may be found in several gel pieces with different pI<br />
values. The differences observed for some other proteins might be caused either by<br />
posttranslational modifications or by discrepancies between experimental and theoretically<br />
calculated pI values. For more detailed studies, the gels can be excised into a higher number<br />
of narrower pieces and the pI values of proteins identified between the locations of pI mar<strong>ke</strong>rs<br />
can be interpolated or a higher number of pI mar<strong>ke</strong>rs can be used. The identifications of the<br />
proteins were confirmed from the stained gel by the same proteomic protocol.<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2006, strana 257
Gel<br />
pos. Name<br />
Protein<br />
Number<br />
pI mar<strong>ke</strong>r<br />
Theor.<br />
pI<br />
No. pI<br />
a Protein Z (Z4) (Major endosperm albumin) P06293 5.70 II 4.3<br />
b Beta-amylase P1<strong>60</strong>98 5.58 III 5.3<br />
Glucan endo-1,3-beta-glucosidase GV Q02438 6.92<br />
c Protein Z (Z4) (Major endosperm albumin) P06293 5.70 IV 5.7<br />
d Granule-bound starch synthase 1 P09842 7.06 VI 7.0<br />
Alpha-amylase/subtilisin inhibitor precursor P07596 7.78<br />
e Glucan endo-1,3-beta-glucosidase GV Q02438 6.92 VII 7.5<br />
f Lichenase II precursor P12257 9.0 IX 8.4<br />
Elongation factor 1-alpha Q40034 9.15<br />
g 26 kDa endochitinase 2 precursor P23951 8.83 XI 9.0<br />
Histone H3 (Fragment) P06353 9.73<br />
h 26 kDa endochitinase 2 precursor P11955 8.54 XII 10.1<br />
Elongation factor 1-alpha Q40034 9.15<br />
Tab.1: The list of proteins identified directly in the same gel piece as the pI mar<strong>ke</strong>r. Standard<br />
proteins are bolded.<br />
CONCLUSIONS<br />
The method is based on separation of intact proteins by gel IEF along with low-molecular<br />
mass pI mar<strong>ke</strong>rs and subsequent sequential determination of molecular masses of separated<br />
compounds by MALDI-TOF/TOF MS. The identification of both pI mar<strong>ke</strong>rs and proteins in<br />
the same gel piece give reliable information about the correct pI values of particular identified<br />
proteins in complex samples. Moreover, it allows omitting protein staining and destaining<br />
procedure, because the Coomassie procedure is in the case of IEF gels complicated by<br />
presence of ampholytes causing dark background. The suggested procedure shortens roughly<br />
by half the time of analysis. The choice of appropriate ampholytes allows to modify the pH<br />
gradient and to study only the certain area of the pH scale.<br />
There are only twelve mainly colored pI mar<strong>ke</strong>rs shown in this work. Nevertheless, the<br />
number of pI mar<strong>ke</strong>rs used may be much higher. The color of pI mar<strong>ke</strong>rs is not relevant for<br />
MS determination. Focused pI mar<strong>ke</strong>rs may cover whole pH range and from gels narrow<br />
pieces can be cut out, which will allow more precise determination of identified proteins.<br />
Utilization of low-molecular mass pI mar<strong>ke</strong>rs with IEF gels was chosen as a first example of<br />
their applicability because the gel process can be easily observed visually. There is a good<br />
chance to use this approach also in other IEF techniques (e.g. in capillaries or on chips).<br />
REFERENCES<br />
[1] Šlais K, Friedl Z. Low-molecular-mass pI mar<strong>ke</strong>rs for isoelectric focusing. Journal of<br />
Chromatography A 1994; 661: 249.<br />
[2] Shevchenko A, Wilm M, Vorm O, Mann M. Mass spectrometric sequencing of proteins<br />
from silver stained polyacrylamide gels. Analytical Chemistry 1996; 68: 850.<br />
[3] Strohalm M, Santrucek J, Hynek R, Kodicek M. Analysis of tryptophan surface<br />
accessibility in proteins by MALDI-TOF mass spectrometry; Biochemical and<br />
Biophysical Research Communications 2004; 323: 1134.<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
Sekce DSP 2006, strana 258
Název: Sborník příspěvků soutěže Studentské tvůrčí činnosti „STUDENT 2006” a doktorské<br />
soutěže „O cenu děkana 2005 a 2006”<br />
Editor: Mgr. Radek Přikryl, Ph.D., doc. Ing. Martin Weiter, Ph.D., prof. Ing. Ladislav Omelka, DrSc.<br />
Vydání: První<br />
Počet stran: 259<br />
Vydavatel: <strong>Vysoké</strong> učení technické v Brně, <strong>Fakulta</strong> <strong>chemická</strong><br />
Výroba: <strong>Vysoké</strong> učení technické v Brně, <strong>Fakulta</strong> <strong>chemická</strong><br />
ISBN: 80-214-3321-3<br />
Publikace nebyla jazykově upravena<br />
Sborník soutěže Studentské tvůrčí činnosti Student 2006 a doktorské soutěže O cenu děkana 2005 a 2006<br />
strana 259