chemical physics of discharges - Argonne National Laboratory
chemical physics of discharges - Argonne National Laboratory
chemical physics of discharges - Argonne National Laboratory

Create successful ePaper yourself
Turn your PDF publications into a flip-book with our unique Google optimized e-Paper software.
)i<br />
1<br />
reesonably complete and accurate synthesis to describe the macroscopic character-<br />
istics. This approach <strong>of</strong> course implies that one has a detailed understanding <strong>of</strong><br />
the processes involves and know tile rates and cross sections for the processes.<br />
While one can use this approach succesfully up to a point, the extreme complexity<br />
4 <strong>of</strong> discharge phenomena forces one also to formulate a macroscopic description<br />
, and work toward a juncture with the microscopic point <strong>of</strong> view.<br />
1<br />
Chemical Physics <strong>of</strong> Discharges<br />
\.lade L. Fite<br />
University <strong>of</strong> Pittsburgk<br />
Introduction<br />
_I___<br />
The gas discharge as a <strong>chemical</strong> tool is <strong>of</strong> interest for the environment<br />
which it provides, an environment which in many respects is very close to and in<br />
others very far awa:: from thermal and <strong>chemical</strong> equilibrium. Since achieving +.his<br />
situation in tne gas discharge is a result <strong>of</strong> the ph:rsical processes occurrir,g to<br />
sustain the electrical characteristics <strong>of</strong> the discharge it is appropriate to<br />
consider sone <strong>of</strong> the 5asic physical aspects <strong>of</strong> gas <strong>discharges</strong> before examining<br />
the <strong>chemical</strong> consequences.
2<br />
I1 Electron Motion and Behavior<br />
Be it supposed that a gas is enclosed in a container and that a free electron<br />
has been produced--perhaps by a cosmic ray, or by the radiation fro4 the experimenter's '<br />
watch dial, or from that little piece <strong>of</strong> uranium glass in a graded seal, or by I<br />
cold cathode emission by the very strong electric fields around a sharp point on<br />
an electrode in the discharge tube.<br />
I<br />
If a dc electric field is imposed, the electron will respond according to \<br />
Newton's law,<br />
-&€<br />
"= nf<br />
and begin to accelerate in free fall. This acceleration will continue until it<br />
has a collision with a gas molecule. If the energy <strong>of</strong> the electron at the time<br />
<strong>of</strong> collision is very low, only elastic scattering will be admitted. At higher<br />
energies large amounts <strong>of</strong> energy can be lost by the electron in exciting the molecule<br />
to high-lying states <strong>of</strong> internal energy and at higher energies yet, ionization can<br />
occur. The latter process is, <strong>of</strong> course, essential to achieve the electron multi-<br />
plication and convert the gas with a single electron in it into a gaseous medium<br />
<strong>of</strong> high electrical conductivity.<br />
Since each type <strong>of</strong> collision has separate consequences for the discharge ve<br />
consider them in turn. The collisions are <strong>of</strong> course statistical and we must<br />
inco_rporate statistics with particle mechanics in their treatment.<br />
A. Elastic Collisions: Transfer <strong>of</strong> Energy<br />
In elastic collisions <strong>of</strong> electrons with heavy gas molecules, two effects are<br />
<strong>of</strong> importance. First there is a redistribution <strong>of</strong> directions <strong>of</strong> travel <strong>of</strong> the<br />
electrons and second there is a very slight loss <strong>of</strong> energy (and therefore speed)<br />
as a very small mount <strong>of</strong> momentum and energy are transferred to the molecule by<br />
the electron.<br />
At low energies, i.e., a few ev and less, electron scattering tends to be<br />
isotropic in angle in the center <strong>of</strong> mass coordinates. This implies that if we<br />
examine many electrons immediately after they have had a collision the average<br />
vector velocity will be zero, but <strong>of</strong> course the average scalar speed will not be<br />
zero. The probability per unit time that an electron will have a collision<br />
(i.e. the collision frequency) is given by<br />
wtiere n is the number density <strong>of</strong> molecules, c is the electron's speed and Q(c)<br />
is the total cross section for elastic collisions. The cross section generally<br />
diminishes with increasing speed, although resonances in scattering impart<br />
considerable structure in the Punctional form <strong>of</strong> the cross section in the case<br />
<strong>of</strong> most gases.<br />
We suppose that an electric field, E, is imposed in the z direction, and<br />
(1)
3<br />
inquire about the component <strong>of</strong> velocity in the z direction, averaged over all<br />
electrons, , which is identical to . Since each electron responds to the<br />
field in the same way, the average acceleration due to the field isthat <strong>of</strong><br />
each electron separately. Further, because the average velocity is 'zero following<br />
a collision, the loss <strong>of</strong> average velocity due to collisions is to good approximation<br />
just the product <strong>of</strong> the average collision frequency and the average velocity.<br />
Thus we can write that for the average velocity in the z direction<br />
When the steady state is achieved the left side vanishes, which implies that the<br />
pickup <strong>of</strong> directed velocity from the field is just balanced by the loss <strong>of</strong> directed<br />
velocity due to the randomization <strong>of</strong> direction <strong>of</strong> motion by the collisions. Under<br />
steady state collisions , then,<br />
where p is designated the mobility.<br />
Equation (4) can be re-written in terms <strong>of</strong> Eq. (2) as<br />
In this expression the second term gives all the information about the interaction<br />
between the electron and the particular gas molecule and the third term contains<br />
the parameters available to the experimenter, i.e., the electron field and the<br />
gas number density. Since the number density is proportional to the gas pressure,<br />
the experimental quantity <strong>of</strong> major relevance is the ratio E/p.<br />
Randomization <strong>of</strong> directions <strong>of</strong> velocity says nothing whatever about average<br />
speeds <strong>of</strong> the particles. For this we turn to other considerations. Conservation<br />
<strong>of</strong> momentum requires that the energy loss from an electron <strong>of</strong> mass m and kinetic<br />
energy W in collision with a heavy particle <strong>of</strong> Mass M is given by
4<br />
where 0 is the angle <strong>of</strong> deflection <strong>of</strong> the electron in the collision. If the<br />
scnttering is isotropic (as it close1.v approximates at low energies) then the<br />
energy loss in a collision averaged over angle is ,<br />
where for convenience we let 2m/M = h.<br />
We would expect a steady state ultimately to be achieved between the electron<br />
energy picked up from the electric field between collisions and the energy transmitted<br />
from the electron tc the heavy particles through the collisions. We proceed to<br />
evaluate these separately. For simplicity we assume that the collision frequency<br />
is independent <strong>of</strong> speed <strong>of</strong> the electrons.<br />
Since the r'--tric field acts only in the z direction, all the increase in<br />
kinetic energy <strong>of</strong> an electron will occur through increasing the z component <strong>of</strong> its<br />
velocity which is given by<br />
where voz is the value <strong>of</strong> vz immediately following the collision and t is the time<br />
elapsed since the last collision and a = (q/m)E. The energy picked up will be<br />
If we now average over all angles <strong>of</strong> direction <strong>of</strong> motion immediately following the<br />
last collision, '-0 so that<br />
If we now average over all collision times, Eq. (10) becomes<br />
I<br />
(9) I ,<br />
1
4<br />
I<br />
I<br />
5<br />
To find (t2)av, we assume that collisions are random events and the collision<br />
frequency is independent <strong>of</strong> speed. We can then write that the probability <strong>of</strong> a<br />
collision occurring in the interval t to t + dt following the precdding collision is<br />
. .<br />
for which the average value <strong>of</strong> t2 is 2@ where< = 112 is the mean collision time.<br />
Eq. (11) then becomes<br />
a&#<br />
t2.<br />
= ma-t<br />
If we also average over all speeds <strong>of</strong> electrons and neglect some <strong>of</strong> the finer pohts<br />
<strong>of</strong> statistics, Eq. (13) becomes<br />
where A is the mean free path and c is the mean speed <strong>of</strong> the electrons.<br />
This quantity, in the steady state, will equal the right hand side <strong>of</strong> Eq. (7)<br />
averaged over all electrons. If we let d.ls 1 -2 c<br />
2<br />
Thus we would expect that the mean speed would be given by<br />
-<br />
and the mean energy <strong>of</strong> the electrons in the steady state by<br />
(13)
6<br />
The remarkable aspect <strong>of</strong> this result comes on the insertion <strong>of</strong> numbers in<br />
Eq. (17). If for example, one has electrons moving through a gas with M 30 AMU,<br />
at pressures giving X ?r Imm (i.e. p 6 1 torr) then the mean energy in ev is around<br />
25 times the field strength in volts/cm. With as little as a few tenths <strong>of</strong> v/cm<br />
fields, mean electron energies are several ev. We can thus understmd the appearance<br />
<strong>of</strong> high electron "tempreatures" in gas <strong>discharges</strong>.<br />
It is beyond the scope <strong>of</strong> this review to go further and question the distribution ,<br />
<strong>of</strong> electrcn energies. It is sufficient to indicate that if one takes assumptions<br />
similar to those nade in this simplified argument and utilizes them in the Boltzmann<br />
ea-uation, one can obtain an approximate solution for the distribution <strong>of</strong> speeds. The t<br />
result is not the I.laxwell-Boltzmann distribution, but rather that known as the<br />
Druyvesteyn distribution whose dependence on speed is given by<br />
This distribution function is similar in shape to the hxwell-Boltzmann distribution<br />
for a given mean speed, except that the most prob2ble speed is slightly higher and the<br />
high energy tail is diminished in the Druyvesteyn distribution. Nonetheless, the<br />
distributions are sufficiently similar that one can, to good approximation, think <strong>of</strong><br />
the electrons as having a temperature in the Maxwellian sense which is much higher than<br />
the neutral gas temperature; i.e. typically 30,000'K vs 300'K. It is this dichotomy <strong>of</strong><br />
temperatures which is perhaps the most striking <strong>of</strong> the non-equilibrium aspects <strong>of</strong> a gas<br />
discharge.<br />
This general effect <strong>of</strong> collisions randomizing the direction <strong>of</strong> motion <strong>of</strong> electrons<br />
which have picked up energy from the field between collisions has another important<br />
manifestation. It is responsible for the operation <strong>of</strong> microwave electrodeless<br />
<strong>discharges</strong>.<br />
E(t) = Eo cos ut, then Newton's law for the electron becomes, in the absence <strong>of</strong><br />
collisions,<br />
which solves to give<br />
If we consider an electron moving in an ac field which is <strong>of</strong> the form<br />
The rate <strong>of</strong> energy pickup from the field i.e. the power, P is given by<br />
tEv<br />
4<br />
<<br />
\
\<br />
vnich, it is noted can be negative as well as positive. If we consider the total<br />
power pickup over a comFlete cycle, the sine-cosine product integrates to zero,<br />
indicatinfi that there is no net transferral <strong>of</strong> energy from the electric field to<br />
the electron. This is <strong>of</strong> course a result <strong>of</strong> the fact that the applied field and<br />
the electron velocity are 90O out <strong>of</strong> pnase.<br />
7<br />
Furthermore, we can note from (20) that the maximum electron velocity will be<br />
a_Cg/nu and the maximum kinetic energy will ue<br />
It is noted that if one has a field oscillating at say 109 cps and a maximum field<br />
strength <strong>of</strong> say 300 volts/cm, equation (22) indicates that the maximum kinetic energy<br />
<strong>of</strong> the freely oscillating electron will be only about 2 ev. "his is insufficient<br />
enera to ionize gases, and yet breakdown will occur for this type <strong>of</strong> field.<br />
It is collisions <strong>of</strong> the type which lead to the Druyvestryn distribution which<br />
are responsible for ionization. An electron will be accelerated by the field during<br />
a portion <strong>of</strong> its cycle and then be deflected in a collision. The energy parallel<br />
to the electric field is thus converted to energy perpendicular to the field.<br />
The field then proceeds<br />
to give the electron additional energy in the direction <strong>of</strong> the field. Energy is<br />
pumped from kinetic energy <strong>of</strong> motion parallel to the field to kinetic energy in all<br />
directions.<br />
If we write Newton's law for an electron, adding a collision term, we are left<br />
with Eq. (3), except now, E is time-dependent. Writing E = Eo cos ut<br />
1<br />
I - p = I - fiz - (24)
Again if this power gain is equated to the loss <strong>of</strong> energy <strong>of</strong> electrons in<br />
elastic collisions with the gas molecules, as would occur in the steady state, then<br />
qEo<br />
where a f -. m<br />
E<br />
8<br />
From (26) one would expect the electron "temDerature" for fixed Eo to diminish<br />
with increasing frequency, but for pressures <strong>of</strong> the order <strong>of</strong> one torr and gas masses<br />
<strong>of</strong> around 30 AMU the electron temperature will remain near the dc value for frequencies<br />
usually employed for gas discharge work.<br />
B. Elastic Collisions, and Diffusion<br />
-<br />
Since in either dc or ac <strong>discharges</strong>, the electrons will have high temperatures,<br />
one can expect that all the manifestations <strong>of</strong> a kinetic temperature will be present.<br />
Particularly important among these is diffusion through elastic collisions. If only<br />
electrons are present in a neutral gas they will tend to diffuse as would any other<br />
gaseous component with a current density<br />
where ne is the number density <strong>of</strong> the electrons and De is the diffusion coefficient <strong>of</strong><br />
the electrons through the gas.<br />
will contain a mobility term as well so that<br />
I<br />
(25)<br />
If a field is also superposed, the current density \<br />
,<br />
where the minus sign in the second term indicates that because <strong>of</strong> the negative charge 4<br />
on the electrons, their motion will try to be in the direction opposite to that Of<br />
the applied field. pe is taken to be a positive number. Whether the electron motion<br />
is diffusion-dominated or field dominated depends on whether the first term<br />
l<br />
is larger<br />
'1
I<br />
7<br />
than or smaller than the second.<br />
In a gas discharge, the electrons will have produced ions and :these also will<br />
tend to diffuse through the neutral gas as well as respond to any electric fields.<br />
The current density <strong>of</strong> ions will be given therefore by a similar equation<br />
--3<br />
s, = -9+ On, 4 “+F+<br />
9<br />
From Eq. (4) the mobility <strong>of</strong> either type <strong>of</strong> particle is & Ifl[m(z>)<br />
and from kinetic theory, the diffusion coefficient is given by D e. KT/t-m).<br />
Since both these parameters have the mass appearing in the denominator the electron<br />
will diffuse and respond to an electric field much more rapidly than will the heavy ions.<br />
The case <strong>of</strong> major interest for discharge <strong>physics</strong> is that where the number density<br />
<strong>of</strong> ions is very nearly equal to that <strong>of</strong> the electrons. Clearly because the electron<br />
mass is very light these will try to diffuse awa:y from the ions. However, as soon as<br />
they do this, an electric field is set up between the electrons and ions so that the<br />
electrons are held back by the ions and the ions are dragged along by the electrons.<br />
We would thus expect that the current fluxes Se and S+ would be equal.<br />
If we set Se = S, 5 S, and ne = n+ t n, to indicate an electrically neutral<br />
plasma, Eqs. (28) and (29) can be combined to eliminate the electric field with the<br />
result that<br />
where<br />
Since<br />
and<br />
or<br />
and b z &<br />
re ,gel<br />
we can write<br />
(29)
LU \<br />
If the electron temperature is equal to the ion temperature, as in the case <strong>of</strong><br />
late in an afterglow, the ambipolar diffusion coefficient is just twice the value<br />
<strong>of</strong> D+. In the active discharge, however, as we have seen Te will be large, perhaps<br />
<strong>of</strong> the order <strong>of</strong> 30,000°K. On the other hand the ion temperature will deviate little<br />
from the neutral gas temperature. The reason is that the ion mass id compwable<br />
to the mass <strong>of</strong> the neutral molecules; thus by arguments similar to those leading<br />
to Eq. (7) the ion will in a single collision be able very effectively to give to<br />
the neutral gas the energy it picks up from the field between collisions. The<br />
temperature <strong>of</strong> the ions will therefore remain very close to the neutral gas temperature.<br />
The ratio Te/T+ will be <strong>of</strong> the order <strong>of</strong> 100 typically in an active discharge<br />
and rapid diffusion <strong>of</strong> the plasma through the neutra1,gas and to the walls <strong>of</strong> the<br />
container will result.<br />
Summary <strong>of</strong> the Remainder <strong>of</strong> the Paper<br />
C. Electron production and loss mechanisms are discussed and the plasma<br />
balance equation is formulated.<br />
D. Excitation to radiating and metaetable states is eumaarized and some <strong>of</strong><br />
their consequences for the operation <strong>of</strong> a discharge are presented.<br />
E. Wall phenomena.<br />
111. Macroscopic phenomena.<br />
A.<br />
Plasma polarization and the Debye length are discussed.<br />
-B. Characteristics <strong>of</strong> certain types <strong>of</strong> <strong>discharges</strong> are reviewed; Glow Discharges,<br />
Arc Discharges and rf <strong>discharges</strong>.<br />
IV. Afterglows.<br />
briefly reviewed.<br />
The effect8 <strong>of</strong> suddenly reducing the electron temperature are
i<br />
I<br />
i<br />
I<br />
I<br />
I<br />
)<br />
)<br />
i<br />
L<br />
,<br />
I<br />
I . INT1:ODtiCTIOfi<br />
1l<br />
THE PRODIXTIOX OF 1\TOi.!S A::D SI:lTLL PJ$I)ICP.LS L< GLOW OISCIIARChS<br />
*<br />
Frederick Kaufman<br />
Department <strong>of</strong> Chemistry, University <strong>of</strong> Pittsburgh<br />
The size and topical variety <strong>of</strong> this symposiun clearly show tliat electrical<br />
<strong>discharges</strong> are finding increasing application in many areas <strong>of</strong> cliemistry ranging<br />
from tile iproduction <strong>of</strong> simple atoniic species such as 11, 0, or i from their diatomic<br />
molecules to the sviitiiesis or specific ueconposition <strong>of</strong> complex ory.anic or inorgsiiic<br />
ComPoUndS. It is unfortuiiately true that our understanding <strong>of</strong> tile chcmistrv <strong>of</strong> dis-<br />
cllarge processes is still in a rudimeoLary state, tiint the field is more an art than<br />
a science, and thus represents one <strong>of</strong> the last f'rontiers <strong>of</strong> chemistry.<br />
There is jiood reason for this unsatisfactory state <strong>of</strong> affairs. Glow disciiar-<br />
Res are complex phenomena in which gases at sul,-atmosplit!ric pressure are undergoing<br />
excitation and ionization by electron impact and so Give rise to hiziily uncquili-<br />
brated steadv-state conditions where the effective temperature <strong>of</strong> free electrons is<br />
typically tens <strong>of</strong> thousands OK, that <strong>of</strong> electronically or vibrationally excited<br />
States may be thousands <strong>of</strong> OK, whereas tiie transl.ationa1 and rotational temperature<br />
will only be teris to hundreds <strong>of</strong> '1; above ambient. It sliould be clear, <strong>of</strong> course,<br />
that apart from the processes occurring at tiie electrodes, energy from the electric<br />
field is coupled to the xas almost eiitirelv through the kinetic energy <strong>of</strong> free<br />
electrons wiiicli, due to their small mass, acquire eiiergv more rapidlv froni the field<br />
and lose it more slowly in elastic collisions (the mean fractional energy loss per<br />
elastic collision equals 2 ci/N in the siiiiplest clasical model where in and X are the<br />
masses <strong>of</strong> the electron and <strong>of</strong> the molecule). In tllis manner, electrons become suf-<br />
ficiently cnercetic to ionize some <strong>of</strong> the neutral species ana thereby balance their<br />
continuous loss by diffusion, attachment, and recombination. As the ionization<br />
potentials <strong>of</strong> most neutral gases are'in the 10 LO 20 ev range (230 to 460 kcal/mole),<br />
an appreciable fraction <strong>of</strong> the electrons has enough energy to produce electronic<br />
excitation (responsible for the emitted glow) and dissociation.<br />
In the following sections, the mechanism <strong>of</strong> dc and ac glow <strong>discharges</strong> will be<br />
briefly described, with emphasis on high frequency electrodeless <strong>discharges</strong> (f = lo6<br />
to 1O1O sec-') and on the simple geometry <strong>of</strong>ten encountered in rapidly pumped steady-<br />
state flow systems at pressures near 1 torr. After a brief discussion <strong>of</strong> tile rates<br />
and energy dependence <strong>of</strong> specific collision and diffusion processes, available ex-<br />
perimental data will be brought to bear on the problem <strong>of</strong> H2, S2, and 02 dissociation<br />
and on the chemistry <strong>of</strong> some more complicated systems.<br />
Although there are several fine monographs available on electron impact plie-<br />
nomena and, discharge <strong>physics</strong>'-', thev contain relatively little information on active<br />
high frequency <strong>discharges</strong> which is pertinent to the problem <strong>of</strong> dissociation and chem-<br />
ical reaction. The electron <strong>physics</strong> <strong>of</strong> microwave <strong>discharges</strong> is discussed in some<br />
review articles. 5*6<br />
XI. BASIC PtiYSICAL PROCESSES<br />
IT. 1. General Mechanism and Frequency Dependence.<br />
Glow <strong>discharges</strong> are typically observed in the pressure range <strong>of</strong> about 0.1 to<br />
10 torr. .At much lower pressures, the electron mean free path is too long for gas<br />
collisions to be important, electrons pick up large amounts <strong>of</strong> energy from the dc or<br />
slow ac field and bomhard the anode or the tube wall which may then fluoresce. At<br />
* This work was supported by tiie <strong>National</strong> Science ?oundation<br />
/I
L<br />
12<br />
much higher pressures, the mean free path is very short, the breakdown field strength<br />
is very high, and when it is exceeded, local, highly ionized, but narrow pathways are<br />
created for the conduction <strong>of</strong> current, i.e. spark filaments are formed. The normal<br />
dc glow discharge in a long cylindrical tube is characterized by many axially distinct<br />
but radially fairly uniform regions <strong>of</strong> quite different optical and electrical proper-<br />
ties such as the Aston Dark Space, Cathode Glow, Cathode Dark Space, Negative Glow,<br />
Faraday Dark Space, Positive Column, Anode Glow, and Anode Dark Space, in this order,<br />
between cathode and anode. The reason for this complexity is that quite different<br />
processes OCCUK in the different regions as is also shown by a very,non-uniform<br />
voltage rise between cathode and anode. Host <strong>of</strong> the potential difference is taken<br />
up in the 'cathode fall' which comprises the first four regions enumerated above, is<br />
dependent on the cathode material as well as on the nature <strong>of</strong> the gas and on the<br />
natural variable E/N (V cm*/molecules) where E is the field strength (V/cm) and N the<br />
total neutral density (molecules/cm3). The latter is a measure <strong>of</strong> the electron<br />
energy under conditions <strong>of</strong> steady-state drift. Equating energy loss and gain per<br />
2m cV 1<br />
electron per second, - - - eEw, where E = - mT2 is the electron energy, Q the mean<br />
M e 2<br />
free path for electron-neutral collisions, e the electronic char e, and w ty<br />
eEQM1$' =- E eM1,'<br />
electron drift velocity which equals 3 % one obtains c = (6m)'f2 N 211uL(3m)n<br />
where Q was replaced by lql/ 211u2N)from simple kinetic theoj.. The 'cathode fall'<br />
and 'anode fall' regions a so have large gradients <strong>of</strong> electron and ion concentrations<br />
and a local imbalance <strong>of</strong> electrical charge. The positive column is simpler in nature<br />
(although striated positive columns are still poorly understood), has a small and<br />
constant axial voltage drop, and only a small imbalance <strong>of</strong> charge carriers, because,<br />
although electrons initially diffuse to the tube wall faster than ions, the resul-<br />
tant radial field prevents further charge separation and forces electrons and ions to<br />
diffuse equally fast. This process is called ambipolar diffusion and is further<br />
discussed below. The voltage drop along the positive column is also independent <strong>of</strong><br />
the total current over a fairly wide range, and since the current, 1, is carried<br />
mostly by the electrons whose drift velocity is about 100 times larger than that <strong>of</strong><br />
the ions, i = n ew = - - n which shows that the electron concentration, ne,<br />
3 mC e'<br />
increases linearly with increasing current because E and J are constant. In its<br />
normal range <strong>of</strong> electron (and ion) concentrations <strong>of</strong> lo8 to 10" ~ m - ~ the , positive<br />
column <strong>of</strong> a dc glow discharge is diffusion-controlled and serves as the electrical<br />
connection between the cathode and anode regions.<br />
In high frequency electrodeless <strong>discharges</strong> the complications <strong>of</strong> the cathode<br />
and anode regions are absent, the entire plasma is approximately neutral and<br />
diffusion-controlled, and the discharge <strong>of</strong>ten resembles the positive column <strong>of</strong> an<br />
equivalent dc discharge. Yet, there are differences in its fundamental mechanism,<br />
especially at microwave frequencies. Free electrons oscillating in an alternating<br />
field can not derive power from the field on the average, because their motion is<br />
90' out <strong>of</strong> phase with the field. They therefore acquire energy only because c01lisions<br />
with neutral molecules change their phase relationship with the field, while<br />
at the same time representing a small fractional loss <strong>of</strong> the energy gained. Under<br />
the assumption that the ac frequency, f, is greater than the elastic collision frequency,<br />
v the maximum displacement, x, <strong>of</strong> an electron due to the high frequency<br />
e'<br />
2eE<br />
field is given by x = where w = 2llf. In an active microwave discharge E is<br />
typically 30 V/cm and x is therefore less than cm when f = 2.5 x lo9 sec-l (as<br />
in the widely used Raytheon Microthem Generator). The corresponding maximum electron<br />
energy acquired during the cycle is eEx, about 0.02 ev, i.e. the electrons<br />
slowly accummulate the energy necessary to undergo inelastic, ionizing CollIsiOnS<br />
and to sustain the discharge. When elastic collisions are approximately accounted<br />
for, it is convenient to define an effective field strength, Ee = Eo ("2 + ,f<br />
where E<br />
.2 )1/2<br />
is the rms value <strong>of</strong> the applied field strength. The power gained from the<br />
9
13<br />
field per electron is and per collision ttrereforc<br />
11. 2. iliffusion <strong>of</strong> Ciiargeci Species.<br />
2 e2L 2 ,<br />
in u<br />
e<br />
rnGT *<br />
In the discliarges <strong>of</strong> interest iiere, tile concentration <strong>of</strong> charged species is<br />
greater ellan and a large fractional separation <strong>of</strong> elrctrong alid positive ions<br />
becomes impossible, because it would set up a very large opposiug field. The currents<br />
<strong>of</strong> electrons and ions reaching the wall must tiicn be equal, i = - i) Vn - nu E'<br />
i+ = - D+Vn + nu E' where n = n<br />
+<br />
IJ the diffusion coefficient, LI the mobility, and E' the field due to tile (small) space<br />
charge. The subscripts e and + refer to clectror. and positive ion. Equating i and<br />
i, and eliminating C' one obtains<br />
r<br />
Dew+<br />
I = -<br />
+ U+we<br />
?+ + Ue<br />
= n+ is the electron density, 011 the densitv gradient,<br />
Vn = 1) Vn<br />
which serves as the definition <strong>of</strong> the anbipolar diffusion coefficient, Da. SubstitukTe<br />
ting P - for i)<br />
e c<br />
and the equivalaut expression for 1)+ otic obtains<br />
v+k<br />
which approximately equals - ('1<br />
c e + T ) or D+(1<br />
+<br />
T<br />
+ ")<br />
T+<br />
because the electron mobility<br />
pee' is much larger than the ionic mobility P+. In active glow <strong>discharges</strong> T /T+ is<br />
typically 20 to 100, whereas in the afterglow the electrons thermalize rapidly ana<br />
Te/T+ = 1.<br />
Thus, D 2 20 to 100 D+ in the active discharge, and 2 D+ in the afterglow.<br />
The disappearance <strong>of</strong> charged species bv ambipolar diffusion in the absence <strong>of</strong><br />
6n<br />
a source term is described by the diffusion equation = uavZn where n is a function<br />
<strong>of</strong> r, 8, z, and t. The well-known solution <strong>of</strong> this equation for the case <strong>of</strong> an infinite<br />
m<br />
cylinder is n(r,t) = Z<br />
-kit<br />
AiJo(% I) e where al is the i th root <strong>of</strong> Jo, the Bessel<br />
i=1 0<br />
a.<br />
2 Da<br />
function <strong>of</strong> zero order and k = ($) . The diffusion length, A, therefore<br />
i<br />
Da =<br />
r 0<br />
equals . The first few zeroes <strong>of</strong> Jo are a1 = 2.405, a2 = 5.520. a3 = 8.654,<br />
'i<br />
a,, = 11.792 which shows that, as diffusion proceeds, tile time decay will be increasingly<br />
governed by kl = (-) Da, the first (lowest) diffusion mode, because the next three<br />
r0<br />
higher modes are damped out more rapidly by factors <strong>of</strong> 5.3, 12.9, and 24. After a<br />
short transient, the diffusion-controlled electron decay or the diffusion controlled<br />
loss under steady-state conditions with a spatially well distributed source term can<br />
therefore be closely approximated by a first-order rate constant, k = 5.78 Da/ro2.<br />
This analysis applies when there are only positive ions and electrons present.<br />
h'hen negative ions are also present, their principal effect is to accelerate the<br />
ambipolar diffusion <strong>of</strong> the electrons, (Da)e, which now becomes approximately<br />
T<br />
Te<br />
(Da)e = (1 + A) D+ (1 + -) + X - 1) which for active <strong>discharges</strong> (Te/T+ >> 1)<br />
*+ T+<br />
can be further approximated by (1 + 2 A) Te/T+, where<br />
ratio <strong>of</strong> negative ions and electrons.<br />
= n-/ne. the concentration<br />
It can be seen that for A >> 1 electrons
.<br />
14 \<br />
will be lost much more rapidly be diffusion to the wall than in the absence <strong>of</strong><br />
negative ions. t<br />
11. 3.<br />
Electron-Ion and Ion-Ion Recombination.<br />
Although several radiative, two-body, and three-body recombination mechanisms<br />
exist, only the fastest ones will be mentioned here. These are the dissociative recombination<br />
<strong>of</strong> electrons and positive molecular ions as exemplified by<br />
NO+ + e + N + 0 (where the products are likely to be electronically’ excited), similar<br />
ion-ion reactions such as 1; + I- + I2 + +<br />
I or 31, NO + NO2- + neutral products, and ,<br />
+ -<br />
three-body ion-ion recombinations such as NO + NO2 + M + neutral products.<br />
All <strong>of</strong> these processes have very large rate constants, due to the long range<br />
coulombic attraction between reactants, if reasonable paths are available for the<br />
dissipation <strong>of</strong> the large exothermic reaction energy (* 10 ev) such as dissociation 1<br />
and electronic excitation. The first <strong>of</strong> these three processes has been studied in<br />
greatest detail, especially by Biondi and coworker^^'^ who found a value <strong>of</strong> I<br />
2.9 5 0.3 x lo-’<br />
3 -1 -1<br />
cm molecule sec for the rate constant <strong>of</strong> Np+ + e * N + N, for 1<br />
example. The generally observed range <strong>of</strong> 1 to 5 x lo-’ (6 x 1013 to 3 x 1014 lit 1<br />
mole-l sec-l) means that at an electron and ion concentration 10l1 which is near)<br />
the upper limit <strong>of</strong> charged particle densities encountered in glow <strong>discharges</strong>, the<br />
effective first order rate constant for electron removal under steady-state conditions,<br />
1<br />
will be 1 to 5 x lo4 sec-l.<br />
Two-body ion-ion recombinations have rate constants in the same general range<br />
although few have been studied in detail, none with precise analysis <strong>of</strong> reactants and<br />
products. Some three-body ion-ion recombinations have recently been studied by Mahan<br />
and coworkersg’ lo who found effective termolecular rate constants in the range<br />
4 x 1U-26 to 3 x<br />
approximate dependence, and at a total pressure <strong>of</strong> 1 torr, such processes<br />
would have effective first-order rate constants <strong>of</strong> ion removal in the 10 to 100 sec-’<br />
range, too slow to be <strong>of</strong> importance.<br />
11. 5.<br />
+<br />
cm6 molecule‘2 sec” for NO + NOp- + M near 300’K. With an<br />
Electron Attachment and Detachment.<br />
Only the fastest <strong>of</strong> the many possible processes need to be discussed here.<br />
Radiative attachment and photodetachment as well as three-body attachment processes<br />
are unlikely to be <strong>of</strong> importance. Dissociative attachment reactions such as<br />
e + 02 + 0- + 0 have rate constants1’ which rise from zero at an electron energy<br />
threshold (4 to 9 ev for the formation <strong>of</strong> 0- from 02, NO, or CO) to a maximum <strong>of</strong><br />
to cm3 molecule-l sec-l for electrons with 6 to 10 ev. For average electron<br />
energies <strong>of</strong> 2 to 3 ev in an active discharge, the effective rate constant must therefore<br />
be lowered about 10 fold to a range <strong>of</strong> to Moreover, several aSSOci<br />
ative detachment reactions such as 0- + 0 + 02 + e, 0- + N + NO + e, 0- + 82 -+ 840<br />
have recently been found12 to be very rapid (k = 1 to 5 x cm3 molecule’’ Sec-’)<br />
under thermal conditions near 3OO0K. This further reduces the likelihood that negati<br />
Ions are important species in rapidly pumped steady-state glow <strong>discharges</strong> <strong>of</strong> diatomic<br />
gases. In this regard, active <strong>discharges</strong> probably differ markedly from their Cor-<br />
responding afterglows in which the electrons are rapidly cooled to ambient temperatux<br />
and will then readily attach to form 02-, NO-, Cog-, Cob-, and other negative ions<br />
even though the electron affinities are quite small.<br />
I<br />
1<br />
><br />
!
.<br />
11. 5. CharRe-Transfer and Ion-Molecule Reactions<br />
Both positive and negative ion-molecule reactions have recently been studied<br />
by a variety <strong>of</strong> experimental methods, and consistent values for many rate constants<br />
have become available. Because <strong>of</strong> the strong ion-dipole or ion-induced dipole interaction,<br />
these reactions usually have little or no activation energy if they are exothermic,<br />
and <strong>of</strong>ten have rate constants near lo-' cm3 molecule-' sect', in accord with<br />
the simple theory based on the polarizability <strong>of</strong> the neutral reactant. Some excep-<br />
tions such as 02+ + Ng +<br />
NO+ + NO which is at least lo6 times slower are probably<br />
due the large energy requirements for bond rearrangement which in the corresponding<br />
neutral four-center reaction gives rise to a very large activation energv. Other unusually<br />
slow reactions such as 0' + h2 -c NO+ + N (k 'L 2 x lo-") are less easily<br />
rationalized, particularly since k rises sharply when the reactant 'L2 is vibrationally<br />
excited.<br />
From the magnitude <strong>of</strong> to lo-' for many <strong>of</strong> the exothermic reactions it is<br />
clear that the effective first-order rate constant for the transformation <strong>of</strong> an ionic<br />
species by reaction with a major neutral constituent (P = 1 torr = 2 x molecules<br />
at the higher temperature <strong>of</strong> the discharge) is 2 x lo6 to 2 x lo7 sec-l, i.e.<br />
such reactions will go to completion in a small fraction <strong>of</strong> the residence time in even<br />
the most rapidly pumped flow systems.<br />
Minor neutral constituents such as atomic<br />
species at 0.1 to 1 male % will transform ionic species witti rate constants <strong>of</strong> lo3 to<br />
lo5 sec-l, still much faster than the rate <strong>of</strong> traversal through most <strong>discharges</strong> whose<br />
average flow velocities are in the range 102 to lo4 cn sec-' and whose lengths are 1<br />
to IO cm.<br />
11. 5. Electron Impact Ionization.<br />
As shown in section 11. 2 above, the electron loss term by ambipolar diffusion<br />
can be approximated by a first-order rate constant, k = 5.78 Da/ro2, which for an<br />
cm, makes<br />
active discharge with Te/T+ "i, 50, U+ % 100 cm2/sec, and ro = 0.5<br />
k 2, 1 x lo5 sec-l. Although it is conceivable that chemi-ionization will occur in<br />
which two electronically excited molecules with 5 to 10 ev energy react to produce<br />
ionization, such processes will normally be <strong>of</strong> very minor importance. Even with a very<br />
large chemiionization rate constant <strong>of</strong> lo-'' cm3 molecule-' sec-l, a concentration <strong>of</strong><br />
5 x 1014 ~ m - ~ 2 , to 3 mole %, <strong>of</strong> such excited molecules would be required to balance<br />
the diffusional loss. It thus seems likely that electron impact ionization is the<br />
major source term for charged species in the discharge. Although this requires more<br />
than the ionization potential <strong>of</strong> the atom or molecule, i.e. electron energies in excess<br />
<strong>of</strong> about 15 ev, the large electron velocity and its very high average temperature,<br />
Te, can easily provide the required magnitude <strong>of</strong> the rate constant.<br />
The ionization cross section <strong>of</strong> most atoms and simple molecules rises sharply<br />
from zero at the ionization potential and comes to a broad maximum <strong>of</strong> about 1 to 5 x<br />
cm*/molecule at electron energies <strong>of</strong> 70 to 120 ev. It normally reaches a value<br />
<strong>of</strong> 1 x about 2 to 4 ev above its ionization potential, i.e. at electron energies<br />
<strong>of</strong> 12 to 17 ev. For average electron energies <strong>of</strong> 2 to 3 ev in active glow <strong>discharges</strong>,<br />
this lends to total effective ionization rate constants <strong>of</strong> IO-" to loe1' cm3 molecule-'<br />
sec-l if a Maxwell distribution is assumed. A somewhat lower range would be obtained<br />
for a Druyvesteyn distribution, but since the ratio E/? <strong>of</strong> required to average energy<br />
is never very large the error due to the assumption <strong>of</strong> a Maxwell distribution should<br />
be fairly small.<br />
An important, though infrequently applicable ionization mechanism is the direct<br />
ionization'by collision with sufficiently energetic, metastable neutral species. This<br />
process, called Penning ionization, can occur only if the excitation energy <strong>of</strong> the<br />
metastable exceeds the ionization potential <strong>of</strong> the other reactant. For He, Ne, and Ar<br />
the excitation energy <strong>of</strong> the lowest triplet state is 19.80, 16.62, and 11.55 ev re-<br />
' spectively, and since the rate constants for Penning reactions are <strong>of</strong>ten gas kinetic,<br />
I
state is bound,but the molecule is formed on the repulsive part <strong>of</strong> its potential energy 1<br />
cume at a point above its dissociation energy, and will therefore dissociate on its first'<br />
' 111. APPLICATION TO THE DISSOCIATION OF SOME DIATOMIC MOLECULES.<br />
111. 1. Summary <strong>of</strong> Some Atom Production and Loss Processes<br />
(1
17<br />
In the followin:: sections much evidence will be cited for catalytic effects<br />
in the production <strong>of</strong> atomic species such as 11, N, or 0 in glow <strong>discharges</strong> <strong>of</strong> t!ieir<br />
diatomic gases. The existence <strong>of</strong> such effects suggests large ctlanzes in the atom<br />
production or loss terms upon small variations in qas composition. Tile-principal<br />
processes are therefore summarized in this section, and approximate ranges given for<br />
their rates. A cylindrical discharge tube <strong>of</strong> 1 cm diameter, average electron concentration<br />
<strong>of</strong> 10l1 ~ m - ~ and , electron energy <strong>of</strong> 2 to 3 ev are assumed corresponding<br />
to known conditions in microwave <strong>discharges</strong> at input power levels <strong>of</strong>1 about 10 to 500<br />
watt.<br />
Production terms: (a) Electron impact dissociation via excited states de-<br />
pends on the existence <strong>of</strong> a dissociating or predissociating state at moderate excita-<br />
tion energy and is therefore quite variable. When the dissociation energy is rela-<br />
tively small, as in €1 and (,,and when states are available at 8 to 10 ev excitation<br />
threshold, an atom production rate <strong>of</strong> 10 to 100 torr/sec can be calculated. For N2<br />
whose dissociation energy is larger, the rate will be at least 10 times lower. It<br />
should be clear that this process must occur in active <strong>discharges</strong>, since electron<br />
impact ionization, which requires appreciably larger electron energies, is the princi-<br />
pal source term for electron production. Therefore, since there are enough electrons<br />
present with 15 to 20 ev energy to balance the rapid ambipolar diffusion (and recom-<br />
bination) loss, there must be appreciably more with 8 to 12 ev for excitation-<br />
dissociation.<br />
(b) Electron-Ion recombination at the wall (following ambipolar diffusion)<br />
or in the:gas phase will also produce atomic species in reactions such as<br />
e + 112' -+ 2 H, e + N2+ -t N + N, etc. -It is clear that: the upper limit to this atom<br />
production term is given by the total rate <strong>of</strong> ionization except for a possible factor<br />
<strong>of</strong> two from the stoichiometry <strong>of</strong> the dissociation. But as this surface recombination<br />
may also lead to the molecular product by the dissipation <strong>of</strong> energy to the surface,<br />
the dissociation rate due to this process is likely to be lower than the corresponding<br />
ionization or equivalent ambipolar diffusion term, i.e. < 0.5 to 5 Torr/scc. The gas-<br />
phase dissociative recombination will produce atoms at a rate <strong>of</strong> 0.1 to 0.3 Torr/sec<br />
at an electron density <strong>of</strong> emq3 and much more slowly at lower densities.<br />
(c) Ion-molecule reactions <strong>of</strong> primary ions may in exceptional cases be sufficiently<br />
exothermic to produce atomic species and ions <strong>of</strong> lower ionization potential<br />
such as in H2+ + H2 * l!3+ + 11, but the equivalent reactions for 02 and fi2 are endothermic.<br />
When such a process is possible, its rate is again limited by the total rate<br />
<strong>of</strong> ionization, but should closely approach it if the neutral molecule is a major species<br />
as in the above ii2 reaction.<br />
(d) Neutral-neutral dissociation reactions may also occur, but little can be<br />
said about them in general. As the kinetic temperature <strong>of</strong> all but the electrons is<br />
fairly low (300 to 800°K, mostly near 6UO°C), two excited, metastable molecules would<br />
have to react to transfer their excitation to a predissociating state. Few such reactions<br />
are known and their rate constants are likely to have upper limits in the<br />
to cm' molecule-l sec-lrange. If that were so, metastable mole fractions<br />
as high as 0.1% would only produce atoms at ratios <strong>of</strong> 0.02 to 0.2 Torr/sec. Such<br />
states would also have to be optically metastable, i.e. have radiative lifetimes longer<br />
than 0.01 sec, and be resistant to collisional quenching, i.e. have deactivation probability<br />
per collision lower tlian<br />
Loss Terms: (a) iiomogeneous sas-phase atom recombinations are three-body<br />
processes vith rate constants near 10- cm6 molecule" sec-' so that even for atom<br />
mole fractions <strong>of</strong> 5X, such loss rates are near 0.01 torr/sec, negligibly small.<br />
(b) Surface recombination is a well-known process, usually kinetically first<br />
order, and characterized by a rate constant, k = E , for a cylindrical tube <strong>of</strong> diameter d.
H.ere Y<br />
18<br />
is the recombination coefficient, the fraction <strong>of</strong> surface collisions leading<br />
to recombination, and E is the average molecular velocity <strong>of</strong> the atomic species. y<br />
which is <strong>of</strong>ten as low as outside the discharge for a well-cleaned or suitable<br />
poisoned surface, is likely to be or larger in the active discharge. For y = 10-3,<br />
atom loss rates <strong>of</strong> 2 to 10 torrlsec can be calculated, and for larger y, these rates<br />
would be larger, but not proportionately so, because large radial concentration gradients<br />
would be produced and diffusion would become rate-controlling. Then, an effective<br />
first order rate constant, k = D/A2 - 5.8 D/r2, would be applicable, similar to ambipolar<br />
diffusion (Section 11.2). but with the smaller, molecular diffpsion coefficient.<br />
For an atom mole fraction <strong>of</strong> 52, the upper limit to the diffusion91 atom recombination<br />
rate will be 100 to 300 torrlsec depending on the atomic species.<br />
(c) Ion-molecule reactions can remove atomic species if the ion is polyatomic<br />
such as in N + Ng+ * N2 + N2+ or the equivalent oxygen reaction. If the polyatomic ion<br />
is a major ionic species, and the reaction is a fast one, its rate at 5% atom mole<br />
fraction will be near 5 torrlsec. It will then $e limited by the rate <strong>of</strong> regenTration<br />
<strong>of</strong> the polyatomic ion. Reactions <strong>of</strong> the type 02 + N * NO+ + 0 or Ng+ + 0 * NO + N<br />
are known to be fast, but as they replace one atomic species by another they need not<br />
be considered here.<br />
(d) Neutral, bimolecular atom-molecule reactions such as N + 02 * NO + 0 or<br />
0 + H2 + OH + H <strong>of</strong>ten have appreciable activation energies. Moreover, they, too, only<br />
shuffle atomic species and can not be considered loss terms. In the few applicable<br />
cases where this does not hold as in 0 + N20*2 NO or N + N20 * N2 + NO, the reactions<br />
are known to be negligibly slow.<br />
111. 2. Hydrogen Discharges<br />
The discussion <strong>of</strong> the electron collisional aspects <strong>of</strong> H2, N2, and 02 <strong>discharges</strong><br />
is greatly aided by the excellent papers by Phelps and coworkers in which elastic and<br />
inelastic collision cross sections are obtained by numerical solution <strong>of</strong> the Boltzmann<br />
transport equation and comparison with all available experimental data on electron<br />
transport coefficients. For H2 and D214, the vibrational excitation cross section<br />
becomes appreciable<br />
cm2) at about 1 ev and peaks near 5 ev at 8 x cm2.<br />
For dissociation, a threshold at 8.85 ev and peak <strong>of</strong> 4.5 x cm2 at 16 to 17 ev<br />
was found to be consistent with the data. A recent measurement by Corriganl’ <strong>of</strong> the<br />
dissociation cross section <strong>of</strong> H2 is in good agreement with the above except for a<br />
larger peak <strong>of</strong> about 9 x crn2 at 16.5 ev. For average electron energies, E <strong>of</strong><br />
3.0 and 2.0 ev, dissociation rate constants, kd, <strong>of</strong> 11 and 2 x 10-10cm3 molecules<br />
kl 1<br />
sec-lcan be calculated by summation <strong>of</strong> Qvf(s) from E = o to m, using energy increments<br />
<strong>of</strong> 2 or 3 ev, where Q is the appropriate cross section, v the electron velocity, and<br />
is the Maxwell distribution function. Such k ‘s correspond to H-atom production rates<br />
d<br />
<strong>of</strong> 200 or 40 torrlsec. An effective ionieation rate constant, k , was similarly<br />
found to be 7 x<br />
( E ~ = 3ev) and 6 x ( E - ~ 2 ev), corresponding to ionization<br />
rates <strong>of</strong> 7 and 0.6 torrlsec, respectively. The latter should be approximately equal<br />
to the ambipolar diffusion loss, 5.8/rO2 D+Te ne/T which equals 9 or 6 torrlsec if<br />
g’<br />
D+.= 700 cm2/sec at 1 torr pressure. This crude calculation suggests that E - 3 ev is<br />
a better choice than 2.0 ev. It neglects the serious deviation from a Maxwelfl distribution<br />
which is to be expected.<br />
More realistic estimates <strong>of</strong> kd and ki were obtained by Dr. Phelps by simul-<br />
taneously adjusting E/N, ck. and using the appropriate rate coefficients to balance<br />
ionization against diffusion losses. This led to values <strong>of</strong> Ek = 3.0 ev.<br />
E/N 1.1 x 10-l’ Vcm2/molecule and ki = 7 x in surprising agreement with the
above. With that EIN, an estimate <strong>of</strong> kd can be obtained from ref. 14, Fig. 8 by multiplying<br />
the excitation coefficient (8 x<br />
(1.5 x lo7 cm/sec) which gives kd = 1.2 x<br />
cm2/molecule) by the drift velocity<br />
also in fortuitously good agreement<br />
with 1.1 x above. This excitation coefficient includes other, non-dissociating<br />
processes, but since application <strong>of</strong> Corrigan's' larger dissociation cross section<br />
would increase3otal excitation coefficient , its subsequent apportioning would not<br />
lead to serious discrepancies. *<br />
I<br />
Thus, a very large atom production term <strong>of</strong> about 200 torrlsec is indicated by<br />
all available estimates. In addition, there is the source term due to electron-ion<br />
recombination at the wall, and due to the fast reaction €12' + Hp -+ H3+ + li. Each <strong>of</strong><br />
these can at most equal the ionization rate, so that their sum will contribute 10 to<br />
20 torr/sec <strong>of</strong> atomic hydrogen. All other source terms are negligible.<br />
Surface recombination provides the only comparably large loss term in the<br />
discharge. For small y, the first order rate constant, k, = yC/d = 2 x 105y, and the<br />
steady-state atom concentration, [HIss is approximately 1 x 10-3/y. This shows that<br />
y must alwasy be relatively large, and that the diffusion controlled<br />
ks = 5.8 D/r2 = 2 to 4 x lo4 sec-' may be applicable which leads to an [Illss<br />
to 1 x lob2 torr, less than 1% mole fraction. Idhen y is lowered to<br />
<strong>of</strong> 0.5<br />
[HIss = 0.1 torr, and under either condition the halflife for its formation is very<br />
short (3 x or 3 x lO-'?sec).<br />
One is thus led to the interesting conclusion that the dissociation yield <strong>of</strong><br />
such H2 <strong>discharges</strong> is mainly controlled by surface recombination and that the surface<br />
in the discharge must be moderately to highly efficient for atom recombination. More-<br />
over, the effect <strong>of</strong> small amounts <strong>of</strong> added gases such as H20 or 02 in increasing the<br />
yield must be through their influence on the surface efficiency, as they can not affect<br />
the principal production term and as no homogeneous loss mechanisms <strong>of</strong> proper magnitude<br />
are available. These conclusions are in general agreement with recent work by Goodyear<br />
and Von Cngel" on radio frequency <strong>discharges</strong> at lower pressure and <strong>of</strong> different<br />
geometry.<br />
The very large body <strong>of</strong> experimental work on the "catalytic" production <strong>of</strong> H<br />
when small amounts <strong>of</strong> Hgi) or 02 are added will not be reviewed here. The effect is<br />
unquestionably real, i.e. factors <strong>of</strong> 10 or so in 11-atom yield when switching from<br />
"dry" H2 to H2 containing 0.1 to 0.3% H20 can be demonstrated routinely and reversibly.<br />
Rony and Hanson18 have recently questioned the existence <strong>of</strong> such an effect at a pressure<br />
<strong>of</strong> 0.075 torr and have reviewed the general subject. Our early experiments do<br />
not bear this out, as they show a large catalytic effect for added H20, but they do<br />
indicate that the effect is less pronounced at pressures below 0.1 torr. This trend<br />
can be expected if the above analysis is correct, because at low pressures, the production<br />
terms are decreased and the loss terms increased. Moreover, for a given<br />
discharge energy input, the surface should be hotter at low pressure, and this may<br />
nullify the deactivation which is probably due to adsorbed water molecules. Several <strong>of</strong><br />
the relevant experiments are now under way in our laboratory by Dr. D. A. Parkes, using<br />
Lyman-! absorption for the do-tream measurement <strong>of</strong> [HI.<br />
111. 2. Oxygen Discharges<br />
Electron collision cross sections have been calculated by Hake and Phelps19<br />
from all available experimental data on electron drift velocity and other transport<br />
coefficients. The cross sections for vibrational excitation are much smaller than in<br />
Hp and that process can probably be neglected. Electronic excitation is described by<br />
* It is Dr. phelps's view that the high E/N portion <strong>of</strong> the enelastic collision frequency<br />
and the corresponding cross sections will have to be modified in view <strong>of</strong> recent work<br />
by Fletcher and Haydon, Austral. J. Phys. 19, 615 (1966).
20<br />
three processes with thresholds at 4.5, 8.0, and 9.7 ev. The latter two are larger<br />
than the first, and should lead to dissociation, since the upper states have shallow<br />
potential energy mimima at larger internuclear distances than the ground state. The<br />
combined cross section for the 8.0 and 9.7 ev excitation shows a broad first peak <strong>of</strong><br />
1.1 x cm2 at 12 to 15 ev, then drops slightly and rises again to a second peak<br />
in the 50 to 100 ev range. With the assumption <strong>of</strong> an average electron energy, Ek, <strong>of</strong><br />
3.0 ev the effective dissociation rate constant, kd, is about 2 x cm3 molecule-’<br />
sec-l.<br />
I<br />
The corresponding ionization rate constant, ki, was calculated to be<br />
1.3 x cm3 molecule-’ sec’l. Fitting the 02 data in a similar way to lip, Dr.<br />
Phelps obtained E/N = 9 x<br />
V cm2/molecule, Ek = 3.4 ev, kd = 1 x lo-’, and<br />
k, = 1 x 10-l’ cm3 molecule-’ sec-l, in fair agreement with the above except for the<br />
smaller ki, but k is strongly dependent on E/N near 10-15Vcm2/molecule, so that a<br />
20% increase <strong>of</strong> ESN would lead to a tenfold increase <strong>of</strong> ki.<br />
An patom production rate <strong>of</strong> about 200 torr/sec by dissociative electron<br />
impact is thus indicated, and all other production terms are negligible by comparison.<br />
Surface recombination <strong>of</strong> 0-atoms is kinetically similar to that <strong>of</strong> H-atoms except for<br />
a lower diffusion coefficient and molecular velocity by a factor <strong>of</strong> 3. Thus, the<br />
effective first order surface recombination rate constant is 6 x lo4 y sec-l for Emdl<br />
r and 5 x 10’ 8ec-l for y approaching unity.<br />
In pure 02, as in Np, there is another loss term which does not arise in H2.<br />
The polyatomic ions Os+ and O4+ can react exothermically and rapidly via<br />
01,’ + + 0 + 03 + 02, 03’ + 0 -+ 02+ + 02 to recombine 0-atoms. To obtain an upper limit<br />
+<br />
for this loss rate one may assume that 01, is a major ion, i.e. [O4+] * lf” ~ m - ~ ,<br />
and that it is therefore regenerated by the bimolecular reaction 02 + 02 + 01,’. The<br />
latter assumption requires that the lifetime <strong>of</strong> the unstabilized OI,+ collision complex<br />
be equal to or longer than the collision time, 3 x sec, which seems excessively<br />
long-for such a simple species. At its (unreasonable) maximum estimate, such a chain<br />
process may recombine 0-atoms at twice the rate <strong>of</strong> @ + 0 -c O3+ + 02, i.e. with an<br />
effective first order rate constant <strong>of</strong> 200 sec-l if both ion molecule reactions have<br />
rate constants <strong>of</strong> cm3 molecule-l sec-’. Even so, it would not seriously limit<br />
0-atom production, since the corresponding [O], = 0.5 torr = 50% mole fraction. Moreover,<br />
Knewstubb, Dawson, and TicknerZ0 saw no Ob+ in their mass-spectrometric study<br />
<strong>of</strong> dc 02 <strong>discharges</strong> at 0.4 torr, though the weaklv bound ion could have dissociated in<br />
the large electric field at the sampling orifice as Schmidtz1 suggests for a in his<br />
mass-spectrometric study <strong>of</strong> nitrogen ions. The above mechanism does have the desirable<br />
property that it is easily quenched by small amounts <strong>of</strong> added gases such as N2 or H2<br />
which are capable <strong>of</strong> transforming the oxygen ions into more stable ions such as NO+ or<br />
EJO’, but it is unlikely to be important here.<br />
The process 0- + 0 -c 02 + e is known to be-fast (k * 1.5 x and it should<br />
follow the dissociative attachment step e 2 O2 + 0 + 0 which has a threshold <strong>of</strong> 4.5 ev<br />
Ins<strong>of</strong>ar as 0 is formed mainly by this reaction ad<br />
and a low maximum near 7 ev.<br />
removed by its reverse, and the concentrations <strong>of</strong> electrons and ions are very much<br />
lower than those <strong>of</strong> neutral species, these steps leave [O] unchanged.<br />
If the above large excitation-dissociation rates are approximately correct, the<br />
O-atom yield, as the H-atom yield in the preceding section, is principally controlled<br />
by surface recombination in the discharge. The smallest calculated [Ol,, (y 1) is<br />
about 0.04 torr, 4% mole fraction, considerably larger than experimental values for<br />
pure 02. A possible explanation <strong>of</strong> this discrepancy may lie in the surface properties<br />
immediately downstream from the discharge region. Active <strong>discharges</strong> have a sharp<br />
boundary as shown by their light emission, because electron loss processes are Very<br />
fast. The corresponding transition <strong>of</strong> the surface from a region where y is<br />
i<br />
near unity<br />
to one where it is less than is likely to be more gradual, and would provide a<br />
f
21<br />
region in w!iich large, nighlv localized surface 10s; terns could qulcLlv reduce tile<br />
atom concentration.<br />
Experimentally, 1ary.e catalytic effects by :;?, SO, or li2 in tile production <strong>of</strong><br />
0-atoms in microwave ciisciiargcs ~iave ~cerl reported.22 Very ptire oxygen qave only u.6~<br />
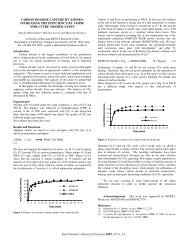
atoms (still ~ O W ~ yields K oi 0.3';; WCKC latcr obtained), !>ut small adtiitions (U.01 to<br />
0.u5;:) <strong>of</strong> Sz, >;20, or !;o produced +atoms at tile rate <strong>of</strong> 4u to 45 per added ;:, and<br />
similar additions <strong>of</strong> 112 produced 160 to 200 +atoms per added 112. Ih terms <strong>of</strong> the<br />
!'resent interpretation, tile large catalytic cffrct may he understandable for ti2 additions<br />
as due to t!20 wall effects, !,ut less so for nitrogen compounds which sliould not<br />
be StKOIlGlY adsorlied at tile surface. (:onceivably, XO+ ir ::02+, stron): Lewis acids, my<br />
be involved in poisoiiiny, the surface. Thus, our understanding <strong>of</strong> 02 discharzes is still<br />
in an unsatisf actory state. Furthcr experiinents are required in wliich particular<br />
attention should he given to tile condition and cliaracterization <strong>of</strong> the surface as vel1<br />
as to the imnedinte downstream rccion.<br />
111. 4. SitroRcn Disc!iarges.<br />
The great complexity <strong>of</strong> "active nitro1:en" is probably due to its larger cross<br />
sections for vibrational excitation anci to tile existcncc <strong>of</strong> metastable electronically<br />
excited states helot) tlie dissociation l i m i t <strong>of</strong> ground-state 1iz. Consequently, extensive<br />
vibrational excitation persists for times much longer than those spent in tlie discharge<br />
zone, and chemiionizatioll is observed in regions such as tlie "pink glow" well downstream<br />
<strong>of</strong> the discharge. The absence <strong>of</strong> the lowest triplet state, A,3Z:, in active nitrogeng3<br />
contaiiiing '.';-atoms indicates that tliesc excited molecules are very efficiently quenched<br />
by S, and that vibrationally liiglily excited ground-s tate molecules arc the principal<br />
carriers <strong>of</strong> excitation to tlie dowiistreai~! reqioii. Engelliardt, Phelps, and Riskz4 have<br />
determined tlie relevant elastic arid inelastic electron collision cross sections. Some<br />
<strong>of</strong> the electronically excited states above tne dissociation limit do not lead to pre-<br />
dissociation, and thcrefore only the state with tlireshold energy <strong>of</strong> 14V was used in<br />
tile estimate <strong>of</strong> dissociation. Assuming an average electron energy. = 3 ev, and a<br />
maxwellian distribution, one obtains an effective dissociation rate constant, kd, <strong>of</strong><br />
3 x lo-'' (bo torr/sec) and a correspondinz ionization rate constant, ki, <strong>of</strong> 6 x lU-l'.<br />
'The latter is larzer (b torr/sec) than the corresponding ambipolar diffusion loss term<br />
(13.5 to 1 torr/sec). lilt more realistic calculation by Dr. Phelps which simultaneously<br />
fits ck, L/:i, and tlie known cross sections to make the ambipolar diffusion loss equalthe<br />
rate <strong>of</strong> ionization gave ck = 2.2 ev, E/?: = 1.2 x cm2/molccule. kd = 3 x 10 "<br />
(6 torr/sec), and ki = 3 x This dissociation rate is very much lower than that<br />
<strong>of</strong> 1iZ or O2 and properly reflects the tiifficulty <strong>of</strong> producing extensive dissociation <strong>of</strong><br />
Hz in glow discharxes. ?io other source terms <strong>of</strong> comparable magnitude are available.<br />
The principal loss processes include atom recombination at the surface which can be set<br />
equal to those <strong>of</strong> oxygen, because the molecul K velocities are similar. The catalytic<br />
a atom loss nechanicm by E;,++ + X + i
1<br />
\<br />
22 .<br />
Experimentally, there is abundant evidence for "catalytic" effects as summarized<br />
by Young et alZ8 who studied the effectiveness <strong>of</strong> 02, NO, and SFg added either<br />
before or immediately after a nicrowave discliarge in highly purified, flowing Xz.<br />
\<br />
All three gases were "catalytic" when added before, but only NO after the discharge, ,<br />
and SF6 added before "produced" 230 !
i<br />
’ understanding:<br />
23<br />
(a) Electron impact ionization and aissociation rates <strong>of</strong> reactants<br />
1 and other major species; (b) Surface recombination ratcs in the discharge: (c)<br />
Ion-molecule reaction rates involving major neutral species; (d) neutral-neutral<br />
reaction rates and their tcmpcrature dependences in and out <strong>of</strong> the disciiarge. ‘[uch <strong>of</strong><br />
) the necessa information for (a), (c), and (d) is becoming available for many systems.<br />
The role OBurface, however, is little understood for electrically neutral reactants,<br />
1<br />
I<br />
, even less so for charged species, and seems to be the present bottleneck.<br />
\<br />
i 1.<br />
I<br />
. \<br />
2.<br />
3.<br />
I 4-<br />
5.<br />
6.<br />
’ 7.<br />
1 8.<br />
Y.<br />
10.<br />
1 11.<br />
’ 12.<br />
’, 13.<br />
1; 14.<br />
15.<br />
I 16.<br />
17.<br />
j 18.<br />
19.<br />
20.<br />
21.<br />
22.<br />
23.<br />
, 24.<br />
1 25.<br />
i 26.<br />
1 27.<br />
28.<br />
, 29.<br />
Acknowledgement :<br />
The author would like to thank UK. A. V. Phelps for many interesting discussions<br />
and for calculations <strong>of</strong> important parameters.<br />
References<br />
H. S. W. Massey and E. H. S. Burhop, Electronic and Ionic Impact Phenomena, Oxford<br />
University Press, 1952.<br />
E. N. McDanel, Collision Phenomena in Ionized Cases, John Wiley and Sons, N.Y. 1964.<br />
P. Llewellyn-Jones, The Glow Discliargc, ?lethuen and Co., London, 1966.<br />
s. c. Brown, Introduction to Electrical Discharees in Gases, John Wiley and Sons,<br />
N.Y. 1966.<br />
L. Goldstein, Advances in Electronics and Electron Physics, Vol. 7, 399-503 (1955).<br />
S. C. Brown, Handhuch der Physik, Vol. 22, 531-575 (1956).<br />
!4- A. Uiondi, Advances in tlectronics, Vol. 18, Academic Press, Inc., N.Y.<br />
‘-4. ii. Kasner and ?4. A. Uiondi, Phys. Kev. 137, A317 (1965).<br />
B. li. Mahan and J. C. Person, J. Cliem. Phys. 40, 392 (1964).<br />
T. S. Carlton and B. li. Xahan, J. Chem. Piiys. a, 3683 (1964).<br />
1963, p.67.<br />
D. Rapp and U. 1). Urif,lia, J. Chem. Phvs. a,<br />
1480 (1965).<br />
F. C. Fehsenfeld, E. t. Ferguson, and A. L. Schmeltekopf, J. Chem. Phys. 44, 1844<br />
(1966).<br />
Y.<br />
A.<br />
Tanaka, F. R. Innes, A.<br />
G. Lngelhardt and A. V.<br />
S. Jursa, and ?I. :
24<br />
Ion-Molecule Reactions in Electric Discharges<br />
3. L. Franklin, P. K. Ghosh and Stanley Studniary<br />
Rice University, Houston, Texas I<br />
Before the development <strong>of</strong> good vacuum equipment and techniques, mass spectrometrists<br />
observed many ions occurring at masses well above those <strong>of</strong> t,he molecules admitted<br />
to the instrument. These interferred with the main interests <strong>of</strong> the experimenters<br />
at that time, although in a few instances it was recognized that reactions<br />
were occurring between primary ions and molecules. When good vacuum facilities became<br />
available in the early 1930s they were adapted to mass spectrometry and for a<br />
number oi years every effort was made to maintain pressures well below torr ,<br />
in order to avoid collisions <strong>of</strong> primary ions with neutrals. However, in 1940, Mann,<br />
Hustrulid and Tate (l), in studying water, observed an ion at mass 19, and by vary-<br />
I<br />
(1) Mann, M.M., Hustrulid, A. and Tate, J. T., -. Rev., 58, 340 (1940)<br />
ing the pressure in their instrument, concluded that it was H30+, probably formed<br />
by the reaction: + +<br />
H20 + H20 + H 0 + OH<br />
3<br />
During the middle 50s, several groups <strong>of</strong> mass spectrometrists became curious as to<br />
the results that would be observed if source pressures were raised sufficiently to<br />
allow a few collisions to occur between ions and molecules. Several ions occurring<br />
at masses above those <strong>of</strong> the parent ion were observed. Because <strong>of</strong> this, a number<br />
<strong>of</strong> systematic studies were made and increasing interest and emphasis on reactions<br />
<strong>of</strong> ions with neutral molecules has developed, until at present some 50 to 100 papers<br />
per year are published on this subject alone.<br />
Perhaps one <strong>of</strong> the strongest reasons for the interest in ion-molecule reactions<br />
is the fact that ions <strong>of</strong> unusual and unsuspected composition were observed. Most<br />
fascinating <strong>of</strong> these has been CH5+, which was completely unexpected, but which,<br />
nevertheless, is now well established as a stable ioc. This ion was first announced<br />
by Tal'roze and Lyumbimova (2), but it was also reported shortly after the Russians<br />
(2)<br />
Tal'roze, V. L. and Lyumbimova, A. K., Dokl. Akad. Nauk SSSR, 86, 909 (1952)<br />
by several groups in this country (3,4,5,9).Studies <strong>of</strong> secondary ions from a large<br />
number <strong>of</strong> molecules followed.<br />
(3) Field, F. H., Franklin, J. L. and Lampe, F. W., 2. Amer. Chem. %., 79, 2419<br />
(1957)<br />
(4) Stevenson, D. P.and Schissler, D. O., J. Chem. PQ., 2, 1353 (1955)<br />
(9) Schissler, D. 0. and Stevenson, D. P., 2. Chem. Phys.,-24, 926 (1956)<br />
(5) Meisels, G. G., Hamill, W. H. and Williams, R. R., Jr., J. E m . -., 2, 79c<br />
(1956)<br />
The earlier studies <strong>of</strong> secondary ions were largely limited to ions <strong>of</strong> masses<br />
greater than that <strong>of</strong> the parent ion, but subsequent studies have shown that many<br />
secondaries <strong>of</strong> lower mass also occur. In most instances, the fact that the ion re-<br />
sulted from collision rather than from impurities was demonstrated by varying the<br />
pressure in the ion source. If the ion intensity increased as a second power <strong>of</strong><br />
the pressure, the ion clearly resulted from a collision.<br />
Having established that an ion was indeed the result <strong>of</strong> a collision, it was<br />
obviously <strong>of</strong> interest to ascertain the primary ion precursor <strong>of</strong> the secondary ion.<br />
TWO methods have usually been used for this purpose. The method most <strong>of</strong>ten used<br />
was to measure the appearance potential <strong>of</strong> the secondary ion and compare it to<br />
appearance potentials <strong>of</strong> various primary ions occurring in the system in question.<br />
-<br />
-<br />
-<br />
-<br />
,'<br />
,<br />
1
Obviously, the secondary ion must have the same appearance potential as its precursor,<br />
and where reasonable agreement <strong>of</strong> primary and secondary ion appearance potentials was<br />
found the identities <strong>of</strong> reactant and product were established. In some instances, it<br />
has been possible to reduce the electron energy to the point where only one or two<br />
Primary ions were present. Under these conditions, it is <strong>of</strong>ten possible by comparing<br />
intensities <strong>of</strong> secondary ions with the disappearance <strong>of</strong> primaries to identify unequivocally<br />
the precursors <strong>of</strong> the various secondaries. Obviously, boqh <strong>of</strong> these<br />
techniques suffer from certain difficulties. Where the primary ion spectrum is sufficiently<br />
complicated, or where the appearance potentials <strong>of</strong> several primary ions are<br />
sufficiently close together, it is very difficult and <strong>of</strong>ten impossible to determine<br />
the precursor <strong>of</strong> a secondary ion satisfactorily.<br />
Further, comparison <strong>of</strong> primary and<br />
secondary appearance potentials usually serves to identify only the precursor <strong>of</strong><br />
lowest appearance potential. Possible precursors <strong>of</strong> higher appearance potential<br />
are obscured and can only be detected by other means. Of course, it is also true<br />
that one cannot always simplify the primary spectrum sufficiently by reducing the<br />
electron energy to permit satisfactory identification <strong>of</strong> the precursor. As a result,<br />
in recent years a few mass spectrometrists following Lindholm (6) have employed a<br />
primary mass sorter to select the primary ion which is then injected into the gas<br />
(6) For a survey <strong>of</strong> Lindholm's work see E. Lindholm, "Ion-Molecule Reactions in the<br />
Gas Phase," Advances in Chemistry Series 58, American Chemical Society, Washington,<br />
D. C., 1966, pl.<br />
with which reaction is desired.<br />
Mass spectrometer ion sources are quite small <strong>chemical</strong> reactors. It can easily<br />
be shown that the reaction time <strong>of</strong> an ion in the source will normally be in the<br />
order <strong>of</strong> a microsecond, and unless the pressure in the source is well over 100 torr<br />
only a small fraction <strong>of</strong> the ions can undergo collision. It early became apparent<br />
that where secondary ions were observed they must, in most instances, have resulted<br />
at almost every collision <strong>of</strong> ion and neutral. Further, in many instances, the<br />
heats <strong>of</strong> formation <strong>of</strong> ions and neutrals were well established, and in all such<br />
instances it was possible to show that the reactions were exothermic. Indeed, it<br />
would be impossible under most circumstances to observe a reaction to form secondary<br />
ions if the reaction were endothermic. Such endothermicity would appear as activa-<br />
tion energy, which would greatly reduce the probability <strong>of</strong> reaction when the ion<br />
and molecule collide. (Later on we mention certain conditions in which this rule<br />
is violated, but under most circumstances it holds rigorously.) This rule is so<br />
seldom violated that it can be used as a means <strong>of</strong> helping to eliminate possible<br />
precursors <strong>of</strong> a secondary ion.<br />
It was mentioned above that the secondary ions first observed occurred at masses<br />
above those <strong>of</strong> the parent. However, there was always an expectation that ions <strong>of</strong><br />
lower mass might also be formed in collision reactions and as the pressure that<br />
could be tolerated in the ion source was increased it became apparent that such was<br />
indeed the case. For example, it was observed by Munson, Franklin and Field (7)<br />
(7) M.S.B.Munson, J. L. Franklin and F. H. Field, J. Phys. Chem. 68, 3098 (1964)<br />
that the C H + ion from ethane and the C3HTf ion from propane, both present in the<br />
primary sp$c?rum+ increased with increasing pressure and indeed, in the case <strong>of</strong><br />
propane the C H ion increased from a very small proportion <strong>of</strong> the primary spectrum<br />
3 7<br />
until it represented some 70% <strong>of</strong> all the ions present. Many secondary ions <strong>of</strong> mass<br />
less than the parent did not show such spectacular increases and other techniques<br />
were sought to establish these. One method <strong>of</strong> especial interest was that due to<br />
He employed electrons having energies too small to ionize in the<br />
Cermak. (a),.<br />
(8)<br />
V. Cermk and 2. Herman, Collection Czechoslov. Chem. Commun. 2, 406 (1962)<br />
ionization chamber. However, he employed a relatively high variable potential<br />
between the ion chamber and the trap anode, so that the electrons were accelerated<br />
-<br />
-
I<br />
26<br />
in the anode region. Some <strong>of</strong> these ionized molecules present in that region and the<br />
ions were repelled by the potential on the anode and drifted back into the ionization<br />
chamber. These primary ions could not be themselves collected, but new second-<br />
ary ions could be unequivocally identified.<br />
Further, by varying the potential on<br />
the anode, it was possible to obtain an appearance potential for the secondary ions<br />
and from this to deduce the identity <strong>of</strong> its precursor.<br />
A large number <strong>of</strong> reactions <strong>of</strong> ions with neutrals have now been identified and<br />
typical examples <strong>of</strong> the various classes <strong>of</strong> such bimolecular reactibns are given in<br />
tables 1 through 7.<br />
Table 1<br />
Atom Transfer Reactions<br />
+<br />
+ Kr + D2<br />
+ Xe + CH4<br />
+<br />
N+ + D2<br />
+<br />
-+ KrD+ + D (9)<br />
+ + XeH + Cis3 (10)<br />
+ -+ N2D + D (9,11)<br />
+ C3H5 (12)<br />
5+<br />
+ 0 + N2<br />
+ 0 + C02<br />
2+<br />
-+ NO + N (14,15)<br />
-+ 0 + CO (16)<br />
+ CH3 (17)'<br />
CH4 + C3H6 + CH +<br />
D20 + nC4HI0 --f HD 0 + C4H9 (13)<br />
+ 2+<br />
I + cH31<br />
-+ I2<br />
(9) Schissler, D. 0. and Stevenson, D. P., J. Chem. Phys.,24, 926 (1956)<br />
(Ip' u>ld, F. H., Franklin, 3. L., 2. &. *m. E. 83, 4509 (1961)<br />
(11)-Stevenson, D. P., J. a. %m.,6l, 1453 (1957)<br />
(12) Frankevich, E. L. and Tal'roze, V. L., Dokl. Akad. Nauk SSSRZ, 1174 (1958)<br />
(13) Lampe, F. W., Field, F. H. and Franklin, J. L., J. 3. Km. E., 79, 6132<br />
- (1957)<br />
(14) Potter, R. F., J. E m . Phys., 23, 2462 (1955)<br />
s=<br />
(15) Fehsenfeld, F. C., SchmeltekopfTA. L. and Ferguson, E. E., Planet. Space Science<br />
13, 219 (1965)<br />
(16) zhsenfeld, F. C., Ferguaon, E. E. and Schmeltekopf, A. L., 2. Chem. Phys. 2,<br />
3022 (1966)<br />
(17) Pottie, R. F., Barker, R. and Hamill, W. H., Radiation Research 2, 664 (1940)<br />
(18)<br />
(19)<br />
(20)<br />
Table 2<br />
Positive Atomic Ion Transfer Reactions<br />
0 ++ H2 + H O++ 0 (18)<br />
H2 2+ + O2 +€SO> + H (9,11) .<br />
HI++ CH31 + CH I + + H (19)<br />
33<br />
C2H: + D20 +€ID 0 + C2H5<br />
+ 3<br />
O2 + N +NO + 0 (21)<br />
0; + C2H2 --t C2H20+ + 0<br />
Hutchinson, D. A., Paper presented at American Chemical Society Meeting,<br />
Minneapolis, Minnesota, September, 1955<br />
Pottie, R. F., Barker, R. and Hamill, W. H., Radiation Research 10, 664 (1959)<br />
Lampe, F. W., Field, F. H. and Franklin, J. L., J. Amer. Chem. s., 2, 6132<br />
(1957)<br />
(21) Golden, P. D., Schmeltekopf, A. L., Fehsenfeld, F. C., Schiff, H. I., and 1<br />
-<br />
Ferguson, E. E., 2. Chem. Phys. 44, 4095 (1966)<br />
(22)<br />
(20)<br />
c<br />
1<br />
\<br />
!I<br />
4<br />
I\<br />
\<br />
,
27<br />
(22) Franklin, J. L., Munson, M.S.B., Tenth Symposium (International) on Combustion,<br />
The Combustion Institute, 1965, p. 561<br />
(23)<br />
(24)<br />
(25)<br />
(26)<br />
(27)<br />
Table 3<br />
Symmetrical Transfer Reactions<br />
+<br />
H; + H2 i H, + H (23,24,11)<br />
CH + CH4 i CH' + CH3 (2,3,4,5) i<br />
4+ 5+<br />
H 0 + H20 +H30 + OH (1)<br />
2<br />
HC1+ + +<br />
HC1 + H2C1 + H (9)<br />
+<br />
CH202+ + CH202 4 CH 0 + HC02 (22)<br />
NH;+ NH3 +NH4 3+2 + NH2 (25,26,27)<br />
I2 + I2 +I + I (28)<br />
3<br />
Eyring, H., Hirschfelder, J. 0. and Taylor, H. S., 2. Chem. Phys. 3, 479 (1936)<br />
Stevenson, D. P. and Schissler, D. O., J. %m. Phys.23, 1353 (1953)<br />
Lampe, F. W. and Field, F. H., Tetrahedron 1, 189 (l"3)<br />
Dorfman, L. M. and Noble, P. C., J. Phys. CEem. 63, 980 (1959)<br />
Derwish, G. A. W., Galli, A., Giardini-G-idoni, A., and Volpi, G. G., J.<br />
- Chem. Phys. 3, 1599 (1963)<br />
and Harkness, R. W., Phys. Rev. 2, 784 (1928)<br />
(28) Hogness, T.<br />
Table 4<br />
~<br />
-<br />
H- and H,- Transfer Reactions<br />
+ L +<br />
C2H5+<br />
C H<br />
+ C3H8<br />
+ C3H8<br />
+ C2H6 + C H<br />
7+<br />
+ C H + C3H7<br />
(29)<br />
(29)<br />
4+ 5+<br />
CH3 + C2H6 + C2H5 + CH4 (34)<br />
+ C3H5 + neo-C 5 H 12 + tC5H:1 + '3f6 (34)<br />
+ C H + C3H8 + C H + C3H6 (29)<br />
2 6<br />
24 C H + iC4H10 -+ C H + + iC4H8 (29)<br />
3 6 3 8<br />
+"<br />
(29) M.S.B.Munson, J. L. Franklin and F. H. Field, J. Phys. Chem. 68, 3098 (1964)<br />
Table 5<br />
Some+$eactions <strong>of</strong> Excited Ions<br />
+ C2H6 + C2Hl+ + C2H5 (29)<br />
C2H6<br />
H2+* + He + HeH + H (30)<br />
N + N2<br />
O2 2+* + O2<br />
+ N -I- + N (21,23,33)<br />
+ 0: + 0 (22,31)<br />
(30) H. von Koch and L. Friedman, J. Chern. Phys. 38, 115 (1963)<br />
(31) R. K. Curran, 2. Chem. Phys. 38, 2974 (1963)-<br />
(32) M.S.B.Munson, F. H. Field and?. L. Franklin, 2. Chem. Phys. 37, 1790 (1962) -<br />
(33) M. Saporoschenko, Phys. Rev. 111, 1550 (1958)<br />
(34) Field, F. H. and Lampe, F. W., J. Amer. Chem. z., 2, 5587 (1958)
C H +<br />
28<br />
Table 6<br />
Charge Transfer Reactions<br />
+<br />
0 +02 -+ 02+ + 0 (35)<br />
N2 + + O2 + N2 + 02+ (36)<br />
+<br />
2+<br />
C2H4 --f C H +<br />
Xe + C2H4<br />
Ar + + C2H4<br />
+<br />
Ar + CH4 4<br />
C2H2 (37)<br />
+<br />
c<br />
4+<br />
C H + Xe (38)<br />
I lH4' + H: + Xe (38)<br />
C2H2+ + H2 + Ar (38)<br />
C:H; + H + A r (38)<br />
CH? + H + Ar (5,39)<br />
F++ CO -+ CO + F (40)<br />
I, C+ + 0 + F (40)<br />
(35) F. C. Fehsenfeld, P. D. Goldan, A. L. Schmeltekopf and E. E. Ferguson, Planet.<br />
Space Sci. 2, 579 (1965)<br />
(36)<br />
F. C. Fehsenfeld, A. L. Schmeltekopf and E. E. Ferguson, Planet. Space Sci. 13,; -<br />
919 (1965)<br />
(37)<br />
(38)<br />
(39)<br />
F. H.<br />
J. L.<br />
G. G.<br />
Field, 2. fi. %m. e. 83, 1523 (1961)<br />
Franklin and F. H. Field, 2. Am. Chem. E. 83, 3555 (1961)<br />
Meisels, W. H. Hamill and R. R. Williams, Jr; 2. Phys. Chem:Z, 1456<br />
(1957)<br />
(40) E. Lindholm, Arkiv Fysik - 8, 433 (1954)<br />
CH4 + O2 -+ CH30 + OH (22)<br />
+<br />
+<br />
Table 7<br />
Xe + C H ~ -+ Xem; + H (10)<br />
t<br />
I<br />
I<br />
1
Tables 1-6 present examples <strong>of</strong> relatively simple reactions. However, reactions<br />
involving quite pr<strong>of</strong>ound changes in structure and bond reorganizations have been<br />
observed by a number <strong>of</strong> investigators. Table 7 shows several <strong>of</strong> these, which are<br />
given as typical examples. Certain <strong>of</strong> the reactions shown result in more than one<br />
set <strong>of</strong> products, all <strong>of</strong> which apparently occur at the same appearance potential<br />
and thus involve the same precursor. Of course, the intensities <strong>of</strong> the product<br />
ions will, in most instances, not be the same. One would expect tha,t reactions<br />
<strong>of</strong> this kind would involve the formation <strong>of</strong> a relatively stable complex, which<br />
breaks up in ways dictated by the energy content <strong>of</strong> the complex. Unfortunately,<br />
no quantitative treatment <strong>of</strong> the break up <strong>of</strong> the complex has yet been published,<br />
so it is not now possible to predict the ratios <strong>of</strong> product ions where more than<br />
one product arises from a single reactant. It has, however, been observed by Lampe,<br />
et al. (41) that the ratios <strong>of</strong> secondary ions from a collision complex will <strong>of</strong>ten<br />
be very similar to those <strong>of</strong> the same fragment ions in the primary mass spectrum<br />
<strong>of</strong> a compound having t e Sam$ composition as the comp ex. Thus, they pointed out<br />
9 4<br />
that the ratio <strong>of</strong> C H /C H in the reaction <strong>of</strong> C2H4 with C H was about the<br />
35. 4 7<br />
same as that observed in the mass spectra <strong>of</strong> the butenes. Thg fiterature contains<br />
(41) F. W. Lampe, J. L. Franklin and F. H. Field, "Progress in Reaction Kinetics,"<br />
Val. 1, Pergamon'Press, New York, 1961, p. 69.<br />
only a few examples <strong>of</strong> simple condensation reactions. This is not surprising in view<br />
<strong>of</strong> the fact that every complex is formed with enough energy to break up in either<br />
the forward or reverse direction. Since most complexes can break up very rapidly<br />
they will generally do so in a time short compared to that required to collect the<br />
ion. In a few instances such complexes have been observed to survive long enough<br />
to be measured. One example <strong>of</strong> this is given in Table 7. It might be mentioned<br />
that several apparently long lived complexes were observed by Field (37) in his<br />
study <strong>of</strong> ethylene, but these in all cases turned out to depend upon the third or<br />
higher power <strong>of</strong> the pressure, and thus were in fact complexes that had been<br />
stabilized by collision or had resulted from the decomposition <strong>of</strong> a complex <strong>of</strong><br />
higher molecular weight.<br />
Although the preceding discussion has largely been devoted to ions formed as<br />
bimolecular reaction products, in the last few years, many examples <strong>of</strong> ions formed<br />
with much higher pressure dependence have been observed. One <strong>of</strong> the earliest studies<br />
carried out at such elevated pressures, i.e., above 100 microns, was the study <strong>of</strong><br />
Field (37) <strong>of</strong> ion-molecule reactions in ethylene. In this study he observed ions<br />
with pressure dependencies as high as about 6, although those exhibiting the highest<br />
pressure dependence were <strong>of</strong> such low intensity that the nature <strong>of</strong> the reactions<br />
involved could not be ascertained. Indeed, above about 3rd order the method <strong>of</strong><br />
appearance potentials becomes completely useless, and the identification <strong>of</strong> precursors<br />
to a given product becomes very tenuous indeed. To form ions at high pressures<br />
in a region from which they can be collected it is usually necessary to employ<br />
electrons <strong>of</strong> several hundred volts rather than the usual 60-70 volts employed in<br />
most mass spectrometric problems. Indeed, because <strong>of</strong> this problem, Kebarle and<br />
Hogg (45) have employed alpha particles <strong>of</strong> high energy to provide primary ions.<br />
(45) P. Kebarle and A. M. Hogg, J. &m. a. 42, 668 (1965); 2, - 449 (1965)<br />
When ions are formed by high energy massive particles it is possible to operate<br />
at much higher pressures and Kebarle (45), Wexler et al. (46) and others have<br />
(46)<br />
S. Wexler, Assa Lifshitz and A. Guattrochi, "Ion-Molecule Reactions in the<br />
Gas phase," Adv. in Chem. Series, Amer. Chem. SOC., Washington, D. C. 1966<br />
p. 193<br />
studied the ions formed at pressures up to about one atmosphere. Naturally, they<br />
have observed ions with a very high pressure dependence, although they have not been<br />
able to establish the order <strong>of</strong> reaction. With such a system, Kebarle et al. (49,50)<br />
-
-_<br />
30<br />
have observed ions having the general formula, H+(H O)n, with n varying from 1 to 7,<br />
and Wexler, et al. (46) have observed polymer ions Prom acetylene having up to 12<br />
carbon atoms.<br />
Recently, studies have been carried out in mass spectrometry sources at pressures<br />
up to several torr. Under these conditions, any primary ions formed will undergo<br />
many collisions and will have ample opportunity to react if they are capable <strong>of</strong><br />
doing so. Field and Munson (47,48) have taken advantage <strong>of</strong> this to carry out some<br />
very intereciting studies <strong>of</strong> reactions <strong>of</strong> highef order. pey observeh that in very<br />
pure methane, the principal secondary ions CH5 and C2H5 reached a plateau and re-<br />
(47) M.S.B.Munson and F. H. Field, 2. &. =m. %. 88, 2621 (1966)<br />
(48) F. ti. Field, M. S. B. Munson and D. A. Becker, "Ion Molecule Reactions in<br />
the Gas Phase," Adv. in Chem. Series, Amer. Chem. SOC., Washington, D. C.<br />
(49)<br />
(1966), p. 167<br />
P. Kebarle and A. M. Hogg, 2. s m . m. 42, 798 (1965)<br />
(50). A. M. Hogg and P. Kebarle, J. Chem. Phys. 43, 498 (1965)<br />
mained constant with increases in pressure beyond about 0.5 torr. They observed<br />
also that if there were any small amount <strong>of</strong> impurities in the methane the intensities<br />
<strong>of</strong> these ions passed through a maximum around a few .loth8 torr and then declined<br />
steadily with further increases in pressure. The primary ions had very small probability<br />
<strong>of</strong> colliding with anything but methane and consequently the disappearance<br />
<strong>of</strong> the secondaries must be due to their reaction with the impurities, since it is<br />
demonstrated that they did not react with methane itself. It was a simple step<br />
from this to the addition <strong>of</strong> small amounts <strong>of</strong> a variety <strong>of</strong> materials to the methane<br />
plasma, with results that proved extremely interesting. When, for example, smell<br />
amounts <strong>of</strong> long chain paraffin hydrocarbons, such as dodecane, were added to the<br />
methane plasma, the spectrum <strong>of</strong> ions from the high molecular weight paraffin was<br />
quite different from that obtained by electron impact. Such high molecular weight<br />
paraffins give only small intensities <strong>of</strong> ions above about the C5 range under<br />
eleqtron impact. However, in the methane plasma, ions <strong>of</strong> the general composition<br />
C H formed at each carbon number from the parent down to C4. No doubt ions<br />
o? tailer mess are also formed, but these are not observable because <strong>of</strong> the interference<br />
from secondary and ternary ions from methane. Further, the largest <strong>of</strong> thfse<br />
ions is the one corresponding to the parent molecules; i.e., with dodecane,C12H25 .<br />
Field and Munson have studied a number <strong>of</strong> compounds by this method, and in<br />
many instances have obtained pr<strong>of</strong>ound changes in the mas spectrum, produced by<br />
"Chemical Ionization," the term which they have given the processes. (47,48)<br />
They have concluded that in the case <strong>of</strong> the methane the principal reactions are<br />
probably as follows:<br />
CH5 + + CnH2w2 + CH4 + H2 +'Cn+H + ,<br />
2#1 .<br />
-<br />
+<br />
4 C H + CnH2#l<br />
2 6<br />
Although the previous discussion has been devoted entirely to reactions <strong>of</strong> positive<br />
ions, negative ions are also known to undergo reactions on collision. Relatively<br />
few <strong>of</strong> these reactions have been studied, however, largely 'because negative<br />
ions present some rather serious difficulties to the investigator.<br />
<strong>of</strong> negative ions, however, have been carried out by Melton, Henglein and others,<br />
and several typical reactions are given in table 9.<br />
-<br />
c<br />
Some reactions
Table 8<br />
Some Ions Formed by Processes <strong>of</strong> Order Higher than 2<br />
Rea c tan t Ionic Product Reference<br />
H2°<br />
m3<br />
H+(H,O), (1
J<br />
32 ',<br />
1<br />
A number <strong>of</strong> investigators have subsequently studied this chemi-ionization reaction <strong>of</strong>\<br />
the rare gas ions and have confirmed and extended Hornbeck and Molnar's observation,<br />
It has now been observed that all <strong>of</strong> the rare gases react with each other to form<br />
.the hetero-nuclear diatomic ions (57). In addition, a number <strong>of</strong> ionic compounds <strong>of</strong> ,<br />
the rare gases with nitrogen, CO, 02, methane, acetylene and others have been report-'<br />
ed. Chemi-ionization reactions are not necessarily limited to rare gases, however. , !<br />
Chemi-ionization products <strong>of</strong> excited mercury atoms with a number <strong>of</strong> compounds have<br />
been observed (59) and nitrogen and CO are known to und r o chemi-ionization with<br />
their own ground state species, forming respectively N -? :nd C20; '(32).<br />
4<br />
1<br />
I<br />
(59) P. Cermak and 2. Herman, Tenth Annual Conference on Mass Spectrometry, New<br />
I<br />
,<br />
Orleans, La., 1962, p. 358. I<br />
,<br />
It is natural that when both reactants and products are readily measurable<br />
c nsiderat'on should early be given to the fate <strong>of</strong> the reaction. With the reaction<br />
4? + S, if M is much larger than A , the feactior rate is psuedo first order'<br />
A + M -%<br />
and the equation expressing the concentration <strong>of</strong> A and B is as follows:<br />
- B+ =<br />
AO<br />
+<br />
-kMt<br />
1-e<br />
-<br />
-1<br />
A Llot <strong>of</strong> B+ against M will yield a straight line whose slope is kt. If a<br />
At relatively low pressures, (a few microns), i<br />
time is thus:<br />
B+<br />
- =<br />
A' + B+<br />
rate constants for second order ion-molecule reactions are given in table 11. It<br />
will be observed that many 2 them are in the order <strong>of</strong> 10 cc/molecule/sec. How-<br />
€3<br />
ever, values as small as 10 cc/mo_l culelsec have been reported for some reactions.<br />
6 The values in the neighborhood <strong>of</strong> 10 cc/molecule/sec represent reactions that must<br />
occur at essentially every collision in that they have cross sectionslSonsiderably<br />
larger than ordinary collision cross sections. The values around 10 represent<br />
or very small.<br />
must not involve appreciable energy <strong>of</strong> activation. Stevenson and Schissler (4,11)<br />
have confirmed this experimentally for a few reactions and the very fact that a<br />
kMt<br />
As was mentioned before, ion-molecule reactions to be observable ,<br />
~ ~~<br />
I
33<br />
velocity <strong>of</strong> the reacting partn rs is sufficie tl y great. Giese and Maier (60) have<br />
f shown that for the reaction Ar + CO 4 Ar + C + 0, which is endothermic by 6.62<br />
+<br />
and 6.44 e\’ for the 2p state <strong>of</strong> Ar , the threshold for the<br />
“co<br />
Their measured values were in agreement with this relation.<br />
(60) C. F. Giese and W. B. Plaier, J. X m . a. 39, 197 (1963)<br />
(61)<br />
(62)<br />
(63)<br />
React ion<br />
1<br />
112<br />
Table 11<br />
Some Second-Order Rate Constants<br />
10 10 k, cc/molecule sec Reference<br />
D2 -3 D3 + D 14.5 4,11<br />
+<br />
CH4 + CH4 + CH<br />
5+ + m3<br />
10 61,62,63<br />
H20 + H20 --f H30 + OH 12.7 1,2,13<br />
+ +<br />
C H + C H + C H +CH4 14.5 9<br />
2 6 3+5<br />
2+3<br />
+ C H + ~ CH o + n 0.126 22<br />
O2 + 33<br />
CH4 + O2 + CH30 + OH<br />
0.257 22<br />
V. L. Tal’roze and E. L. Frankevich, J. Phys. Chem. USSR 34, 1275 (1960)<br />
C. W. Hand and H. von Weyssenh<strong>of</strong>f, Can. J. Chem., 42, 195, 2385 (1964)<br />
J. L. Franklin, Y.<br />
2353 (1966)<br />
Wada, P. Natalis and P. M. Hierr J. Phys. Chem. 70, -<br />
All reaction rate studies have not been limited to extremely low pressures.<br />
When the pressure in the ion source becomes sufficiently great the reactions follow<br />
the psuedo first order rate laws over rather wide ranges <strong>of</strong> pressure, unless subse-<br />
quent reactions interfere. Thus, Field et al. (51) found the disappearance <strong>of</strong> CH4<br />
in methane to obey first law kinetics over a pressure range <strong>of</strong> about 0.1 to 400 Torr.<br />
However, as the pressure is raised, in certain systems, reactions <strong>of</strong> a higher order<br />
do occur. Field, (37) studying ethylene, has reported some reactions having apparent<br />
orders as high as about 6. These, <strong>of</strong> course, are not truly 6th order reactions, but<br />
simply represent a succession <strong>of</strong> perhaps five secondary reactions which show dependence<br />
upon the 6th power <strong>of</strong> the pressure. Rates <strong>of</strong> reactions <strong>of</strong> such high order do<br />
not yield satisfactory rate constants, but it has been possible to determine rate<br />
constants for reactions <strong>of</strong> 3rd order, and Field and several others have made approximate<br />
measurements <strong>of</strong> the rate constants for such third order processes. As is the<br />
case with the second order processes, these reaction rates are relatively high. For<br />
example, in ethylape eld deter ined several third order rate constants to be<br />
F3 2 -1<br />
in the order <strong>of</strong> molecule- sec .<br />
Although most <strong>of</strong> the measurements <strong>of</strong> rate constants in the literature were<br />
obtained with the source operating in a continuous mode employing a variation in<br />
pressure to establish the rate, some studies have been made in which retention time<br />
in the source was varied.<br />
Such measurements were originally made by Tal’roze and<br />
Frankevitch (61), but subsequent measurements have been made by Hand and von Weissenh<strong>of</strong>f<br />
(62) and Franklin, Natalis, Wada, and Hierl (63). In order to obtain a satisfactory<br />
variation <strong>of</strong> time, a pulsed mode is employed. In such an operation a pulse<br />
<strong>of</strong> electrons is fired through the gas. After it is stopped, the resulting ions<br />
can be retained in the source for a controlled period <strong>of</strong> time and then rapidly<br />
extracted by a pulse <strong>of</strong> high energy. By varying the delay time, the time <strong>of</strong> retention<br />
in the source is varied, and rate constants determined in the manner more usually<br />
employed by chemists in rate studies. Results obtained in this way generally agree<br />
-<br />
I<br />
+
.<br />
34<br />
rather well with those obtained by the pressure method, although some differences<br />
have been observed. One difference that may be <strong>of</strong> significance is that the ions in<br />
the pulsed mode will generally have approximately thermal energies, whereas those<br />
reacting in the continuous mode will have variable energies, depending upon the point<br />
<strong>of</strong> their reaction in travelling from the electron beam to the exit slit.<br />
The question <strong>of</strong> the effect <strong>of</strong> ionic energy on reaction rate has been one <strong>of</strong><br />
considerable interest and the subject <strong>of</strong> a number <strong>of</strong> investigations, both theoretical<br />
and experimental. In their early work on ion-molecule reactions, hranklin, Field and<br />
Lampe (3) observed that when they varied the field strength in the ion source in<br />
order to vary the retention time, the rate constants that they calculated for their<br />
ion-molecule reactions varied considerably. In general, they seemed to drop as<br />
the field strength increased, suggesting that the reaction rate constant decreased<br />
with the relative velocity <strong>of</strong> ions and neutrals. Other investigators have made<br />
similar observations. However, it appears that not all reactions show such reduction<br />
in rate constants with increasing relative velocity. Attempts to explain this have<br />
been made by a number <strong>of</strong> investigators. Field, Franklin and Lampe (3) attempted<br />
to obtain the theoretical relations based upon a balance <strong>of</strong> polarization and<br />
centrifugal forces. Gioumousis and Stevenson (64) derived a more precise expression<br />
(64) Gioumousis, G. and Stevenson, D. p., 2. Chem. Phys.,g, 294 (1958)<br />
(65) P. Lannevin. Ann. Chim. Phvs. 5. 245 (1905)<br />
for the collision rate based upon LaiTgevin's(65) treatment for polarizable systems.<br />
Gioumousis and Stevenson found the collision cross section to be:<br />
where a is the polarizability <strong>of</strong> the neutral, v<br />
i<br />
is the velocity <strong>of</strong> the ion,<br />
lJ is the reduced mass and e the charge on the electron. Since<br />
1'<br />
(I will vary as Further, since k = uv<br />
and thus is independent <strong>of</strong> velocity or energy. This, unfortunately, did not agree<br />
with the observed rate behavior <strong>of</strong> a number <strong>of</strong> reactions, although it appears to<br />
hold for some. In fact, Field, et al. (3) studied the effect <strong>of</strong> field strength upon<br />
rate constant and found that for several reactions k decreased with increasing field<br />
strength. Hamill and his associates (66,67) have shown that ion-molecule reactions<br />
cannot be accurately treated as involving point particles. By considering the de-<br />
(66) N. Boelrijk and w. H. Hamill, J. &. %m. =. 84, 730, (1962)<br />
(67) D. A. Kubose and W. H. Hamill, J. &. X m . e. 85, 125 (1963) -<br />
formable neutral to exhibit a hard core to high energy collisions while being deformable<br />
in low energy collisions, these workers showed that for small ion energies<br />
u obeped the Gioumousis-Stevenson relation (equation 5), but for large energy<br />
uk E which agrees with experimental results. Theard and Hamill (68) and Moran<br />
and ffamill (69). have extended their treatment to ion-molecule reactions involving<br />
(68) L. P. Theard and W. H. Hamill, J. Am. -m. E. 84, 1134 (1962)<br />
(69) T. F. Moran and W. H. Hamill, J. Chem. Phys. 39, z13 (1963)<br />
neutrals with permanent dipoles. They showed that at law relative velocities a<br />
=ne,<br />
cross section for the ion-permanent dipole interaction, ~~<br />
-<br />
Et<br />
(4)<br />
(5)
35<br />
must be added to the Langevin cross section. Here is dipole moment and Etis the<br />
translational energy <strong>of</strong> the reacting system in center <strong>of</strong> mass coordinates.<br />
NO attempt will be made here to review all <strong>of</strong> the studies <strong>of</strong> the effect <strong>of</strong> energy<br />
upon the rates <strong>of</strong> ion-molecule reactions. However, several investigators who have<br />
made especially important contributions should be mentioned. In addition to the<br />
work <strong>of</strong> Hamill discussed above, important studies have been made by Futrell and<br />
Abramson (70), Giese and Maier (71), Friedman (72,73) and Light and Horrocks(74).<br />
- -- --<br />
(70) J. H. Futrell and F. P. Abramson, "Ion-Molecule Reactions in the Gas Phase,"<br />
Adv. in Chem. Series 58, Amer. Chem. SOC., 1966, p. 107.<br />
(71) C. F. Giese and W. B.xaier, J. Chem. Phys. 39, - 197, 739 (1963)<br />
(72) T. F. Moran and L. Friedman, J. Chem. Phys. 39, 2491 (1963); 62, 2391 (1965) - -<br />
(73) F. S. Klein and L. Friedman, J. Chem. Phys. 4l, 1789 (1964)<br />
(74) J. C. Light and J. Horrocks, Proc. Phys. 527 (1964)<br />
It should be pointed out that if the relative velocities <strong>of</strong> ions and neutrals<br />
become sufficiently high other reactions begin to OCCUL as a result <strong>of</strong> the different<br />
forces coming into play. Thus a fast moving ion passing a molecule with sufficient<br />
velocity may simply strip <strong>of</strong>f a peripheral atom, leaving the partially denuded entity<br />
behind. Such stripping reactions have been studied by Henglein (75) and Koski<br />
(76), who found that they obey quite different rules from those above. It thus<br />
(75) A. Henglein, "Ion Molecule Reactions in the Gas Phase," &. in Chem. Series<br />
- 58, Amer, Chem. SOC., Washington, D. C.,' p. 63<br />
-<br />
(76) M. A. Berta, B. Y. Ellis and W. S. Koski, ibid., p. 80<br />
appears that a complete theory <strong>of</strong> the rates <strong>of</strong> ion-molecule reactions has not been<br />
developed, but there is little doubt that to a first approximation the equation <strong>of</strong><br />
Gioumousis and Stevenson givsfairly good results. It is also true that the<br />
reactions tend to be quite fast, and this becomes a matter <strong>of</strong> overriding importance<br />
in many processes involving ions. Thus is has been shown by Lampe (77,78) and by<br />
Futrell (79) that ion-molecule reactions are the controlling factor in certain<br />
(77) F. W. Lampe, Radiation Research lo, 671 (1959)<br />
(78)<br />
(79) J. H. Futrell, J. &I. -m.<br />
82, 1551 (1960)<br />
=E<br />
*. 8l, 5921 (1959)<br />
-<br />
F. W. Lampe, 2. &. k m . z.,<br />
-<br />
radiation induced processes involving paraffin and olefin hydrocarbons.<br />
An electric discharge <strong>of</strong> course involves ions and electrons and the ions present<br />
can be sampled and analyzed by mass spectrometry. Such studies have been undertaken<br />
by a number <strong>of</strong> investigators and some progress is being made toward understanding<br />
the <strong>chemical</strong> behavior <strong>of</strong> ions in various <strong>discharges</strong>. The problem is complicated by<br />
the fact that there are several kinds <strong>of</strong> electric discharge, each <strong>of</strong> which has its<br />
own physical and <strong>chemical</strong> characteristics. Of these, the type most <strong>of</strong>ten studied is<br />
the direct current glow discharge, but corona and high frequency and micro-wave<br />
<strong>discharges</strong> have also received some attention.<br />
In order to analyze the ionic content <strong>of</strong> a discharge it is necessary to trans-<br />
fer the ions from the discharge into the mass analyzer. While this is not a par-<br />
ticularly serious problem at pressures below 10 microns, the difficulty becomes<br />
more acute as the pressure increases. This is attributable to the fact that elec-<br />
trons and ions diffuse to the walls at different rates, so that an electric gradient<br />
is established which alters the distribution <strong>of</strong> energy <strong>of</strong> the various charged species.<br />
Ordinarily a sheath <strong>of</strong> ions is formed at any surface, including that <strong>of</strong> a sampling<br />
probe and ions or electrons must have, or be given enough energy to pass through<br />
this sheath in order to be sampled. However, if the energy is sufficiently high<br />
some ions may be decomposed by collision. The result then is that there is <strong>of</strong>ten<br />
-
36<br />
considerable undertainty as to the quantitative correspondence <strong>of</strong> the ion distribu-<br />
tion reported by the mass spectrometer with the actual distribution in the discharge,<br />
Further, while there is little doubt that the ions observed were actually present,<br />
there is always some question as to the presence <strong>of</strong> ions that might be expected, but<br />
that are not observed.<br />
An added complication that must be taken into account, especially in glow dis-<br />
charges, is that different portions <strong>of</strong> the discharge have different characteristics,<br />
Thus the negative glow and positive column have quite different el'ectric fields, the<br />
ions and electrons present have different energies and the distribution <strong>of</strong> ions in<br />
the two regions is different.<br />
In spite <strong>of</strong> these reservations, considerable information has been obtained<br />
concerning the ions present in certain <strong>discharges</strong>~,and some understanding <strong>of</strong> the re-<br />
actions occurring is beginning to develop.<br />
' In the glow discharge, the regions most studied have been the negative glow and<br />
the positive column. Both are regions <strong>of</strong> nearly equal positive and negative ion<br />
concentration, although the ion concentration in the negative glow is usually greater<br />
(<strong>of</strong>ten 10-100 times) than that in the positive column. Further, the electric field<br />
in the negative glow is considerably greater than that <strong>of</strong> the positive column, through<br />
which the ions drift with relatively small energies.<br />
Discharges in the rare gases, <strong>of</strong> course, always contain atomic ions and, at<br />
sufficiently high pressures, diatomic ions as well. This latter can be produced<br />
by two possible reactions, typified by helium:<br />
*<br />
He + He --f He: + e (6)<br />
+ He + 2He + He2+ + He<br />
(7)<br />
Since the formation <strong>of</strong> He* is an excitation process it will occur over a relatively<br />
small energy range with electrons <strong>of</strong> energy close to the ionization potential <strong>of</strong> He.<br />
Thus, it would be expected to predominate in the positive column <strong>of</strong> a glow discharge.<br />
This has been observed by Morris (80) and by Pahl (81,82).<br />
(80) D. Morris, Proc. Phys. E. (London) s, 11 (1955)<br />
(81)<br />
(82)<br />
M. Pahl and U. Weimer, z. Naturforsch-G, 753 (1958)<br />
M. Pahl, 2. Naturforsch, &, 239 (195r<br />
-<br />
In order for reaction 7 to be observed relatively higQ pre5sures are required.<br />
-0 2<br />
The third order rate constant will probably not exceed 10 cc /m lecule second<br />
-9<br />
and the timt <strong>of</strong> the ion in the plasma will probably not exceed 10 seconds. Thus,<br />
+ for He2 /He to be approximately 0.1 by reaction 7 the pressure must be approximately<br />
5 Torr. Thus the formation <strong>of</strong> the diatomic ion by the three-body process will<br />
decrease very rapidly with decreasing pressures and will be negligible below 0.1 Torr.<br />
Knewstubb and Tickner (83) have studied the ions <strong>of</strong> the rare gases in both the negative<br />
(83) P. F. Knewstubb and A. W. Tickner, J. Chem. Phys. 36, 674, 684 (1962)<br />
=<br />
glow and the positive column <strong>of</strong> a dc glow discharge. They find the ratio Ar +/Ar+<br />
to be much less in the negative glow than in the positive column, and conclude that<br />
the diatomic ion is formed principally by the three body process in the negative<br />
glow, but that the chemi-ionization reaction predominates in the positive column..<br />
Similar considerations apply to the other rare gases.<br />
I<br />
1<br />
I<br />
I
37<br />
In our laboratory a microwave discharge was generated in helium by a 100 watt<br />
Raytheon microtherm generator and sampled through a pinhole leak at the apex <strong>of</strong> a<br />
+ .<br />
conical probe into a quadrupole mass filter. The intensity <strong>of</strong> the He ion dropped<br />
exponentially in the range studied from a relative intensity ,<strong>of</strong> unity at 0.1 Torr<br />
to about .002 at 0.5 Torr.<br />
+ .<br />
In the same pressure range Hez increased in intensity from O.,OOZ at 0.1 Torr<br />
to a broad maximum around 0.3 Torr and then declined rapidly at higher pressures.<br />
+.<br />
If the He2 1s forme g8by she three- ody process (equation 7) the rate constant would<br />
9<br />
have to be about 10 cc /molecule /sec. which seems excessive. Ne conclude then<br />
that the diatomic ion is probably formed principally by the chemi-ionization process.<br />
(equation 6).<br />
Of perhaps greater interest are the ionic processes occurring in more complex<br />
gases. Thus, glow <strong>discharges</strong> in hydrogen (84,85,) and in H,-D, mixtures (86) showed<br />
..-<br />
(84) C. J. Braesfield, Phys. e. 2, 52 (1928)<br />
(85) 0. Luhr, J. Chem. Phys. - 3, lLib(1935)<br />
(86) H. D. Beckey and H. Dreeskamp, 2. Naturforsch 9.; 735 (1954)<br />
t<br />
the formation <strong>of</strong> H3t or H - D mixtures, which increased in concentration with<br />
pressure at the expense 01 the aiatomic ion. In an effort to interpret the behavior<br />
<strong>of</strong> the discharge in hydrogen, Eyring, Hirschfelder and Taylor (87) considered the<br />
(87) H. Eyring, J. 0. Hirschfelder and H. S. Taylor, 2. Chem. Phys. 4, 479 (1936)<br />
reaction forming H3t to be:<br />
Hz + + Hz +H: + H<br />
They took the activation energy for the reaction to arise from the balance <strong>of</strong> cen-<br />
trifugal force and polarization attraction acting in opposite directions. The result-<br />
ing rate constant for the reaction is:<br />
k = 2 TI e kl”<br />
i.e., the same as equation 5. Subsequent studies <strong>of</strong> this reaction in a mass spec-<br />
trometer ion source (4) have established this reaction beyond doubt, and have shown<br />
that the reaction rate is given approximately by the above relation <strong>of</strong> Eyring, et al.<br />
Recent studies by Ortenburger et al. (88) employing a high frequency discharge have<br />
(88) I. B. Ortenburger, M. Hertzberg and R. A. Ogg, J. Chem. Phys. 2, 579 (1960)<br />
shown that reaction 8 occurs under these conditions as well.<br />
Studies <strong>of</strong> ions in the negative glow and Faraday dark space <strong>of</strong> a glow discharge<br />
in water vapor at 0.4 Torr have been made by Knewstubb and Tickner (89). They<br />
(89) P. F. Knewstubb and A. W. Tickner, J. X m . Phys. 8, 464 (1963)<br />
-<br />
fouqd the maximum ion intensity to occur in the negative glow. There was little<br />
H 0 present, but a series <strong>of</strong> solvated protons was observed having the general composition<br />
2 .<br />
H (H O)n with n varying from one to five. Mass spectrometer studies have<br />
established t?ie reaction: (1,2, 13)<br />
+ +<br />
H 0 + H20 + H30 + OH<br />
2<br />
(9)<br />
and more recent studies have detected the more highly solvated species. (49,50) The<br />
results <strong>of</strong> the discharge studies showed the first four water molecules to be more<br />
strongly bonded to the proton than succeeding molecules and this has been substantiated<br />
by the work <strong>of</strong> Kebarle (49,50) previously discussed.<br />
-<br />
-<br />
(8)
36<br />
Similar studies have been made <strong>of</strong> ions in a glow discharge in ammonia (90)<br />
(90) P. H. Dawson and A. W. Tickner, J. Chem. Phys. 40, 3745 (1964)<br />
w'th similar results. The negative g)ow at 0.4 Torr was found to contain the ions<br />
+ H (NH3), with n from 1 to+5, with NH being formed in highest concentration in the<br />
negative glow, but with H (NH ) preiominating in the Faraday dark space. The<br />
3 4<br />
multiply solvated proton has also been observed in the ion source <strong>of</strong> a mass spectrometer<br />
at elevated pressure. (91, 92)<br />
I<br />
(91) A. M. Hogg and P. Kebarle, J. -. m. 43, 449 (1965)<br />
-<br />
(92) A. M. Hogg, R. M. Haynes and P. Kebarle, J. $. E m . z. 88, 28 (1966)<br />
(93)<br />
Knewstubb (93) also mentions the observation <strong>of</strong> ions in a glow discharge in<br />
P. F. Knewstubb, "Mass Spectro~metry <strong>of</strong> Organic Ions," Academic Press, New<br />
York, 1963, p. 284<br />
methane in which the ions C H + and CH5+ predominated, and in which Some 40% <strong>of</strong> the<br />
2 5<br />
ions present contained three or more carbon atoms. This is quite different from<br />
the results obtained by Munson and Field (47, 48) for studies <strong>of</strong> methane at elevated<br />
pressurTs. They found that with quite pure methane at pressu es above about 1 Torr<br />
f<br />
the CH5 and C2H5 ,ions were present in the same ratio as CH4 and .I: (the precursors)<br />
in the primary mass spectrum <strong>of</strong> methane, and that ions having more than two<br />
carbon atoms were present in only minor proportions. This suggests that the ions<br />
<strong>of</strong> higher mass reported by Knewstubb (93) originated either from impurities in the<br />
methane employed or, more probably, from molecules such as acetylene or ethylene<br />
formed by the action <strong>of</strong> the discharge on methane.<br />
Nitrogen has been the subject <strong>of</strong> several investigations employing both mass<br />
spectrometer ionization chambers and dis har es for the production <strong>of</strong> ions. The<br />
F io s <strong>of</strong> greatest interest are N + and N4 . g The mass spectrometer studies have shown<br />
P 3<br />
to be formed by the reaction:<br />
N3 -<br />
N2 +* + N p + N:+N<br />
where N +* imp ies an excited ion having an appearance potential <strong>of</strong> about 21-22 eV.<br />
i.<br />
(31,32,33) N4 has been found to result from the reaction:(94)<br />
+ ,N2 + 2N2 + -+ N4 + N2<br />
(94)<br />
G. Junk and H. J. Svec, 2. &. 2. E. 80, 2908 (1958)<br />
In addition, Munson et al. (32) showed that under certain conditions N4+ is formed<br />
by the chemi-ionization reaction:<br />
* +<br />
N2 + N2 -+ N4 + e<br />
(12)<br />
Both ions have been obs rved in electric <strong>discharges</strong> in nitrogen. Luhr (95) and<br />
-f<br />
Dreeskamp (96) found N3 , but it appeared to be formed only in the drift space<br />
(95)<br />
(96)<br />
-<br />
0. Luhr, w. E. 44, 459 (1933)<br />
H. Dreeskamp, 2. Naturforsch G, 876 (1958)<br />
-<br />
following the discharge. It was thought to result from the reaction<br />
+ N + 2N2 + + N3 + N2<br />
(13)<br />
formed in a glow discharge at 0.3 Torr in nitrogen<br />
+<br />
Shahin (97) has reported N3<br />
(97) M. M. Shahin, "Ion-Molecule Reactions in Gases," Advances in Chemistry Series<br />
No. 58, American Chemical Society, Washington, D. C.. 1966 p. 315<br />
-<br />
-
+ .<br />
4 . r<br />
39<br />
as well as N in a cor0 a discharge in a mixture <strong>of</strong> nitrogen and water vapor. The<br />
relative intensity <strong>of</strong> Nq passed through a maximum at about 10 Torr, then slowly<br />
declined at higher pressures. Shahin attributed the formation <strong>of</strong> the ion to the<br />
\ reaction:<br />
><br />
N3 + + N3 + N C<br />
'$ In the same system he observed H 0 , H 0 , H 0; fnd N H+ ions, all apparently<br />
2<br />
rresulting, at least in part, from reac2ion 02 N* wit6 water.<br />
In this laboratory nitrogen has been passed through a microwave discharge at<br />
pressures <strong>of</strong> 0.01 to 0.3 Torr and the plasma saypled into a quadrupole mass filter<br />
where the io s were separated and analyzed. was not observed at fny condition<br />
studied . P<br />
N4<br />
N3<br />
was formed in very small concentrations (about 1% <strong>of</strong> N2,) at 0.01<br />
Torr, but increased rapidly in intensity, passing through a broad maximum at about<br />
0.15 - 0.20 Torr, and the decreasing at higher pressures. In the same pressu e<br />
f<br />
range the intensity <strong>of</strong> N: dropped precipitously from 90 to 1.5+ and that <strong>of</strong> N<br />
remained constant at about 9, all in arbitrary units. At the N3 maximum all <strong>of</strong> the<br />
ions were <strong>of</strong> nearly equal intensity. A brief study <strong>of</strong> the variation <strong>of</strong> the intensi<br />
ies <strong>of</strong> these ions with nominal power input at a pressure <strong>of</strong> 0.15 Torr showed<br />
4 + and N to decrease in intensity with decreasing power until the discharge was<br />
N2 .<br />
extinguished at about 40% <strong>of</strong> maximum. These curves were reminiscent <strong>of</strong> an ionizetion<br />
efficiency curve, and strongly fuggest the average electron energy decreased<br />
with decreasing power input.<br />
power input up to a broad maximum between 75 and 65% <strong>of</strong> maximum power, after which<br />
it again decreased. This suggests that an excited state is formed by electron<br />
impact with elec~trons <strong>of</strong> broad energy spread. Such an energy spread has been found<br />
in our studies, and will be reported separately. Further, we find that the electrons<br />
possess an approximately Maxwell-Baltzmann distribution <strong>of</strong> energy, and that the<br />
average energy decreases as the pressure increases, in accordance with our observa-<br />
+ tion <strong>of</strong> the va iation <strong>of</strong> N3 with pressure. This also accounts in part for the<br />
f<br />
decrease in N2 with pressure.<br />
+<br />
(14)<br />
The N3 ion increased in intensity with decreasing<br />
The near c nstancy <strong>of</strong> N with increasing pressure is difficult to understand.<br />
?<br />
The number <strong>of</strong> N ions formed by direct electron impact up0 N or N must be relatively<br />
small and can hardly account for+the intensity <strong>of</strong> the N' ogserved. It is possible<br />
that at the higher pressures most N is formed by the reaction:<br />
N;+N + N + + N2<br />
(15)<br />
but this is, <strong>of</strong> course, speculative.<br />
+<br />
The absence <strong>of</strong> N4 is puzzling, since it has been observed in some <strong>discharges</strong>.<br />
It is possible the ion is d stroyed in passing through the sampling probe, This seem<br />
8 unlikely, however, since N4 is held together by a bond <strong>of</strong> about 1.5 eV energy.<br />
This is about the same strength as the bond in He:, which we observe. Conceivably,<br />
the ion may appear at higher pressures, but some alterations in our sampling probe<br />
will be necessary to achieve this.<br />
+<br />
Seve a1 investigations <strong>of</strong> ions in glow <strong>discharges</strong> in oxygen have been reported.<br />
0 and 0: were reported by Knewstubb (93), Luk (98) and Dickenson and Sayers (99).<br />
L<br />
(98) Luhr, O., m. g. 38, 1730 (1931)<br />
(99) PT-H. G. Dickenson andJ. Sayers, Proc. Phys. =. (London) G, 137 (1960)<br />
~~<br />
+<br />
In addition, 03+ and O4 (99) have been reported.<br />
4<br />
D<br />
In OUK stud es <strong>of</strong> orygen In the pressure range 0.01-0.3 Torr, using a microwave<br />
discharge only 0 and O2 were found. Both ions dropped in intensity exponentially +<br />
with increasing pressure, but the 0' dropped slightly less rapidly than did O2<br />
t
+<br />
The drop in 0<br />
with 0 by charge exchange.<br />
2<br />
40<br />
with increasing pressure may be due to the fact that 0' can react<br />
+ +<br />
0 +02 -4 o2 + o<br />
This would account for the drop in 0' intensity at conditions at which N+ (which<br />
cannot un ergo loss by charge exchange) remains constant. In the same pressure<br />
9<br />
range, O2 drops in intensity to about the same extent as does N +, which can<br />
disappear by charge exchange with N. 2(<br />
In a mass spectrometer ion chamber 0 + and 04+ are found in rather smell in-<br />
3<br />
tensities, apparently formed by the reactions:<br />
--f +* +<br />
0; 2Tu +02 c o4 -3 o3 + o<br />
+*<br />
04 + o2 + 04+ + o2<br />
The latter is very faint, however. The ionization potential <strong>of</strong> 0 is slightly<br />
greater than that <strong>of</strong> O2 (100,101), so O3 can react with 0 as fo?lows:<br />
2<br />
o3 + + o2 + 0,' + o3<br />
(19)<br />
(100) J. T. Herron and H. I. Schiff, 2. Chem. Phys. 24, 1266 (1956)<br />
-<br />
(101) R. K. Curran, 2. Chem. Phys. 35, 1849 (1961) -<br />
+<br />
No doubt this accounts for the failure to observe O3 .<br />
~ ~~<br />
~ -~<br />
The absence <strong>of</strong> 0; may be<br />
merely a matter <strong>of</strong> sensitivity. t<br />
In our laboratory we have also studied the ions formed in a microwave discharge<br />
in a mixture <strong>of</strong> nitrogen and oxygen at a constart pressure <strong>of</strong> about 0.1 To r,<br />
AS+<br />
might be expected, the intensities <strong>of</strong> Npf and N decreased and those <strong>of</strong> 0: and 0<br />
kngreased as the proportion <strong>of</strong> nitrogen decreased and that <strong>of</strong> oxygen increased.<br />
NO was very intense ovfr th$ range $f 10% to 75% oxygen in the m xture.+ This is<br />
not surprising, since N , 4<br />
N2 and N, reqcting with 0 or 0 and 0 and 0 reacting<br />
with N or N are capable <strong>of</strong> producing NO .<br />
and no doubg is ionized by electron impact.<br />
(16)<br />
Further, &O is produced in ghe discharge<br />
+<br />
Small amounts o f NO2 ions were observed in all <strong>of</strong> the mixtures studied, and<br />
small amounts <strong>of</strong> N 0 were found in the nitrogen rich mixtures, but disappeared<br />
2<br />
when the proportion <strong>of</strong> nitrogen in the mixture dropped below 75%. The manner <strong>of</strong><br />
their formation is not known, but from their very small intensity we infer that they<br />
are probably formed by third order processes. It is surprising that no N: ion was<br />
observed when oxygen was present. Presumably it is capable <strong>of</strong> reacting in several<br />
ways with 0 or 0, and so is destroyed as fast as it is formed. The system OZ-N2<br />
2<br />
is <strong>of</strong> great interest and will be studied further.<br />
This research was supported by Project SQUID <strong>of</strong> the Navy, under Grant NOM<br />
3623 S-21, which we acknowledge with gratitude.<br />
1<br />
,<br />
J<br />
I<br />
\I<br />
i<br />
\I
ION-MOIECULE REACTION RATES MEASUFIED IN A DISCHARGE AFERGLOW<br />
E. E. Ferguson, F. C. Fehsenfeld, and A. L. Schmeltekopf<br />
Institute for 'Peleccanmunication Sciences and Aeronomy<br />
Environmental Science Services Administration Boulder., ' Colorado<br />
ABSTRACT<br />
The application <strong>of</strong> a flowing afterglow reaction technique to the measurement <strong>of</strong><br />
thermal energy ion-molecule reactions is briefly described. The flowing afterglow<br />
system allows the measurement <strong>of</strong> reactions <strong>of</strong> ions with such unstable neutrals as 0,<br />
H, N, and 03. The reaction O+ + CQ - Od+ + CO appears to be important in Cod dis-<br />
charges, as Q+ has been found to be a dominant ion in this case.<br />
The Of, c+, and<br />
CO+ ions rapidly react to produce either Q+ or COa'$ the dominant ions observed in<br />
a COz discharge. In an argon-hydrogen discharge, lb would be an important ion, ,<br />
since it i s the most stable ion in that system and a reaction sequence leading to Ib.'<br />
production is fast. Several associative-detachment reactions such as 0- + 0 - 02 +e<br />
and H- + H - Hz + e have been found to have large rate constants and such reactions<br />
may be important in determining the negative ion concentrations in <strong>discharges</strong>.<br />
I. Introduction and Experimental<br />
A flowing afterglow system has been utilized for the past several years in the<br />
ESSA Laboratories in Boulder, Colorado, for the measurement <strong>of</strong> reaction rate constants<br />
at 300' K for bot2 ositive and negative ions reacting with both stable and<br />
unstable neutral species"'. Figure 1 illustrates one version <strong>of</strong> the flowing afterglow<br />
tube which has been utilized. A tube <strong>of</strong> about 1 m length and 8 cm diameter<br />
serves as the reaction vessel. A gas, usually helium, is introduced at one end <strong>of</strong><br />
the tube and exhausted at the other end at a rate <strong>of</strong> around 100 atm cc/sec, the<br />
helium pressure being typically - 0.3 torr. The helium is ionized by a pulsed dc<br />
discharge producing about 10'" He' ions and He(2S) metastable atoms per cc. Positive<br />
ions are produced in most cases by adding a relatively smll concentration <strong>of</strong><br />
neutral gas into the helium afterglow by means <strong>of</strong> a small nozzle. The positive ions<br />
are products <strong>of</strong> He+ and He(2-S) reactions with the added neutral. The reactions <strong>of</strong><br />
these ions with a second neutral added at a second downstream nozzle are then<br />
measured. The ion composition <strong>of</strong> the afterglow is monitored by means <strong>of</strong> a frequency<br />
scanned quadrupole mass spectrometer covering the mass range 1 - 100 mu. The rate<br />
<strong>of</strong> disappearance <strong>of</strong> a reactant ion with neutral reactant addition leads directly to<br />
a reaction rate constant.' Cur estimate <strong>of</strong> the reliability <strong>of</strong> the rate constants so<br />
determined is f 30$ in favorable cases. Cur experience in comparing our rate constants<br />
with other measured rate constants generally supports this estimate.<br />
This experimental scheme has many variations. We have, for example, successfully<br />
used Pyrex and quartz reaction tubes as well as stainless steel, and microwave<br />
and electron beam ionization as well as the dc discharge. We sometimes find it<br />
desirable to produce reactant ions by adding a suitable gas through the discharge<br />
with the helium rather than downstream in the afterglow, particularly in the case <strong>of</strong><br />
certain negative ions such as 0- and H-, which are readily created by dissociative<br />
attachment by fast electrons in the discharge. We sometimes use carrier gases other<br />
than helium, particularly argon.<br />
Some <strong>of</strong> the important features <strong>of</strong> the flowing afterglow experimental technique<br />
are the follodng: (1) The reactant ions in many cases axe known to be in their<br />
ground states, either because <strong>of</strong> the reaction which produces them or by virtue <strong>of</strong><br />
superelastic electron collisions in the plasma prior to neutral reactant addition.<br />
Stable neutral reactant species are added without being subjected to discharge or
42<br />
RMTS<br />
REAC'TANT /<br />
GAS MASS SPECTROMETER I<br />
Figure 1. Flaring Afterglow Reaction System.<br />
..
43<br />
excitntion 2onditions so that they can be assumed to be neither vibrationally nor<br />
electronically excited. On the other hand, some selective excitation is possible.<br />
For example, the reaction<br />
o+(~s) + N,('E) - NO+(~Z) + N( 4 s) + 1.1 eV<br />
(1)<br />
0<br />
has been measured as 3 function <strong>of</strong> vibrational temperature from 300 - 5000 K.<br />
(2) It is possible to add <strong>chemical</strong>ly unstable neutral reactanJs into the<br />
afterglow so that reactions such as<br />
I!*+ + 0 - NO+ + N, (2) 02+ + N - NO + 0,<br />
(3)<br />
0- + O -. o2 + e,<br />
have been studied in this system.<br />
(4) and 0- + o - o - + o<br />
3 3<br />
(3) The difficulty <strong>of</strong> resolving concurrent reactions does not arise, as it<br />
does in mass spectrometer ion sources, and we have measured the reaction<br />
without complication from the reaction<br />
H2 i + C02 - C02H+ + H,<br />
+<br />
(5)<br />
C02+ + H2 - C02H+ + H (6)<br />
since H is not ionized in the flowing afterglow arrangement.<br />
2<br />
11. Thermal Energy Charge -Transfer Reactions<br />
The flowing afterglow system is well suited to exothermic charge-transfer<br />
reaction measurements, since the neutral reactant, necessarily <strong>of</strong> lower ionization<br />
potential, does not go through the ionization region and hence is not selectively<br />
ionized as would be the case in same experimental arrangements used for ion-molecule<br />
rezction studies. For positive ions, either atomic or molecular, charge-transfer to<br />
molecular neutrals is usually fast (barring occurrence <strong>of</strong> a competitive exothermic<br />
rearrangement reaction). Very many such examples (several dozen) have been observed<br />
and very few exceptions, notably the reaction between He+ + Hz, which is observed<br />
not to have a rate constant as large as<br />
cm3/ sec. For example, Ar', CO', COz',<br />
Na+, N+, and ILOY-all charge-transfer with Oa to produce 4' with rate constants<br />
greater than lo--- cm'/sec (or cross sections greater than 20 8").<br />
This result contradicts most theoretical predictions which had assumed that<br />
charge-transfer would be slow except in unusud cases inwlving fortuituous energy<br />
resonances.<br />
The situation appears the same for negative ion charge-transfer although much<br />
less data is available in this case. We have found that the reactions<br />
H- + NO2 N&- +H (8) OH- + NQ2 +<br />
NOa- + OH (9)<br />
0- + - 03- + 0 (10)<br />
and several other negative ion charge-transfer reactions have rate constants greater<br />
than lo-' cm /set n_t 300° K and previously Curran4 had observed a number <strong>of</strong> fast<br />
negative ion charge-transfers to NOa, and Henglein and Muccini' observed a fast<br />
negative ion charge-transfer with Sa. In the absence <strong>of</strong> experimental data one<br />
would certainly predict that exothermic charge-transfer to a molecular neutral will<br />
be fairly efficient.<br />
111. Ion-Atom Interchange Reactions<br />
The most cornonly studied ion-molecule reactions have involved changes in<br />
molecular configuration. Typical examples are<br />
c+ + o* - co+ + 0,
44<br />
whose rate constant, 1.1 x 10- cm'/sec, from flowing afterglow experiments2 agrees<br />
with an earlier value 9 x lo-' cm3/sec measured in a mass spectrometer ion SOUpCe<br />
by Franklin and Munson"; the reaction<br />
+ +<br />
0 + co2 - o2 + co (12)<br />
with a rate constant <strong>of</strong> 1.2 x lo-' cm3/sec from both flowing afterglow' and mass<br />
spectrometer ion source measurements7 ; and<br />
C+ + co2 - co+ + co I (13)<br />
with a rate constant 1.9 x lo-' cm3/sec.<br />
Such reactions are more <strong>of</strong>ten fast than slow. One <strong>of</strong> the slowest exothermic<br />
ion-molecule reactions (again barring cases where charge-transfer comptes) is<br />
o+ + N2 -t NO+ + N, (14)<br />
with a rate constant - m3/sec3. Elis rate constant increases3 to about<br />
3 x cm3/sec for an N2 vibrational temperature <strong>of</strong> 5000' K and also increases<br />
with 0' kinetic energy"'.<br />
No case <strong>of</strong> a fas? ion-atom interchange reaction involving the breaking <strong>of</strong> two<br />
bonds has so far been reported. The exothermic reaction<br />
02+ + N2 +<br />
has a rate constant less than 1O-l' cm3/sec, for example.<br />
NO+ + NO (15)<br />
IV. Associative Detachment Reactions<br />
A number <strong>of</strong> associative detachment reactions have been recently measured to be<br />
fast in the flowing afterglow system (i.e. k > lo-' an3/ see), including:<br />
and<br />
0- + 0 + O2 + e ~<br />
OH- + o -t H O ~ + e<br />
0- + H2 - H20 + e<br />
(16)<br />
H- + H - H2 + e (17)<br />
(18)<br />
(20)<br />
OH- + H - H20 + e<br />
0- + co - C O + ~ e<br />
Phelps and Moruzzil' have been making similar measurements in drift tube expriments<br />
at Westinghouse and have measured reactions, (20), (21), and (22). The flaring<br />
afterglow and drift tube results agree within better than a factor <strong>of</strong> two in each<br />
case. Several exothermic associative detachment reactions do not occur at measurable<br />
rates (k < 10-l' cm3/sec) , an example being<br />
0- + N2 - N20 + e + 0.2 eV. (23) :<br />
V. Ion-Molecule Reactions in Discharges<br />
he obvious application <strong>of</strong> measured rate constants to the qualitative interpretation<br />
<strong>of</strong> the ion composition <strong>of</strong> a gas discharge is the case <strong>of</strong> the COZ discharge ion<br />
composition studied by barnon and Ticher''. Dawson and Tickner observed the d d -<br />
nant ions in a glow discharge in COZ to be Oa+ and COZ+. This is quite reasonable in 1<br />
xLew <strong>of</strong> the known occurrence <strong>of</strong> reactions (ll), (12) , and (13) above, together with<br />
the fast charge-transfer <strong>of</strong> CO+ vith C& to produce COZ'.<br />
The results <strong>of</strong> these<br />
reactions are graphically illustrated in Fig. 2 (from Fehsenfeld, et al., J. Chem.<br />
Rys. 5 23 (1966)), which shars that dl <strong>of</strong> the ions, C+, O+, and CO+, do convert<br />
to 02+ and Cb+ by reaction with COa in the - 6 milliseconds reaction tbne in the<br />
PlOWhg afterglow. If molecular oxygen were added, one would expect the dominant<br />
ion to become Oa+ alone, since CoZ+ is horn to charge-transfer rapidly with &la.<br />
(19)<br />
(21)<br />
,' '<br />
,
45<br />
In 2 like manner Fig. 3, showing ion composition in an Argon-& afterglod3 ,<br />
suggests that the ion-molecule chemistry is such that the dominant ion is K3+, and<br />
this would very likely be true for certain active discharge conditions as well.<br />
An example <strong>of</strong> the possible importance <strong>of</strong> associative detachment reactions in<br />
<strong>discharges</strong> was noted by Masse$', who speculated that the low negative ion density<br />
'in oxygen dischzrges (relative to iodine <strong>discharges</strong> for example) might be due to<br />
,electron detachment by reaction (16). In iodine the analogous reactionlI- + I -<br />
L + e is endothermic. In view or the subsequent finding that reaction (16) is<br />
indeed fast (kl; = 1.9 x lo-'" cm2/sec), Massey's suggestion takes on renewed interest.<br />
The same argument could be applied to Hz <strong>discharges</strong> in view <strong>of</strong> the rapidity<br />
i <strong>of</strong> reaction (17), kl- - lo-' cm'-/sec.<br />
VI. Conclusions<br />
The growing body <strong>of</strong> quantitative ion-molecule reaction rate data now available<br />
should allow in favorable cases prediction and in many cases correlation <strong>of</strong> observed<br />
1 ion compositions <strong>of</strong> gas <strong>discharges</strong> with known ion-molecule chemistry.<br />
\<br />
Ach.owledgement: This work has been supported in part by the Defense Atomic Support<br />
Agency.<br />
1<br />
/<br />
1<br />
I<br />
1<br />
i<br />
I<br />
'><br />
I<br />
,<br />
I<br />
\<br />
REFERENCES<br />
1. F. C. Fehsenfeld, P. D. Golden, A. L. Schmeltekopf, H. I. Schiff, and E. E.<br />
Ferguson, J. Chem. Phys. 4087 , 4095 (1966).<br />
2. F. C. Fehsenfeld, A. L. Schmeltekopf, and E. E. Ferguson, J. Chem. Phys. 44,<br />
3022, 4537 (1966); 45, 23, 404, 1844 (1966).<br />
3. A. L. Schneltekopf, F. C. Fehsenfeld, G. I. G i b , andE. E. Ferguson, Planet.<br />
Space Sci. in press; J. Chem. Phys., to be published.<br />
4. R. K. Curran, Phys. Rev. x, 910 (1962).<br />
5. A. Henglein and G. A. Muccini, J. Chem. Phys. 15, 1426 (1959).<br />
, 6. J. L. Franklin 2nd M. S. B. Munson, 10th Combustion Symposium, The Combustion<br />
Institute, Pittsburgh, Pa. (1965) p. 561.<br />
: 7. J. L. Paulson, R. L. Mosher, and F. Dale, J. Chem. Phys. 44, 3025 (1966).<br />
8. R. F. Stebbings, B. R. Turner, and J. A. Rutherford, J. Geophys. Res. II_, 771<br />
(1966) -<br />
9. C. F. Giese, 152 Am. Chem. SOC. Meeting, New York, 1966.<br />
' 10. J. L. Moruzzi and A. V. Phelps, J. Chem. Phys., in press.<br />
11. P. H. Dawson and A. GI. Tickner, Proc. Sixth Int. Conf. on Ionization Phenomena<br />
> in Gases, VO~. 3 p. 79 (1963) Paris.<br />
> 12. R. B. Norton, E. E. Ferguson, F. C. Fehsenfeld, and A. L. Schmeltekopf, Planet.<br />
Space Sci. &, 969 (1966).<br />
, 13. F. C. Fehsenfeld, A. L. Schmeltekopf, and E. E. Ferguson, J. Chem. Phys. in<br />
press.<br />
' 14. H. S. W. Massey, "Negative Ions" , Second ed. , Cambridge Univ. Press, Cambridge<br />
(1959) -
46<br />
. ...... , .<br />
k
47<br />
Absorption Spectra <strong>of</strong> Transient Species<br />
in a Single-Pulse Microwave-Discharge<br />
by A.B.Csllear<br />
Department <strong>of</strong> Physical Chemistry,<br />
University <strong>of</strong> Cambridge, Lensfield Road, Cambridge.<br />
ABSTRACT<br />
An apparatus was described for production <strong>of</strong> an intense and<br />
short duration microwave pulse discharge in various gases. At<br />
power levels in the 50 kw range, the microwave energy was coupled<br />
to the gas with a high Q tuned cavity. For higher power levels<br />
up to 1 mw peak, a glass tube containing the gas was placed inside<br />
a wave guide, in the central region <strong>of</strong> maximum field. Thereby<br />
intense <strong>discharges</strong> were produced in metre long columns <strong>of</strong> gas, to<br />
provide ideal conditions for kinetic absorption spectroscopy.<br />
Some simple applications <strong>of</strong> the techni ue were described, including<br />
measurement <strong>of</strong> energy transfer from Beq23s1 to atomic neon,<br />
exchange <strong>of</strong> vibrational energy from nitric oxide to D2S, and the<br />
production <strong>of</strong> diatomic free radicals in various gases.<br />
Flash photolysis ie proving to be <strong>of</strong> considerable value in the<br />
study <strong>of</strong> structure and kinetics. Our first object in developing<br />
the microwave technique, in which the photolytic flash is replaced<br />
by a powerful microwave pulse, was to explore alternative means <strong>of</strong><br />
achieving eubstantial electronic excitation <strong>of</strong> gases. It was<br />
believed that the method would complement flash photolysis and would<br />
also extend the direct techniques <strong>of</strong> flash spectroscopy to more<br />
diverse systems, for example gases which only abeorb light in the<br />
extreme vacuum ultravioletl. Thus to mention a few as yet unattained<br />
and perhaps ambitious objects, we hope to follow dimer ormation<br />
in pure helium and the other inert gases, to study N2 A 4 xi by<br />
absorption spectroscopy, to investigate CH formation In CH (which<br />
has now been achieved by vacuum ultraviolet flash photolysh *),<br />
and to detect both positive and negative ions by absorption<br />
spectroscopy.<br />
A microwave field will couple only with free electrons, and<br />
direct change <strong>of</strong> translational energy <strong>of</strong> gases is unimportant<br />
(because <strong>of</strong> the mass restriction) compared to the energy appearing<br />
as electronic and vibrational excitation. Described here are<br />
results obtained with a microwave-pulse generator <strong>of</strong> peak power<br />
-50 loa, and also the development <strong>of</strong> a more powerful apparatus with<br />
a capability to deliver power to a gas at just under 1 m. Resulte<br />
from the second machine will be available by the early spring <strong>of</strong><br />
1967.<br />
I<br />
$merbental an d Discussion<br />
The first apparatus was constructed to establish the feasability<br />
?<br />
I
49<br />
-<br />
Of ProCucing transients at concentrations detectable by absorption<br />
spectroscopy. An English Electric M561 Idagnetron (3046 Ec/sec)<br />
was powered with a 1 JJ F capacitor charged to 15 kV, and the<br />
pulse duration could be varied in the range 2 - 50 JJsec with<br />
English Electric ~ ~ 2 thyratrons. 5 0<br />
Via .a conventional 'door-knob'<br />
Coupler and waveguide section (without an isolator), the povier Was<br />
transmitted to a cylindrical cavity with a Q <strong>of</strong> 380. This is<br />
illustrated by the photograph shown in figure 1, and the :main ,<br />
details have already been describ.ed. Although a IC psec pulse<br />
corresponds to 0,8 joules, only a minute fraction <strong>of</strong> this was,<br />
successfully delivered to the gas. However, a number <strong>of</strong> encouraEinE<br />
observations were made, ana some energy transfer rate coeff'icients<br />
were measured, as described below.<br />
Yore recently, R.E.M.Hedges, J.G.Guttridge and I have completed<br />
the construction <strong>of</strong> a powerful microwave .generator, which .'<br />
incorporates the English El.ectric M578 magnetron, with a rated<br />
peak-poi;ler <strong>of</strong> 900 kw. Into a 'matched load', we have produced<br />
single pulses at 500 kw, duration 5 bsec.,.which is close to the<br />
maxinum power available per pulse. The H.T. arrangement is similar<br />
to tnilt employed in the prototype equipment, with two thyratrons<br />
delivering a potential up to 30 kV. This particular magnetron turns<br />
out to be rather susceptible to arcing on single pulse operation,<br />
and in this respect is especially sensitive to reflected power.<br />
This has. necessitated the inclusion <strong>of</strong> an isolator between the<br />
magnetron and the load; to obtain satisfactory functioning up to<br />
25 kV (as evidenced by the pr<strong>of</strong>ile <strong>of</strong> the current pulse), it is<br />
necessary to condition the magnetron cathode by somewhat laboriously<br />
pulsing with the applied potential increased by small increments<br />
from 20 kV upwards. . .<br />
It was anticipated that the same cavity that had been employed<br />
in the early experiments would also be suitable for the high energy<br />
equipment. With the assistance <strong>of</strong> hlr. F.J.!Veaver <strong>of</strong> the English<br />
Electric Valve Comgany, the size <strong>of</strong> the hole coupling the cavity<br />
to the waveguide was carefully optimised to give a stsnding-wave<br />
ratio <strong>of</strong> 1.2 and a Q <strong>of</strong> about 800. With such a tuned cavity, a<br />
discharge can be produced in heliumat 1 atmos. pressure; hoijever,<br />
the discharge was still quite feeble and it was obvious that only<br />
a minute fraction <strong>of</strong> the total energy was being coupled to the gas.<br />
Although the tuning and matching was near perfect under low power<br />
test conditions, for various reasons the cavity detunes as soon as<br />
the gas strikes.<br />
A41tnough a study <strong>of</strong> the cavity discharge may prove to be <strong>of</strong><br />
some value, interest was directed at the problem <strong>of</strong> achieving iuen-e<br />
more efficient coupling <strong>of</strong> the microwave pulse to the experimental<br />
gas. In fact we have now achieved conditions under which it appears<br />
that practically the entire pulse may be absorbed by the gas.<br />
This innovation is a very simple one; a thin walled glass tube<br />
contqining the gas is placed inside the waveguide in the region <strong>of</strong>
50<br />
maximum fiela. In this manner, an intense discharge can be<br />
proaucea in metre long columns <strong>of</strong> gas, to provide ideal conditions<br />
for absorption spectroscopy. R.E.?d.Sedges is presently<br />
assembling the optical components to carry out spectroscopic Studies<br />
under these conditions. The beauty <strong>of</strong> this discovery lies in its<br />
inherent simplicity; power is coupled to the gas at low Q and<br />
matching problems are virtually eliminated.<br />
Results<br />
This section relates only to the low power equipment, with a<br />
tuned cavity discharge. Figure 2(a) shows the formation <strong>of</strong><br />
He(23S ) in a single pulse <strong>of</strong> IO Msec duration, and its decay<br />
followhg the pulse. Also bserved in pulsed helinm were the<br />
conparatively short lived 2 3 P and 2IS states. A simple quantita- ‘<br />
tive ap lication shown in figbe 2(b),’is the enhanced rate <strong>of</strong> decay<br />
<strong>of</strong> He(2 3 S ) in the presence <strong>of</strong> a trace <strong>of</strong> neon,<br />
By means <strong>of</strong><br />
photometr3 <strong>of</strong> plates similar to that illustrated in figure 2, the<br />
rate <strong>of</strong> the energy transfer process<br />
He(23S1) + Ne(2’So) -> He(llSo) + Ne(2s)<br />
2 2<br />
reported<br />
by Javan, Bennett and-Herriott 3. The helium system is perhaps<br />
the simplest <strong>of</strong> all electric <strong>discharges</strong> and, as mentioned above, we<br />
hope to make more detailed observations <strong>of</strong> the formation <strong>of</strong> diatomic<br />
helium. All four <strong>of</strong> the Is metaables were observed in pulsed neon lo<br />
was recorded as 0.35 + 0.02 2 , in agreement with 0.37 2<br />
z’igure 3 illustrates vibrational excitation <strong>of</strong> nitric oxide,<br />
and its decay following the pulseo It was concluded that excitation<br />
occurs by direct collision <strong>of</strong> nitric oxide molecules with electrons,<br />
because <strong>of</strong> the ex reme weakness <strong>of</strong> fluorescence from electronically<br />
excited molecules .t The total yield <strong>of</strong> vibrationally excited NO<br />
produced with a microwave pulse, is about 10 - I00 fold higher than<br />
the yield <strong>of</strong> electronically excited species or free radicals, in<br />
all the systems which have thus far been investigated. The technique<br />
may therefore prove to be generally ap licable to the study <strong>of</strong><br />
vibrational energy transfer.<br />
Figure 3fb) shows the catalysis <strong>of</strong> the<br />
NO relaxation, by a trace <strong>of</strong> D2S, which has a vibrational frequency<br />
close to that <strong>of</strong> NO, A number <strong>of</strong> rate coefficients for V-V<br />
processes <strong>of</strong> this type have recently been published,<br />
Finally we mention the formation <strong>of</strong> <strong>chemical</strong> intermediates,<br />
produced by microwave pulses in polyatomic gaseso Thus far, these<br />
aspects <strong>of</strong> the chemistry <strong>of</strong> electric <strong>discharges</strong> have only been I<br />
investigated in CS (CN) H 0 and H S. In each case, diatomic<br />
free radical inter6;diateg’wege obseraed. Some <strong>of</strong> these are<br />
included in figure 4.<br />
I<br />
.f
Before<br />
n. d.<br />
IO.~/AS~<br />
22.4<br />
31.6<br />
42.0<br />
52.8<br />
66.0<br />
78, I<br />
90-9<br />
113<br />
I33<br />
I64<br />
206<br />
243<br />
312<br />
'366<br />
Before<br />
n, d,<br />
22.&sec<br />
31-6<br />
42-0<br />
52.8<br />
66.0<br />
78, I<br />
90-9<br />
113<br />
(a) Formation and decay <strong>of</strong> He (2 3S,) In 5 mm .<strong>of</strong> He,<br />
'(b) Deczy <strong>of</strong> He (2 34) In 5 mm He + I.? x IOe2,, Ne.<br />
Figure 2
52<br />
3<br />
%<br />
%-<br />
%-<br />
5-<br />
a-<br />
7<br />
h<br />
W<br />
P<br />
H<br />
0<br />
L<br />
v)<br />
a<br />
P<br />
o w<br />
L<br />
0<br />
+<br />
E!<br />
c<br />
n<br />
i3<br />
W<br />
al<br />
=:<br />
.-<br />
E<br />
3<br />
(2<br />
rr\<br />
+<br />
0<br />
b=l N<br />
E<br />
e<br />
XJ<br />
3<br />
0<br />
b<br />
+<br />
3<br />
z<br />
a<br />
E<br />
rn<br />
n<br />
P<br />
U
'3 (A'ncX'Z')<br />
53<br />
OH (A2Zf + X2n)<br />
>'is+ Formation ana decay <strong>of</strong> (a) cs : 2mm C S ~<br />
+ 60 mm He;<br />
(b) OH : 5 mm %O + 35 mm HE; (C) SH : 20 mm H28 + 100 mm He.<br />
0.9
References I<br />
I . A.B.Callear, J.A.Green and G.J.Williams, Trans.Faraday Soc.,<br />
2.<br />
3.<br />
54<br />
I<br />
(1965), 61, 1831. f<br />
W.Braun, K.H.Welge and A.M.Bass, J.Chem.Phys., (1966), a, 2650.<br />
A.Javan, W.R.Bennet and D.R.Herriott,<br />
6, 106.<br />
Phys.Rev.Let.
55<br />
The Collection <strong>of</strong> Positive Ions and Electrons by a<br />
I , Screened Probe in the Neon Negative Glow*<br />
I M.J. Vasilet and R.F. Pottie,<br />
\<br />
i<br />
Division <strong>of</strong> Applied Chemistry,<br />
<strong>National</strong> Research Council,<br />
Ottawa, Canada.<br />
i I<br />
I INTRODUCTION<br />
3' The collection <strong>of</strong> ions and electrons by small probes placed<br />
iithin the plasma <strong>of</strong> a gas discharge has received considerable<br />
ttention over the past four decades. A major advance in measurement<br />
?echnique was reported by Boyd' in 1950 with the introduction <strong>of</strong> a<br />
'mall screened flat probe.<br />
The new technique made it possible to<br />
.eparate the collected currents into the ion and electron components.<br />
?s a result it became possible to measure their concentrations<br />
,eparately and to extend the range <strong>of</strong> measured electron velocities to<br />
Ieyond the ionization potential <strong>of</strong> the discharged gas with no inter-<br />
'erence from positive ion current. The method was extended by Boyd<br />
\nd co-workers2 and by Pringle and Farvis3 to the measurement <strong>of</strong> ions<br />
nd electrons in both the positive column and negative glow in various<br />
!as,, . Despite the apparent success <strong>of</strong> these studies, little or no<br />
Ise has been made <strong>of</strong> this technique by other workers in the field.<br />
I<br />
i Previous work in this lab~ratory~,~ had established the<br />
isefulness <strong>of</strong> a mass spectrometer to obtain relative ion concentrations<br />
in the various regions <strong>of</strong> glow <strong>discharges</strong>. The work reported here is<br />
in extension <strong>of</strong> these earlier studies and represents an attempt to<br />
la) measure the absolute concentrations <strong>of</strong> ions and electrons in<br />
conjunction with the mass spectrometric studies and (b) to determine<br />
;he electron energy distributions, particularly in the negative glow<br />
;o assist in the interpretation <strong>of</strong> processes that result in the<br />
1.<br />
qroduction <strong>of</strong> ions.<br />
EXPERIMENTAL<br />
Apparatus<br />
f a fine wire grid that was spot welded to a 5 x 5 mm platinum frame<br />
3<br />
hich was mounted on a Pyrex plate. A 1/8" circular hole in the glass<br />
~late defined the current reaching the platinum collector (.010" thick)<br />
iounted below it. A mica insulator shielded the collector from the<br />
ischarge. The glass plate was 1 mm thick, and the grid was a stainless<br />
$tee1 mesh (0.001'' diameter wires) with an optical transparency <strong>of</strong> 42%.
._ . _-.. - , , . . ,. .<br />
The Pyrex ‘discharge tube56as 50 cm long and 5.5 cm in<br />
diameter. The electrodes were polished stainless steel discs, 5 cm<br />
in diameter and the cathode could be moved magnetically over a<br />
distance <strong>of</strong> 30 cm. The entire discharge tube, including side-arms, .<br />
could be baked to greater than 35OoC. Metal bellows valves were used<br />
for all openings into the discharge tube and the normal background<br />
pressure <strong>of</strong> 2 x Torr was achieved with a silicone oil diffusion<br />
PUP *<br />
The gases were <strong>of</strong> assayed research grade and pkriodic checks<br />
were made for impurities by mass spectrometry. The operating gas was<br />
admitted to the discharge tube via a Granville-Phillips variable leak<br />
valve, the pressure being measured with a Decker diaphragm gauge. A<br />
small flow was maintained through the discharge tube during operations<br />
by means <strong>of</strong> a needle valve to the pumps. The exit pressure was about<br />
Torr. Preliminary <strong>discharges</strong> <strong>of</strong> about one hour duration were<br />
always carried out before measurements were taken. The tube was then<br />
evacuated and a fresh gas sample was admitted for the experiment.<br />
All results reported here were taken with a gas pressure <strong>of</strong><br />
0.28 Torr and a discharge current <strong>of</strong> 0.250 ma.<br />
A schematic diagram <strong>of</strong> the discharge tube and electrical t<br />
circuit is given in Fig. 1. The potentials <strong>of</strong> the grid and collector<br />
relative to the grounded anode could be controlled independently so<br />
that either one could be positive or negative with respect to the anode.<br />
The collector current passing through precision resistors (0.05%) was<br />
measured with a 1 megohm impedance nanovoltmeter which was isolated from<br />
ground. The minimum detectable current was 1O‘loA. The potential <strong>of</strong><br />
1<br />
. the collector was measured with a vacuum tube voltmeter, while a<br />
~ and<br />
digital voltmeter was used for measurement <strong>of</strong> the grid potential. Grid<br />
and-discharge currents were monitored by dc micrometers. Discharge<br />
power’was provided by a commercial dc power supply.<br />
B. Results<br />
(1) Collection <strong>of</strong> low energy electrons. Fig. 2(a) is a typical semilonarithmic<br />
plot <strong>of</strong> the electron current, showing two straight line<br />
segments for- the thermal and secondary electrons respectively. The<br />
space potential was obtained from extrapolation <strong>of</strong> the saturation<br />
current to the rising portion <strong>of</strong> the curve. The gas was pure neon and<br />
the probe was near the position <strong>of</strong> maximum electcon intensity in the<br />
negative glow. The electron temperatures <strong>of</strong> the MaxwellIan distributions<br />
were 0.271 and 3.67 eV for the thermal and secondary electrons, and<br />
their concentrations were 158 x 10’ 3/m3 and 2.9 x 10’ 3/m3 respectively,<br />
assuming collection <strong>of</strong> the random current at the space potentia16s7.<br />
The small electron residual current that was always observed at higher<br />
retardation potentials was subtracted from the total electron currents.<br />
The collector potential was +7O V with respect to the anode.<br />
2) Collection <strong>of</strong> positive ions. The collector potential was<br />
maiAtained at -70 V and the grid voltage was varied from -70 V to Well<br />
above the space potential for the collection <strong>of</strong> positive ions. The<br />
method <strong>of</strong> calculation <strong>of</strong> the random positive ion current differed<br />
somewhat from previous approaches and is given briefly below. We<br />
assume that positive ions are accelerated from the sheath to the grid<br />
and are further accelerated by the full potential between the COlleCtor<br />
the grid. The current rises until all the random Ion current<br />
arriving at the sheath edge has been collected. Further collection <strong>of</strong><br />
Ion current is the result <strong>of</strong> sheath expansion, edge effects and<br />
potential penetration <strong>of</strong> the sheath. If we treat the ion currents<br />
. .<br />
. .<br />
I<br />
,‘<br />
1<br />
,A<br />
1<br />
!<br />
I<br />
1
I<br />
\<br />
\<br />
I<br />
i<br />
h<br />
FIG. 1<br />
I<br />
NVY<br />
57<br />
ANODE<br />
I<br />
e00<br />
io00 .OB<br />
IOOK 1%<br />
IOOK in<br />
Apparatus and schematic circuit diagram.
58<br />
0 0 0<br />
VI<br />
J<br />
><br />
I<br />
cv .<br />
u<br />
H<br />
a
elow the saturation limit we find59<br />
in which n is the random ion density, q’ the electronic charge<br />
and Vf thePvelocity <strong>of</strong> the ions arriving at the collector. This is<br />
given by:<br />
in which Vo is the initial ion velocity and Vacc is the potential<br />
difference between the plasma and the collector. Thus:<br />
Therefore, a plot <strong>of</strong> i+ versus Vacc1/2 should yield a straight line<br />
whose slope is<br />
+ np q(?)’’2 .<br />
The positive ion currents, plotted in this manner always yielded a<br />
straight line to a break point which was assumed to be the saturation<br />
limit. A plot <strong>of</strong> this type is shown in Fig. 2 (b); the small residual<br />
ion current has been subtracted from the experimental points. From<br />
this slope we calculate np = 145 x 1013/m3 in good agreement with the<br />
electron concentration <strong>of</strong> Fig. 2 (a) for the same probe position.<br />
(3) Axial variation <strong>of</strong> ions and low energy electrons. The observed<br />
ion and electron concentrations as a function <strong>of</strong> axial distance from<br />
the cathode are given in Fig. 3 for the probe facing the anode. There<br />
is a slight displacement between the maxima for the two distributions<br />
and this may reflect a slight penetration <strong>of</strong> the field from the cathode<br />
dark space. However the results show that a true plasma is present in<br />
the negative glow, since to a gbod approximation ne = np.<br />
electron current was relatively insensitive to position <strong>of</strong> the probe<br />
and gave an average concentration <strong>of</strong> 3 ? 1 x 10 3/m3 with electron<br />
temperatures in the range <strong>of</strong> 3.5 - 4.0 eV. The rising portion <strong>of</strong> the<br />
secondary electron curve marks the anode edge <strong>of</strong> the negative glow. At<br />
this point the electric field begins to accelerate thermal electrons<br />
to the secondary electron velocities. For example at d = 7.2 cm, the<br />
secondaries account for almost 50% <strong>of</strong> the total electron current.<br />
(2)<br />
(3)<br />
The secondary<br />
(4) Residual electron current. Small residual electron currents<br />
were observed ,for all probe positions and orientations. These are<br />
summarized in Fig. 4 for pure neon. These minimum currents were<br />
ccnstant for a range <strong>of</strong> -35 to -50 V in grid potential. For grid<br />
voltages in excess <strong>of</strong> -50 V there was a gradual rise in the current,<br />
presumably as a result <strong>of</strong> grid emission by the bombardment <strong>of</strong> positive<br />
ions. We find in Fig. 4 that the residual current decreases<br />
exponentially throughout the negative glow with increasing distance<br />
from the cathode for both orientations <strong>of</strong> the probe surface. Additionally,<br />
there is a reduction by almost a factor <strong>of</strong> 5 for the probe<br />
facing the anode versus cathode position.<br />
We have also observed no<br />
variation in the residual current as a function <strong>of</strong> radial position from<br />
the center to a point only 0.6 cm from the wall. The current increases<br />
slowly with axial distance in the Faraday dark space (d > 5 cm) and<br />
reaches a plateau in the positive column.
.... .<br />
0 2<br />
e<br />
I /<br />
m<br />
0<br />
H<br />
F4<br />
:<br />
1
62<br />
The effect <strong>of</strong> adding O.O* xenon to neon is seen in Fig. 5.<br />
The residual current drops and there is a steeper slope to the exponentti<br />
part <strong>of</strong> the curve. In addition a smaller increase is observed in the<br />
Faraday dark space.<br />
(5) Energy distribution <strong>of</strong> secondary electrons. From Fig. 2 (a) we<br />
see that the energy distribution <strong>of</strong> secondary electrons is essentially<br />
Maxwellian at the indicated position <strong>of</strong> the probe. However, as the<br />
axial distance is increased we find a successively greater loss <strong>of</strong><br />
electrons whose energies exceed about 10 eV, although thd distribution<br />
is not radically altered from Maxwellian. The addition <strong>of</strong> 0.07% xenon<br />
results in almost complete depletion <strong>of</strong> electrons above 12 eV and there \I<br />
are considerable deficiencies beginning only 5 eV above the space<br />
potential.<br />
The data are summarized in Fig. 6. The distribution curves<br />
for the mixture were calculated from the experimental measurements<br />
according to the method <strong>of</strong> Medicus8, and comparison plots <strong>of</strong> the ideal<br />
Maxwellian distribution are given. The two Curves in each case were<br />
normalized to those regions along the potential axis which gave a I<br />
straight line on the semi-logarithmic plot. The fluctuations in the<br />
curves for the mixture were reproducible and are similar in appearance ‘I<br />
to the structural features that were observed by Twiddyg in the cathode<br />
region <strong>of</strong> rare gas <strong>discharges</strong>. It is apparent that xenon effectively<br />
reduces the higher energy secondary electrons, even at 0.07% concentration.<br />
Twiddy’O has shown that there is a similar loss <strong>of</strong> energetic<br />
electrons in the positive column <strong>of</strong> an argon-neon mixture.<br />
1<br />
(6) Residual positive ion current. The ratio <strong>of</strong> residual currents<br />
for ie m i d i + min was 5:1 with the probe facing the cathode and about<br />
1O:l with the probe facing the anode. These ratios were constant for<br />
all axial positions <strong>of</strong> the probe in the negative glow and in the Faraday<br />
dark space. The ratios for the 0.07% xenon-neon mixture were the same<br />
as for pure neon.<br />
DISCUSSION<br />
Since the production <strong>of</strong> ions and low energy electrons in<br />
the negative glow is governed largely by the rate <strong>of</strong> arrival <strong>of</strong> high<br />
energy electrons from the cathode dark space it is obvious that the<br />
direct observation <strong>of</strong> these high energy electrons <strong>of</strong>fers an important<br />
method for interpreting the processes <strong>of</strong> ionization in the negative<br />
glow. A possible means <strong>of</strong> making such a measurement is t o consider the<br />
negative residual currents collected by the screened probe when it faces<br />
the cathode. These currents could arise from several processes:<br />
(a) Direct collection <strong>of</strong> high energy electrons.<br />
(b) Grid emission by high energy electrons.<br />
(c) Grid emission by bombardment <strong>of</strong> positive ions.<br />
(d) Grid emission by metastable atom impact.<br />
(e) Grid emission by photon impact.<br />
Rotation <strong>of</strong> the probe to face the anode caused a 2/3 redyctio4<br />
in the-residual current. One would not e)tpect processes (c), (d), and<br />
(e) to depend markedly on probe orientation so that these processes 1<br />
probably account for less than 1/3 <strong>of</strong> the total residual negative<br />
Current when the probe faces the cathode. Since neither- the positive ‘1<br />
ion density nor the visible photon intensity decays exponentially,<br />
Processes (c) and (e) are similarly rejected as a major source <strong>of</strong> curred<br />
for the anode orientation. We conclude that the major causes <strong>of</strong> the<br />
negative residual current in the negative glow are processes (a) and (b) i<br />
,<br />
),<br />
t<br />
1
\<br />
0.9<br />
0.8<br />
0.7<br />
06<br />
0.5<br />
0.4<br />
0.3<br />
0.2<br />
01<br />
0.8<br />
0.7<br />
0.6<br />
0.5<br />
04<br />
0.3<br />
0.2<br />
0.1<br />
0.9<br />
0.2 02<br />
0.1 01<br />
I c)<br />
2 4 6 8 IO I2 14 . 16 18 2C<br />
ELECTRON ENERGY I V-VSP~CE)<br />
FIG. 6 Energy distribution function <strong>of</strong> secondary elec-<br />
trors compared with the Naxwellian distribution.<br />
(a) pure neon, d = 2.3 cm(b) mixture .07$<br />
Xenon in neon, d = 2.2 cm(c) mixture, d = 3.0 cm.<br />
Probe facing anode.
64<br />
for the probe facing the cathode. We Cannot distinguish between (a)<br />
and (b), nor is it possible to calculate the density <strong>of</strong> such high<br />
energy electrons without knowledge <strong>of</strong> their velocities.<br />
The observed exponential decrease in ie min through the<br />
negative glow <strong>of</strong> pure neon (Fig. 4) can be written as:<br />
id = io exp(-a n(d-do))<br />
I<br />
in which io is the residual current at do cm from the cathode, d is<br />
the distance <strong>of</strong> the probe surface from do and n is the concentration<br />
<strong>of</strong> neon atoms per unit volume. Thus a has the dimensions <strong>of</strong> cm2 and<br />
is an experimental cross section for loss <strong>of</strong> high energy electrons, as<br />
a first approximation. From the slope <strong>of</strong> the exponential curve in<br />
Fig. 4 we calculate that a = 8.7 x cm2. This is a reasonable<br />
value since the cross section for ionization <strong>of</strong> neonll by 100 eV<br />
electrons is 7.58 x cm2.<br />
The addition <strong>of</strong> 0.07% xenon brings about a 30% reduction in I<br />
the negative residual current and the exponential slope increases so<br />
that the experimental a = 1.03 x cm2. This cannot be accounted /<br />
for by assuming that the only additional loss is that ar sing from the<br />
ionization <strong>of</strong> xenon. At a concentration <strong>of</strong> only 7 x atom %, this<br />
would imply a cross section for ionization <strong>of</strong> xenon equal to 228 x 10-'6 ,<br />
cm2 - about a factor <strong>of</strong> 40 higher than the known <strong>of</strong> xenonll for 100 eV<br />
electrons. A more reasonable inference is that the loss <strong>of</strong> neon metastables<br />
by the Penning reaction:<br />
Ne*(3P2,0) + Xe * Xe+ + Ne + e- (5)<br />
is responsible for the lower values <strong>of</strong> ie min, and that the change in<br />
slope more nearly reflects the actual stopping power <strong>of</strong> neon for high<br />
energy electrons. Since excitation as well as ionization can result<br />
from impact <strong>of</strong> a high energy electron, a cross section <strong>of</strong> the order <strong>of</strong><br />
1 x cm2 is <strong>of</strong> the expected magnitude for an electron <strong>of</strong> approx-<br />
imately 100 eV.<br />
It would be expected that the addition <strong>of</strong> a small amount <strong>of</strong><br />
xenon would not significantly change either the cathode fall or the<br />
number <strong>of</strong> directed high energy electrons passing into the negative<br />
glow. Thus, if one assumes that the reduction in the residual electron<br />
current is caused by reaction (5) it is possible to obtain an approx-<br />
imate value for the Concentration <strong>of</strong> metastables in the beginning <strong>of</strong><br />
the negative glow.<br />
Referring to Fig. 4, for d = 2 cm, -Ai, the reduction in<br />
the residual current is 0.02 VA. If the neon metastables have thermal<br />
velocities, and if one considers a reasonable yield <strong>of</strong> about 0.03 for<br />
the grid emission (geometric factor x efficiency), then a minimum<br />
concentration <strong>of</strong> 1 x 10i3/m3 is obtained for the metastables. It is<br />
then possible to calculate the reactive collision frequency between<br />
Ne* and Xe by assuming the same cross section for deactivation as<br />
Sholette and Muschlitz12 observed for the He* - Xe Penning reaction,<br />
namely 12 x cm2. This results in a calculated lifetime for the<br />
neon metastables <strong>of</strong> 0.5 m sec. This Is somewhat shorter than the<br />
lifetime <strong>of</strong> 0.875 m sec measured by Blevis et a1.13 for the decay <strong>of</strong><br />
Ne(3P2) in pure neon.<br />
We conclude that xenon can intercept the<br />
metastable neon atoms in the required time and that our assignment <strong>of</strong><br />
the Penning reaction is reasonably justified as a major contributing<br />
(4)<br />
I<br />
1<br />
i(
65<br />
' cause <strong>of</strong> the reduction in the residual negative current. Similar<br />
argUmentS can be used to account for the.decrease in the residual<br />
current when the probe faces the anode. The effect is even more<br />
' marked in this case, particularly at the beginning <strong>of</strong> the positive<br />
column.<br />
The treatment <strong>of</strong> the positive ion residual current is not<br />
' as straightforward, since there arises the additional complication<br />
7 ,<strong>of</strong> electron impact ionization in the space between the grih and the<br />
collector. To a rough approximation we can account for about 60% <strong>of</strong><br />
i' the current with the probe facing the cathode by this process, but<br />
{ the remainder can be a combination <strong>of</strong> secondary emission from the<br />
? collector by photons, by metastable atoms and by energetic electrons.<br />
' The very small currents observed for the probe facing the anode suggest<br />
that the release <strong>of</strong> electrons by metastable impact is much less than<br />
9 would be expected for the metastable density obtained above. Apparently<br />
i the impact <strong>of</strong> metastables with the platinum surface produces secondary<br />
electrons with a much lower efficiency than does the stainless steel<br />
1 grid. The reason is not clear, but a contributing cause may be due to<br />
the higher work function <strong>of</strong> platinum.<br />
\<br />
I<br />
1<br />
4<br />
1.<br />
'~ 2.<br />
' 3.<br />
; 4.<br />
~ 5.<br />
6.<br />
; 7.<br />
\<br />
1. 8 .<br />
t 9.<br />
REFERENCES<br />
N.R.C. No. ...., 'N.R.C. Postdoctorate Fellow.<br />
R.L.F. Boyd, Proc. Roy. SOC. m, 329 (1950).<br />
R.L.F. Boyd and N.D. Twiddy, Nature 173, 633 (1954).<br />
D.H. Pringle and W.E.J. Farvis, Proc. Phys. SOC. =, 836 (1955).<br />
P.F. Knewstubb and A.W. Tickner, J. Chem. Phys. 36, 674 (1962).<br />
P.F. Knewstubb and A.W. Tickner, J. Chem. Phys. 36, 684 (1962).<br />
I. Langmuir and H. Mott-Smith, Jr., G.E. Review 2J,449 (1924).<br />
H. Mott-Smith and I. Langmuir, Phys. Rev. a, 727<br />
G. Medicus, J. Applied Phys. 27, 1242 (1956).<br />
N.D. Twiddy, Proc. Roy. SOC. E, 379 (1961).<br />
.;' 10. N.D. Twiddy, Proc. Roy. SOC. a,<br />
338 (1963).<br />
(1956).<br />
i 12. W.,P. Sholette and E.E. Muschlitz, Jr., J. Chem. Phys. 36,<br />
11. D. Rapp and P. Englander-Golden, J. Chem. Phys. '13, 1464 (1965).<br />
3368 (1962).<br />
I<br />
i<br />
13. B.C. Blevis, J.M. Anderson, and R.W. McKay, Can. J. Phys. 35,<br />
941 (1957).
66<br />
ATTACHMENT OF MOLECULES WITH HIGH PERMANENT<br />
DIPOLE MOMENT TO IONS IN GASES<br />
AND INDUCED<br />
P. Kebarle, G.J. Collins, R.M. Haynes and J. Scarborough<br />
Department <strong>of</strong> Chemistry, University <strong>of</strong> Alberta, Edmonton, Canada<br />
ABSTRACT I<br />
The mass spectrometric study <strong>of</strong> clustered ions in function <strong>of</strong> polai<br />
gas pressure, temperature and electric field strength provides informa-<br />
tion not only on what ionic species might be present, but also on the 1<br />
strength <strong>of</strong> the ion-dipole interactions. Studies <strong>of</strong> the reaction<br />
H30+.(n-1)H20 + H20 = H30+.nH20 in a Nier type ion source with a 100 ke-c<br />
proton beam are compared with earlier work on an alpha particle mass .<br />
spectrometer. The residence time <strong>of</strong> the ions in the two sources are a<br />
few psec and a few msec resp. Similarity <strong>of</strong> results shows that cluster,<br />
ing equilibrium is established within psec for water clusters above 0.5<br />
torr. The presence <strong>of</strong> electric fields reduces the cluster size. 50 1<br />
volts/cm (at 1 torr) strip the clusters to H 0'. Experiments on the cor<br />
parative attachment <strong>of</strong> different molecules S~OW that the stability <strong>of</strong> tl<br />
attachment increases in the order H20, NH3, CH30H, CH3N02 for first she!<br />
attachment. At larger distances from the ion (second shell) water be-<br />
comes the strongest attaching. These results are explained by differenc<br />
<strong>of</strong> dipole moments and polarizabilities.<br />
'I<br />
/!<br />
/.<br />
I<br />
I<br />
,,-
INTRODUCTION<br />
I' Attachment <strong>of</strong> polar molecules to ions in the gas phase occurs in a<br />
mber <strong>of</strong> systems: gases under high energy irradiation, gas <strong>discharges</strong>,<br />
IlameS, etc. The polar molecules may be present as an accidental imkrity<br />
or as a deliberate addmixture. The attachment has a number <strong>of</strong><br />
$Portant effects such as change <strong>of</strong> mobility, change <strong>of</strong> rektivity with<br />
2gard to ion-molecule reactions, change <strong>of</strong> the positive (negative) ion<br />
tlectron) recombination coefficient and change in the nature <strong>of</strong> the<br />
-0ducts resulting from the positive negative ion recombination. The<br />
+dy <strong>of</strong> ion-polar molecule interactions in the gas phase can also pro-<br />
:de information on ionic solvation since the formation <strong>of</strong> ion clusters<br />
2nstitutes the first and most important step in the solvation <strong>of</strong> anion.<br />
; A systematic mass spectrometric study <strong>of</strong> ion-polar molecule inter-<br />
:tiOnS was started in this laboratory some five years ago.8-12<br />
The mass spectrometric gas phase studies are based on measurement<br />
! the relative concentrations <strong>of</strong> the clustered ionic species: A+.nS,<br />
+.(n+l)S, etc. The measurement <strong>of</strong> the relative concentrations is obfined<br />
by bleeding a probe <strong>of</strong> the gas into an ion mass analysis system,<br />
.,e. a vacuum chamber attached to a mass spectrometer. In the vacuum<br />
hamber .> the gas is pumped out while the ions are captured by electric<br />
ields, accelerated and focused and then mass analysed by some convenional<br />
means (magnetic separation quadrupole filter, etc.). After mass<br />
qalysis, the ion beam intensities are detected as electrical currents.<br />
Several types <strong>of</strong> solvation studies can be undertaken if the relaive<br />
concentrations <strong>of</strong> the ionic species are known.<br />
1. Solvation Enthalpies and Entropies <strong>of</strong> Individual Solvent Mole-<br />
ule Additions Steps: Consider the ion A+ produced in the gas phase by<br />
3me form <strong>of</strong> ionizing radiation or thermal means. If the atmosphere<br />
urrounding the ion contains the vapor <strong>of</strong> a polar molecule (solvent S),<br />
number <strong>of</strong> clustering reactions will occur.<br />
i<br />
A+ + S + A+.S (0,1)<br />
I A+.S + S + A+.2S (1,2)<br />
,~<br />
~ where<br />
+<br />
A+.(n-l)S + S + A+.nS (n-l,n)<br />
At equilibrium the following relations will hold<br />
AFo = AFo + AFo 1,2 ... + AFon-l,n<br />
O,n 0,1<br />
(1)<br />
AFon-l , n<br />
= -RT In<br />
'A+. nS<br />
'A+. (n-1) s-'s<br />
= -RT In Kn-l,n (11)<br />
P, is the partial pressure <strong>of</strong> X.<br />
Thus knowledge <strong>of</strong> the equilibrium concentrations <strong>of</strong> the clustered<br />
pecies A+.nS obtained from experiments at different pressures <strong>of</strong> s w ill<br />
llow the determination Of Xn-1 n and AFn-l,n. Such measurements done<br />
k different temperatures will lead to the evaluation <strong>of</strong> AHn-l<br />
and<br />
Sn-lIn. The availability <strong>of</strong> such detailed information will, #or In-<br />
,tance, reveal the shell structure since a discontinuous change <strong>of</strong> the<br />
Hn-1 n and ASon-l,n values will occur whenever a shell is completed.<br />
,inally, the total heat <strong>of</strong> solvation <strong>of</strong> the ion can also be obtained<br />
rom the expression 111, with equations <strong>of</strong> the same form holding for the<br />
/ m
free energy and entropy change <strong>of</strong> solvation.<br />
It is evident from equation I1 that only the relative concentrations<br />
<strong>of</strong> the ionic species are required. Thus AFon,l and Kp can be obtained<br />
from equation 11 by assuming that the mass<br />
'<br />
spechometrically measured<br />
ion intensities are proportional to the equilibrium parti 1 pressures <strong>of</strong><br />
the ions in the ion source. The solvation <strong>of</strong> NHa by NH3 and H3O+ by<br />
H20 l2 represent examples <strong>of</strong> studies <strong>of</strong> this type.<br />
I /<br />
2. Comparative Solvation <strong>of</strong> Two Different Ions by the Same Solvent:<br />
There are three variants <strong>of</strong> this type <strong>of</strong> study. 11<br />
+<br />
Comparative solvation <strong>of</strong> ions A , C+ by solvent S.<br />
(a)<br />
The two ions are produced in the same system which also contains<br />
vapor <strong>of</strong> the solvent S. In general, depending on the effective radius<br />
and structure <strong>of</strong> the ion, the relative concentration <strong>of</strong> clusters A+.ns<br />
will be different from that <strong>of</strong> C+.rnS. Thus, for example, in the average<br />
n may be larger by 1 or 2 units than m. This will reveal a stronger interaction<br />
<strong>of</strong> A+ with S. Comparison <strong>of</strong> n and m can be done at different<br />
temperatures and pressures (<strong>of</strong> S) so as to compare the interactions in<br />
the inner shell or in the outer shells. An example <strong>of</strong> this type <strong>of</strong> study<br />
is the system ~30+, NHa fyd Na+ in H20 vapor which is described in a<br />
forthcoming publication.<br />
(b)<br />
Comparative solvation <strong>of</strong> ions A+ and B- by solvent S.<br />
An example <strong>of</strong> this type <strong>of</strong> study would be the system K+ and C1'<br />
with H20. Since the orientation <strong>of</strong> the water dipoles,is reversed in the<br />
solvation <strong>of</strong> positive and negative ions, such a comparative study is <strong>of</strong><br />
great interest, particularly for isoelectronic pairs a8 the one quoted<br />
above. We have not yet done studies on such isoelectronic pairs. However,<br />
such studies are perfectly possible. The pair K+ and C1' could be<br />
produced in water vapor. By reversing the mass spectrometer controls,<br />
measurements on the positive and negative ions in the system could be<br />
done within minutes <strong>of</strong> each other.<br />
(c) Comparative solvation <strong>of</strong> two negative ions by S.<br />
An example <strong>of</strong> this type <strong>of</strong> study for the ions Cl', BCl-, and B2C1'<br />
by H20 is given in a forthcoming p~b1ication.l~<br />
3. Competitive Solvation <strong>of</strong> Ion A+ (or B-) by Solvent Molecules <strong>of</strong><br />
Solvents S and S : A comparison <strong>of</strong> the solvating power <strong>of</strong> two different<br />
solvents cb-tained by measuring the composition <strong>of</strong> ion clusters<br />
when two different solvents are present at known partial pressur 8. An<br />
example <strong>of</strong> this type <strong>of</strong> study is the competiti e solvation <strong>of</strong> NIig by HqO<br />
and NH3 which has been reported on elsewhere.2C Some more recent studies<br />
will be described in the present paper.<br />
These concern the competitive<br />
solvation <strong>of</strong> the proton by water and methanol. The water methanol system<br />
is <strong>of</strong> interest since the dipole moment <strong>of</strong> water is slightly higher than<br />
that <strong>of</strong> methanol while the polarizability <strong>of</strong> methanol is considerably<br />
larger than that <strong>of</strong> water. Thus it might be expected that at close range<br />
to the central ion methanol might be preferentially solvating. I<br />
' ,<br />
1<br />
1<br />
'<br />
I<br />
1
I<br />
69<br />
EXPERIMENTAL<br />
The mass spectrometric study <strong>of</strong> ion solvent molecule interactions<br />
requires mass spectrometric apparatus which can sample ions originating<br />
in ion sources at relatively high pressures. Three somewhat different<br />
arrangements are presently in use in this laboratory.<br />
1. Alpha particle mass spectrometer<br />
11<br />
A recent version <strong>of</strong> this apparatus has been described elsewherel’<br />
in detail. The gas, supplied from a conventional gas handling system,<br />
is irradiated in the ionization chamber. The radiation is supplied<br />
from an enclosed 200 mc polonium alpha source. The irradiated gas bleeds<br />
through a leak into the evacuated electrode chamber. There the ions<br />
carried by the gas are captured by the electric fields while the gas is<br />
pumped away. The ions are focused, accelerated and then subjected to<br />
mass analysis and electron multiplier detection in a 90° sector field<br />
analyzer tube.<br />
2. Electron beam mass spectrometer<br />
6<br />
In a more recent apparatus an electron beam is used as ionizing<br />
medium. The ion source is identical to that used with the alpha source<br />
with the exception that th former alpha source port contains only one<br />
very thin nickel foil (lo-‘ inches) window through which the electrons<br />
enter the source. The electrons are created by an ordinary electron<br />
gun housed in a sidearm <strong>of</strong> the vacuum chamber opposite the nickel window.<br />
The electron filament is kept at some -25000 volts while the ion<br />
source is near ground potential. Absence <strong>of</strong> radioactive contamination,<br />
high intensity (- 10 microamps) and possibilities for pulsing (for determination<br />
<strong>of</strong> ionic lifetimes) are some <strong>of</strong> the advantages <strong>of</strong> the electron<br />
beam source. A disadvantage is the much greater scattering <strong>of</strong><br />
electrons at high ion source pressures which makes beam collimation a<br />
problem. A quadrupole mass analyzer is used with this instrument.<br />
5<br />
3. Proton beam mass spectrometer<br />
A 100 kev proton beam obtained from a Walton-Cockr<strong>of</strong>t accelerator<br />
is used as ionizing medium. The ion source is not like the previous ones<br />
but <strong>of</strong> the conventional design, i.e. a rectangular box with repeller<br />
and narrowed-down ion exit slit. The proton beam enters and exits the<br />
ion source through very thin nickel foil windows (10-5 inches).<br />
ion optics are <strong>of</strong> the conventional Nier type and magnetic analysis is<br />
used. A proton beam (preferably <strong>of</strong> even higher energy than used by us<br />
since at energies below 100 kev charge exchange converts appreciable<br />
fractions <strong>of</strong> the proton beam into an H atom beam) seems to be the most<br />
convenient ionizing medium for high ion source pressures since it pro-<br />
vides high intensity, possibility for pulsing, and little scattering at<br />
high pressure. The proton beam can also be deflected electrostatically<br />
before entering the ion source. This permits variation <strong>of</strong> the proton<br />
beam - exit slit distance in the ion source. The cost <strong>of</strong> Walton-<br />
Cockr<strong>of</strong>t accelerators is relatively low.<br />
A high pressure mass spectrometer using Mev rotons from a Van de<br />
Graaff accelerator has been described by Wexler. 13<br />
The<br />
? -
70<br />
RESULTS AND DISCUSSION<br />
+<br />
1. Competitive solvation <strong>of</strong> CH3E2 by water and methanol I<br />
A study <strong>of</strong> the ions in methanol vapor in the pressure ran e 1-10 t<br />
torr showed that most <strong>of</strong> the ions belonged to the series CH30Hq.nCH30H.<br />
This sug ested the possibility <strong>of</strong> observing the competitive solvation<br />
<strong>of</strong> CH30H$ by water and methanol molecules. Figure 2 shoys the relative r<br />
intensities <strong>of</strong> the mixed clusters obtained when irradiating mixtures <strong>of</strong><br />
5% methanol: 95% water and 20% methanol: 80% water, both at 5 torr totall<br />
pressure and 5OoC ion source temperature. In the mixed methanol-water<br />
clusters the question <strong>of</strong> the structural assignment arises. Thus the<br />
ion <strong>of</strong> mass 119 could be assigned as H+.2CH30H.3H20 or CH OH3.CH30H.3H20,<br />
or H30+.2CH30H. 2H20. We have selected the notation CH30Hi.CH30H. 3H20<br />
(or CH30H3.mM.wW, for the general cluster where M and W stand for CH30H ,<br />
and H20 and m and w for the number <strong>of</strong> methanol and water molecules). [,<br />
The proton was assigned to the methanol oxy en ion since the proton af- ,<br />
finity <strong>of</strong> methanol is some 10-20 kcal/mole q3 higher than that <strong>of</strong> water<br />
This assignment is also consistent with the observation that even in<br />
mixtures where water predominates, i.e. 5% methanol, the pure h drate '<br />
H30+.wW was not observed, while the pure methanol cluster CH30H2.m<br />
was observed.<br />
The clusters obtained with 5% methanol (Figure 1) contain, on the<br />
average, considerably more methanol than water even though the partial<br />
pressure ratio <strong>of</strong> water to methanol is 19:l. Thus methanol is the<br />
stronger solvent in the observed clusters, i.e. clusters containing up<br />
to six solvent molecules. We shall be able to understand the meaning<br />
<strong>of</strong> this result better after a more detailed treatment <strong>of</strong> the data. It<br />
can be shown that the distribution <strong>of</strong> water and methanol in the observed<br />
clusters follows quite closely a probability distribution. Calling the<br />
probabilities for inclusion <strong>of</strong> water and methanol w and p, for a cluster<br />
with a total <strong>of</strong> II solvent molecules the probability distribution will<br />
be given by the binomial expansion <strong>of</strong> the term (w+p) E. For example if<br />
a probability distribution is followed, the cluster containing three<br />
solvating molecules CH30HZ.mM.wWl where II = m + w = 3, should show the<br />
following relative intensities: sH30H5.3W : CH30H;. 2WM : CH3OH5.WZM :<br />
CH30H2.3M = m3 : 3~ p : 3wp2 : p . We have obtained values for u and<br />
by fitting binomial expansions to the experimentally observed distri-<br />
bution. The calculated intensities shown in Figure 2 demonstrate that<br />
a relatively good fit <strong>of</strong> the experimental data can be obtained. In<br />
order to express the preference for inclusion <strong>of</strong> methanol and water per<br />
unit methanol and water pressure we define y = (p/p~)/(w/P~) as the<br />
factor for preferential take up <strong>of</strong> methanol, PM and Pw being the p a r d<br />
pressures <strong>of</strong> methanol and water present in the ion source. The y's tal-<br />
culated in this manner are given in Figure l. The results at 5 and 20%<br />
methanol show that the yls for a cluster <strong>of</strong> a fixed size (i.e. e = COnt)<br />
are approximately independent <strong>of</strong> the methanol-water pressure ratio.<br />
This independence was confirmed in a number <strong>of</strong> other runs with 2, 4, 5,<br />
8, 20, 50% methanol at 2 and 5 torr total pressure. The y's are found<br />
to decrease as e increases. Thus methanol is taken up preferentially<br />
by a factor <strong>of</strong> 55, 21 and 8 for clusters containing 3, 4 and 5 solvent<br />
molecules. Figure 2 shows a log y plot versus the number <strong>of</strong> solvating<br />
molecules. The plot is almost linear and allows an extrapolation to<br />
log y = 0 or y = 1. This occurs when the cluster contains seven Sol-<br />
vating molecules. For e > 7, y becomes less than unity, i.e. water be-<br />
gins to take precedence. Figure 2 also shows results obtained with the<br />
proton beam mass spectrometer. The total pressure in these runs was<br />
much lower (0.6 torr) and the reaction time much shorter (a few micro- ,<br />
seconds versus a few milliseconds in the alpha particle ion source).<br />
'5<br />
.!
+<br />
Figure 1 Mixed water and methanol content in CH30H2.mCH30H.wHZ0<br />
clusters. Ion source temperature 5OOC.<br />
2 .o<br />
1.5<br />
0.5<br />
0<br />
2 3 4 5 6 7<br />
n= Number <strong>of</strong> Solvating Molecules 1<br />
Figure 2 Plot <strong>of</strong> log y versus number <strong>of</strong> solvating molecules.<br />
y is factor for preferential take up <strong>of</strong> methanol over<br />
+<br />
water into ion CH30H2.mCH30H.wH20, where m + w - tl.
72<br />
One might suspect that under these conditions clustering equilibrium<br />
might not be achieved. However the results are, on the whole, quite<br />
1<br />
similar to those obtained with the alpha ion source. This might be<br />
1<br />
taken to mean that clustering equilibrium establishes very rapidly and<br />
that the proton beam results approach equilibrium.<br />
In the interpretation <strong>of</strong> the present results considerable help can<br />
be obtained from the "electrostatic theory" for metal-ion coordination<br />
complexes.2 It is recalled that this theory, using simdle electrostatic '<br />
concepts, allows the calculation <strong>of</strong> the binding energies <strong>of</strong> metal complexes<br />
in the gas phase and that the results <strong>of</strong> such calculations have<br />
been in many cases very successful. In general, the potential energy<br />
I<br />
<strong>of</strong> a complex ion is built up <strong>of</strong> four terms. These are due to the attrac- ''<br />
tion between the ion and the permanent and induced dipole <strong>of</strong> the ligands,<br />
the mutual repulsion <strong>of</strong> the dipoles, the energy required to form the<br />
induced dipoles and the van der Waals repulsions between the ligands<br />
and the central ion. Comparing the potential energies <strong>of</strong> an ion having<br />
water or methanol molecules as ligands, it is found that the first term<br />
is the decisive one. The first term contains the sum <strong>of</strong> the permanent<br />
dipole and the polarizability. The dipole moments <strong>of</strong> water and met anol<br />
are 1.85 and 1.69 D while the polarizabilities are 1.48 and 3.23 b. 9<br />
The potential energy <strong>of</strong> an ion dipole interaction varies inversely with<br />
the square <strong>of</strong> the distance while the polarizability interaction depends<br />
on the fourth power. It follows that the methanol molecules with their<br />
slightly lower dipole but considerably higher polarizability will be<br />
more strongly solvating than water at close range to the ion. The experimentally<br />
observed preference for methanol is thus to be understood<br />
as resulting from the higher methanol polarizability.<br />
It can be shown that the possibility <strong>of</strong> fitting the observed clusters<br />
with a given II by a probability distribution suggests that for<br />
!? < 6 all solvent molecules belong to an inner solvation shell. Suppose<br />
that for !? = 5 some <strong>of</strong> the molecules went into an inner shell (fully<br />
occupied) and the rest into an outer shell. The preference for methanol<br />
over water in the inner shell will be very different from that in the<br />
outer shell. It might even be expected that water will be preferentially<br />
taken up in the outer shell. It follows that the water-methanol<br />
distribution in a cluster containing inner and outer shell molecules<br />
could npt be fitted by a single probability distribution <strong>of</strong> the type<br />
(W + p) but that the inner and outer shell would have to be fitted<br />
separately. Such a case is in fact observed in the Eqmpetitive sOlVation<br />
<strong>of</strong> NH$ by H20 and NH3 which was studied earler. Since the watermethanol<br />
clusters <strong>of</strong> !? = 3, 4, 5 can each be fitted with a probability<br />
distribution we can conclude that these clusters do not contain molecules<br />
which occupy a distinct outer shell, i.e. that the inner shell<br />
' contains at least five solvent molecules.<br />
The ability to fit a cluster <strong>of</strong> constant k with a probability distribution<br />
is, to a certain extent, surprising even if all molecules<br />
belong to the same solvation shell. A probability distribution means,<br />
for example, that in the five cluster the preference for methanol over<br />
water is the same whether all the remaining four ligands are water Or<br />
methanol or a mixture <strong>of</strong> them. Obviously this can not be strictly true-<br />
The meaning <strong>of</strong> the experimental result must be that the nature <strong>of</strong> the<br />
other occupants is , in the first approximation, not important.<br />
While y remains approximately constant for a cluster distribution<br />
with k = const, it was observed that Yp=COnst decreases from II = 3 to<br />
J<br />
k = 5. This can be understood if one assumes that whenever II is increased<br />
by one unit the effective radius <strong>of</strong> the (inner) shell increases. This<br />
causes the polarizability to become less important and leads to a decrease<br />
<strong>of</strong> the preference for methanol. An increase <strong>of</strong> the effective<br />
I<br />
i<br />
14<br />
i<br />
I
73<br />
' , radius might be expected because <strong>of</strong> the mutual repulsion due to dipole<br />
and van der Wall's forces between the ligands.<br />
i Experiments on the competitive solvation <strong>of</strong> CH3NO2 and H20 have<br />
also been made but this work is not yet completed. The available results<br />
show that CH3NO2 is taken up preferentially at close range <strong>of</strong> the central<br />
( ion (NH; and H3O+ gave similar results). Thus at room temperature the<br />
\ preference factor is - 200 for three ligands, 60 for four Ligands, 40<br />
for five, etc. The results also seemed to show that nitromethane is<br />
always taken up preferentially in an incomplete.shel1 (inner or outer)<br />
but that it is ultimately expelled from inner shells.<br />
1<br />
,<br />
2. Effect <strong>of</strong> electric fields on cluster size<br />
One may expect that the presence <strong>of</strong> electric fields will lead to a<br />
decrease <strong>of</strong> the average cluster size. This effect is illustrated in<br />
Figure 3. The data were obtained with the proton beam mass spectrometer<br />
which has a Nier type ion source, i.e. ion exit slit and repeller. The<br />
distance from proton beam to ion exit slit was II 0.4 cm. E, is the repeller<br />
field strength. In preliminary experiments at very low repeller<br />
fields it could be shown that at H20 pressures higher than about 0.3<br />
torr the cluster composition obtained with the proton mass spectrometer<br />
was very similar to that obtained with the alpha particle instrument.<br />
Since the estimated ion residence times are - 10 micron for the proton<br />
mass spectrometer and - 1 millisec for the alpha instrument these results<br />
were taken to mean that in both instruments clustering equilibrium<br />
or near equilibrium is achieved. Figure 3 shows that increase <strong>of</strong> the<br />
electric field decreases the cluster size. This is a gradual process<br />
as shown by the succession <strong>of</strong> maxima. The maximum composition shifting<br />
successively from four water to zero water content <strong>of</strong> the H~O+ ion.<br />
Possibly plots <strong>of</strong> the type given in Figure 3 could be used for determination<br />
<strong>of</strong> the enthalpy <strong>of</strong> attachment, i.e. AHn-l,n.<br />
-f LITERATURE CITED<br />
I<br />
1. A.P. Altshuller, J. Am. Chem. SOC. 77, 3480 (1955).<br />
2. F. Basolo and R.G. Pearson, Mechanisms <strong>of</strong> Inorganic Reactions, p.64<br />
(John Wiley & Sons, New York, 1958).<br />
' 3. R.P. Bell, The Proton in Chemistry (Cornel1 University Press, Ithaca,<br />
t New York, 1959).<br />
z<br />
4. M. Born, 2. Physik, 1, 45 (1920).<br />
5. G.J. Collins and P. Febarle, to be published.<br />
6. D.A. Durden and P. Kebarle, to be published.<br />
7. W.J. Hamer (Edit.) "The Structure <strong>of</strong> Electrolytic Solutions" Chapter<br />
5 (John Wiley & Sons, New York, 1959).<br />
8. A.M. Hogg and P. Kebarle, J. Chem. Phys. 43, 449 (1965).<br />
9. A.M. Hogg, R.M. Haynes and P. Kebarle, J.X. Chem. SOC. 80, 28 (1965).<br />
, 10. P. Kebarle and E.W. Godbole, J. Chem. Phys. 39, 1131 (196n.<br />
,11. P. Kebarle, R.M. Haynes and S.K. Searles, Advances in Chemistry, 58,<br />
, 210 (1966).<br />
\12. P. Kebarle and A.M. Hogg, J. Chem. Phys. 42, 798 (1965).<br />
13. P. Kebarle, Advances in Chemistry: Applications <strong>of</strong> Mass Spectrometry<br />
to Inorganic Chemistry (to be published) (American Chemical Society).<br />
)14. M.S.B. Munson, J. Am. Chem. SOC. 87, 2332 (1965).<br />
15. S. Wexler, Advances in Chemistry,TL, 193 (1966).<br />
I<br />
I<br />
J<br />
1-
z<br />
0 c<br />
a<br />
60<br />
2 40<br />
0<br />
2<br />
U<br />
+<br />
2<br />
5 20<br />
M<br />
0<br />
Figure 3<br />
74<br />
036 torr HzO<br />
0 10 20 30 40 50<br />
ION ENPRGY, E,I (volts)<br />
Effect <strong>of</strong> electric field on cluster composition.<br />
Ere equals voltage between repeller and ion source exit slit.<br />
Temperature <strong>of</strong> ion source 3OOC.<br />
Water pressure 0.36 torr.<br />
Experiments taken with 100 kev proton beam mass spectrometer.<br />
!<br />
I<br />
II
75<br />
Negative Ion Mass Spectra <strong>of</strong> Some Boron Hydrides<br />
D. F. Munro, J. E. Ahnell, and W. S. Koski:<br />
Department <strong>of</strong> Chemistry, The Johns Hopkins University, Baltimore, Maryland 21218<br />
Abstract I ,<br />
Questions <strong>of</strong> the role <strong>of</strong> negative boron hydride ions frequently arise in<br />
problems concerning energetics <strong>of</strong> electron impact processes, discharge tube reactions,<br />
and radiation chemistry. In view <strong>of</strong> the fact that the boron hydrides are<br />
electron deficient compounds, it might be expected that they are more prone to<br />
negative ion formation than hydrocarbons; however, very few studies <strong>of</strong> such ions<br />
have appeared in the literature.<br />
This paper gives some qualitative results obtained<br />
on boron hydride negative ions produced by electron bombardment <strong>of</strong> diborane, tetraborane,<br />
and pentaborane-9. A pulsed electron beam, a quadrupole mass analyzer, and<br />
an electron multiplier were used for this work. In diborane, the most inteise ions<br />
observed were BzH3- pd BHQ-.<br />
B*H-, Bz-, BH3-, BH2 , BH-, and B were also readily detectable. Similarly, the -<br />
negative ions observed in tetraborane - included the relatively high intensity B3H7<br />
and lower intensities <strong>of</strong> BzHg-, B2H4 , BHI, ,,etc. In addition, indications were<br />
obtained for B4Hg . Similar results were obtained with pentaborane-9.<br />
Lover and varying intensities <strong>of</strong> BzH4-, B2H3 , B2H2 ,<br />
Introduction<br />
For many years, mass spectroscopists have dealt almost exclusively with processes<br />
involving positive ions; ionization, fragmentation, and ion-molecule reactions.<br />
Recently, however, despite problems <strong>of</strong> low intensity, an increasing amount<br />
<strong>of</strong> work has been done on negative ions. A number <strong>of</strong> species have been character-<br />
ized,' and a few reactions have been studied.2s3<br />
In addition, at least one investi-<br />
gation on the role <strong>of</strong> negative ions in flames has been carried out, using mass<br />
spectroscopic techniques.<br />
The boron hydrides are traditionally classified among the electron deficient<br />
compounds, and might well be expected to form negative ions. whennore, some work<br />
has been done with the boron hydrides in gaseous <strong>discharges</strong> and in radiationchemistry<br />
where these ions might conceivably play a role.5<br />
Thus, it was <strong>of</strong> interest to us to<br />
study the negative ions <strong>of</strong> the boron hyhrides with the eventual hope <strong>of</strong> providing<br />
information to permit the evaluation <strong>of</strong> the role these ions might play in <strong>discharges</strong><br />
and other associated phenomena.<br />
We have under construction in our laboratory a tandem mass spectrometer for the<br />
study <strong>of</strong> ion-molecule reactions. In the course <strong>of</strong> testing the first stage <strong>of</strong> the<br />
instrument, it became apparent that the spectrometer might be suited for the observation<br />
<strong>of</strong> negative ions. The ions from SF6 and 02 were detected with relative ease.<br />
It was decided to investigate vhat negative ions could be observed in the simpler<br />
boron hydrides; diborane, tetraborane, and pentaborane.<br />
Because the spectrometer is still in the construction stage, some <strong>of</strong> the re-<br />
finements normally found on conventional instnunents are lacking, so that the<br />
results obtained are necessarily <strong>of</strong> a qualitative nature. Nevertheless, it was felt<br />
that the observations were sufficiently interesting to warrant reporting.<br />
Experimental<br />
The important features <strong>of</strong> the basic experimental arrangement are shown diagremmatically<br />
in Fig. 1. The gas to be analyzed flows from a bulb at roan temperature<br />
into the ion source through a Granville-Phillips leak. The ion source, which is<br />
similar in design to one described by Von Zahn,6 has an electron beam energy
76<br />
variable from 0 - 100 volts. There are no provisions for measuring pressure,<br />
temperature, or ionizing current in the ion source. The ions are focused and<br />
accelerated to 50 - 100 volts, and mass analyzed by a quadrupole mass filter with a<br />
length <strong>of</strong> 10.0 inches and an ro <strong>of</strong> 0.27 inches. Mass scanning is accomplished by<br />
varying the RF and DC voltages to the quadrupole. The resolution <strong>of</strong> the quadrupole<br />
is electronically variable. The ions are then accelerated to 600 volts and detected<br />
by a Bendix Channeltron continuous electron multiplier.<br />
Because <strong>of</strong> the absence <strong>of</strong> slits in the mass spectrometer, the lrelatively open<br />
nature <strong>of</strong> the ion source, and the high sensitivity <strong>of</strong> the detector, it is necessary<br />
to reduce the background from scattered electrons reaching the detector. This is<br />
accomplished in two ways:<br />
the electrons but not the ions; and second, by pulsing the electron beam. The<br />
ionizing beam is pulsed at 10 kc by applying a 40 volt pulse to the first lens <strong>of</strong><br />
the electron beam. The beam ''on'' time is 10 microseconds. After a delay <strong>of</strong> 5<br />
microseconds, the second pulser sends a one volt pulse to turn on the transistor-<br />
ized "AND" gate for 20 - 30 microseconds.<br />
first, by establishing a weak magnetic field to deflect<br />
Only those pulses which arrive at the<br />
gate during this interval are sent on to the ratemeter. At the voltages used, most<br />
<strong>of</strong> the electrons arrive at the detector vithin one or two microseconds; the flight<br />
time <strong>of</strong> the negative ions, however, is <strong>of</strong> the order <strong>of</strong> 10 - 20 microseconds. Thus,<br />
the signal to the ratemeter is primarily due to ions, with a drastic reduction in<br />
the electron background. The output <strong>of</strong> the ratemeter is plotted versus the DC<br />
voltage on the quadrupole with an XY recorder. A signal <strong>of</strong> five ions per second<br />
can readily be observed. A typical plot, shown in Fig. 2, is the lover portion <strong>of</strong><br />
the mass spectrum <strong>of</strong> the negative ions <strong>of</strong> pentaborane.<br />
All the boron hydride samples were purified by distillation in a vacuum<br />
system. The diborane was trapped out at -154OC. The tetraborane was purified in<br />
three steps: pumping <strong>of</strong>f hydrogen and diborane while trapped at -130, passing<br />
through a trap at -95 to condense the pentaborane and the higher boron hydrides,<br />
and finally condensing at -196. The pentaborane was similarly purified: pumping<br />
<strong>of</strong>f diborane and tetraborane at -98, passing through a trap at -79, and final trapping<br />
at -196.<br />
Results and Discussion<br />
The results are shown in the bar graph in Fig. 3. The narrow bars represent<br />
peaks which have been multiplied by a factor <strong>of</strong> ten. All measurements are for 70<br />
volt ionizing electrons. The monoisotopic mass spectrum was computed assuming the<br />
natural BlO/B11 distribution <strong>of</strong> 20/80, and also assuming that there was no isotope<br />
effect in loss <strong>of</strong> hydrogen from B10 versus B11. In nearly all cases, the Ykak<br />
heights from which the monoisotopic spectra were calculated were an average <strong>of</strong> at<br />
least three measurements, with separate measurements in agreement within 10%. In<br />
calculating the monoisotopic spectrum, the system is "over-determined," :.e., there<br />
are more equations than there are unknowns. This provides a means <strong>of</strong> checking the<br />
internal consistency <strong>of</strong> the results. For every case except tvo, the self-consistency<br />
deviation was less than 2%, and in all cases less than 10%. On the basis Of<br />
these consi_derations, relative intensities within a group <strong>of</strong> peaks, e.g., BgHg ,<br />
B5He , B5H7 , etc., should be accurate to 2 10%.<br />
In comparing peaks between<br />
different groups, however, it is more difficult to assign limits <strong>of</strong> error, for the<br />
spectrometer may favor the observation <strong>of</strong> low masses over high masses in certain<br />
cases. One can observe this "discrimination" in the positive ion spectra, and it<br />
appears to be primarily a function <strong>of</strong> the resolution <strong>of</strong> the quadrupole. It is<br />
possible, however, that the continuous electron multiplier and the ion source also<br />
mass discriminate, perhaps differently for positive and negative ions.<br />
Since the<br />
signal from the multiplier is analyzed as pulses rather than current, mass discrim-<br />
ination by the detector should be minimized. The exit holes in our ion source are<br />
rather large, and it would be expected that discrimination here would also be<br />
negligible. Thus, if it is assumed that the discrimination effects are the same<br />
r<br />
i,<br />
f<br />
1<br />
L<br />
r<br />
,<br />
4
A b 1 1<br />
I H I<br />
I I<br />
FILAMEN T<br />
CONTINUOUS<br />
I<br />
COWPR<br />
SOURCE ION ' I QUAORUPOLE I<br />
ELECTRON<br />
SUPPLY I<br />
AfULTIPLIER<br />
I I<br />
I<br />
+<br />
-<br />
b<br />
ELECTRON f QU*oauPoLE x-v POWER SUPPLY<br />
ION OCTICS POWER SUPPLY RECORDER I PULSE<br />
COWER SUCCLV f MASS SCAN AMPLIFIER<br />
T<br />
1 1 ><br />
A -<br />
A + t + 1<br />
PULSCR I OSCl LLOSCOPE RATEME TER -<br />
TRIGGER 4<br />
OELAV<br />
--------_<br />
i CULStR 2<br />
--_<br />
i<br />
GATE<br />
4 4<br />
FIGURE I<br />
-
z"<br />
0-<br />
a?<br />
Q<br />
zc<br />
c<br />
rr) 'I
79<br />
for positive and negative ions, approximate corrections can be made. At worst, the<br />
relative intensities throughout the whole, spectra are probably correct within 225%.<br />
Although reasonable precautions were taken to assure the purity <strong>of</strong> the sample<br />
in the diborane spectrum, peaks corresponding to B3H7 and B4Hg were observed.<br />
These could have arisen either from ion-molecule reactions, from pyrolysis <strong>of</strong> the<br />
diborane as the sample flowed into the source, or from impurities i7 the diborane.<br />
The latter is improbable, for in a positive ion spectrum <strong>of</strong> the same sample in the<br />
same spectrometer, the tetraborane peaks could be observed only with difficulty.<br />
It was estimated on the basis <strong>of</strong> the positive ion spectrum that the tetraborane<br />
impurity could be no greater than one part in five-hundred. In the negative ion<br />
spectrum the ?ea& corresponding to B3H7- and B2H3 were <strong>of</strong> almost equal intensity.<br />
It should be noted, however, that in comparing the diborane and tetraborane samples,<br />
it .was much easier to observe the negative ion peaks from tetraborane, which were<br />
larger by sbmbnut two orders <strong>of</strong> magnitude.<br />
There are several mechanisms by which negative ions can be formed. The tradi-<br />
tional processes are as follows:<br />
1. Ion pair. formation:<br />
2. Resonance attachment: XY + e -+ XY<br />
3. Resonance attachment with dissociation: XY + e -- X + Y-<br />
XY + e -+ X+ +-Y- + e<br />
In addition,a process somewhat like the third mentioned above can be imagined, in<br />
which the neutral species is left in an excited state which in turn decays to an<br />
ion pair. Also, a negative ion may fragment, giving rise to a neutral species and<br />
a new negative ion.<br />
4. Excited state decay: XYZ + e +. XY* + Z-<br />
5. Fragmentation: XY- -+ X + Y-<br />
XY* -+ x+ + Y-<br />
+ Using the above scheme, and assuming that the fragments most easily lost are<br />
BHg , BH3 , and hydrogens, most <strong>of</strong> the more intense peaks in the negative ion boron<br />
hydride spectra can be accounted for rather well in the following manner.<br />
Possible Modes <strong>of</strong> Fragmentation <strong>of</strong> Diborane<br />
The underlined peaks are the four most intense in the diborane mass spectrum.<br />
The other peaks come from loss <strong>of</strong> hydrogens, either singly or in pairs, from the<br />
above species.<br />
Possible Modes <strong>of</strong> Fragmentation <strong>of</strong> Tetraborane<br />
This scheme accounts for all <strong>of</strong>-the peaks with an observed intensity <strong>of</strong><br />
greater than 1%, except for the B2H3 peak, whose intensity is 1.1%, and which could<br />
be due to a diborane impurity.
80<br />
Possible Modes <strong>of</strong> Fragmentation <strong>of</strong> Pentaborane<br />
etc.<br />
+<br />
In support <strong>of</strong> the above mechanism, the data in Table 1 can be cited<br />
BH2+<br />
BH3+<br />
Table 1<br />
Abundances <strong>of</strong> BH2+ and BH3+ in Some Boron Hydrides<br />
Diborane7 Tetraborano8 Pentaboraneg<br />
19.1<br />
.59<br />
(These ion intensities were Cbtained with 70 volt electrons, and are given in<br />
percent relative to the most intense peak in the mass spectrum for the cmpound in<br />
quest ion. )<br />
+<br />
Thua, it can be argued that the loss <strong>of</strong> BH2 is much more likely than the loss <strong>of</strong><br />
BH3 . The loss <strong>of</strong> BH3, however, at least from neutral species, is fairly well<br />
established in a number <strong>of</strong> reactions." It should not be too surprising if BH3 can<br />
also be readily fragmented from negative ions.<br />
- It is tempting to question the necessity <strong>of</strong> invoking the excited state decay<br />
process (4) described above. For example, can the formation <strong>of</strong> the B3H7- not be<br />
simply written as a resonant attachment process with the dissociation <strong>of</strong> BH3?<br />
Although this is certainly possible, one or two points argue against this mechanism.<br />
First, at the electron energies used, a resonant process <strong>of</strong> this type would not be<br />
expected to be <strong>of</strong> primary importance. Secondly, one might inquire why the pair<br />
ionization process,<br />
~<br />
+<br />
4 + e ~ + B 1 ~ H ~ ~ + - B H ~<br />
is not observed, especially in view <strong>of</strong> the fact that the analogous process seems to<br />
be the predominant one in diborane. Indeed, the pair ionization process is the one<br />
which is expected; it is disturbing that it is not seen in view <strong>of</strong> the stability <strong>of</strong><br />
B3Hg in solution and in the solid phase. The structure <strong>of</strong> B3Hg- is known frran<br />
x-ray Work, and there is ample theoretical and experimental justification for its<br />
existence.'l On the other hand, we have found no evidence in the literature for<br />
B3H;. The question arises as to whether an error can be made in the mass assignments.<br />
The marker compounds used were 02 and H20, giving rise to the 02 , OH-, and<br />
0- peaks, which were always present in the background. Considerable care was taken<br />
in making the mass assignments, so that unless some unknown instrumental or <strong>chemical</strong><br />
processes were occurring <strong>of</strong> which we are unaware, the given assignments are<br />
correct. Therefore, granting that the reported observations are valid, and accepting<br />
the notion that pair ionization should be $he dominant process at higher electron<br />
enprgies, and further noting that the BH2 ion is much more predominant than<br />
the BH3 in the positive ion spectrum, it seems necessary to invoke some mechanism<br />
analogous to (4). Perhaps further weight can be given to this argument by noting<br />
that the parent peak is not observed in tetraborane, suggesting that a loss <strong>of</strong><br />
hydrogen is a strongly favored process in the electron impact situation.<br />
The present work suggests several further experiments which should be carried<br />
7.0<br />
.6<br />
!<br />
12.2<br />
--
51<br />
out to answer some <strong>of</strong> the questions which have been raised here. An investigation<br />
Of metastable negative ions on a magnetic sector instrument should prove useful in<br />
confirming or negating the fragmentation scheme postulated above. A careful study<br />
<strong>of</strong> the diborane spectrum, either in a tandem instrument, or with an ion source in<br />
which one could be sure that pyrolysis is not taking place would be necessary to<br />
remove the ambiguity in the obseraation <strong>of</strong> tetraborane-in the diborane sample;<br />
whether the peaks above diborane are due to impurities, or whether they do in fact<br />
result from ion-molecule reactions. Perhaps most useful initially would be an<br />
investigation <strong>of</strong> the boron hydride spectra at low electron energies, where resonance<br />
attachment becomes the dominant process. Ideally, one should study the spectra as<br />
a function <strong>of</strong> energy; the clastogram technique" might prove valuable in sorting<br />
out the various processes occurring. We made some attempt to work at low electron<br />
energies, but largely without success.<br />
Since we are unable to measure the ionizing<br />
current, i.e., keep it constant as a function <strong>of</strong> energy, we would have little con-<br />
fidence in an appearance potential measurement, particularly since the background<br />
from electrons again becomes a problem at low electron energies, when they can be<br />
pushed out <strong>of</strong> the ionization chamber by the repeller.<br />
Reese, Dibeler, and Mohler have reported on the spectrum <strong>of</strong> pentaborane, in<br />
which they observed the resonant attachment process in the parent ion.13 Although<br />
their relative intensities are different from those which we observed, this is<br />
probably not surprising, in view <strong>of</strong> the different energies <strong>of</strong> the ionizing electrons<br />
in the two cases. Curiously, however, Dibeler does not report any <strong>of</strong> the lower<br />
fragments for pentaborane.<br />
In conclusion, it is evident that the lower boron hydrides do form negative<br />
ions. In certain circumstances, the abundance <strong>of</strong> these ions mag be great enough so<br />
that they would have to be taken into consideration in mass spectral, radiation, or<br />
gaseous discharge phenomena. It is also clear, however, that more than a mere qual-<br />
itative study <strong>of</strong> these negative ions will be necessary for the complete understand-<br />
ing <strong>of</strong> their roles in these situations.<br />
Acknowledment- This work was done under the auspices <strong>of</strong> the United States Atomic<br />
Energy Commission.<br />
Figure Captions<br />
Fig. 1. Block diagram <strong>of</strong> the mass spectrometer.<br />
Fig. 2. Lower portion <strong>of</strong> the mass spectrum <strong>of</strong> the negative ions from pentaborane.<br />
Fig. 3. The monoisotopic relative abundances for the negative ions frm diborane,<br />
tetraborane, and pentaborane.<br />
References<br />
1. F. Fiquet-Fayard, Actions Chimiques et Biologiques des Radiations 11, p. 44<br />
2.<br />
(1965 1.<br />
J. F. Paulson, Ion Molecule Reactions in the Gas Phase, p. 28, American Chemical<br />
3.<br />
4.<br />
Society Publications (1966).<br />
G. Jacobs and A. Henglein, Advances in Mass Spectroscopy 2, p. 28, The Institute<br />
<strong>of</strong> Petroleum (1966).<br />
M. F. Calcote and D. E. Jensen, Ion Molecule Reactions in the Gas Phase, p. 291,<br />
American Chemical Society Publications (1966).<br />
5.<br />
6.<br />
V.<br />
V.<br />
V. Subbanna, L. H. Hall, and W. S. Koski, J. Am.<br />
Von Zahn, Rev. Sci. Inst. 34, 1 (1963).<br />
Chem. SOC. 86, 1304 (1964).<br />
7. MBS No. 333 Mass Spectra Data, American Petroleum Institute Project 44 (1949).<br />
8. T. P. Fehlner and W. S. Koski, J. Am. Chem. SOC. &, 1905 (1963).<br />
9. NBS No. 33b Mass Spectra Data, American Petroleum Institute Project 44 (1949).<br />
10. T. P. Fehlner and W. S. Koski, J. Am. Chem. SOC., 86, 2733 (1964).<br />
11. W. N. Lipscomb, Boron Hydrides, W. A. Benjamin, Inc. (1963).
82<br />
12. R. W. Kiser, Introduction to Mass Spectrometry and Its Applications, p. 191,<br />
Prentice-Hall, Inc. (1965). f<br />
13. R. M. Reese, V. E. Dibeler, and F. L. Mohler, J. Research NBS 57, 367<br />
I<br />
I .<br />
(<br />
/ I<br />
1956). ''<br />
\I<br />
J
93<br />
INTERACTIONS OF EXCITED SPECIES AND ATOMS IN DISCHARGES**<br />
*<br />
Robert A. Young and Gilbert A. St. John<br />
Stanford Research Institute<br />
Menlo Park, California<br />
ABSTRACT<br />
The influence <strong>of</strong> atomic nitrogen on the concentration <strong>of</strong> N2(A3C:) excited in<br />
nitrogen by a weak discharge has been studied. It was found that (1) a high-order<br />
Process involving atomic nitrogen destroys N?(A3Ct), (2) a process utilizing<br />
species, other than atomic nitrogen, created in the discharge removes Nz(A%$), and<br />
(3) a process involving atomic nitrogen and another excited species produces<br />
N~(A~c;).<br />
It was also found that NO rapidly quenches N2(A3Ct)<br />
with a rate coefficient<br />
<strong>of</strong> 3 X 10-l' cm3/sec. A quarter <strong>of</strong> the N2(A3E:) destroyed excites NO to the<br />
A~E+ VI = o level.<br />
INTRODUCTION<br />
Nitrogen subjected to an electrical discharge at low pressure has been a subject<br />
<strong>of</strong> study for over 70 years. In general, phenomena occurring in the discharge itself<br />
have been studied in static systems, while phenomena caused by long-lived species<br />
generated by a discharge have been studied in fast-flow apparatus.<br />
In all these phenomena there are apparently two classes <strong>of</strong> reactants involved,<br />
atomic species and excited molecules. Recombination <strong>of</strong> atoms accounts for most <strong>of</strong><br />
the long-lived afterglow phenomena, while the latter are necessary for much that<br />
occurs during the discharge and shortly (2 10 m sec) after its termination.<br />
In both the discharge and the long-lived afterglow,' molecular nitrogen in the<br />
A3E$ state is suspected <strong>of</strong> playing an important role. This is because it is<br />
metastable, it is the lowest electronic state <strong>of</strong> N2, it is supposedly easily<br />
excited in a discharge, and it is a necessary result <strong>of</strong> atom recombination. However,<br />
emission from this state <strong>of</strong> N2, the Vegard-Kaplan bands, is exceedingly difficult<br />
to observe in these phenomena. In fact the Vegard-Kaplan bands have never been<br />
observed in the long-lived afterglow, and only under very special circumstances in<br />
the discharge or in the short afterglow.<br />
.) Recently' we attempted to observe the Vegard-Kaplan bands in the long-lived<br />
(Lewis-Rayleigh) nitrogen afterglow. This afterglow is almost entirely due to the<br />
first positive bands <strong>of</strong> N2 which terminate on the A3Ct state. The flux <strong>of</strong> this<br />
radiation represents a minimum rate <strong>of</strong> production <strong>of</strong> N2(~3C+). From our failure<br />
to observe the Vegard-Kaplan bands, from the radiative lifethe <strong>of</strong> the A3E: state<br />
(5 10 sec) and from the sensitivity <strong>of</strong> detection, an upper limit to the actual<br />
lifetime <strong>of</strong> N2(A3C$) in the afterglow <strong>of</strong> 5 x 10-4 sec was derived.<br />
This result implied that collision processes were removing N2(A3Ct). However,<br />
it is known that N2 does not quench N2(A3Et). Aside from N2, atomic nitrogen<br />
is the largest constituent present, and we conjectured that it was responsible for the<br />
short lifetime <strong>of</strong> N2(A3Ct).<br />
**<br />
This work was done under Contract DA-31-124-ARO-D-434.<br />
* Visiting Fellow, Joint Institute for <strong>Laboratory</strong> Astro<strong>physics</strong>, 1966-67.
84<br />
This paper describes experiments aimed at investigating the hypothesis that<br />
atomic nitrogen as well as several other compounds (NO, CO, NH3, H2), effectively<br />
destroy N2(A3Et).<br />
EXPERIMENTAL - ATOMIC NITROGEN INTERACTION<br />
Nitrogen was excited by a pulsed or continuous 150 kc, 10 kV oscillator (<strong>of</strong> the<br />
type used in high-voltage supplies) in a 5-liter quartz bulb with qimple one-eighth<br />
inch tungsten rod electrodes separated by the diameter <strong>of</strong> the bulb. A one-half-meter<br />
Jarrel-Ash Seya-Namioka monochromator was used to scan the discharge spectrum or to<br />
isolate bands for decay measurements. Signal levels were very low, and an<br />
Enhancetron signal integration unit' was used to abstract the decaying signal from<br />
the noise.<br />
Prepurified nitrogen was passed through a liquid nitrogen trap, then through<br />
heated titanium-zirconium and two additional liquid nitrogen traps, past a microwave<br />
excitation section, an NO titration inlet, and finally into the 5-liter observation<br />
bulb, from which it was exhausted by a mechanical vacuum pump at approximately 100<br />
cm3/sec. A filtered photomultiplier downstream from the bulb observed the N2 first<br />
positive bands resulting from atom recombination.3 This permitted the atom concentration<br />
to be computed after calibration against an NO titration.<br />
RESULTS INVOLVING ATOMIC NITROGEN<br />
Figure 1 shows spectrometer scans <strong>of</strong> the discharge. In Fig. la the N2 second<br />
positive and NO Y bands predominate below 3000 A. The 6 bands were the only<br />
other system <strong>of</strong> NO detected, and these were very weak. If the microwave discharge<br />
is adjusted to produce a small quantity <strong>of</strong> atomic nitrogen, the NO bands can be<br />
suppressed almost entirely (Fig. lb). Although the Vegard-Kaplan bands are also<br />
reduced, the second positive bands remain relatively unchanged. Addition <strong>of</strong> NO<br />
cacses a large increase in the intensity <strong>of</strong> the NO y bands (Fig. IC).<br />
Figure 2 shows the decay <strong>of</strong> the (0.5) Vegard-Kaplan band and the (0,3) NO<br />
band with the addition <strong>of</strong> atoms, &, when the upstream microwave discharge was on.<br />
The decay <strong>of</strong> the NO Y bands is identical to that <strong>of</strong> the Vegard-Kaplan bands.<br />
Figure 3 demonstrates the buildup and decay <strong>of</strong> the (0,5) Vegard-Kaplan band<br />
when the 150 kc discharge is turned on and <strong>of</strong>f; the rise and fall <strong>of</strong> this band<br />
involved the same exponential.<br />
Figure 4 indicates the rate <strong>of</strong> decay <strong>of</strong> the Vegard-Kaplan bands as a function <strong>of</strong><br />
pressure without added nitrogen atoms.<br />
Figure 5 shows the dependence <strong>of</strong> the Vegard-Kaplan decay rate on [NI2 derived<br />
from the downstream photomultiplier.<br />
Figure 6 illustrates the pressure dependence <strong>of</strong> the rate coefficient for<br />
de-excitation <strong>of</strong> N2(A3Z$) by atomic nitrogen.<br />
Figure 7 is a plot <strong>of</strong> the growth rate constant <strong>of</strong> the Vegard-Kaplan bands,<br />
after the discharge is turned on, as a function <strong>of</strong> [NI2.<br />
Figure 8 shows (Io/I)vk as a function <strong>of</strong> atomic nitrogen, where IoVk IS the<br />
intensity without added atoms and where Ivk is the intensity vith atoms.<br />
Figure 9 indicates the decreased number <strong>of</strong> nitrogen atoms leaving the quartz<br />
bulb due to the discharge.<br />
.
L<br />
4<br />
I*C z<br />
0'2 =<br />
a,<br />
a<br />
U x<br />
I<br />
m<br />
4<br />
m<br />
0<br />
a,<br />
CI<br />
1'0 =<br />
m<br />
0'1 =<br />
C'C =<br />
1'2 =<br />
I'I =.<br />
0'0 =<br />
t'Z = -3<br />
C'I =<br />
2'0 =<br />
S't z<br />
C'I =<br />
C'O =<br />
C'O -<br />
L'E z<br />
9'0 =<br />
6'1 5<br />
x<br />
0<br />
8 M<br />
r<br />
U<br />
d<br />
e<br />
71<br />
d<br />
w<br />
d
Pig. 2<br />
z<br />
w<br />
l-<br />
4<br />
W<br />
a<br />
1.0<br />
0.9<br />
0.8<br />
0.7<br />
0.6<br />
0.5<br />
0.4<br />
0.2<br />
0.1<br />
-.<br />
- PRESSURE 0.8m m Hg<br />
-<br />
-<br />
-<br />
-<br />
VEG ARD-K APL A N<br />
0toms/cm3 add(<br />
I I I<br />
0 10 20 30<br />
TIME- rnsec<br />
10-452522-40<br />
_-<br />
The intensity <strong>of</strong> the (0.5) Vegard-Kaplan band and the (0.31.~ band.as a<br />
function <strong>of</strong> time for various amounts <strong>of</strong> added atomic nitrogen.<br />
0<br />
0
I<br />
L<br />
I,<br />
;<br />
><br />
><br />
E<br />
Fig. 3<br />
I50<br />
200<br />
150<br />
100<br />
50<br />
. .<br />
s;<br />
0<br />
msec 0 25 50 75 100 125<br />
TA- 45 2 5 2 2- 4 2<br />
Buildup and decay <strong>of</strong> the (0,5) Vegard-Kaplan band when the 150-kc discharge<br />
is turned on and <strong>of</strong>f. The curve represents integration <strong>of</strong> approxirtely<br />
1000 cycles.<br />
-1<br />
-<br />
0 " 0 '<br />
1<br />
I<br />
0 0.5 I .o 1.5 2.0 2.3<br />
1<br />
i<br />
,<br />
? .<br />
PRESSUSE-nmHg<br />
..<br />
0<br />
TB-452522-41<br />
P Fig. 4 The dependence <strong>of</strong> the rate <strong>of</strong> decay <strong>of</strong> the Vegard-Kaplan bands as a<br />
function <strong>of</strong> pressure without added n.itrogen atoms.<br />
I<br />
A*><br />
I<br />
i
1' &p<br />
400 2.0 mm<br />
300<br />
100<br />
0 4 0 " 5 10 15 20<br />
ATOMS [NI2 x (atoms/cm3)<br />
ADDED<br />
T0- 452522-47 ~<br />
Fig. 5 The rate <strong>of</strong> decay <strong>of</strong> the Vegard-Kaplan bands as a function <strong>of</strong> the square<br />
<strong>of</strong> the nitrogen atom concentration. 4<br />
-<br />
20<br />
15<br />
IO<br />
5<br />
0 0 0.5 1.0 . 1.5 2.0<br />
PRESSURE-~~H~<br />
TA-452522-44<br />
Fig. 6 The ressure dependence <strong>of</strong> the rate,coefficient for atom destruction <strong>of</strong> the<br />
N3(A s Et) state.<br />
2<br />
-- -<br />
i<br />
<<br />
/<br />
i<br />
1
I 300<br />
r<br />
1<br />
- 200<br />
i io<br />
;' a<br />
cn<br />
I I.<br />
0 5 IO 15 20<br />
I . -<br />
Fig. 7 The buildup rate <strong>of</strong> the Vegard-Kaplan bands during the excitation discharge<br />
as a function <strong>of</strong> the square <strong>of</strong> the atomic nitrogen concentration.<br />
4<br />
\<br />
I<br />
I .<br />
2.50<br />
2.00<br />
1.50<br />
[N] (otoms/crn3)<br />
0 0.5 I .o 1.5 2.0 2.5~10'~<br />
- I I I I -<br />
-<br />
1.00<br />
2 3 4 5 x1026<br />
/ [NI2 (a toms /c m3I2<br />
-<br />
T8-452522-49<br />
The. normalized inverse intensity <strong>of</strong> the Vegard-Kaplan bands during the<br />
excitation discharge as a function <strong>of</strong> added atomic nitrogen. The discharge<br />
was run continuously.
E<br />
E
where<br />
DISCUSSION OF XESULTS PERTAINING TO ATOMIC NITROGEN<br />
The observations indi,cate that duriyg decay<br />
1 1<br />
- = -<br />
T<br />
0<br />
+ K [NI2 [MI<br />
with K 2 4 x cm9/sec. Neither or can be the radiative lifetime <strong>of</strong><br />
N2(A3X3 'which is 210 sec.<br />
The differential equation describing the time dependence <strong>of</strong><br />
3+<br />
d[N2(A Eu) 1<br />
dt<br />
= ' P - L. [N2(A 3 Eu)] +<br />
I<br />
3+<br />
[N2(A Zu)] is<br />
3+<br />
where L contains all factors other than [N2(A XU)] such as collision partner<br />
concentrations, rates <strong>of</strong> radiation or diffusion, s., that are involved in processes<br />
removing N2(A3Et), and where P is the rate <strong>of</strong> production. Substituting Eq. (1)<br />
in Eq. (3) gives<br />
or<br />
Then either<br />
3 + -t/.'o 1<br />
[N2(A Eu) lo e (L--) = P .<br />
(4)<br />
'a<br />
1<br />
L = -<br />
Ta<br />
P<br />
1 3+<br />
(L - [N2(A Xu) lo<br />
a<br />
(3)<br />
and P = O (5)<br />
-t/Ta ,<br />
= e (6)<br />
-t/Ta<br />
In the latter eventuality L > l/ia, since neither P nor e can be negative.<br />
If L is comparable to 1/~~, it must be essentially constant in time,' while if it<br />
is large compared to l/ia,<br />
Since all constitutents <strong>of</strong> the system probably decay when the power input is<br />
stopped, would be characteristic <strong>of</strong> the decay <strong>of</strong> a reactant concentration<br />
ia<br />
involved in P to a higher power than in L, while other reactant concentrations<br />
are either constant or common to both P and L. The previous statement would be<br />
true if a product <strong>of</strong> reactant concentrations replaces a single reactant concentration.<br />
Since 1 / changes ~ ~ by almost a factor <strong>of</strong> 10 over the range <strong>of</strong> [N] used,<br />
either L >> 1/~, for small [N] and hence P/L [N2(A3E;)lo = e-t/Ta, or L
92<br />
closely follows 1 / in ~ dependence ~ on [N]. *, L a l/Ta. Discussion <strong>of</strong> the<br />
~<br />
last possibility will be deferred. In the former case L must be very large indeed<br />
and [N2(A3xt)] must be essentially in a quasi-steady state with its production<br />
rate, & T~ refers to the decay <strong>of</strong> P. Since [N2(A3Et)] is not strongly<br />
deactivated by pure N2, L would certainly not increase when the energy <strong>of</strong> the<br />
system dissipates as would be necessary if P did not decay. Because the decay <strong>of</strong><br />
[N2(A3c:)] appears to remain exponential for all values <strong>of</strong> [N] and because Ta<br />
is a smooth function <strong>of</strong> [N] then L > 1/T (max) if the data are; interpreted as<br />
the decay <strong>of</strong> P with [N2(A3xi)] in a quasf-steady state. For this to be true<br />
some species capable <strong>of</strong> deactivating N2(A31t) must be generated in the discharge<br />
and remain constant for times long compared to those where [N2(A3C$)] is detectable.<br />
The most abundant <strong>chemical</strong> species generated by the discharge is N; the most<br />
abundant excited molecule is Nz(A3L:) -- except possibly for the 3A state <strong>of</strong> N2 I<br />
which has not yet been directly detected. Only atomic nitrogen appears to be a<br />
realistic choice for the species which decays slowly and would keep L >> l/Ta.<br />
i.<br />
I<br />
. I<br />
However, a difficulty arises in the interpretation with L >> l/Ta if we consider<br />
the observed behavior <strong>of</strong> [N2(A3 :>I when the discharge is turned on. The rise <strong>of</strong> 1<br />
[N2(A3xz)] to a steady state is describable by<br />
If this is substituted into Eq. (3). we obtain<br />
3 +<br />
If -1/~~ = I,, then P = L [N2(A E,)] = Po, a constant in time. When t = 0,<br />
P = Po = ([N2(A3~~)]o/Ta) if L # (19~~); and if P = Po + P', we obtain<br />
If L >> 1/~~, P' is positive; and if L > 1 / ~ 1 ~<br />
is the only one tenable during the excitation discharge. 4<br />
Since Po follows the discharge transients, it must become zero when the dis-<br />
charge is turned <strong>of</strong>f, so that P changes rapidly compared with Ta from P + P' 1<br />
to P'. But this is inconsistent with the interpretation <strong>of</strong> the decay <strong>of</strong> [82(A3$)1 ,<br />
previously given on the assumption that L >> l / because ~ ~ this rapid fall in P<br />
would be reflected in a rapid fall <strong>of</strong> [N2(A3C$)] which is not observed. Hence this<br />
contradiction forces L = l/ta and P = Po, a constant, during the discharge and I<br />
L = l/ra and P = 0 after the discharge.<br />
During the excitation discharge a steady state Is reached, and<br />
j<br />
><br />
I<br />
!<br />
I<br />
4
93<br />
where the bar indicates values constant in time. Then indexing all quantities with<br />
a subscript 1 when atoms are not added and with N when they are added, we obtain<br />
where the last equality uses the results <strong>of</strong> Fig. 8. Hence 4 involves [N] to a<br />
higher power than does sN. Thus T~ cannot refer to the decay <strong>of</strong> [Nl. This is<br />
consistent with the usual slow decay <strong>of</strong> [N].<br />
Since 4 is essentially proportional to [NI2, these results imply that FN<br />
is proportional to [N]. If this additional production <strong>of</strong> N2(A3Z$) utilizes<br />
nitrogen atoms on a one-for-one basis, [N] must decrease in passing through the<br />
discharge, &,<br />
where At is an average residence time, where L[NlOut represents the rate <strong>of</strong> atom<br />
removal, and where mixing is rapid compared with reaction. This leads to<br />
or<br />
which is the general form observed.<br />
From Fig. 9 we can obtain L (using At %50 sec), and from Fig. 8 with a slope '<br />
<strong>of</strong> (KbPo[N2]/L), we can also obtain L if KT,P,[N~] can be found. A comparison<br />
<strong>of</strong> L derived from Fig. 9 and Fi . 8 is then possible. Since P = N2(A3L;fi>]/~,,<br />
absolute measurements <strong>of</strong> [N2(A3$)] derived from I(VK) = ( [N2(A3~)]/Tr) L where<br />
is the radiative lifetime <strong>of</strong> the excited state give T~P.<br />
T~<br />
The absolute intensity <strong>of</strong> the 0,5 Vegard-Kaplan band in the discharge bulb can<br />
, be roughly obtained by measuring its signal and calibrating the spectrometer response<br />
using the NO y (0,3) band excited by the chemiluminescent association <strong>of</strong> N and 0<br />
in conjunction with measurements <strong>of</strong> [N] and [O] and the known rate coefficient for<br />
I these pro~esses.~ In general, the comparisons <strong>of</strong> L derived from Figs. 8 and 9 show<br />
' that if nitrogen atoms are consumed in the excitation <strong>of</strong> N2(A3c), then several<br />
I hundred times more atoms need to disappear than in fact are destroyed. This implies<br />
I that nitrogen atoms are not consumed but catalyze the production <strong>of</strong> N2(A3~t) in<br />
conjunction with other species generated in the discharge.<br />
> A similar argument can be used to show that atomic nitrogen is not consumed in<br />
1 destroying N2(A3c). For example, there are <strong>of</strong>ten as many N~(A3zt)~ $olecules as<br />
\ N atoms; these would be seriously depleted during the decay <strong>of</strong> N2(A zU) and thus<br />
would lead to a nonexponential behavior.<br />
,<br />
i<br />
'1<br />
3 +<br />
The short lifetime <strong>of</strong> N2(A &,> in the discharge (Fig. 4) cannot be due to atoms<br />
because their concentration, as inferred from downstream chemiluminescence measurement,<br />
' is too small. Furthermore, the pressure dependence <strong>of</strong> T~ is incompatible with atom
94<br />
deactivation. Hence, some species is produced in the discharge in addition to atomic<br />
nitrogen which is capable <strong>of</strong> rapidly deactivating N2(A3X$). This species persists<br />
for times long compared to T . '1<br />
The results <strong>of</strong> these experiments indicate that 3everal unusual processes are<br />
occurring: (1) a high-order process destroying N2(A 1:) which involves atomic<br />
nitrogen; (2) a process utilizing species, created in a discharge other than atomic<br />
nigrogen, which removes N2(A3X$); and (3) a process involving atomic nitrogen and<br />
another species generated in the discharge which produces hi2 (A31i)'. These processes .<br />
are in addition to electron excitation and radiative destruction <strong>of</strong> N2(A3C$). Several<br />
eossible reaction schemes can be constructed which are consistent with our observations.<br />
However, without positive identification <strong>of</strong> all the species created in the discharge<br />
and the istermediaries necessarily involved in high-order processes, these reaction<br />
schemes are speculative. It seems best to postpone discussions <strong>of</strong> these until more<br />
experimental information is available.<br />
EXPERIMENTAL REACTIONS INVOLVING NO<br />
For experiments on the effect <strong>of</strong> NO on N2(A<br />
3 Zh), three photomultipliers were<br />
installed downstream from the RF excitation bulb to observe chemiluminescence. One<br />
photomultiplier had an interference filter centered on the strong bands <strong>of</strong> N2 at<br />
5800 A, a second (called the yellow detector) observed radiation at wave lengths<br />
longer than 5200 A, and a third (called the ultraviolet detector) observed emission<br />
between 3800 A and the pyrex cut<strong>of</strong>f. The first two photomultipliers are sensitive<br />
to the N2 first positive bands excited by recombination <strong>of</strong> atomic nitrogen and to<br />
the NO2 continuum excited by reactions <strong>of</strong> 0 with NO, while the ultraviolet<br />
photomultiplier is mainly sensitive to the NO, 6 bands excited in the association <strong>of</strong><br />
atomic nitrogen and oxygen.<br />
These photomultipliers were calibrated by generating<br />
nitrogen atoms in the microwave discharge and titrating them with NO. It should be<br />
remembered that all emission observed in the downstream bulb is <strong>of</strong> chemiluminescent<br />
origin.<br />
RESULTS<br />
Figure 10 shows the decay rate when the exciting discharge is turned <strong>of</strong>f from the<br />
0,3 NO y band, and the 0,5 Vegard-Kaplan band as a function <strong>of</strong> atomic nitrogen added<br />
by the upstream microwave discharge.<br />
Figures 11 and 12 show the intensity <strong>of</strong> the 0,5 Vegard-Kaplan band, the 0,3 NO y<br />
band and the 0,6 NO 6 band in the discharge bulb as a function <strong>of</strong> the NO concentration<br />
that would have existed in the stream if no destruction <strong>of</strong> NO occurred. Also shown<br />
are the responses <strong>of</strong> the yellow photomultipler and the ultraviolet photomultiplier,<br />
both <strong>of</strong> which are located at the downstream bulb.<br />
Spectrometer scans <strong>of</strong> the discharge without adding NO and on the plateau <strong>of</strong><br />
Figs. 11 and 12 are shown in Fig. 1.<br />
Figure 13 is a plot <strong>of</strong> the area intensity ratio <strong>of</strong> 0,3 NO y/0,5 Vegard-Kaplan<br />
bands as a function <strong>of</strong> NO added beyond the null and as a function <strong>of</strong> the relative<br />
response <strong>of</strong> the yellow phototube to the NO2 continuum. Figure 14 is similar to Fig-<br />
13 but involves the 1,lO Vegard-Kaplan band and the 3,O NO y band.<br />
DISCUSSION<br />
The simultaneous decay <strong>of</strong> N2(A<br />
3<br />
Xu)<br />
+<br />
and Iys the intensity <strong>of</strong> the NO y bands,<br />
suggests that NO is excited in the process5<br />
NO(X 2 ll) + N2(A32:) -f NO(A22+) + N2(X12) .<br />
1<br />
,<br />
i
'I<br />
i<br />
'I<br />
0 2. 4 6 8<br />
2<br />
[ N]* x' I 0-26 (otorns/crn3)<br />
"<br />
IO<br />
TB-- 45 2 5 22- 37<br />
Fig. 10 The dependence <strong>of</strong> the rate <strong>of</strong> decay <strong>of</strong> N2(A 3 xu) + and NO(A 24- I ) as a function<br />
<strong>of</strong> the square <strong>of</strong> the atomic nitrogen concentration.<br />
IO0<br />
80<br />
GO<br />
40<br />
20<br />
0<br />
0 IO 20 30 40 50 '60 70 80<br />
[NO] x IO-" (molecules/cm3)<br />
TO-452522-39<br />
11 The dependence <strong>of</strong> emission intensities on NO concentration: a- the NO B<br />
bands, A - the N2 first positive bands, 0 - the NO2 continuum -- all excited<br />
in the downstream observation bulb; 0 - the 0,3 NO y band, A - the 0,6 NO 6<br />
band, and 0- the 0,s Vegard-Kaplan band excited in the Tessla discharge<br />
bulb *
[NO] x IO-" (molecules/cm3) .<br />
18- 4 52 5 22-48<br />
. . .<br />
---<br />
Fig. 12 The dependence <strong>of</strong> emission intensities on NO concentration: m - the NO B<br />
bands, 0 - the NO2 continuum -- both excited in the downstream observation<br />
bulb; 0 - the 0,3 NO y band, A - the 0.6 NO f3 band; and 0- the 0,5<br />
Vegard-Kaplan band excited in the Tessla discharge bulb.<br />
[NO]x IO-" (rnolecules/cm3)<br />
20 22 24 26 28 30 32 34 36<br />
I500<br />
PRESSURE 1.8 mmHg<br />
".<br />
0 O-<br />
A 0<br />
" 5<br />
H<br />
2,<br />
Fig. 13<br />
0<br />
SLOPE = I. I 10-9~m3<br />
7 21 35 49 63 77 91 105 119<br />
RELATIVE RESPONSE YELLOW PHOTOTUBE (NOZcontinuum)<br />
-<br />
78-452522-36<br />
The relative photomultiplier response to the NO y (0,3) and N2(0,5) Vegard-<br />
Kaplan bands as a function <strong>of</strong> both [NO] and the response <strong>of</strong> the yellow<br />
photomultiplier.
Fig. 14<br />
IO<br />
8<br />
6<br />
4F<br />
[NO] x IO-" (mole c u I es /c m3)<br />
14 16 18 20 22 24 26<br />
I<br />
SLOPE = I x 10-11cni3<br />
0 IO 30 50 70 90 110 130<br />
RE L AT I V E RES PO N S E YE L LOW P H OTO T U B E ( N O2 con t i n u u m )<br />
T8-452522-51<br />
The relative photomultiplier response to the NO y (3,O) and N2(1,10)<br />
Vegard-Kaplan bands as a function <strong>of</strong> both [NO] and the response <strong>of</strong> the<br />
yellow photomultiplier.
O-‘<br />
The initial decrease <strong>of</strong> the signals from the yellow downstream photomultiplier<br />
is due to NO titration <strong>of</strong> nitrogen atoms produced in the exciting discharge, &,<br />
The null point represents the complete conversion <strong>of</strong> nitrogen atoms t;o oxygen atoms.<br />
The concentration <strong>of</strong> these atoms derived from the amount <strong>of</strong> NO added to reach this<br />
null is consistent with the intensity <strong>of</strong> the first positive bands before NO addition.<br />
The linear rise <strong>of</strong> the yellow sensitive photomultiplier after null is due to<br />
and indicates that 0 is essentially constant.<br />
NO + 0 -+ NO2 + hv (18)<br />
There is a slow increase <strong>of</strong> the NO y and f3 bands in the excitation bulb because<br />
<strong>of</strong> the slow increase in the actual steady-state concentration <strong>of</strong> NO which is main-<br />
tained by<br />
and reaction (16), a,<br />
N + 0 + M -+ NO + M<br />
NO+O+M+N02+M<br />
NO + 0 -+ NO + O2<br />
When [N] goes to zero, this steady state cannot be maintained, and NO in the stream<br />
increases as expected for no reaction <strong>of</strong> NO. In the reaction time available reaction<br />
(20) does not significantly alter either [O] or [NO].<br />
As [NO] increases sharply after the removal <strong>of</strong> N, the NO y and f3 bands increase<br />
sharply in the discharge and the N2 Vegard-Kaplan bands decrease rapidly. Since<br />
I Y = [NO(A2E+)]/r,<br />
for excitation in the discharge bulb, and since<br />
(22)
for chemiluminescent excitation in the downstream bulb, reaction (16) implies<br />
49<br />
3+<br />
I = kl6[NO1 [N2(A Zu) 1 = k16[NOl Ivk~vk =<br />
Y<br />
2 +<br />
if NO(A C ) is only deactivated by emission.<br />
Since 0 is constant,<br />
This is true, as Figs. 13 and 14 show.<br />
but relative.<br />
k161N02 'vkTvk<br />
In these graphs intensities are not absolute,<br />
To obtain k from Eq. (26), we must experimentally relate the emission<br />
intensities more agrectly to the concentration <strong>of</strong> the emitting state. If Iy(0,3)<br />
represents the absolute intensity <strong>of</strong> the 0,3 y band <strong>of</strong> NO and similarly for the<br />
Vegard-Kaplan bands, then Eq. (11) becomes<br />
where fy(0,3) is the fraction <strong>of</strong> all emission from the v' = 0 level which terminates<br />
on the v" = 3 level and similarly for f (0,5) and where T is the radiative lifetime<br />
<strong>of</strong> the A32+ state6 and 6 representsvhe fraction <strong>of</strong> allv% (A313 which are in<br />
v' = 0. Only vy = 1 and 0 are detected by their emission, an2 6 is found to be<br />
one half and constant. Then<br />
The parameters in parentheses depend only on the internal characteristics <strong>of</strong> the<br />
molecules and have previously been measured6 as<br />
fy(0,3) = 0.14. Hence<br />
T vk = 20 sec, fvk(0,5) = 0.17, and<br />
0.03 I (0,3)<br />
k16 = [NO]Iv:(0.5) '<br />
To obtain k , only the relative intensity <strong>of</strong> Iy(0,3) and IVk(O,5) are<br />
required. Since kkese bands occur very near each other in the spectra <strong>of</strong> the discharge,<br />
their relative intensity is equal to their relative signal level. Hence k16 is 0.03<br />
times the slope <strong>of</strong> the line in Fig. 13, &,<br />
k16 = 0.03 (1.1 x lo-') = 3 x cm3/sec . (31)<br />
7<br />
This rate is much larger than that reported by Dugan.
100<br />
The existence <strong>of</strong> the 6 bands and emission from higher vibrational levels <strong>of</strong> the<br />
NO(A2Z+) state indicate that this rate is a lower bound to the rate <strong>of</strong> energy transfer<br />
from N2(A3Z$) to NO(X2JI). There are several states <strong>of</strong> NO<br />
energy exchange involving N2(A3C+) -- the a4n, b4n, b4Z-,<br />
accessible in an<br />
as well as the A2Z+<br />
and B2n which are revealed by deir emission.<br />
The total rate <strong>of</strong> de-excitation <strong>of</strong><br />
3+<br />
N2(A Zu) can be estimated, since we know,<br />
in the absence <strong>of</strong> NO, its deactivation rate is 1/~ = 50/sec, and the intensit<br />
<strong>of</strong> the Vegard-Kaplan bands is reduced by half when [NO] % 7 x 10l1 &olecules/ cm 3 ,<br />
i.e., 50 = ki6(7 x 1011); in other words<br />
ki6 = 7 x cm3/sec<br />
3+<br />
or essentiallya quarter <strong>of</strong> all the N2(A 1 )<br />
U<br />
v' = 0 level <strong>of</strong> the NO(A2Z+).<br />
deactivated leads to excitation <strong>of</strong> the<br />
2<br />
It is apparent from the spectra that excitation <strong>of</strong> the A Z state <strong>of</strong> NO by<br />
Nz(A3Z:) decreases in efficiency as v' increases in the NO molecule, becoming<br />
very small for v' = 3 which is the highest level observed. This level can only be<br />
excited by Nz(A3Zt v = l), while the v' = 2, 1, and 0 levels <strong>of</strong> the NO(A2C+)<br />
state can be excited by<br />
the rate coefficient <strong>of</strong><br />
N2(A3C$ v = 0,l). Using the data in Fig. 14, we can obtain<br />
as<br />
1.<br />
2.<br />
3.<br />
4.<br />
5.<br />
6.<br />
7.<br />
1 2 +<br />
N2(A31: v' = 1) + NO(X2n) -+ N2(X Z) + NO(A Z v = 3)<br />
k (1,3) = 10-l~ cm3/sec. .<br />
16<br />
REFERENCES<br />
R. A. Young, Canad. J. Chem. 46, 1171 (1966). This paper contains additional<br />
references. K. Wray independently arrived at similar conclusions in J. Chem.<br />
Phys. 44, 623 (1966).<br />
Enhancetron Nuclear Data Inc.<br />
R. A. Young and G. Black, J. Chem. Phys. 44, 3741 (1966). This paper has references<br />
leading back to the description <strong>of</strong> most <strong>of</strong> the phenomenon associated with dis-<br />
charged nitrogen.<br />
R. A. Young and R. L. Sharpless, Discussions Faraday SOC. 33, 228 (1962).<br />
A. B. Collear and I. W. M. Smith, Trans. Faraday SO~. 6l, 2383 (1965). .,-<br />
W. R. Brennan, Studies in active nitrogen, Ph. D. Thesis, Dept. <strong>of</strong> Chemistry,<br />
Harvard University, Sept. 1964, and references quoted here; R. W. Nicholls,<br />
J. Res. Natl. Bureau Std. (U.S.) m, 451 (1961); R. A. Young and R. L. Sharpless,<br />
I<br />
J. Chem. Phys. 39, 1071 (1963); R. A. Young and R. L. Sharpless, J. Chem. Phys.<br />
- 39, 1971 (1963); R. A. Young and G. Black, J. Chem. Phys. (in press). These<br />
references contain an extensive review <strong>of</strong> the pertinent chemiluminescent reactions. !<br />
C. H. Dugan, An experimental study <strong>of</strong> metastable atoms and molecules, Thesis,<br />
Division <strong>of</strong> Engineering and Applied Physics, Harvard University, June 1963;<br />
C. H. Dugan and N. P. Carleton (in press).<br />
(33)<br />
I<br />
1
101<br />
Chemiluminescent Reactions <strong>of</strong> Excited Helium with' Nitrogen and Oxygen<br />
Mark Cher .<br />
North American Aviation Science Center, Thousand Oaks, California 91360<br />
Abstract<br />
Active sp,ecies created in a fast flov <strong>of</strong> helium by a micrdwave discharge<br />
react outside the $ischar e with nitrogen or oxygen'causing the emission <strong>of</strong><br />
9<br />
visible bands <strong>of</strong> N. and 0 . .<br />
The emission intensity <strong>of</strong> thesebands decays '<br />
exponentially with'distanze,' and the decay coefficient depends on both the<br />
flow rate <strong>of</strong> helium and the flow rate <strong>of</strong> nitrogen or oxygen. A t constant<br />
flow rate <strong>of</strong> helium the decay coefficient increases with increasing flow rate<br />
<strong>of</strong> added gas, but the dependence is less than linear. A theoretical analysis<br />
<strong>of</strong> the flow system predicts the exponential decay and a linear dependence<br />
between the decay coefficient and the flow rate <strong>of</strong> added gas. Comparison <strong>of</strong><br />
the experimental and theoretical results permits the calculation <strong>of</strong> approximate<br />
values <strong>of</strong> the rate constants for the reactions populating the emitting<br />
states and the diffusion coefficients <strong>of</strong> the excited helium species. A<br />
plausible explanation and a means for removing the discrepancy between the<br />
experimental and theoret.ica1 results are suggested.<br />
In t r oduc t i on<br />
Long lived reactive species, produced when helium gas is subjected to an<br />
electrical discharge, react with many other gases and generate characteristic<br />
emissions <strong>of</strong> visible light. The nature <strong>of</strong> some <strong>of</strong> these che i uminescent<br />
reactions'has been discussed recent.ly in a number <strong>of</strong> papers. e-3 The reaction<br />
with nitrogen gas produces+an intens2 !right blue flame consisting <strong>of</strong> the<br />
2 +<br />
first negative system. <strong>of</strong> N (B-C X C ). With oxygen a bright yellow-green<br />
flamt is obeerved due to tze ex!itati<strong>of</strong>i <strong>of</strong> both the+fisst neggtiv,e system <strong>of</strong><br />
Oi(b 2- -. a n ) and the second negativ,e system <strong>of</strong> 0 (A n -. X n 1. In this<br />
paper Be repoyt the results <strong>of</strong> our measurements <strong>of</strong> the sFecial gariation <strong>of</strong><br />
the emission intensity in a fast flow system for various flow conditions,and<br />
show how these measurements can be used to evaluate the rate constants for<br />
the reactions populating the emitting states.<br />
Apparatus. The reaction cell and associated equipment are shown schematically<br />
in Fig. 1. The reaction cell consisted <strong>of</strong> a pair <strong>of</strong> concentric<br />
pyrex tubes 13 and 25 mm 0.d. Helium flowed through the inner tube. The<br />
titrating gas was introduced into the helium stream via the outer tube<br />
through eight 1 mm diameter holes located sytmetrically in the wall <strong>of</strong> the<br />
inner tube. High velocity flows <strong>of</strong> about 10 cm/sec were maintained by a<br />
mechanical vacuum pump rated at 425 liters/sec. The flow rates <strong>of</strong> the gases<br />
were measured using a pair <strong>of</strong> calib ated critical velocity orifice flowmeters<br />
2<br />
described by Andersen and Friedman. The pressure in the reaction cell was<br />
taken as the average value measured by two oil manometers located approximately<br />
20 cm upstream and downstream from the flame zone. The temperature<br />
was determined in separate experiments by means <strong>of</strong> a fine wire thermocouple<br />
sealed from the outside with black wax. The observed temperature was determined<br />
primarily by the pressure <strong>of</strong> helium in the discharge, and was nearly<br />
independent <strong>of</strong> the nature and flow rate <strong>of</strong> the added gas.<br />
The discharge $n the helium stream was excited by a 2450 Mc/sec cavity<br />
<strong>of</strong> the Evcnson type, powered by a 125 W Raytheon diathermy unit. The cavity<br />
was located 85 mm upstream <strong>of</strong> the injection holes. A metal shield excluded<br />
the active discharge from extending into the flame zone.<br />
Spectra and intensity measurements were made with a 500 mm Jarrell-Ash<br />
Ebert spectrophotometer using an.RCA 1P21 photomultipl.ier tube. A pair.<strong>of</strong><br />
large condensing lenses served to focus the image <strong>of</strong> a cross-section <strong>of</strong> the<br />
flame on the entrance slit. The spectrophotometer was mounted on a table<br />
which could be moved at constant speed parallel to the axis <strong>of</strong> the tube. In<br />
this way the intensity <strong>of</strong>-the flame was recorded as a function <strong>of</strong> distance<br />
along the tube. i
1.8<br />
0.21<br />
I<br />
Titrotion with Np 01 3915A<br />
9.30<br />
MOlor diiven loble<br />
I I I I 1 I<br />
0 0.5 1.0 1.5 2.0 2.5 3.0<br />
F,,~ I io6 (rnoleslsec)<br />
5<br />
c<br />
1
103<br />
All gases were taken directly from the commercially available high pressure<br />
cylinders. The stated concentration <strong>of</strong> impurities in the helium tanks<br />
was 50 ppm, with N2 being the major contaminant.<br />
Interpretation <strong>of</strong> Data<br />
We assume that an excitTd heliyp) species X* reacts with N or O2 to give<br />
the electronically excited N or O2 , and the Latter immediately decays by<br />
emitting a quantum <strong>of</strong> light,2as indicated by reactions (1) and (2)<br />
8 +*<br />
X + N2 - N2 + x<br />
Na* - N+ + hv (2)<br />
2<br />
At this point the id ntity <strong>of</strong> X remains unspecified. X could be for<br />
example metastable 2 9 S helium atom, in whi$h case X represeqta a ground state<br />
He atom plus an electron.<br />
Alternatively X could be the He2 molecule, and X<br />
would then represent two ground state helium atoms. For simplicity we assume<br />
that under any one set <strong>of</strong> conditions only one excited helium species is<br />
dominant, although we can gxpect+geveral processes occurring simultaneously.<br />
The natural lifetime <strong>of</strong> N+ or 0 is short compared with the time scale <strong>of</strong><br />
the experiment, and thus ?he emigsion intensity is proportional to the rate<br />
<strong>of</strong> reaction (I). If the concentration <strong>of</strong> nitrogen is uniform and constant<br />
along the tube, which should be the case after some distance <strong>of</strong> travel to<br />
effect complete mixing, the decgy in intensity with distance should parallel<br />
the decay in concentration,<strong>of</strong> X . ne therefore need to derive an expression<br />
f,or the concentration <strong>of</strong> X as a function <strong>of</strong> position.<br />
Ue consider a semi-infinite cylinder <strong>of</strong> radius r . Let n be the concentration<br />
<strong>of</strong> X and N the concentration <strong>of</strong> the addadogas, say N2. We assume<br />
tbat the two major mechanisms responsible for the decay <strong>of</strong> n are diffusion <strong>of</strong><br />
X to the walls where n=O and the <strong>chemical</strong> reaction represented by Eq. (1).<br />
Under these conditions the rate <strong>of</strong> change <strong>of</strong> n is given by<br />
u = Dv2, - kNn (3)<br />
ax<br />
where u is the stream flow velocity, x is the axial coordinate, D is the dif-<br />
fusion coefficient <strong>of</strong> X , and k is the specific rate constant for reaction (1).<br />
Since transport along the axial direction by convcctio~~ is much greater than<br />
by diffusion, we need only consider radial diffusion, and for this case the<br />
solution <strong>of</strong> the axial part <strong>of</strong> Eq. (3) is<br />
A, the characteristic diffusion length given by A = r /2.405, is obtainedfrom<br />
the solution <strong>of</strong> the ra la1 part <strong>of</strong> Eq. (3) using the gssumed boundary condi-<br />
tion n=O at the walls. The radial dependence <strong>of</strong> n(x) is taken care by<br />
the experimental arrangement, since the photomultiplier tube views an entire<br />
cross-section <strong>of</strong> the flame, and its response is proportional to the average<br />
intensity. The flow velocity u and the concentration N are calculated by<br />
Eqm. (5) and (6)<br />
u = RTZF/pWro 2<br />
(5)<br />
N = (F,/IF)(p/RT) (6)<br />
where p is the pressure, T is the temperature, FN is the flow rate <strong>of</strong> the<br />
added gas in molee/sec, and ZF is the total flow rate, which in these experi-<br />
ments is essentially equal to-the flow rate <strong>of</strong> helium. If the emitted inten-<br />
aity I(x) is proportional to n(x), it follows that a plot <strong>of</strong> An I c. x should<br />
be a straight line with slope<br />
2<br />
(Dopa) (2.405)2n pr<br />
--zs din1<br />
dx = 760 XFRT + kW &&> FN (7)<br />
~ ~.<br />
10 Eq (7) we make use <strong>of</strong> the fact that D is inversely proportional to pressure<br />
and (D p ) is the diffusion coefficient at pressure 1 mm Hg. Using Eq (8) it<br />
0 0<br />
-
should be possible to evaluate k and D p from the variation <strong>of</strong> S with flow<br />
0 0<br />
rate <strong>of</strong> added gas.<br />
Results<br />
veasurements <strong>of</strong> intensity VS. distance were recorded for nitrogen at<br />
3915 A and for oxygen at 5587 G n d 4116 8. These wave length? are the heads<br />
<strong>of</strong> intense vibrational bands <strong>of</strong> $he first negative system <strong>of</strong> N and the first<br />
2 .<br />
and second negative systems <strong>of</strong> 02. Typical flames were 2-10 cm In length,and<br />
over this distance the intensity varied by nearly a factor <strong>of</strong> 100. Plots <strong>of</strong><br />
the logarithm <strong>of</strong> intensity x. distance demonstrated a linear bqhavior over a<br />
50 fold change in intensity, as predicted from Eq (7). Negative deviations<br />
from linearity occurred in the region within 0.5-1.0 cm from the injection<br />
ports, this no doubt resulting from incomplete mixing <strong>of</strong> t he reactive gases.<br />
The slopes <strong>of</strong> the linear portions were plotted against the flow rate <strong>of</strong> the<br />
added gas, and the results are shown in Figs. 2-4. It is evident that theae<br />
curves are not linear over the range <strong>of</strong> flow rates covered, as expected from<br />
Eq. 8, but rather show a pronounced downward curvature. We have provisionally<br />
taken the extrapolated intercepts and the initial slopes and used them to-<br />
gether with Eq. (7) to compute the reaction rgte constant as well as the<br />
diffusion coefficient <strong>of</strong> the excited species X . The results are shown in<br />
Table I. The magnitude <strong>of</strong> the rate constants fns the nitrpgen-fnd oxygen<br />
reactions are comparable and in the range <strong>of</strong> 10 cc mole sec . These rate<br />
constants are very fast indeed being <strong>of</strong> the order <strong>of</strong> the colli6ion frequency.<br />
The diffusion coefficients ?re <strong>of</strong> the right order <strong>of</strong> magnitude for any one <strong>of</strong><br />
the excited helium species, but the observed temperature dependence is<br />
clearly too steep.<br />
Discussion<br />
The procedure <strong>of</strong> using initial slopes to evaluate the rate constants in<br />
Table I is admittedly questionable in view <strong>of</strong> the fact that the curves in<br />
Figs. 2-4 do not fit well the model embodied in Eq. (8). Although the intensity<br />
<strong>of</strong> the flames does decay exponentially with distance, and the decay<br />
coefficient increases with increasing flow rate <strong>of</strong> added gas, the increase is<br />
not 1-inear. Deviations from linearity appear to be more pronounced at higher<br />
pressures (i.e. higher flow rates <strong>of</strong> helium), presumably because <strong>of</strong> slower<br />
mixing <strong>of</strong> the reactive streams. This suggests that incomplete nixing and<br />
non-uniform concentration <strong>of</strong> added gas may be responsible for the breakdown<br />
<strong>of</strong> the model, even though the method <strong>of</strong> injecting the second gas into the<br />
helium stream with an initial radial component <strong>of</strong> velocity should encourage<br />
fast mixing. At the lower flow rates <strong>of</strong> added gas the intensity <strong>of</strong> the flame<br />
decays less rapidly, and consequently intensity measurements are made farther<br />
away from the inlet <strong>of</strong> the second gas. Under these conditions we expect<br />
mixing to be mora nearly complete and this is in fact the rationale for using<br />
the initial part <strong>of</strong> the curves for evaluating the rate constants. Ye can<br />
check whether incomplete mixing is indeed the source <strong>of</strong> our difficulties by<br />
changing the size <strong>of</strong> the inlet holes and seeing whether the intensity pr<strong>of</strong>iles<br />
are affected. We are currently carrying out these experiaentm.<br />
we have not consideref so far the identity <strong>of</strong> the excited hflium specie8<br />
x Collins and Robertson have shown that the upper state <strong>of</strong> N giving ri e<br />
to the blye omission is populated by reaction <strong>of</strong> N with both ae$astable 2’s<br />
2<br />
He and He . +Similarly they have shown that the upper states <strong>of</strong> both band<br />
systems <strong>of</strong> Oz are populated by reaction <strong>of</strong> 0 with 2 3S He,+and in addition<br />
2<br />
the upper state <strong>of</strong> the 5587R system is also populated by HeZ. Difference8 in<br />
the O2 titration curves for the two band systems, paTticularly with regard to<br />
tho diffusion coefficients at high pressures when He2 is dominant, may result<br />
from the fact that different species give rise to the emission. More accurate<br />
measurements <strong>of</strong> the diffusion coefficient are necessary to resolve this point.<br />
There are very few measurements <strong>of</strong> the rate constants for th se reactions<br />
8<br />
with which we may compaft our resu ts. Pehsenfeld and co-worRers report a<br />
rate constant <strong>of</strong> 4 x 10 cc -1<br />
mole sec for reaction (8)
10;<br />
He: + N2 -. 2He + h': (8)<br />
whi!? Sholette and Muschlitz' iive values <strong>of</strong> 5-x 1013 and 11 x 1013 cc mole--'<br />
sec for the reactions (1) and (10)<br />
He(23S) + N2 - He + N+ + e-<br />
2<br />
He(Z3S) + O2 -.. He + 06 + e-<br />
(9)<br />
( 10)<br />
Our preliminary results are in quite reasonable agreement. 1<br />
References<br />
1. C. 8. Collins and W. W. Robertson, J. Chem. Phys. 2, 701 (1964); 40, 202<br />
(1964); lo, 2208 (1964).<br />
2. A. L. Schmcltekopf and H. P. Broida, J. Chem. Phys. 2, 1261 (1963).<br />
3.<br />
4.<br />
5.<br />
E. E. Ferguson, F. C. Fehsenfeld, P. D. Goldan, A. L. Schmeltekopf, and<br />
H. I. Schiff, Planetary Space Sci. 2, 823 (1965).<br />
J. W. Andersen and R. Friedman, Rev. Sci. Just. 2, 61 (1949).<br />
F. C. Fehsenfeld, K. M. Evenson, and H. P. Broida, Rev. Sci. Just. 96,<br />
294 (19651.<br />
6. E. W. HcDaniel, Collision Processes in Ionized Gases (John Wiley & Sons,<br />
Inc., New York, 1964), p. 503.<br />
7. Ibid, p. 516.<br />
8. F. C. Fehsenfeld, A. L. Schmeltekopf, P. D. Goldan, H. I. Schiff, and<br />
E. E. Ferguson, J. Chem. Phys. 5, 4087 (1966).<br />
9. W. P. Sholette and E. E. Huschlitz, J. Chem. Phys. 2, 3368 (1965).<br />
a.<br />
Run<br />
0<br />
Table I<br />
Titration with N2 at 3915 A<br />
4<br />
x 10<br />
FHc<br />
moles/sec<br />
P<br />
mm Hg<br />
T<br />
OK<br />
k x<br />
cc mole-lsec-l<br />
2<br />
(cm scc )(mm Hg)<br />
1 6.93 2.70 363 2.1 374<br />
2 9-30 3.46 376 2.0 480<br />
3 16.1 ' 5.41 413 2.8 696<br />
4 30.6 9.64 480 4.2 2140<br />
5 39.6 12.1 508 4.4 3310<br />
b. Titration with O2 at 5587 (First negative system <strong>of</strong> 0') 2<br />
6 7.94 2-99 370 6.7 40 3<br />
7 16.1 5-36 409 8.4 543<br />
8 30.8 9.58 475 9.0 1660<br />
9 45.6 13.8 515 8.3 4030<br />
c. Titration with O2 at 4118 (Second negative system <strong>of</strong> O*)<br />
2<br />
10<br />
11<br />
12<br />
13<br />
8.00<br />
16.1<br />
30.5<br />
45.5<br />
3.11<br />
5.40<br />
9.58<br />
14.0<br />
3 66<br />
418<br />
488<br />
5 26<br />
5.7<br />
5.3<br />
6.4<br />
5.1<br />
33 1<br />
643<br />
1020<br />
2140<br />
f
THE MATHEMATICS OF STEADY-STATE DIFFUSION AND FLOW TUBE SYSTEMS<br />
11. Measurement <strong>of</strong> Discharge-Zone Rate Parameters<br />
Peter R. Rony<br />
Central Research Department<br />
Monsanto Company<br />
St. Louis, Missouri '<br />
I. INTRODUCTION<br />
The techniques used to measure the rate <strong>of</strong> a <strong>chemical</strong> reaction or to<br />
determine the physical characteristics <strong>of</strong> a <strong>chemical</strong> compound are dictated by<br />
the physical and <strong>chemical</strong> properties <strong>of</strong> the compound, particularly by its lifetime<br />
and reactivity in relation to its <strong>chemical</strong> environment. This environment<br />
can be chosen to maximize the interactions between the compound and other<br />
materials (a reaction environment) or else to minimize such interactions (an<br />
isolation environment). Progress in chemistry has been dependent upon the<br />
skill chemists have shown in selecting the appropriate reaction or isolation<br />
environments.<br />
The problem <strong>of</strong> developing suitable isolation environments has been a<br />
difficult one in the study <strong>of</strong> flames, shock waves, explosions, and electrical<br />
<strong>discharges</strong>, where the <strong>chemical</strong> intermediates -- ions, electrons, atoms, free<br />
radicals, excited atoms, excited molecules, excited ions, and metastable mole-<br />
cules -- are highly reactive and have extremely short lifetimes. Notable<br />
advances along these lines have been the matrix isolation technique developed<br />
by Pimentel and co-workers, contacting the intermediates with a very cold<br />
surface, and the use <strong>of</strong> materials that deactivate surfaces against the recom-<br />
bination <strong>of</strong> atoms and free None <strong>of</strong> these techniques have yet<br />
been successful in significantly prolonging the lifetime <strong>of</strong> an ion.<br />
This problem can be circumvented by the expedient <strong>of</strong> placing a source <strong>of</strong><br />
the <strong>chemical</strong> intermediates in close proximity to a sink and optimizing the rates<br />
<strong>of</strong> loss and transport <strong>of</strong> the highly reactive species between these two regions.<br />
The most popular source-sink system for the study <strong>of</strong> gas-phase kinetics <strong>of</strong> neutral<br />
molecules is the diffusion or flow tube, which usually employs an electrodeless<br />
discharge as the source.s,io<br />
Such systems have also been used with considerable<br />
success in the fieId <strong>of</strong> plasma chemistry for the experimental measurement <strong>of</strong><br />
associative detachment and ion-neutral reaction rates.11,12<br />
The simplicity <strong>of</strong> these systems is deceptive -- they are usually quite<br />
complex. To illustrate this point, Table I gives a non-exhaustive list <strong>of</strong><br />
rate processes occurring within the system shown in Fig. l.24 It is clear that<br />
a comprehensive mathematical description <strong>of</strong> such systems is desirable not only<br />
to aid in the choice <strong>of</strong> experimental conditions and assessment <strong>of</strong> experimental<br />
data, but also to guide those who use diffusion or flow tubes or the measured<br />
rate parameters.<br />
Most theoretical descriptions <strong>of</strong> a diffusion or flow tube have ignored<br />
the source <strong>of</strong> reactive species -- the electrical discharge -- by the assumption<br />
<strong>of</strong> 'a specified value for the reactive specie concentration at the discharge-zone<br />
boundary.13-17 Tsu and Boudart were the first to inc<strong>of</strong>porate the discharge zone<br />
in the derivation and the author has elaborated upon this type <strong>of</strong> theoretical<br />
I<br />
,
description for both diffusion and flow<br />
108<br />
In the present paper, it is shown that these new equations provick the<br />
missing link between the theoretical expressions characterizing the elecwon and<br />
ion concentration distributions within a discharge zone and the expressions<br />
typically used for describing the gas-kinetic reactions <strong>of</strong> the discharge pro- /<br />
d u c t ~ . ~ % ~ - Suggestions ~ ~ , are given on how averaged first-order bte oonstants<br />
for the production <strong>of</strong> discharge products can be measured and how they can be used<br />
I<br />
to calculate the steady-state concentrations <strong>of</strong> neutral species at any point<br />
within the reaction tube. In certain cases, relatively modest equiptitent can be<br />
used to perform such measurements.<br />
I<br />
2<br />
The numbering <strong>of</strong> equations in the present paper continues a previous<br />
sequence. le<br />
11. RATE EXPRESSIONS<br />
A. Neutral Species<br />
Consider a binary system <strong>of</strong> atoms and the parent molecules pr-ent within<br />
a long cylindrical tube that has two zones -- a discharge zone and a reactor zone<br />
(Fig. 1). If the walls <strong>of</strong> the tube are not too active, if there are no threebody<br />
recombination reactions, and if the atom concentration is less than l’$, the<br />
mass- balance equations for the atoms become”<br />
- diffusion convection atom production first-order<br />
atom loss<br />
for the discharge zone and<br />
diffusion convection first-order<br />
atom loss<br />
for the reactor zone. These equations can be<br />
and<br />
rewritten in dimensionless form,<br />
In a previous paper, a total <strong>of</strong> 36 equations summarized the operation <strong>of</strong><br />
a flow OK diffusion tube, with or without a first-order atom loss process in the<br />
reactor zone, for three different discharge- zone boundary condidiona and three<br />
different end-plate boundary conditions.le The solutions to Eq. (7) were mb-<br />
I<br />
1<br />
,‘<br />
I<br />
1
divided into four c.ases:<br />
Case I. Diffusion tube (B = 0) with inactive reactor zone<br />
walls (1/p2 = 0);<br />
Case 11. Diffusion tube (p = 0) with active reactor-zone(<br />
walls (1/p2 p 0);<br />
Case ID, Flow tube (p # 0) with inactive reactor-zone Walls<br />
(1/p2 = 0); and<br />
Case IV. Flow tube (B # 0) with active reactor-zone walls<br />
(Up2 # 0).<br />
The eight most useful solutions are listed in Table 11. The most general<br />
solution for the reactor zone, from which the 35 others can be obtained by<br />
appropriate simplifications, is Eq. (60). The original paper should be consulted<br />
for further information concerning the derivation '<strong>of</strong> the mass-balance equations,<br />
the boundary conditions, or the other theoretical solutions. Definitions <strong>of</strong><br />
dimensionless groups are given in the Appendix.<br />
TABLE 11. Pertinent Solutions to Equation (7).<br />
L+M<br />
Discharge-zone boundary condition: = 0 at X = -<br />
dX R<br />
Case I. Case 11. Case 111. Case IV.<br />
= 0 at A = -m Eq. (27) Eq. (36) Eq. (27) Eq. (54)<br />
dJr = at X = 0 Eq. (33) Eq. (42) Eq. (51) Eq. (60)<br />
Jr = a262 (27)<br />
--
B. Ions and Electrons<br />
I<br />
Consider a ternary system <strong>of</strong> electrons and two different positive ions .<br />
present within the discharge zone shown in Fig. 1. If it is assumed that .<br />
Cl+
VI. I+<br />
= 2 [ 1 - p2 ]<br />
4<br />
111<br />
when: I. l/Si2 = 0; 11. the first root <strong>of</strong> Eq. (115) is dominant; 111. the first<br />
root <strong>of</strong> Eq. (Us) is dominant; IV. p = 0, T = 0, and I,(%) is large in Eq. (118);<br />
V. 3 = 0; VI. a21+ = 0 and l/6;2 = 0; and VII. p = 0 and 10(1/6$ is large<br />
3 F<br />
in Eq. (120). Carslaw and Jaeger should be consulted for other solutions to<br />
these types <strong>of</strong> equations.25<br />
An average value for the concentration <strong>of</strong> the minor ionic specie, q+,<br />
can be obtained by the following integration,<br />
dpdT<br />
Unfortunately, the integral jiIo(anp)dp cannot be evaluated explicitly.<br />
111. CONSEQUENCES<br />
A. Previous Results<br />
For the sake <strong>of</strong> brevity, a number <strong>of</strong> topics pertaining to the discharge<br />
zone given previously will not be repeated here. The reader is referred to the<br />
following sub-sections <strong>of</strong> Sec. VI in reference (19): A. Choice <strong>of</strong> Steady-State<br />
Systems; B. Complexity <strong>of</strong> Solutions; C. Velocity and Mass Flux; D. Residence<br />
Time; E. Radial Concentration Gradients; F. Comparison <strong>of</strong> Rate Processes; I.<br />
Back Diffusion; and M. Surface Reaction vs Atom Production.<br />
B. Measurement <strong>of</strong> @<br />
1. Comparison <strong>of</strong> Rate processes<br />
The behavior <strong>of</strong> atoms in the system shown in Fig. 1 is characterized<br />
by eight different rate processes: convection, radial diffusion to the walls,
'<br />
112<br />
axial diffusion through the discharge one, axial diffusion through the reactor<br />
zone, production, recombination on the end plate, recombination in the discharge<br />
zone, and recombination in the reactor zone. Thus, the interaction between the<br />
rate <strong>of</strong> production and the other rate processes can be conveniently characterized<br />
by seven dimensionless groups: 11<br />
-<br />
U2<br />
=<br />
RI n<br />
2-<br />
RIL -<br />
rate <strong>of</strong> atom production in the discharge zone = 'e2<br />
rate <strong>of</strong> radial diffusion to the wall D12<br />
rate <strong>of</strong> atom production in the discharge zone<br />
p M<br />
or &R<br />
rate <strong>of</strong> atom recombination within the discharge zone k: YVl<br />
rate <strong>of</strong> atom production in the discharge zone<br />
P !Go,%&!<br />
rate <strong>of</strong> atom recombination within the reactor zone kl yV1<br />
rate <strong>of</strong> atom production in the discharge zone - W R<br />
rate <strong>of</strong> atom recombination on the end plate - Y'F1<br />
rate <strong>of</strong> atom production in the discharge zone E k&<br />
rate <strong>of</strong> axial convection VZ<br />
rate <strong>of</strong> atom production in the discharge zone -kdRM<br />
rate <strong>of</strong> axial diffusion through the discharge zone D12<br />
rate <strong>of</strong> atom production .in the discharge zone *L I<br />
rate <strong>of</strong> axial diffusion through the reactor zone. D12<br />
These dimensionless groups can be further condensed to,yield just two<br />
groups relating the overall rates <strong>of</strong> atom production, transport, and loss:<br />
= rate <strong>of</strong> atom production = U'<br />
loss rate <strong>of</strong> atom loss 115' + 116Iz + l/p<br />
-<br />
( 124 I<br />
@loss - rate <strong>of</strong> atom loss - l/k' + 1/612 + 1/u2 r<br />
-<br />
Otransport<br />
- R/L + RIM + $ + 1<br />
In a steady-state experiment, there must be a competition between two<br />
rate processes -- a source and a sink. If the source and sink are spatially<br />
separated, then a transport process may further complicate the results. In<br />
the present case, the rate <strong>of</strong> atom production can be measured only if<br />
rod<br />
1, for if the source is much greater than the sink, no concentration<br />
@loss<br />
gradient can exist.<br />
2. Significance <strong>of</strong> k&<br />
For purposes <strong>of</strong> discussion, assume that the rate parameter Id represents<br />
the rate <strong>of</strong> production <strong>of</strong> atomic hydrogen(c;) via the following ion-electron<br />
recombination reaction,<br />
H$ + e' A H + H<br />
(125)<br />
!<br />
f
I<br />
I<br />
1<br />
I<br />
I<br />
I \<br />
Thus,<br />
kA(c - c;) = 2 = 2kr(H:)(e')<br />
defines the parameter k6. If atomic hydrogen is also produced when water is<br />
present via an ion-molecule reaction,<br />
H20+ + H2<br />
k; changes to<br />
kim<br />
> H,O+ + H<br />
It is clear from the above that kt, is a composite parameter which rarely<br />
can be subdivided into its component parameters from discharge product measurements<br />
alone. Furthermore, as shown in Sec. 11. B., the ion and electron concentrations<br />
are not uni'form throughout the discharge zone, so an average value,<br />
-1<br />
k,, is observed,<br />
Thus, the measurement <strong>of</strong> absolute yields <strong>of</strong> discharge products is not a<br />
fruitful technique for studying discharge-zone kinetics. An exception to this<br />
statement occurs when the discharge is initially well characterized and per-<br />
turbations are made on this initial state. For example, in the absence and pre-<br />
sence <strong>of</strong> water, the ratio <strong>of</strong> the above rate parameters becomes<br />
+<br />
The quantity kim(H20 )AV can be determined provided that the ratio <strong>of</strong><br />
is measurable and the other quantities are known.<br />
3. Interaction Between Discharge and Reactor Zones<br />
values<br />
The activity <strong>of</strong> the reactor-zone walls and the location <strong>of</strong> the catalytic<br />
end plate have a measurable influence upon the atom concentration within the<br />
discharge zone and at the discharge-zone boundary. This phenomenon can be<br />
used to advantage for the measurement <strong>of</strong> absolute or relative values <strong>of</strong> the<br />
rate constant <strong>of</strong> atom production k;.<br />
In the absence <strong>of</strong> reactor-zone sinks, Eqs. (27) and (45) apply,<br />
The atom.production rate constant k: can be determined from Eq. (130) provided<br />
that (a) the amount o f atom recombination on the discharge-zone walls is<br />
known, (b) it remains constant during the measurement process, and (c) it is<br />
the dominant atom loss process. . In an electrode discharge, metal sputtering on
the tube walls occurs continuously and the above conditions are not fulfilled.<br />
With other types <strong>of</strong> <strong>discharges</strong>, a period <strong>of</strong> wall aging may be required.<br />
If an end plate is present, the concentration <strong>of</strong> atoms at the discharge<br />
boundary (1 = L/R) is given by Eq. (33),<br />
When L/R + 5' >> M,<br />
then<br />
62R L<br />
then J, = a26' as previously. However, if 9<br />
>> E + e',<br />
The rate constant k& can be determined if the recombination coefficient <strong>of</strong> the<br />
end plate or the diffusion coefficient <strong>of</strong> the atoms is known.<br />
If only the reactor-zone walls are active, Eq. (36) applies at the<br />
discharge-zone boundary (z = 0),<br />
JI = a262 1 + C R<br />
UM<br />
(E - < 1/4) .<br />
The only new result occurs when ER >> 1, and leads to<br />
CIM<br />
which is no improvement over Eq. (132). Equations (42), (5l), (54), and (60)<br />
lead to progressively more complicated solutions.<br />
(131)<br />
(133)<br />
In the preceding paragraphs we have been preoccupied with the measurement<br />
<strong>of</strong> g. The equations derived above are equally applicable if k;, is already know<br />
and show how such data can be used to predict steady-state atom concentration<br />
levels in diffusion and flow tubes.<br />
As an example, consider the effect <strong>of</strong> an end plate on the atom concentration<br />
within the discharge zone. In the absence <strong>of</strong> the end plate, the concentration<br />
throughout the reaction tube is given by Eq. (130). When the end plate is<br />
present, either Eq. (33) OK Eq. (131) applies for the reactor zone and Eq. (135)<br />
applies for the discharge zone (z assumes negative values only between 0 and -MI.<br />
cosh (g)<br />
sinh M/6R<br />
M .<br />
6R<br />
+ 5' + 6 coth -<br />
With R = 2 cm, Di2 = 1.25 * lo4 cm2/sec (atomic hydrogen at 100 mTorr and 25'C),<br />
L = 10 cm, 51 = 2.5 * lo5 cm/sec (atomic hydrogen), M = 8 cm, vz = 0 cm/sec,<br />
(135)<br />
. ..
Ywalls;s lo-', y'end plate = and kh = 0.100 sec-', the average atom concentration<br />
in the discharge zone is reduced from 14% to 0.19%. It is assumed<br />
here that the end plate has no effect on the electron and ion concentration levels<br />
within the discharge.<br />
k. Mass-Flux Devices<br />
As shown above, the measurement <strong>of</strong> the absolute value <strong>of</strong> k,!, requires a<br />
knowledge <strong>of</strong> either 7, M and D12, or M and 7'. A simpler procedure applicable<br />
to the study <strong>of</strong> atoms requires the use <strong>of</strong> a mass-flux device such as an isothermal<br />
calorimeter, which measures the atom flux at a given point within the<br />
reactor zone. l9 Equation (131) becomes<br />
Jls (atoms/cm2 sec) = - D12 "I aZ<br />
=2+2&9L<br />
z=L =O<br />
62R<br />
= 0262<br />
CD~~I<br />
R<br />
Ti+!'+- L 6"R<br />
M<br />
cy2 %2<br />
R<br />
= k&c'<br />
when M >> [k + E) . Thus, the rate <strong>of</strong> atom production can be determined bv a<br />
mass- flux m\easur&nment provided that the value <strong>of</strong> M is known and the calori:<br />
meter is the dominant atom sink.<br />
5. Relative Measurements<br />
I<br />
(136)<br />
Provided that certain parameters remain constant and the appropriate<br />
inequality relationships hold, the measurement <strong>of</strong> relative values <strong>of</strong> k;, is the<br />
easiest measurement to perform in diffusion or flow tubes. The most important<br />
requirement is that
E2R<br />
M '<br />
$6"
117<br />
V. NOTATION<br />
Del operator (vector)<br />
Infinity<br />
Braces indicating molar concentration <strong>of</strong> enclosed species<br />
Total molar concentration <strong>of</strong> gaseous species<br />
I<br />
Molar concentration <strong>of</strong> atoms in reactor zone<br />
Molar concentration <strong>of</strong> atoms in discharge zone<br />
Molar concentration <strong>of</strong> minor positive ionic specie<br />
Molar concentration <strong>of</strong> major positive ionic specie<br />
Molar concentration <strong>of</strong> high-energy electrons<br />
Molar concentration <strong>of</strong> low-energy electrons<br />
Binary diffusion coefficient for atom-molecule system<br />
Ambipolar diffusion coefficient<br />
Exponential<br />
Represents quantity related to specie i<br />
Modified Bessel function<br />
Represents quantity related to specie j<br />
Molar flux <strong>of</strong> atoms to end plate<br />
First-order rate constant for loss <strong>of</strong> atoms in reactor zone<br />
First-order rate constant for loss <strong>of</strong>-atoms in discharge zone<br />
First-order rate constant for production <strong>of</strong> atoms in discharge zone<br />
Averaged first-order rate constant<br />
Second-order ionization rate constant<br />
Second-order ion-electron recombination rate constant<br />
Second-order ion-molecule reaction rate constant<br />
Length <strong>of</strong> reactor zone to end plate<br />
Length <strong>of</strong> discharge zone (or half the length for a symmetrical system)<br />
Radius <strong>of</strong> reaction tube<br />
Maass-average velocity in z direction (axial direction)<br />
Velocity <strong>of</strong> an atom<br />
Distance from discharge-reactor zone boundary to end plate (+ direction)
1.<br />
2.<br />
3.<br />
4.<br />
5.<br />
6.<br />
7.<br />
8.<br />
9-<br />
10.<br />
u.<br />
-<br />
12.<br />
13.<br />
14.<br />
15 *<br />
16.<br />
17 *<br />
18.<br />
19.<br />
20.<br />
21.<br />
22.<br />
23.<br />
24.<br />
25 *<br />
26.<br />
27<br />
28.<br />
29.<br />
30.<br />
118<br />
REFERENCES<br />
Whittle, E., DOWS, D. A., and Pimentel, G.C., J. Chem. Phys. 22, 1943 (1954).<br />
Norman, I., and Porter, G., Nature 174, 508 (1954).<br />
Pimentel, G. C., in Fdnnation and Trapping <strong>of</strong> Free Radicals, (Academic<br />
Press, New York, 1960), p. 69. I<br />
Rice, F. J., and Freamo, M. J., J. Am. Chem. SOC. 73, 5529 (1951).<br />
Rice, F. J., and Freamo, M. J., J. Am. Chem. SOC. 15, 548 (1953).<br />
Goldenberg, H. M., Kleppner, D., and Ramsey, N. F., Phys. Rev. a, 530 (1961).<br />
Berg, H. C., and Kleppner, D,, Rev. Sci. Instr. z, 248 (1962).<br />
Shaw, T. M., in Formation and Trapping <strong>of</strong> Free Radicals, (Academic Press,<br />
New York, 1960), p. 47.<br />
Dickens, P. G., Linnett, J, W., and Palczewska, W., J. Catalysis &, 140 (1965).<br />
Evenson, K. M., and Burch, D. S., J. Chem. Phys. 9, 2450 (1966).<br />
Fehsenfeld, F. C., Schmeltekopf, A. Lo, and Ferguson, E. E., J. Chem. Phys.<br />
44, 4537 (1966).<br />
Fehsenfeld, F. C., Ferguson, E. E., and Schmeltekopf, A. L., J. Chem. Phys.<br />
g, 1844 (1966).<br />
Wise, H., and Ablow, C. M., J. Chem. Phys. 2, 634 (1958).<br />
Wise, H., and Ablow, C. M., J. Chem. Phys. z, 10 (1963).<br />
Dickens, P. G., Sch<strong>of</strong>ield, D., and Walsh, J., Trans. Far. SOC. s, 1338 (1959).<br />
Schulz, W. R.,<br />
Morgan, J. E.,<br />
and Le Roy, D.<br />
and Schiff, H.<br />
J., Can. J. Chem. 40, 2413 (1962).<br />
I., J. Chem. Phys. 2, 2631 (1963).<br />
Tsu, K., and Boudart, M., Can. J. Chem. 2, 1239 (1961).<br />
Rony, P. R., "The Mathematics <strong>of</strong> Steady-State Diffusion and Flow Tubes.<br />
Factors Affecting Rate Measurements," 2nd Annual Chemical Engineering<br />
Symposium, Stanford, California, 1965. (Copies are available).<br />
Rony, P. R, I. & E. C. (to be submitted).<br />
Biondi, M. A., and Brown, S. C., Phys. Rev. a, 1700 (1949).<br />
Phelps, A. V., and Brown, S. C., Phys. Rev. 86, 102 (1952).<br />
Gray, E. P., and Kerr, D. E., Ann. Phys. lJ, 276 (1959).<br />
McDaniel, E. W., Col1isio.i Phenomena in Ionized Gases (John Wiley & Sons,<br />
Inc., New York, 1-<br />
Carslaw, H. S., and Jaeger, J. C., Conduction <strong>of</strong> Heat in Solids (Clarendon<br />
Press, Oxford, England, 1959), Section 8.3, p. 2l7.<br />
Jolly, W. L., "The Use <strong>of</strong> Electric Discharges in Chemical Synthesis,"<br />
Lawrence Radiation <strong>Laboratory</strong> ~~~~-9591<br />
(1960). (unpublished).<br />
Gould, R. F., Free Radicals in Inorganic Chemistry (her. Chem. SOC.,<br />
Washington, D. C., 1962).<br />
Kondrat'ev, V. N., Chemical Kinetics <strong>of</strong> Gas Reactions (Addison-Wesley<br />
Publishing Company, Ind., Reading, Massachusetts, 1$4), p. 467.<br />
Kana'an, A. S., and Margrave, J. L., Advances in Inorganic Chemistry and<br />
Radiochemistry 6, (Academic Press, New York, 1964), p. 143.<br />
Hurt, W. B., J. Chem. Phys. 9, 3439 (1966).
L - 2<br />
A = - R<br />
MI2 - L<br />
T = a+- R<br />
v R<br />
@=A<br />
D12<br />
a2<br />
D12<br />
119<br />
APPENDIX<br />
bt2 = 2D12(1<br />
RYTl y'22 (surface phase)<br />
p2- p<br />
klR2<br />
' d<br />
'I+ = c<br />
(gas phase)<br />
(gas phase)<br />
2k R<br />
y = &<br />
V1<br />
1 1<br />
= u2+- 6'2<br />
= n~(2n + 11<br />
an M
120<br />
. Dlschorge zone Reactor zone<br />
1: L Diffusion or<br />
flow tube<br />
Measurement<br />
' device<br />
I I<br />
I<br />
I<br />
I<br />
I<br />
Fig. 1. Schematic drawing <strong>of</strong> a typical "reaction tube," showing<br />
the location <strong>of</strong> the discharge zone, the reactor zone, the<br />
discharge-zone boundary (at A = L/R), and the end plate<br />
(at - 0).<br />
Table 1. 'Rate processes occurring within a typical reaction tube.24<br />
Ions and Electrons<br />
in Discharge Zone<br />
PRODUCTION: Ionization<br />
Photoionization<br />
Ion-molecule Reactions<br />
Electron Attachment<br />
Charge Transfer<br />
Metastable Atom Reactions<br />
Loss : Ion-electron Recombination<br />
Ion-ion Recombination<br />
Wall Recombination<br />
Loss to Measurement Device<br />
Electron Detachment<br />
TRANSPORT: Convection<br />
Diffusion<br />
Ambipolar Diffusion<br />
Drift<br />
Cyclotron Resonance<br />
X=O<br />
MU. 3 5708<br />
Neutral Species in<br />
Discharge and Reactor Zones<br />
Ion-electron Recombinat ion<br />
Ion- ion Recombinat ion<br />
Ion-molecule Reactions<br />
Electron Attachment<br />
Wall Recombination <strong>of</strong> Ions<br />
Gas-kinetic Reactions<br />
Recombination on Walls<br />
Recombination on End Plate<br />
Loss to Measurement Device<br />
Convect ion<br />
Diffusion
c<br />
i<br />
I<br />
I<br />
t<br />
\<br />
121<br />
I<br />
Radiation Chemistry and Electric Discharge Chemistry:<br />
Comparison and Contrast.<br />
Milton Burton and Koichi Funabashi<br />
Department <strong>of</strong> Chemistry and the Radiation <strong>Laboratory</strong>"<br />
University <strong>of</strong> Notre Dame, Notre Dame, Indiana 46556<br />
* The Radiation <strong>Laboratory</strong> <strong>of</strong> the University <strong>of</strong> Notre Dame is<br />
operated under contract with the U. S. Atomic Energy Commission.<br />
This is AEC document number COO-38-511.
122<br />
In radiation chemistry the customary situation is that a charged<br />
particle with energy in the MeV range is slowed down in matter and, in<br />
the course <strong>of</strong> such deceleration, excites some molecules and ionizes others.<br />
The ultimate fate <strong>of</strong> the initial charged particle is <strong>of</strong> little consequence<br />
compared to the many processes which it initiates but that initial particle<br />
is actually responsible for initial excitations and ionizations which amount<br />
to about one-quarter <strong>of</strong> the total number, <strong>of</strong> such processes. The corollary 1<br />
statement is that about three-quarters <strong>of</strong> all the effects observed are<br />
I<br />
attributable to the action <strong>of</strong> secondary (and, to a very small extent, tertiary) /<br />
charged particles. Roughly, these particles have initial energy, ti, averaging -,<br />
about 75 eV.<br />
The secondary charged particles can cause further ionizations (in<br />
addition to excitations) only so long as they have energy, L, in excess <strong>of</strong><br />
the ionization potential, I, <strong>of</strong> the species which they traverse. A convenient<br />
value for I is about 10 eV. When E becomes < I, the only processes which<br />
can occur significant for radiation chemistry are electronic excitations to<br />
singlet or triplet states. When 6 becomes < 5 eV the major portion <strong>of</strong><br />
the excitations are optically disallowed (i. e., mainly triplet). For radiation<br />
chemistry about ? x or 5% <strong>of</strong> all the phenomena are initiated by particles<br />
4 75<br />
with about that amount <strong>of</strong> energy.<br />
In electric discharge processes, irrespective <strong>of</strong> the voltage employed<br />
the electrons start with about zero energy and rarely attain an energy in<br />
excess <strong>of</strong> 5 eV and much more rarely in excess <strong>of</strong> the ionization potential.<br />
It has been one <strong>of</strong> the important problems <strong>of</strong> electric discharge chemistry<br />
to determine how molecules are excited to <strong>chemical</strong>ly active levels and<br />
how ionized species are produced. According to some theoretical views<br />
most <strong>of</strong> the electrons cause optically forbidden excitations on the first<br />
opportunity and it might appear that <strong>chemical</strong> effects simply could not be<br />
observed. Nevertheless, they are observed and the fact must be that a<br />
significant number <strong>of</strong> molecules are excited to sufficiently high states so<br />
that chemistry results. Furthermore, some <strong>of</strong> them, in number adequate<br />
for maintenance <strong>of</strong> the discharge, are excited up to ionization potentials.<br />
Thus, in this case, ~i is initially approximately zero and might appear not<br />
to exceed very low energies; e. g., E ~ , corresponding to the lowest triplet<br />
state. Yet, high excitations and ionizations do occur.<br />
Two views may be employed to account for this dilemma. The theory<br />
presented is oversimplified and electrons in significant quantity attain<br />
energies in excess <strong>of</strong> 7 eV. Alternatively, it was suggested initially by<br />
Magee and Burton' that electric discharge chemistry is characterized by<br />
a series <strong>of</strong> successive excitations by low-energy electrons in optically<br />
forbidden steps to higher and higher levels with ultimate attainment <strong>of</strong><br />
levels adequate for production <strong>of</strong> a <strong>chemical</strong> effect - or even <strong>of</strong> ionization.<br />
The role <strong>of</strong> thermal energy is somewhat different in the two fields.<br />
In discharge chemistry a local temperature may be raised as high as<br />
1000°C, whereas the usual dosage level used in the basic research <strong>of</strong><br />
radiation chemistry is so low that the temperature increase can be <strong>of</strong> the<br />
order <strong>of</strong> a few degrees C even if the entire energy input is converted to<br />
thermal energy. This difference is not a trivial one, because thermal<br />
2.<br />
I<br />
<<br />
I<br />
I<br />
l
123 3.<br />
energy and electronic.energy .(in t,he forms <strong>of</strong> excitation or ioniz,ation) do<br />
not exist as independent sourc-es <strong>of</strong> energy for, <strong>chemical</strong> reactions; the !<br />
,<strong>of</strong> energy is not a ,, .. .<br />
effect arising from a combination <strong>of</strong> the two.,forms<br />
simple sum <strong>of</strong> the two effects when each source acts alone ... $'he detail<br />
Of the interaction between thermal and electronic energies is not known.<br />
The qualitative picture, however, is fairly well understood. Namely, the<br />
rate <strong>of</strong> energy conversion from the electronic to the thermal form (radiationless<br />
transition or energy degradation) increases as the temperature<br />
increases. Thus, the process has the characteristics <strong>of</strong> an explosive<br />
chain reaction.<br />
The main reason for this difference is attributable to the difference<br />
in the spacial distribution <strong>of</strong> excited and ionized species initially formed<br />
in each field. In radiation chemistry the distribution is more or less<br />
uniform as a natural consequence <strong>of</strong> the high penetrating power .<strong>of</strong> the<br />
impinging radiation and highly random nature <strong>of</strong> the energy loss processes.<br />
The local inhomogeneities <strong>of</strong> energy deposition are submicroscopic in size<br />
and cannot be conveniently conceived as local regions <strong>of</strong> abnormal<br />
temperature. The "hot spot' li idea has little significance in modern<br />
theories <strong>of</strong> the usual radiation chemistry. On the other hand, in a<br />
discharge tube, electronic excitation and ionization are somewhat confined<br />
to certain parts <strong>of</strong> the tube (negative glow and positive column). Since<br />
electronic excitation processes are generally accompanied by vibrational<br />
excitation (because <strong>of</strong> the Franck-Condon.principle), the temperatures <strong>of</strong><br />
these regions are.also expected to be higher than those <strong>of</strong> the rest <strong>of</strong> the<br />
system. The direct transfer <strong>of</strong> electron energy to vibrational or<br />
translational motions <strong>of</strong> molecules can be ignored.<br />
On the other hand, the coexistence <strong>of</strong> high temperature and high<br />
density <strong>of</strong> excitation is not really unique to electric-discharge chemistry.<br />
Under extremely high dose rate with certain charged species (particularly<br />
heavy particles such as fission fragments), it is probable that a similar<br />
situation has significance in the radiation chemistry <strong>of</strong> unusual systems.<br />
A rather obvious feature <strong>of</strong> radiation chemistry, which cannot be<br />
found in discharge chemistry is the fact that the impinging energy is<br />
theoretically sufficient to excite the molecule to a level where multiplebond<br />
dissociation and multiple ionization are possible. These, effects<br />
have indeed been observed, but their contribution to the total <strong>chemical</strong><br />
effect <strong>of</strong> high-energy radiation turns out to be negligible. The role <strong>of</strong><br />
highly excited states or excited ionized states in the physical aspect <strong>of</strong><br />
radiation chemistry (energy or charge transfer processes) is far from<br />
being negligible. By contrast, it would appear that lower excited states<br />
alone are the initial products <strong>of</strong> excitation in electric-discharge chemistry.<br />
In the latter case higher states appear to result from successive excitation<br />
exclusively and, under certain conditions <strong>of</strong> <strong>chemical</strong> reactivity, any<br />
presumptive substantial role <strong>of</strong> such states in the chemistry <strong>of</strong> discharge<br />
processes can be completely excluded - at least, .on a speculative basis.<br />
Radiation chemistry may be studied in systems <strong>of</strong> any degree <strong>of</strong><br />
aggregation from highly attenuated matter (as in interstellar space) to<br />
liquids or solids under high compression. Studies in electric-discharge
124 4.<br />
chemistry seem limited essentially to gases or to interface.s involving<br />
gaseous systems. In radiation chemistry a major,question. is why, in<br />
spite <strong>of</strong> the initially high energies involved, the <strong>chemical</strong> effects are as<br />
specific as observed. In e1ectri.c-discharge chemistry, by contrast, a<br />
very important question is why <strong>chemical</strong> effects are at all observed and<br />
why they are observed in the high yields reported.<br />
1.<br />
Refe rence s<br />
Milton Burton and John L. Magee, J. Chem. 'Phys., 23, 2194 (1955).<br />
2. F. Dessauer, 2. Physik, 20, 288 (1923).<br />
'<br />
. .I
Inorganic Synthesis with Electric Discharges<br />
William 'L . Jolly I<br />
Department <strong>of</strong> Chemistry <strong>of</strong> the<br />
University <strong>of</strong> California and the<br />
Inorganic Materials Research Division <strong>of</strong> the<br />
Lawrence Radiation <strong>Laboratory</strong><br />
Berkeley, California 94720<br />
Electric discharge reactions which yield thermodynamically<br />
unstable products are <strong>of</strong> interest to synthetic chemists because such<br />
products are <strong>of</strong>ten difficult to prepare by other methods. Many<br />
fascinating compounds <strong>of</strong> unusual structure have been isolated from<br />
discharge reactions. Although such syntheses usually have low<br />
efficiencies, the general techniques are nevertheless <strong>of</strong> interest to<br />
chemists who hope to discover new types <strong>of</strong> compounds.<br />
In this paper,<br />
we shall consider some important types <strong>of</strong> reactions which can be<br />
effected with the aid <strong>of</strong> electric <strong>discharges</strong>, together with some<br />
recent examples <strong>of</strong> these reactions.<br />
Conversion <strong>of</strong> Simple Molecules into<br />
Higher Homologs<br />
When simple volatile hydrides are passed through an ozonizertype<br />
discharge tube near atmospheric pressure or through a glow<br />
discharge at low pressure, fairly good yields are obtained <strong>of</strong> the<br />
higher homologs <strong>of</strong> the hydrides. For example, both silane and germane<br />
can be converted to their respective higher hydrides; silanes as high<br />
as SieHle and germanes as high as Ge9Hzo are formed. Similarly,<br />
arsine may be converted to diarsine. A wide variety <strong>of</strong> boron hydrides,<br />
including BgH15, B1&16, B2$16, B&12 and B&2, has been prepared<br />
in electric <strong>discharges</strong>.<br />
In all these reactions, hydrogen is a by-<br />
product, and there is no tendency for back-reaction <strong>of</strong> the hydrogen<br />
with the product molecules.<br />
When the volatile halides, SiC14, GeC14, and BC13, are passed<br />
through a glow discharge at low pressure, small yields <strong>of</strong> the higher<br />
homologs, Si2Cl6, GenCl6 and B2C14, are obtained if provision is made<br />
to separate the products from the by-product chlorine by fractional<br />
condensation. (Chlorine reacts readily with the product molecules<br />
to reform the starting materials.)<br />
improved if a reducing agent which can react with chlorine is included<br />
in the discharge zone or immediately after the discharge zone.<br />
Mercury and copper wool have been found to be very effective reducing<br />
agents for this purpose.<br />
However, the yields are much<br />
In fact, P2C14 can be obtained from a<br />
discharge in H2 + Pcl3 or from a Pc13 discharge followed by copper<br />
wool, whereas no P2C14 has been obtained in the absence <strong>of</strong> the<br />
reducing agents.
126<br />
Conversion <strong>of</strong> Mixtures into<br />
More Complicated Molecules<br />
mien mixtures <strong>of</strong> relatively simple hydrides are passeq through<br />
an electric discharge, higher molecular weight ternary hydrides are<br />
formed. Thus by using appropriate mixtures <strong>of</strong> SiH4, GeH4, PH3, and<br />
AsH3, it has been possible to prepare, among other things, SiH3GeH3,<br />
SiH3PH2, SiHdsH2, GeHsPH2, and GeHdsH2. By using a silent electric<br />
discharge, which does not cause drastic fragmentation and rearrange-<br />
ment, it is possible, within limits, to tailor-make molecules. Thus<br />
a mixture <strong>of</strong> SiH3PH2 and SiH4 yields (SiH3)2PH, and a mixture <strong>of</strong><br />
Si2E6 and Ph3 yields SizH5PH2.<br />
Passage <strong>of</strong> diborane-acetylene mixtures through an electric<br />
discharge yields carboranes as the major volatile products, including :<br />
1, 5-C2B3H5, 1,6-C2B4H6, 2, ~ - C E B ~ and H ~ at ~ least six B-methylated<br />
derivatives <strong>of</strong> these plus C,3-(CH3)2-1,2-C2B3H3. The reaction <strong>of</strong><br />
SiH, and (CH3)zO in a silent electric discharge yields mainly a<br />
series <strong>of</strong> inseparable mixtures, but a fair yield <strong>of</strong> SizHsCH3 can be<br />
isolated.<br />
A fascinating series <strong>of</strong> oxygen fluorides (02F2, 032, 04F2,<br />
O5F2 and 06F.7) has been prepared by subjecting mixtures <strong>of</strong> oxygen<br />
and fluorine to electric discharge at very low temperatures. All<br />
these compounds are very unstable, and, on warming, they decompose<br />
to oxygen and fluorine. Nitrogen trifluoride, AsF5, and F2 when<br />
subjected to a glow discharge at low temperatures yields NF4AsF6.<br />
This material is a relatively stable material which is fairly well<br />
characterized.<br />
The only reported method for the preparation <strong>of</strong> a xenon compound<br />
that does not involve the use <strong>of</strong> elementary fluorine or some<br />
fluorinating agent almost as difficult to handle as fluorine, is<br />
the electric discharge synthesis <strong>of</strong> XeF2 from Xe and CF4. The<br />
carbon-containing by-products <strong>of</strong> the reaction were not identified.<br />
Controlled Reactions <strong>of</strong> Atoms and Radicals<br />
with Other Species '<br />
In the discharge methods discussed above, all the reactant<br />
species are passed through an electric discharge. However many<br />
synthetically useful reactions can be carried out in the absence <strong>of</strong><br />
a discharge by allowing a stream <strong>of</strong> atoms or radicals (prepared in<br />
an electric discharge) to impinge on various compounds. For example,<br />
atomic hydrogen is a very reactive species which has been used for<br />
the preparation <strong>of</strong> hydrides and for carrying out reductions.<br />
promising field <strong>of</strong> study is the reaction <strong>of</strong> atomic hydrogen with<br />
aqueous solutions. In basic solutions, the hydrogen atoms yield<br />
electrons, and the method furnishes a convenient method for studying<br />
the reducing effects <strong>of</strong> the aqueous electron.<br />
A<br />
I
i<br />
\<br />
’.<br />
I<br />
i<br />
t<br />
127<br />
Atoms arid radicals act as electrophilic reagents. The atoms<br />
0, C1, Br, I, and N favor attack at polarizable donor atorps. Thus<br />
atomic nitrogen reacts with divalent sulfur compounds (HzS, Cs2,<br />
OCS, Sa, S2Cl2 and SC12) to yield sulfur-nitrogen compounds, whereas<br />
no sulfur-nitrogen compounds are formed in the reaction <strong>of</strong> N atoms<br />
with SOe, SOC12, and SO>. The reaction with s2C12 gives fair yields<br />
<strong>of</strong> the interesting molecule NSC1.<br />
Equipment<br />
Most <strong>of</strong> the readily-available laboratory equipment for producing<br />
electric <strong>discharges</strong> is suitable only for relatively small-scale<br />
operation, involving only a gram or two <strong>of</strong> material. Such equipment,<br />
and the corresponding low yields, is satisfactory for the preparation<br />
<strong>of</strong> new compounds in amounts satisfactory for identification purposes.<br />
But in order for electric discharge syntheses to be useful for the<br />
preparation <strong>of</strong> mole quantities <strong>of</strong> materials, as are the usual synthetic<br />
methods, it will be necessary to have a marked break-through in the<br />
development and marketing <strong>of</strong> these instruments.
Hydrazine Synthesis in A Silent dlectrical<br />
Dischame.<br />
Thornton J.D., Charlton W.D., SPeddina; P.L.<br />
Department <strong>of</strong> Chemical bngineering, ,<br />
University <strong>of</strong> Newcastle-upon-Tyne, Merz<br />
Court, Claremont Road, Newcastle-upon-<br />
Tyne 2. dngland.<br />
IIU!HODlr CT ION.<br />
The eynthesis <strong>of</strong> hydrazine from ammonia ursing the<br />
eilent electric discharge was firet demonstrated by Beeson<br />
in 1911 (1). Subsequent investigation ehowed that both<br />
the electrical energy yield and percentage conversion<br />
obtained were very low (2,3) and interest in trie procese<br />
waned. Further work was introduced in the early 1950's<br />
enabling substantial improveuents in yields to be obtained<br />
in certain circumstaxcea and a better understanding <strong>of</strong> the<br />
kinetic mechanisms in the discnarge to emera. Devine and<br />
Burton (4) showed that significant hydrazine fornation only<br />
took place in the positive column <strong>of</strong> a D.C. discharge. \<br />
Moreover, they found that yields could be substantially<br />
increased if tne atomic hydrogen concentration in the dis-<br />
charge could be reduced by recombination. These general<br />
observations were later confirmed b: Hathsack (5). As a<br />
reeult mechanisms were proposed for hydrazine syntheeie baaed<br />
I on the underlying premise that the hydrazine was fket<br />
formed in the discharge and then degraded by back reactions.<br />
kchi (6) enowed that yielde could be increased by reducing<br />
the residence time <strong>of</strong> the hydrazine in the diechargo in<br />
agreement with the general premise <strong>of</strong> hydr85ine dyradation<br />
in the discharge. Subeequent work nas not been at variance<br />
with thie finding (7,83.<br />
Recently, I.C.I. <strong>of</strong> the U.K. have reported (9) that<br />
removal <strong>of</strong> product hydrazine in a liquid absorbent give0 a<br />
subetantial increase in yield and an improvement In p ep<br />
centage conversion. Thia ie, <strong>of</strong> cour~e, a modification <strong>of</strong><br />
the general premise <strong>of</strong> hydrazine degraation in the diecharge<br />
proposed by Devine and Burton (4) and Oucbi (6). Furthermore,<br />
following on the pioneering work <strong>of</strong> Ouchi (61, and other.<br />
(lo), I.C.I. apparently were able to achieve even better<br />
yields by euitable modification <strong>of</strong> the waveform characterietice<br />
<strong>of</strong> the discharge. In order to confirm their clair<br />
and to help clarify the mechanisms taking place in the diecharge,<br />
work wae commenced on this eyetem at the Univereity<br />
<strong>of</strong> Newcastle-upon-Tyne in 1965. Thie ie a prelimiPary<br />
statement <strong>of</strong> the reaulte obtained to date.<br />
BXPrnIrt&rn&.<br />
The main ah <strong>of</strong> thie work was to attempt to hCI=eaEe<br />
hydrazine yielde by reducing the residence tine <strong>of</strong> the<br />
product in the discharge. A concentric barrier dimcharge<br />
reactor was employed with and without trie uee <strong>of</strong> a liquid<br />
abeorbent. Reactant flow rate wae increaeed UP to the
+xiip.ni +xmi.ng ca iscity <strong>of</strong> trie appai,atus after W-LiCii, tile<br />
Ti. ?.I.. e b::s reducer:,'.p;- chaibing the electrode area<br />
r .to, give a .f.urther-.reauc!,ion in product residence<br />
t.irJiei. %'i+ly a d.C. '. paral'lel electroae react& was we'd '.<br />
in ,:w,yicn t.le discharge waveform characteristicg;,werd<br />
altered'. so that only a s:iort activating pulse.; was 'supplidd<br />
, to 'the reactarit in tne eiectrode gap as it passed throqh;,<br />
.. . the ., reaat,or.<br />
,in .a:<br />
flowing gas train. Commercially 'pure amon5d.uas fed ihto<br />
the raeasurinf. section <strong>of</strong> the ap::aratus via a'reduction<br />
valve, &d- a reigxlating needle .vzlve. The flow rjte: was '.<br />
measured on a rotaJ:ieter wriich had been previsusly calibrated<br />
under operating coilditions by us- a soap film manometer.<br />
Gas te:.lperatures an6. 1.ressures.. also were uie:?.sL.red before the<br />
dischar,,;e reactor. The hycirazine formed in t iie discharge<br />
was absorbed in ethylerie glycol either in situ or in a<br />
separate absorrition trLin. Hydrazine was determined using<br />
the spectrophoiietric method <strong>of</strong>' W a t t ' and 'cLr?sp (11).<br />
Vacuum control. 'was acuie'ved by a Cartesian manostat located<br />
..<br />
before the 'vacuuh-.puunp.-<br />
The A.C. radio frequency power (1.2meg c/s) to-the<br />
A- and B-type reactors was supplied by a modified C-12<br />
Radyne generator -<strong>of</strong> law rated output. 3easurement <strong>of</strong> the<br />
power dissipat,e.d.. in the. aischarge was acnieved by firstly<br />
'.determining t.h,e, power factor directly. on%uitable osc.il10-<br />
'sc3pe. This value in conjunction with the direct<br />
-readings <strong>of</strong> an X.iG.S. voltmeter (Airmec 314) and a radio '<br />
. frequency ammeter- (.Cambriuge , Unipivot ) . enabled a reasonably<br />
accurate .d,etermination <strong>of</strong> the power in the discharge .to be<br />
-achieved. The D.C. power for the C-type reactor was<br />
provided by a specially engineered 8KW generator. Measure- .<br />
:merit <strong>of</strong> the'actual discharge power was made using a<br />
,combination <strong>of</strong> an oscilloscope trace and the appropriate<br />
meter .r.eadinga.' , . . ,<br />
The vaxioue types <strong>of</strong> diqcharke reactors used 'in this<br />
work are illustrated schen&%ically in Fig.1. Other essent.ia1<br />
geopetric details <strong>of</strong> these reactors and their operating data<br />
are given In Table 1. The A-type seriqs <strong>of</strong> reactors<br />
consisted <strong>of</strong> a precision silica tube (which acted as the<br />
capacitive barrier) with the high tension electrode attached.<br />
wound the outeide. The inper electrode was a spinning<br />
cylinder so constructed that ,absorbent liqukd could be<br />
eprayed onto the ineide <strong>of</strong> the tube to flow-down through the<br />
m,ular diecharge gap. Co-current gas and liquid flow was<br />
'. 'reactor 'set brtueen measur mg and analysing sec%,i.ons<br />
: , -;'<br />
\ ..<br />
The apparatus conshod esse:;tially <strong>of</strong> t-b discharge , : '<br />
employed; : . .<br />
The B-type eerie6 <strong>of</strong> reactors were <strong>of</strong> -similar. conetruction<br />
except tllat a central wire electrode vae used and the<br />
absorbent liquid was fed into the incornin@; gas a8 a epray.<br />
Dieperpion <strong>of</strong>, the liquid vas, achieyed ylt,raeonically using a<br />
vibratory gemrator. The apray was fed into fhe kae atream<br />
through on annular orifice placed at a euffieient dietanbe<br />
, ,, _. ..<br />
,. . '
{;I .<br />
-<br />
. .. . - .., . . .. _- .. ..., ,, : .......- . .:,.: - . , : ,,; - . .. . :;:.:.:.. .:..:.:. ._.__ . ..... .- .:.. ... - :: .<br />
Flg. 1 kh.motlc dlqmm <strong>of</strong> Factors urd for hyamln<br />
E = E1octrod.r L=UqJd Inlet Q--QI Rorr<br />
S-Sllko Borrlw U=UltramlC Vlbmta<br />
4 6 0 10 12 14 16 18 20 22 24 26 28<br />
Pcwu h i t y KW /ccx103<br />
FI) 2 Hydrotln. y W tor th. cwx~ol rmctor A1 wlth L without WA<br />
!
I<br />
I<br />
t<br />
I,<br />
'\<br />
I<br />
'a<br />
downstream from the discharge to ensure proper dispersal <strong>of</strong><br />
spray in the electrode gap. The C-type reactor employed a<br />
pair <strong>of</strong> rectwar electrodes so set in the gas flow as to<br />
avoid reactant by-passing. "ne use <strong>of</strong> D.C. power,<br />
necessitated stabilization <strong>of</strong> the discnarge by means <strong>of</strong> a<br />
resistive load in the electrical circuit. Provision was<br />
made to admit liquid spray into the inlet gas stream using<br />
the same aerosol generator used in tile type-B reactors.<br />
Table 1.<br />
Reactor units employed.<br />
Type<br />
Reactor Flow Area Discharge Discharge Uilectic<br />
CoGe cm 2<br />
I idth rjarr ie r<br />
cm c1n cm<br />
Tubular 81<br />
Centrifwal A2<br />
Fi3.m ileactor A3<br />
A4<br />
2.203<br />
II<br />
II<br />
n<br />
1.270<br />
0.254<br />
0.127<br />
0.040<br />
2.798<br />
0.560<br />
0.280<br />
0.089<br />
0.15875<br />
II<br />
II<br />
II<br />
rubular<br />
Spray Reactor<br />
B1<br />
62<br />
1.089<br />
II<br />
la270<br />
0.254<br />
1.383<br />
0.277<br />
It<br />
II<br />
Used<br />
Reactor<br />
c1 1.4U 0- 635 0.896 0<br />
"O1Y "hicme ss<br />
gmJLTs:.<br />
Hydrazine yields for the various reactor geometries and<br />
the operating variables employed are given in Figs. 2 tm 6.<br />
From the data presented in rig 2 to 4, it is evident that the<br />
yield varies inversely as an exponential function <strong>of</strong> the<br />
*lower density at pressures under l0Umm <strong>of</strong> mercury. The effect<br />
<strong>of</strong> pressure also follows a Regative exponentid variation<br />
therefore a general equation <strong>of</strong> the form<br />
Y = a exp (-W-cv)<br />
adequately describes the results. Of course this correlation<br />
<strong>of</strong> the results does not in any sense give a complete<br />
description <strong>of</strong> the underlying pnysical chemistry involved in<br />
the synthesis. Ideally,it would have been preferable to<br />
develop separate rate equations in terms <strong>of</strong> the partial<br />
pressures <strong>of</strong> tne various components in the overall hydrazine<br />
1 synthesis. This would have necessitated a complete product<br />
analysis and since,in the present case,these data are lacking<br />
the phenomenological approach had to be used. The constants<br />
the equation for tne various reactors acd operating<br />
conditions used were evaluated using the method <strong>of</strong> least<br />
squares (12). These are tabulated in Table 2. It should be<br />
noted in this Table tnat certLin pressure exponential values<br />
were bracketed. here the relevant data were lacking for<br />
c&cdat&n and ,therefore, the dependence <strong>of</strong> energy yield upon<br />
preesurefdeactors A2 to A4 and B2 were assumed to be the<br />
eane RS those found experimentally for Reactors Al and B1.
1<br />
\<br />
\<br />
\<br />
,<br />
\ ,<br />
\<br />
\<br />
Pmer hnrity K.W. /ccxiD)
. a b c liqqid<br />
*iea.ctor L.!n/iLWH ~3a-l c c/Kwat t absorbent<br />
Al .l3.14 O.OUB7 73.81 firm<br />
81 6.33 3.02j6 118.74 none<br />
A2 10.70 (O.OU87) 19.43 f iliil<br />
I1<br />
I1<br />
A3 9.56<br />
12-93<br />
11<br />
A4 7.28 3.73<br />
II<br />
B1 12.99 0*0098 73.58 spray<br />
32 9.26 (0.0098) . 13.69 II<br />
B2 8.88 (U.0236) 35.15 none<br />
This is a reasoliable procedure within any one<br />
1,erticular series <strong>of</strong> reactors such as A1 to 4, but it is<br />
OiQY Eiproximate witii iiiffering reactor series( e.g. Reactor<br />
series A and B)due to dissimilarities in discharge geometries.<br />
JJi,c;uSSI #If.<br />
The i?ost notable overall feature <strong>of</strong> the results is the<br />
.qrogresuive increase in hydrazine yield obteined by tite use<br />
<strong>of</strong> a liqu-id absorbent and by pihing tne discharge. The<br />
himest averase. jield was obtained wit!i tile pulsing<br />
technique with or without liqcdd absorbent aid was approxinietely<br />
15 gms if H /KMH. Isolated values as nigi? as 20 @as/<br />
W h were obtaine8 %ut further experimental work is required<br />
before these values can be made reproducible. It is<br />
noteworthy that a yield <strong>of</strong> 15 pis/KMH is beginning to look<br />
comercially attractive although it must be remenbered that<br />
the fiiL;,.l. product cost will depend to a great extent on the<br />
cost <strong>of</strong> the product purification process downstream <strong>of</strong> the .<br />
reactor.<br />
There are several interesting features shown by this<br />
work which bear discussion. From the results set out in<br />
Fig. 2 to 4 it is evident that the hydrazine yield decreases<br />
with increasing discharge power intensity despite a corresponding<br />
increase in the decomposition <strong>of</strong> ammonia. The only<br />
reasonable explanation for this which 118s been advanced is<br />
that hydrazine is formed in the discharge bj a complex<br />
reaction mechanism and is subsequently decomposed by electron<br />
bombardment or other collision phenomena (4,6 ) .<br />
The use <strong>of</strong> a liquid absorbent resdts in a decrease in<br />
the slope <strong>of</strong> the energy yield-power density plot. This is<br />
well illustrated in both Pig. 2 and Fig.4 by comparing the<br />
slope <strong>of</strong> the plots with and drithout the use <strong>of</strong> liquid absorbent.<br />
This indicates that hydrazine is being reinoved from the<br />
diecharge by the liquid absorbent instead <strong>of</strong> being degraded.<br />
If the hydrazine were completely removed so that degradation<br />
did not occur the elope <strong>of</strong> this plot would be wither<br />
independent <strong>of</strong> discharge power or possibly positive. The very
.<br />
I operating<br />
fact that the sloGe is still negetive with tne use <strong>of</strong> liquid<br />
absor.bent nay indicate that a sitnif icant amount <strong>of</strong> hydrazine<br />
is &till being degraded in the discharge in this case.<br />
Therefore, tiie use <strong>of</strong> a more efficient absorptlon process would<br />
reasonably be expected to reco.i;er more <strong>of</strong> the hydragice being<br />
degraded and thus increase yields still further. IuIoreover, the<br />
observed uecreasing effect <strong>of</strong> pressure on the yield with the<br />
iise <strong>of</strong> liquid absorbent (shown in E'ig.2)adds weight to this<br />
sv.gge s t ion.<br />
As tile 3ressur.e was decreased beloit 1001!m <strong>of</strong> mercury the<br />
yield <strong>of</strong> hydrazine increased steadily while the s!.ope <strong>of</strong> -$he<br />
,;;-ield-power de:>.sity curve remained co:istaiit for similar reactor<br />
coiiaii;ions such as tiie use <strong>of</strong> a liquid absorbent.<br />
A reasonable Pxplaiation for this beliaviour is that at the<br />
lower pressures tne eiectrons passing into the discharge ire<br />
less likely to suffer collisions in tne immediate vicinity <strong>of</strong><br />
the electrode so that the average enerLy <strong>of</strong> tiie electrons will<br />
be -high just prior to the required activating collisions.<br />
Therefore, activating collisions are more likely to occur and<br />
yields to iiicrezse corresgondingly. Degradation <strong>of</strong> product by<br />
electrons is. by' the same arguement, more likely under these<br />
conditions but other product degrading discharge collisional<br />
phenomena e.g. the reaction with hydrogen atoms, are reduced<br />
because <strong>of</strong> the greater mean free path at the lower operating<br />
pressure. The overall result is that hydrazine yields increase.<br />
Secondary effects become increasingly important at higher<br />
' pressures and it is likely that other variztions in the effect<br />
<strong>of</strong>.gressure on yield may well occur. One such variation ie<br />
reported to occur at about 5mm pressure where the energy yield<br />
passes through a maximum (8).<br />
Product yields were increased by removing hydrazine in a<br />
liquid absorbent ad 'it was considered that a more efficient<br />
absorption technique should lead to even greater yields. In<br />
order to obtain a more intimate gas liquid disperrknn a spray<br />
reactor was used df the general design Bhown-for Reactors<br />
B1 and 2 in Fig.1. The results (Pig.4) show that yields were<br />
slightly below that <strong>of</strong> the film reactor. In practice there<br />
were difficulties caused by dissimilarities in the construction<br />
<strong>of</strong> Reactors A and B which led to radically different discharge<br />
condition8 being obtained.<br />
The film reactor (U) had a more<br />
uniform discharge density because it was <strong>of</strong> an apnular<br />
construction where the annulus width was small compared to the<br />
reactor diameter. The spray reactor, on the other hand,<br />
employed a central wire electrode and coneequently there was .<br />
a non-uniform field in the discharge gap with a higher .local<br />
diacharge density in the vicinity <strong>of</strong> the wire electrode.<br />
Because the yield is known to depend on an inverse function<br />
<strong>of</strong> the power demity,it is only reasonable to expect that the<br />
reactor deeign :B1 would give eomewhat lower yield than the<br />
.Reactor design Al, under. similar operating conditione.<br />
This means that the more intimate gas-liquid contact in the<br />
epray reactor had no measurable effect on the product yield.<br />
-
On the other hand, if hydrazine is formed and degraded<br />
ullifolvly in the activating section <strong>of</strong> tlie discharge this<br />
result is difficult to explain. It is suggested from this<br />
that,under these conditions, the absorption process is no<br />
lolyer the controlling factor since an increase in, the<br />
overall potential absorption rate,tiirou(jh an increase in the<br />
illterfacial area ,produces no corresponaing rise in Warazine<br />
yields. I~ mag be that liquid surface activation phenomena<br />
are t*&ing place and the increased hydrazine yield which is<br />
observed with the use <strong>of</strong> an absorbent liquid is caused by<br />
hydrazine formed by otlier mechanisms in which the liquid<br />
surface plays an important role. If this were the case the<br />
liquid film, wnich presents a complete barrier to the discharge,<br />
would naturally give naximal yield and tile spray reactor<br />
would only tend to tllis value in tile limit.<br />
It was found experimentally that a decrease in the<br />
residence time <strong>of</strong> the recictant in the discharge is accompanied<br />
by an increase in energy yield <strong>of</strong> ivarazine and a corresponding<br />
fall in percent%e conversion. . The results are detailed in<br />
Blg.5 and appear to be in general agreement with those <strong>of</strong><br />
kchi (6). When a liquid,absorbent is used the change <strong>of</strong><br />
yield and percent conversion is steady but without the use <strong>of</strong><br />
an Crbs'orbent the effect becoines very marked at low residence<br />
times. Ouchi (6) has concluded from his results that soae <strong>of</strong><br />
the. hyQatinO formed in the diecharge is preserved from<br />
degradation by rapid physical removal out <strong>of</strong> the discharge.<br />
This would adequately explain, in general terms, the increase<br />
in energy yield with reduced residence time. The degree <strong>of</strong><br />
activation <strong>of</strong> species in the discharge uust fall as the<br />
reactaqt throughput is increased. This would result in a<br />
decrease in percent conversion as residence time was reduced.<br />
Furthermore, as the concentration <strong>of</strong> hydrazine governs the<br />
rate <strong>of</strong> the degrading reaction t!ie smaller. percent conversion<br />
achieved at high flow rates will result in a.higher overall<br />
energy yield being achieved. With the use <strong>of</strong> a liquid<br />
absorbent soize effect on tile yield may well arise beczuse<br />
<strong>of</strong> changes in the absorbtion rates due to i;.;creased turbulence<br />
and a decrease in the gas liquid cmtact time.<br />
Another point which .mst be borne in mind is that physical<br />
removal <strong>of</strong> the product or even,for trlat matter, the use <strong>of</strong><br />
the pulsed uischarge tec'mique cannot give better convers,ions<br />
,than that dictated by tne equilibrium concentration <strong>of</strong><br />
hydrazine for tne basic reactions t?-king place in the discharge.<br />
Tie use <strong>of</strong> the liquid absorbent, on the other hand, permits<br />
nigner conversion to be achieved because <strong>of</strong> tile inability <strong>of</strong><br />
tne uiscliarge reackion to reach equilibriun as ludraziae is<br />
being continuously renov4 after it is formed in tile discharge.<br />
The negative sloDe <strong>of</strong> tne ener$y yield-residence the<br />
plot (Pig. 5), yhen liquid absorbent was used, implies that it is<br />
oossible to increase yields Still furtlier by so:ue otiier<br />
suitable LnoGification <strong>of</strong> technique.. .<br />
'
_-<br />
<strong>of</strong><br />
' .<br />
8. -<br />
7<br />
6,<br />
Residence Time x105 hr<br />
Fig.5 Variation <strong>of</strong> hydrazine yieldwith flow rate tor<br />
reactor A1 ..<br />
X P<br />
&<br />
e V<br />
d<br />
7<br />
v A A<br />
10 -.-- -- I<br />
-\<br />
'\.<br />
! 1<br />
10 20 30 40<br />
Fig.6 Hydrazinc yield for pulsed D.C. reactor C1<br />
-<br />
A A<br />
0<br />
,
i<br />
137<br />
A'ae rcsu'l'cs, lor tiie ca.
135<br />
Ionic Reactions in Corona Discharges <strong>of</strong> Atmospheric Gases<br />
M. M. Shahin<br />
Xerox Corporation, Rochester, New York 14603 .<br />
INTRODUCTION<br />
Mass spectrometric studies <strong>of</strong> electrical <strong>discharges</strong> have been cdrried out during<br />
the past decade by several workers (1-6) in an attempt to identify the ionic precursors<br />
<strong>of</strong> a variety <strong>of</strong> neutral by-products which are formed in these systems. Such studies<br />
are expected to reveal the detailed chemistry <strong>of</strong> the many reactions that follow the<br />
formation nf primary ions through the impact <strong>of</strong> energetic electrons on the neutral gas<br />
molecules. and end with the neutralization <strong>of</strong> the final form <strong>of</strong> the ion either in the<br />
gas-phase or at the electrodes or the walls <strong>of</strong> the discharge tube. Thus, the identi-<br />
fication <strong>of</strong> these ionic reactions will, in many cases, demonstrate the mechanism <strong>of</strong><br />
the production <strong>of</strong> the free-radicals which are the source <strong>of</strong> the neutral by-products.<br />
Further. such studies would be expected to provide information on the mechanisms <strong>of</strong><br />
catalytic or inhibitory effects <strong>of</strong> trace quantities <strong>of</strong> certain compounds (e.g. water<br />
vapor) on the formation <strong>of</strong> these products. Processes such as charge-exchange or ion-<br />
molecule reactions which occur with large cross-sections and have been suspected to<br />
be responsible for such effects will thus be easily identified.<br />
The complexity <strong>of</strong> electrical <strong>discharges</strong> however, <strong>of</strong>ten makes the interpretation<br />
<strong>of</strong> mass spectrometric data very difficult. Specifically, the variations <strong>of</strong> electric<br />
field along and across the discharge tube in certain commonly used <strong>discharges</strong> (e.g.<br />
glows) <strong>of</strong>ten affect the abundance distribution <strong>of</strong> the various ionic species which are<br />
observed at the mass spectrometer (2,3). This is mainly due to the complex dependence<br />
<strong>of</strong> the cross-sections <strong>of</strong> both charge-exchange and ion-molecule reactions on ion<br />
energy as determined by the electric field within the discharge tube. Further com-<br />
plication arises from the formation <strong>of</strong> ion-sheaths around the walls <strong>of</strong> the discharge<br />
tube. through ambipolar diffusion. These may strongly influence the sampling <strong>of</strong> the<br />
discharge by the mass spectrometer. It is therefore essential that the system under<br />
investigation be well understood before the interpretation <strong>of</strong> the data is attempted.<br />
In this paper, the results <strong>of</strong> mass Spectrometric investigations on low pressure<br />
positive corona <strong>discharges</strong> established between two coaxially placed electrodes will<br />
be discussed. This form <strong>of</strong> discharge has been chosen primarily because its electrical<br />
properties are relatively simple and well understood. Further, the ion-sheath effects<br />
at the point <strong>of</strong> sampling are minimized because <strong>of</strong> the low level <strong>of</strong> ionization in<br />
these systems.<br />
EXPERl MENTAL<br />
Detailed description <strong>of</strong> the apparatus has already been given (6). Figure 1<br />
shows the schematic diagram <strong>of</strong> the apparatus. Briefly, a coaxial discharge tube is<br />
operated through a high-voltage d.c. power supply and the current is stabilized<br />
through the use <strong>of</strong> an external current limiting resistor, R. Electrons moving through<br />
the high field region present only at very close distances to the anode wire will<br />
gain energy from the field and cause ionization <strong>of</strong> the gas molecules in this region.<br />
Because <strong>of</strong> the symmetry <strong>of</strong> the electrodes, therefore, the system closely behaves as<br />
a line source <strong>of</strong> positive ions which are continuously regenerated. These ions will<br />
then move through the gas, perpendicular to the axis, undergoing various interactions<br />
before reaching the cathode. A smal 1 sampl ing port (IO to 100 microns in diameter)<br />
at the cathode allows a small portion <strong>of</strong> the ions to escape the discharge tube and be<br />
analyzed by a quadrupole mass spectrometer.<br />
The form <strong>of</strong> the electric field within the discharge tube is rectangular hyper-<br />
bolic before the discharge is established. As the discharge is formed, however,<br />
the presence <strong>of</strong> the positive space charge distorts this initial field, causing the
139<br />
latter to attain a constant value for the major distance between the electrodes (7).<br />
Differential pumping <strong>of</strong> the sampling region and the mass spectrometer allows<br />
operation <strong>of</strong> the discharge tube between pressures ranging from less than 1 torr to<br />
atmospheric. An electron gun placed before the mass spectrometer serves as a indepen-<br />
dent ionizing source for monitoring the composition <strong>of</strong> the neutral discharge gas when<br />
no ions are extracted from the discharge tube. For such analysis, calibration curves<br />
made on prepared gas mixtures, showing mass discrimination due to the specific flow<br />
condition are used. A rapid flow <strong>of</strong> gas is maintained through the discharge tube in<br />
order to avoid appreciable accumulation <strong>of</strong> neutral by-products <strong>of</strong> the discharge. For<br />
this purpose a linear flow velocity is maintained such as to completely renew the<br />
gas within the tube in two seconds. In systems where the concentration <strong>of</strong> the electro-<br />
negative gas (e.9. oxygen) is low, an external source <strong>of</strong> ionization through the use<br />
<strong>of</strong> a weak radioactive source (e.g. P0210) is used to stabilize the discharge.<br />
The general problem <strong>of</strong> mass spectrometric sampling from high pressure sources<br />
has recently been discussed by several workers (8). In systems where condensable<br />
gases are present, the temperature drop following the adiabatic expansion <strong>of</strong> the gas<br />
through the sampling nozzle may cause condensation if such gaseous components are<br />
present in sufficient concentrations. In the present experiments, the water content<br />
<strong>of</strong> the gases under investigation has been chosen below 5 x 10-2 mole %. Under these<br />
conditions, it can be demonstrated" that such condensations make negligible contri-<br />
bution to the results.<br />
RESULTS AND DISCUSS IONS<br />
Earlier experiments (6) on corona <strong>discharges</strong> in air at atmospheric pressure<br />
clearly demonstrated the important role <strong>of</strong> trace quantities <strong>of</strong> water vapor in these<br />
systems. In nitrogen, oxygen and their mixtures, where the water content exceeded<br />
4 to 5 x mole %, the dominant ionic species observed at the mass spectrometer<br />
were those corresponding to hydrated proton clusters, (H20),H+. These species were<br />
apparently formed from the interaction <strong>of</strong> the primary ions with the water molecules<br />
in the system as they passed through the gas to the cathode. In order to determine<br />
the role <strong>of</strong> various reactions which lead to such clusters, low pressure experiments<br />
were designed with individual gaseous components (e.g. 02, Ng) and their corresponding<br />
mixtures with various quantities <strong>of</strong> water vapor. These experiments were<br />
expected to reveal the formation <strong>of</strong> intermediate species and their final conversion<br />
to hydrated protons.<br />
Discharqe in Nitrogen:<br />
Experiments with nitrogen at low pressures, containing various concentrations<br />
<strong>of</strong> water vapor show several intermediates which are formed through the reactions <strong>of</strong><br />
nitrogen ions with water. The results <strong>of</strong> a typical experiment showing the relative<br />
abundances <strong>of</strong> various species reaching the mass spectrometer as the pressure <strong>of</strong> the<br />
discharge is varied, are shown in Fig. 2. In this experiment, it can be observed<br />
that as the pressure is increased, the abundance <strong>of</strong> the primary ion, N2+, is sharply<br />
reduced while those <strong>of</strong> the intermediate ions, namely, N4+, N2@ and H20+ rise to a<br />
maximum and then decrease as the tertiary ion H30+ begins to appear. This latter<br />
ion also rises to a maximum at higher pressures as its more highly hydrated forms<br />
appear. Thus the two reactions:<br />
+<br />
N2 + H20 N2H+ + OH, di = -0.6ev<br />
(1)<br />
and<br />
"Calculations based on the maximum number <strong>of</strong> collisions <strong>of</strong> an ion with water mole-<br />
cules present up to IOe1 mole % in an expanding gas, assuming cross-sections <strong>of</strong><br />
10-13cm2, show negligible contribution to the total collisions that such an ion<br />
and molecule will undergo within the discharge system. These calculations will be<br />
published elsewhere.
GAS OUT<br />
GAS IN<br />
i.ho<br />
QUADRUPOLE<br />
MASS SPECTROMETER<br />
TO 4"PUMP<br />
Fig. I . Schematic diagram <strong>of</strong> the discharge tube and mass spectrometer;<br />
I, platinum wire anode; 2, cylindrical discharge tube; 3, ext ac ion<br />
electrode; 4, mass spectrometer ionization chamber; 5, focuss "9<br />
electrodes; 6, electron multiplier; 7, etectron-gun assembly.<br />
"<br />
5 IO IS 20<br />
PRESSURE IN TORR<br />
Fig. 2. Variation <strong>of</strong> the relative abundance <strong>of</strong> different ions with pre: ure<br />
in.a positive corona discharge in nitrogen containing 2.2 x IO 9 mole %<br />
<strong>of</strong> water vapor.
N2 + + H20 d H20 19 + N2, c?H = -3.0ev<br />
appear to provide species which can further react with water through reactions (3)<br />
and (4) to form the hydrated proton:<br />
-<br />
N2H+ + H20 H O+ + N2,<br />
3<br />
H~O+ + H o H o+ + OH,<br />
2 3<br />
&i = -3.4ev<br />
AH = -lev<br />
I<br />
(3)<br />
(4 1<br />
The third and the major intermediate, namely N4+ which is formed through reaction <strong>of</strong><br />
N2+ with neutral nitrogen molecule (9) apparently also undergoes reactions with ,water<br />
similar to reactions (1) and (2) to form N2H+ and H20+ ion. These reactions are also<br />
expected to be exothermic. The relative abundance <strong>of</strong> N4+ ion is found to be strongly<br />
dependent on the field strength within the discharge tube. Specifically, the formation<br />
<strong>of</strong> a space-charge around the corona wire is found to strongly influence the<br />
relative abundance <strong>of</strong> N4+ and N2+ ions in the system as this space-charge substantially<br />
reduces the field strength within the discharge tube. In the experiments shown in<br />
Fig. 2, the magnitude <strong>of</strong> E/P, the pressure reduced electric field for the major distance<br />
between the two electrodes, was calculated to be about 7 volt/cm. mm Hg.<br />
TWO other intermediate species have been observed in this system. These ions,<br />
not shown in Fig. 2, are N4H+ and NjH+. They apparently arise as a result <strong>of</strong> ionmolecule<br />
reactions involving N4+ and N3+ ions in the system with water molecules and<br />
are expected to undergo proton transfer reactions similar to reaction 3,<br />
H~O+.<br />
to yield<br />
Other ions observed were at m/e = 32 and 46. These ions are believed to arise<br />
from trace quantity <strong>of</strong> oxygen which is inevitably present in this system as a result<br />
<strong>of</strong> the decomposition <strong>of</strong> water.<br />
m/e = 46 is NO2+.<br />
The ion <strong>of</strong> m/e = 32 is apparently 02+ while that <strong>of</strong><br />
In a system <strong>of</strong> pure nitrogen where the water concentration was kept below<br />
5 x 10-3 mole %, under similar discharge conditions as those presented in Fig. 2,<br />
it was observed that the intensity <strong>of</strong> N4+ ion rapidly increased as the gas pressure<br />
was increased, reaching a plateau after a few mm Hg pressure. This plateau corresponded<br />
to 95% <strong>of</strong> total ion intensity, with the remaining 5% being mainly due to<br />
N3+ ion. This result supports the earlier reference to the presence <strong>of</strong> a low E/P<br />
between the two electrodes (9) and the lack <strong>of</strong> any appreciable ion-sheath near the<br />
cathode.<br />
Discharge in Oxyqen:<br />
Figure 3 shows the pressure dependence <strong>of</strong> the relative abundance <strong>of</strong> various<br />
ionic species in a corona discharge in oxygen. This experiment shows several marked<br />
differences from those shown for nitrogen in Fig. 2. The most striking feature is<br />
the presence <strong>of</strong> large abundances <strong>of</strong> hydrated forms <strong>of</strong> the primary ion, namely 02+(H20)1,~<br />
and their dependence on pressure. That the abundances <strong>of</strong> these ions appear to rise<br />
to a maximum and then decrease at higher pressures with the formation <strong>of</strong> hydrated protons<br />
strongly suggests their role in the intermediate processes. This evidence together<br />
with the appearance <strong>of</strong> (H20)2+ ions in the system, and the variation <strong>of</strong> its abundance<br />
with pressure, i.e. rising to a maximum and then reducing at higher pressures, in-<br />
dicates the operation <strong>of</strong>.an entirely new mechanism in this system for the formation<br />
<strong>of</strong> hydrated protons. Moreover, the appearance <strong>of</strong> m/e = 37, (H20)2H* before the first<br />
member <strong>of</strong> the series suggests that a reaction other than the hydration <strong>of</strong> H30+, is<br />
responsible for the fermation <strong>of</strong> this species. Since the ionization potential (I.P.)<br />
<strong>of</strong> oxygen (12.07ev) is lower than that <strong>of</strong> water (I.P. = 12.56ev), a charge-exchange<br />
reaction similar to reaction 2, for nitrogen does not appear probable. Nor is it<br />
llkely that a reaction similar to that <strong>of</strong> reaction I, to form 02p would occur, as it<br />
appears to be endothermic. It is therefore probable that the hydrated form <strong>of</strong> the<br />
primary ion, especially the species containing two water molecule may be a precursor<br />
in the formation <strong>of</strong> the hydrated proton through the reactions:
-<br />
+<br />
( ~ ~ + 0 H ) ~ O ~ ( H~o)~H+ + OH<br />
(H~o)~+ + o2 (H,o)H+ + OH + o2 (7)<br />
Reaction 5 is expected to be exothermic owing to the heat <strong>of</strong> hydration <strong>of</strong> H20+ ion.<br />
It may also be possible, though unlikely, that any required energy fqr the reaction<br />
be supplied through the electric field in the discharge.<br />
One other ionic species which may play a role in this system is 0 . However,<br />
since the I.P. <strong>of</strong> atomic oxygen (13.61ev) is greater than that <strong>of</strong> oxygen, this species<br />
is not observed in this system as it most probably undergoes charge exchange with<br />
molecular oxygen at pressures used in these experiments. The reaction <strong>of</strong> OC with O2<br />
to give rise to 0 , if it occurs at all, is not observed in these experiments since<br />
ozone has also a ?:gher I.P. (12.8ev) than that <strong>of</strong> oxygen and therefore 03+ i s similarly<br />
expected to undergo a charge-exchange reaction with 02.<br />
Three other ions were observed in minor relative abundance in this system. These<br />
ions are OH+, H20+ and H202+. The relative yield <strong>of</strong> the O p ion is shown in Fig. 3<br />
while the yields <strong>of</strong> H20+ and H202+, being somewhat smaller, are not indicated. It is<br />
believed that the first two <strong>of</strong> these ions arise from an ion-molecule reaction and a<br />
charge-exchange process between O+ and water molecule respectively, and are detected<br />
only at higher discharge pressures where such reactions compete with direct charge<br />
transfer <strong>of</strong> O+ to oxygen molecules. H202+ ion, however, may appear as a result <strong>of</strong> an<br />
efficient charge transfer process between 02+ ion and the trace quantities .<strong>of</strong> H202,<br />
apparently formed in the system by the recombination <strong>of</strong> hydroxyl free radicals, or the<br />
reaction, H02 + H2 H202 + H, hydrogen being supplied through the decomposition<br />
<strong>of</strong> water.<br />
In a system <strong>of</strong> pure oxygen, where the waier content was kept below 5 x mole %,<br />
only two ions, 02' and O4+ were observed within the pressure range investigated. The<br />
abundance <strong>of</strong> O4+ ion rises sharply as the pressure is increased while that <strong>of</strong> 02+ decreases<br />
and both reach a plateau beyond a pressure <strong>of</strong> 20 Torr in the discharge tube.<br />
The relative abundance <strong>of</strong> O4+ in this plateau region was found to be about 62%.<br />
Discharge in Nitrogen Containing 0.12 mole % Oxygen:<br />
In order to determine the role <strong>of</strong> oxygen in air <strong>discharges</strong> in the absence <strong>of</strong><br />
water vapor, a number <strong>of</strong> experiments were carried out in nitrogen containin 0.12 mole %<br />
<strong>of</strong> oxygen. The lower limit <strong>of</strong> water vapor in these experiments was 5 x IO' 3 mole %.<br />
Fig. 4 shows the results <strong>of</strong> the relative abundance <strong>of</strong> various ionic species which are<br />
detected at the cathode as the pressure <strong>of</strong> the discharge is increased. These experiments<br />
clearly trace the history <strong>of</strong> the primary ionic species, namely N2" as it is<br />
converted to N4+ and the latter undergoes charge-exchange with the trace quantity <strong>of</strong><br />
oxygen present in this system. Thus, beyond a pressure <strong>of</strong> 20 Torr in the discharge<br />
tube the charge carriers in this system are almost all 02+. The trace amount <strong>of</strong> N3+<br />
observed in the system is also all removed beyond a pressure <strong>of</strong> 15 Torr.<br />
It is significant to note that no nitric oxide ion is observed in this system,<br />
indicating that neither the reaction N4+ + O2 d NO+ + NO + N2, nor 0 + + N2<br />
NO+ + NO, occur to any appreciable extent, in spite <strong>of</strong> the fact that bott these re-<br />
actions are expected to be exothermic.<br />
Experiments in which water was not excluded showed the appearance <strong>of</strong> many ionic<br />
intermediates <strong>of</strong> both pure oxygen and nitrogen and their mixtures. All these ions<br />
could be traced and their final conversion to hydrated protons followed.<br />
Discharge in Air:<br />
Experiments in air in the absence <strong>of</strong> water vapor (less than 5 x mole %)<br />
were carried out in order to determine the importance <strong>of</strong> ionic species <strong>of</strong> oxides Of<br />
nitrogen in these systems. The results <strong>of</strong> these experiments are shown in Figure 5.<br />
+
I<br />
Ii<br />
ZT<br />
PRESSURE IN TORR FOR 02+ ION<br />
IO 15 20<br />
I<br />
25<br />
I<br />
30 35<br />
.08<br />
4-<br />
.3<br />
.2<br />
.I<br />
0<br />
-<br />
-<br />
-<br />
I<br />
5<br />
/<br />
/<br />
/X(H20)2+ '<br />
IO I5 20 25 30<br />
PRESSURE IN TORR<br />
- .06<br />
\<br />
-.04 lI<br />
Fig. 3. Variation <strong>of</strong> the relative abundance <strong>of</strong> different ions with pressure<br />
in a positive corona discharge in oxygen containing 2.0 x 10-2 mole %<br />
<strong>of</strong> water vapor. The pressure scale for 02+ ion is shown on the top<br />
<strong>of</strong> the figure.<br />
PRESSURE IN TORR<br />
Fig. 4. Variation <strong>of</strong> the relative abundance <strong>of</strong> different ions with p<br />
in a positive corona discharge in nitrogen containing 1.2 x<br />
<strong>of</strong> oxygen and less than 5 x 10-3 mole % <strong>of</strong> water vapor.<br />
essure<br />
0-1 mole %
144<br />
i<br />
I , , , , .<br />
+<br />
1- u<br />
3c<br />
w<br />
Inu<br />
c c<br />
00<br />
U<br />
.-<br />
U L<br />
c w<br />
wc,<br />
ks!<br />
u-<br />
u-T .- CI<br />
0 .-<br />
w- 3<br />
01<br />
u-u<br />
nJ<br />
- m<br />
w e<br />
L:<br />
w o .<br />
L u*<br />
U<br />
a w<br />
u- >-<br />
0 .- 0<br />
W E<br />
c .- 0 Inn<br />
*- 0 I<br />
u a0<br />
n J -<br />
.-<br />
9<br />
L<br />
m c<br />
X<br />
> .- VI<br />
m<br />
.- m<br />
LA<br />
i<br />
1
As expected from the previous sections, O$4?on appears as the most abundant ion in<br />
this system. Also as expected, 04+ ion appears in the system and as the pressure is<br />
increased, it becomes an important charge carrier and its relative abundance eventually<br />
reaches a plateau. The appearance <strong>of</strong> the ion 02+(H20), hydrated form <strong>of</strong> molecular<br />
oxygen ion, in this system, indicates the high affinity <strong>of</strong> this ion for hydration as<br />
the gas contained less than 5 x 10-3 mole % water in the discharge tube.<br />
Nitric oxide ion was the only oxide <strong>of</strong> nitrogen found in the system under the<br />
experimental conditions used here. Its relative abundance remainedlconstant as the<br />
discharge pressure was varied between 2 to 40 Torr. The presence <strong>of</strong> NO+ in these experiments<br />
and its absence in experiments carried out in nitrogen containing 0.1 mole %<br />
<strong>of</strong> oxygen, indicates that these ions are probably formed through the reaction<br />
N ~ + o2 + NO+ + NO, AH = -4.5ev<br />
+<br />
Assuming that 02' arises eiti4er through charge-exchange with Ng ion or by<br />
direct electron impact on neutral oxygen molecule, one can use the data in these experiments<br />
to obtain a relative ratio <strong>of</strong> the rate-constants for the charge-exchange<br />
reaction <strong>of</strong> N2+ with oxygen to that <strong>of</strong> ion-molecule reaction (8). This ratio is found<br />
to be equal to 8, a value which is lower than the ratio <strong>of</strong> the published values <strong>of</strong><br />
these rate-constants (IO). This indicates that possibly other reactions such as N+ +<br />
02 + NOf 0 and O+ + N2<br />
NO+ + N or others involving neutral atomic species<br />
also contribute to the total yield <strong>of</strong> NO+ ion.<br />
CONCLUSIONS<br />
Mass spectrometric studies <strong>of</strong> low pressure positive d.c. corona <strong>discharges</strong> in<br />
atmospherrc gases containing trace quantities <strong>of</strong> water vapor show a complex series<br />
<strong>of</strong> reactions with each component leading to the formation <strong>of</strong> hydrated protons in the<br />
system. In the case <strong>of</strong> nitrogen, intermediate species N2* and H20+ are presumably<br />
formed through ion-molecule reaction and charge-exchange <strong>of</strong> N2+ and N4+ with water<br />
molecules. These species later form the hydrated proton through proton transfer reactions<br />
in subsequent collisions with water molecules. In moist gaseous oxygen, it<br />
appears that the hydrated form <strong>of</strong> the primary ion 02+(H20)2, plays an important role<br />
in the conversion <strong>of</strong> the charge carriers to hydrated protons. It is suggested that<br />
this transformation may occur through the formation <strong>of</strong> the intermediate (H20)2+ which<br />
has been found in this system. In experiments where water vapor is excluded from the<br />
system, a concentration <strong>of</strong> 1.2~ 10-1 mole % oxygen can transform, through chargeexchange<br />
reactions, all ionic species <strong>of</strong> nitrogen to 02+ at a discharge pressure <strong>of</strong><br />
20 Torr, and that ion-molecule reactions leading to the formation <strong>of</strong> oxides <strong>of</strong><br />
nitrogen are by far less probable within the pressure range investigated.<br />
ACKNOWLEDGMENTS<br />
The author is indebted to Dr. W. Roth for his invaluable comnents during the<br />
course <strong>of</strong> this work and to A. Friske for his assistance in experimental work.<br />
References<br />
Knewstubb, P. F., Tickner, A. W., J. Chem. Phys. 36, 684 (1962).<br />
Knewstubb, P. F., Tickner, A. W., J. Chem. Phys. 36, 674 (1962).<br />
Knewstubb, P. F., Tickner, A. W., J. Chem. Phys. 11, 294 (1962).<br />
Shahin, M. M., J. Chem. Phys. 3, I798 (1965).<br />
Shahin, M. M., Advances in Chemistry Series, No., 58, Ion Molecule Reactions in the<br />
Gas Phase, p. 315 (1966).<br />
Shahin, M. M., J. Chem. Phys. 45, 2600 (1966).<br />
vonEngel, Ionized Gases (Oxford University Press, London, 1965).<br />
Green, F. T., MiIn, T. A., J. Chem. Phys. 39, 3150 (1963); Leckenby, R. E.,<br />
Robbins, E. J., Treval ion, P. A., Proc. Roy. SOC. (London) G, 409 (1964);<br />
Anderson, J. B., Anders, R. P., Fenn, J. B., Advances in Atomic and Molecular<br />
Physics, p. 345; edited by D. R. Bates, Academic Press, New York 1965.<br />
Varney, R. N., J. Chem. Phys. 2, I314 (1959).<br />
Ferguson, E. E., Fehsenfeld F. C., Golddan, P. D., Schmeltek<strong>of</strong>f, A. L.,<br />
J. Ceophys. Res. 70, 4323 (1965).
146<br />
SYNTHESIS OF ORGANIC COIGhTDS BY TKE ACTION OF ELECTRIC<br />
DISCHARGES IN SIMULATED PRIMITIVE ATMOSPHERES<br />
C. Ponnamperuma, F. Woeller, J. Flores, M. Romiez, and W. Allen<br />
Exobiology Division<br />
<strong>National</strong> Aeronautics and Space Administration<br />
Ames Research Center<br />
M<strong>of</strong>fett Field, California<br />
In the study <strong>of</strong> <strong>chemical</strong> evolution we are interested in the path by which<br />
molecules <strong>of</strong> biological significance could have been formed on the primitive<br />
earth in the absence <strong>of</strong> life. It is generally accepted that the primitive<br />
atmosphere <strong>of</strong> the earth consisted mainly <strong>of</strong> methane, a-monia, and water. Various<br />
forms <strong>of</strong> energy such as ultraviolet light frm the sun, electrical <strong>discharges</strong>,<br />
heat, and ionizing radiation acting on this atmosphere must have given rise to<br />
a wide variety 3f organic substances.<br />
Table I gives a summary <strong>of</strong> the sources <strong>of</strong> energy 03 the earth's surface<br />
today. (1) It is probable, therefore, that solar energy must have made the<br />
principal contribution to the synthesis <strong>of</strong> organic compounds in primordial<br />
times. Next in importance are electric <strong>discharges</strong>, such as lightning and corona<br />
<strong>discharges</strong> from pointed objects. These occur closer to the earth's surface and,<br />
hence, would have more effectively transferred the organic matter synthesized<br />
toths primitive oceans. This paper describes attempts to simulate some <strong>of</strong> the<br />
reactions which may have taken place on the prebiotic earth, through the action<br />
<strong>of</strong> electric <strong>discharges</strong>.<br />
While extensive work has been done on the effect <strong>of</strong> electric <strong>discharges</strong> on<br />
various organic molecules, relatively few experiments have been performed to<br />
elucidate its role in che.mica1 evolution. Some <strong>of</strong> the earliest such investiga-<br />
tions were carried out by the chemist Haber.<br />
Beutner, in his book entitled "Life's<br />
Beginning on the Earth," recalls how Haber performed numerous experiments in which<br />
electrical <strong>discharges</strong> were sent through carbon containing gases like methane,<br />
carbon dioxide, etc., with the aim <strong>of</strong> obtaining sugars. (2) Although traces<br />
<strong>of</strong> some sugars were formed, a large number <strong>of</strong> various other substances were<br />
also synthesized. Haber thus came to the conclusion that by means <strong>of</strong> electrical<br />
<strong>discharges</strong> through carbon containing gases, "practically any substance known to<br />
organic chemistry can be found."<br />
Perhaps the most celebrated experiment in this field was performed by Stanley<br />
Miller in Urey's laboratory in 1953. (3) Miller submitted a mixture <strong>of</strong> methane,<br />
ammonia, and water in the presence <strong>of</strong> hydrogen to electrical <strong>discharges</strong> from tesla<br />
coils. A large number <strong>of</strong> organic compounds were formed. Among these, four amino<br />
acids were identified. Miller postulated two alternative possibilities for the<br />
mechanism <strong>of</strong> synthesis <strong>of</strong> amino acids. According to the first, aldehydes and<br />
hydrogen cyanide are synthesized in the gas phase by the spark. These aldehydes<br />
and hydrogen cyanide react in the aqueous phase to give amino and hydroxynitriles.<br />
These nitriles are, in turn, hydrolyzed to amino and hydroxy acids. The mechanism<br />
is essentially a Strecker synthesis. A secmd suggestion made was tkat the amino<br />
and hydroxy acids were synthesized in tk gas phase by ions ad radicals produced<br />
in the electrical discharge. MiUer's subsequent work has shown that the first<br />
mechanism is the one most likely to have produced the amino acids. (4) The rate<br />
<strong>of</strong> production <strong>of</strong> aldehydes and hydrcgen cyanide by the spark and the rate <strong>of</strong><br />
hydrolysis <strong>of</strong> the aminonitriles were sufficient to account for the total yield<br />
<strong>of</strong> amino acids.<br />
I
\<br />
. 147<br />
In Our experimental work, we have endeavored to study the mechanism by which<br />
various molecules <strong>of</strong> biological interest could have been formed by the action <strong>of</strong><br />
electrical <strong>discharges</strong>. A series <strong>of</strong> experiments was outlined in which the starting<br />
b materials were vaxied. Four different classes <strong>of</strong> experiments have been performed;<br />
with methane; with methane and ammonia; with methane, ammonia, and water; and with<br />
methane and water.<br />
1 -<br />
I<br />
The effect <strong>of</strong> a semi-corona discharge, a low intensity arc discharge, and a<br />
, high intensity arc discharge on gaseous methane was first investigated. The current<br />
through the discharge was measured by the voltage drop across a resistor with the<br />
( Cell. For the semi-corona discharge, the cell current was 0.4 m. amp, 0.5 m. amp<br />
, for the low arc, and 10 m. amp for the high arc discharge. Gas chromatography and<br />
mass spectrometry were used for the analysis <strong>of</strong> the end products. Comparative ret<br />
sults <strong>of</strong> the analysis <strong>of</strong> hydrocarbons up to C5 are shown in Table 11. In the semicmona<br />
most <strong>of</strong> the methane remained unreacted after a 24-hour discharge. Some ethane<br />
and propane were formed. There were small amounts <strong>of</strong> ethene, propene,-and substituted<br />
paraffins. In the case <strong>of</strong> the low and high arcs, ethylene and acetylene were also<br />
> present.<br />
The analysis <strong>of</strong> the hydrocarbons fram Cg - Cg reveals that the semi-corona<br />
gave unsaturated substances while the arc discharge gave rise to aromatic compounds.<br />
i The semi-corona cell'yielded a colorless distillate, the gas chromatogram <strong>of</strong> which<br />
was poorly resolved. The high intensity arc gave a yellow fluid, the chromatogram<br />
\ <strong>of</strong> which had well spaced peaks. Benzene was the most abundant with toluerenext in<br />
order <strong>of</strong> magnitude. The peaks from the semi-corona chromatogram were identified by<br />
, the use <strong>of</strong> mass spectrometry as: 2,2-dimethyl butane, 2-methyl pentane, +methyl<br />
I pentane, 2,b-dimethyl hexane, 3,4-dimethyl hexane. In figure 1 the chromatogram<br />
:I <strong>of</strong> the semi-corona discharge products (low) has been superinposed on that from the<br />
~ high intensity arc (high). The results presented here show that the character <strong>of</strong><br />
compounds in the range 3f interest appears to be determined by the type <strong>of</strong> discharge<br />
1 more than by any factor.<br />
(5)<br />
1 We have also examined the composition <strong>of</strong> the hydrocarbons above C9 in the products<br />
<strong>of</strong> the semi-corona discharge. The gas chromatogram is very unresolved (Fig. 2).<br />
No normals 31' branched-chain isoproprenoid hydrocarbons were identified. Analysis<br />
\ <strong>of</strong> the mixture by mass spectrometry shows that the compounds are possibly cyclic<br />
1 in structure. (6)<br />
The effect <strong>of</strong> an arc discharge on anhydrous methane and ammonia was next inves-<br />
1<br />
tigated for two reasons. Firstly, such a study would help us to understand the<br />
' pathways by which some organic compounds such as amino acids can be synthesized.<br />
Secondly, reactions <strong>of</strong> this type would simulate, to some extent, conditions which<br />
may exist on the planet Jupiter.<br />
'' In this investigation, we have used reaction vessels <strong>of</strong> about a liter in<br />
' vglume containing an equimolar mixture anhydrous methane and ammonia up to a pres-<br />
),sure <strong>of</strong> 0.5. The electrdes consisted <strong>of</strong> gold wires about 1 cm apart. A typical<br />
reaction lasted for about 1.5 hours. The current passing through the- system was<br />
about 0.5 mA. The end products consisted <strong>of</strong>: (1) gases, (2) a colorless distillate,<br />
,'\and (3) a ruby colored residue.<br />
1<br />
1 distillate.<br />
In the present study, our attention was prinarily directed to the colorless<br />
The volatile products were vacuum distilled into a U-trap at -78Oc and<br />
analyzed by gas chromatography (Fig. 3). The fractions corresponding to each peak<br />
The GLC retention time,<br />
'' were collected for subsequent mass spectrometric amlysis.<br />
1 the mss spectrometric fragmentation pattern, and the NMR spectrum established the<br />
k<br />
\<br />
.)<br />
Y,<br />
t<br />
I<br />
1<br />
I<br />
i
mi5<br />
a-<br />
dz<br />
CI<br />
n<br />
I<br />
a<br />
I<br />
n<br />
+4 - 0<br />
-I- k<br />
7<br />
0 -<br />
v)<br />
Q)<br />
t<br />
3<br />
c<br />
.-<br />
E<br />
a<br />
u<br />
I<br />
\o<br />
u<br />
V<br />
k<br />
Ld<br />
0<br />
Ld<br />
2<br />
0<br />
0<br />
V<br />
I<br />
Ti<br />
$<br />
d<br />
hD<br />
.r-<br />
F
i<br />
I<br />
/ '<br />
I<br />
",<br />
m<br />
a 2<br />
k<br />
F4<br />
a<br />
I<br />
cu
0<br />
z<br />
cu<br />
I<br />
I<br />
I
151<br />
identity <strong>of</strong> each cf the fractions separated by gas chromatography. Ammonium cyanide,<br />
methyl cyanide, ethyl cyanide, a-minoacetonitrile and its C-methyl and N-methyl<br />
., homologues were identified. The u-aminonitriles on hydrolysis give rise to a-amino<br />
acids. These nitriles my prcvide a reasonable pathway for the origin <strong>of</strong> amino<br />
' acids under prebiological corditions.<br />
In sane cf our discharge experiments, we turned to the question o the origin<br />
<strong>of</strong> lnonocarboxylic acids under prebiotic conditions. If we assume that pre-existing<br />
abiotically synthesized fatty acids were necessary for the functioning <strong>of</strong> selective<br />
menbranes, sone rnechanisn must have existed for their formation. The reaction be-<br />
> tween methane and ammonia appears to provide such a pathway. When a mixture <strong>of</strong><br />
methane and water exposed to a semi-corona discharge and the end products examined<br />
l after saponification, the monocarboxylic acids from C2 - CQ were identified. (7)<br />
\<br />
The volatile acids C1 - C8 were examined as their free acids by gas chromatography.<br />
A resulting chromatogram is illustrated in Figure 4. The. individual peaks<br />
' were then trapped and their identity confirmed by mass spectrometry. 'Acids contain-<br />
\ ing seven or more carbon atoms were analyzed as methyl esters. The methyl esters,<br />
' after chromatography, were exarnined by mass spectrometry. Of eleven major peaks<br />
obtained by gas chromatography (Fig. 5) only one appears as the normal methyl ester.<br />
Presumably, the renaining peaks represented branched-chain isomers.<br />
~<br />
'<br />
While it is clear that in the case <strong>of</strong> the longer chain fatty acids several<br />
isoners have been produced, only a few <strong>of</strong> the innumerable possible compounds are<br />
realized. A preferential synthesis <strong>of</strong> some type appears to be favored. Theoretically,<br />
the branching <strong>of</strong> carbon chains, which is favored in free radical reactions, I<br />
may be repressed by steric restrictions when the lengthening carbon chains are<br />
\ absorbed on monolayers. An attempt to favor the for-mation <strong>of</strong> straight chain acids<br />
by placing the aqueous phase in close proximity with the discharge zone did not<br />
produce any change in our results.<br />
1<br />
In the study <strong>of</strong> prebiotic organic synthesis, perhaps the most relevent experinents<br />
involve the use <strong>of</strong> all the main constituents <strong>of</strong> the presumed primitive earth<br />
' atmosphere. We have therefore exposed a mixture <strong>of</strong> methane, ammonia, and water to<br />
a discharge from tesla coils simulating lightning on the primitive earth. At the end<br />
' 3f a 24-hour discharge, the gas phase analysis has shown that over 90$ <strong>of</strong> the start-<br />
'1 ing methane has been converted into organic ccmpounds. Of this, about 45$ is found<br />
in the water fraction. 188 <strong>of</strong> the water soluble material is in the form <strong>of</strong> cyanide.<br />
- The formstion <strong>of</strong> cyanide in this reation is significant in the light <strong>of</strong> the multiple<br />
role played by hydrogen cyanide in organic synthesis. (8)<br />
The analysis <strong>of</strong> the end products <strong>of</strong> this reaction by pper chromatography re-<br />
, veals that a large number <strong>of</strong> organic compounds were formed but none <strong>of</strong> these cor-<br />
responded to the commonly occurring amino acids.<br />
A certain amount <strong>of</strong> material<br />
\ appeared at the origin. However, when the reaction products were hydrolyzed with<br />
6N HC1 for 24 hours and then analyzed, a large number <strong>of</strong> amino acids were formed<br />
\ (Fig. 6). Among those identified me nine which are commonly found in biological<br />
materials: glycine, alanine, aspartic, glutamic, threonine, serine, isoleucine,<br />
leucine, and phenylalanine. The results obtained by ion exchange analysis were<br />
2 confirmed by gas chromatography. The evidence, thus, points to the fact that the<br />
:lamino acids were already polymerized in the solution <strong>of</strong> end products. Separation<br />
1 by the use <strong>of</strong> a biogel-P column gave us a fraction having a molecular weight in the<br />
range 186 to about 2,000 and whose 1-dimethylaminonapthalene -5 sulphonyl chloride<br />
' (DNS) derivative showed a single band on electrophoresis. When this fraction was<br />
hydrolyzed, the aMno acids aspartic, serine, glutamic, glycine, and alanine were<br />
' obtained.<br />
1,<br />
)<br />
\<br />
1:<br />
T
I-<br />
I - t<br />
d<br />
m<br />
a<br />
.rl<br />
Y<br />
x<br />
c,<br />
t-'<br />
d<br />
k
0<br />
0<br />
AI<br />
r<br />
0<br />
z<br />
a<br />
rc)<br />
I<br />
Z<br />
c.<br />
*<br />
I<br />
0<br />
z<br />
0<br />
W<br />
(3<br />
E<br />
a<br />
I<br />
0<br />
m<br />
5<br />
z<br />
0<br />
K<br />
LL<br />
0<br />
Z<br />
z<br />
-<br />
a<br />
3NllVA<br />
-<br />
N- u? AI- . . 0<br />
*a -0<br />
C<br />
cu-<br />
3N13031<br />
((IlJVdNVlS<br />
lVNL131NI)<br />
3NINOIH13H I<br />
3NllSA3<br />
-a -0<br />
. .<br />
AI-<br />
i<br />
i<br />
f
,<br />
155<br />
This result is significant in the context <strong>of</strong> <strong>chemical</strong> evolution. It has<br />
generally been thought, that amino acids had first to be synthesized and then<br />
cmdensed together into a polymer. The synthesis <strong>of</strong> a polypeptide in an electric<br />
discharge experiment reveals that such a sequence <strong>of</strong> r-actions may not have been<br />
necessary. If a suitable condensation agent is present the polymer appears to be<br />
formed as sson as the acids are s-ynthesized. In our case, the condensation agent<br />
is probably hydr-gen cyanide.<br />
The presence <strong>of</strong> 18$ hydrogen cyanide in the reaction<br />
mixture combined with the fact that in previous experiments we have beeh able to<br />
condense bases and sugars with cyanide support this hypothesis.<br />
The different experlments that have been described so far reveal that impor-<br />
1 tant biclogical molecules can be synthesized by the use <strong>of</strong> a form <strong>of</strong> energy which<br />
existed -n the primitive earth. These conditions may be considered to be genuinely<br />
abiotic since the materials used are the constituents <strong>of</strong> the presumed prinitive<br />
, earth atmosphere, the conditions are aqueous, and the form <strong>of</strong> enera is on2 that<br />
is likely to have sccurred on the earth before the appearance <strong>of</strong> life.<br />
\<br />
References<br />
> 1. S. L. Miller and H. C. Urey, Science ', a, 245 (1959)<br />
2. R. Beutner, Life's Beginning on The Earth, Williams and Wilkins, Baltimore, 1938<br />
3. S. L. Miller, J. Am. Chem. SOC. a, 2351 (1955)<br />
4. S. L. Miller, Bichim. et Biophys. Acta 23, 480 (1957)<br />
5. C. Ponnqperuva and F. Woeller, Nature m3, 272 (1964)<br />
6. C. Ponnamperuma and K. Pering, Nature 2- 979 (1966)<br />
7. W. A. Allen and C. Ponnamperuma, Currents in Modern Biology (in press) (1967) I<br />
8. R. Sanchez, J. Ferris, and L. E. Orgel, Science 153, 72 (1966)<br />
. TAELE I<br />
SOURCE -<br />
Ultraviolet light ( 2500 i)<br />
Electric <strong>discharges</strong><br />
Radio activity<br />
Volcanoes<br />
TABLF: I1<br />
High Arc<br />
Total hours <strong>of</strong> current flow 1- 5<br />
Electrode voltage<br />
Cell current (m. amp)<br />
$ L ss CHq/h<br />
End prsducts ($)<br />
Ethane<br />
Propane<br />
Ethene<br />
Pr=pene<br />
Acetylene<br />
1400<br />
4.0<br />
6.6<br />
32<br />
2<br />
32<br />
27<br />
--<br />
ENERGY<br />
(in cal em+ p-1)<br />
570<br />
4<br />
0.8<br />
0.13<br />
Low Arc<br />
40<br />
2500<br />
0.5<br />
1.1<br />
20<br />
5.2<br />
2.4<br />
1.9<br />
42.5<br />
Semi -Corona<br />
48<br />
9400<br />
0.3<br />
0.6<br />
58. a<br />
36.8<br />
1.5<br />
0.6<br />
0.0
156<br />
The Dissociation <strong>of</strong> Metal Halides in Electrical Discharges<br />
F o KO MCTaggart<br />
Mdsion <strong>of</strong> Mineral Chemistry, C.S.I.R.0. Box 124, Port<br />
Melbourne, Victoria, Australia.<br />
I<br />
Introduction.<br />
in electrical <strong>discharges</strong> in dissociating gae molecules is well known,<br />
and is the basis <strong>of</strong> the <strong>chemical</strong> reactions that a H observed in plasnrs<br />
systems. Since gases such a8 02, H2, B2dC1 9 88 well a8 vapours <strong>of</strong><br />
organic and other compounds have been diesociazed to atoms ad/m<br />
radicals, it appears Logical to suppose that many other molecules would<br />
behave similarly. This paper describes the diesociation <strong>of</strong> the halides<br />
<strong>of</strong> the Croup I, I1 and 111, and rare earth metals.<br />
&m-imental. Ideally, the halides should be introduced Into<br />
the discharge tube in the form <strong>of</strong> vapoure at suitable pressures. Due<br />
to the wide range <strong>of</strong> volatilities occurring in the above compounds and<br />
to other erperimental difficulties it proved more convenient to we<br />
carrier gmee, and to volatilize the halides from small boats contained<br />
wlthin the reaction tube by heating these boats to suitable temperatures.<br />
It was usually possible to make use <strong>of</strong> the heat generated in the plasma<br />
itself for this purpose. Samples were placed upstream from,or within<br />
the central plasma region,ln positione where heat sufficient to give the<br />
desired rate <strong>of</strong> volatilization was produced.<br />
The effect <strong>of</strong> the energetic electrons produced<br />
Since heating <strong>of</strong> solib<br />
due to atomic recombination ie largeu a surface effect it was also<br />
possible to verg the sublimation rate <strong>of</strong> samples by changing the Lmrfece<br />
area exposed to the plasma.<br />
Both inert gases (in particular He and H ) and hydrogen have been<br />
used and certain reactions which are discussed befow appear to involve E<br />
atoms aa well a dissociative effects. These gases were <strong>of</strong> coomerciel<br />
purity, the He and 5 being purified by passing them over zirconim ponder<br />
heated to 800°C; although no essential differences between the cylinder<br />
gases and the purified gases were observed. Pressures <strong>of</strong> the carrier<br />
gaaes were varied between 0.5 ad 2.3 mm. Halides wed were <strong>of</strong> A.B. grade,<br />
but in certain cases impure compounds were deliberately used in order to<br />
determine the effects <strong>of</strong> impurities on the dissociation and on the purity<br />
<strong>of</strong> the metal obtained.<br />
Ertenslve use was made <strong>of</strong> a 2450 mC magnetron generator (Mullard<br />
m2/205A) which although capable <strong>of</strong> an output in exce88 <strong>of</strong> 2 M continuow<br />
wave power, was seldom operated at inputs higher than 600 watts. B&P<br />
frequency energy from the magnetron was fed via a 50 ohm air dielectric<br />
coaxial line, to a section <strong>of</strong> waveguide operated ea a resonant cavity by<br />
means <strong>of</strong> a sliding plunger inserted into the open end. The discharge<br />
tube passed tranevereely through the guide at a point <strong>of</strong> m a r i m electrostatic<br />
field. h e investigatlons, Involving the chlorides <strong>of</strong> Li, Be,<br />
and a, which dissociate very readily, were made using au 9-f" generator<br />
consintlng <strong>of</strong> a 4-125A vacuum tube at a frequency <strong>of</strong> 30 mC, the power <strong>of</strong><br />
which could be varied from close to zero to about 400 watts. In these
Compound Bond<br />
he rgy<br />
Li I 82<br />
UBr 102<br />
LICl 115<br />
Lilr 138<br />
157<br />
cases the reaction tube passed through the "tank coil" <strong>of</strong> the output<br />
circuit . Conventional methods <strong>of</strong> analysis were used to determine 8/ the<br />
SmOunt <strong>of</strong> metal deposited, b/ the undissociated halide sublimed onto the<br />
reaction tube, and c/ the halogen or hydrogen halide collected in the<br />
liquid sir trap which followed the reaction tube in the flow1 syatem.<br />
- Results.<br />
From all the halides studied It was poesible to<br />
separate metals, <strong>of</strong>ten in good yields.<br />
1<br />
The lithim -UP cornpoonds dissociated in both inert carrier gmes and<br />
in H to gLve metals d the relative reaction rates were detedned for<br />
the fodides, bromides, chlorides and fluorides <strong>of</strong> Li, Ha, K end Cs. Son18<br />
Of these are ehown in Table 1. It rill be noted that for any one metal<br />
- Table 1<br />
Relative Reaction Ratee for the Hdidee <strong>of</strong> LI and la<br />
Re10 React. Compound Bond Bel, Bead.<br />
Bate -rgJ Bate<br />
232.5<br />
101.5<br />
66.5<br />
19.0<br />
BaI<br />
BaBr<br />
HaCl<br />
BaF<br />
72<br />
89<br />
99<br />
108<br />
26 e0 15.0<br />
IOIO<br />
5 *o<br />
I
Table 2<br />
Dissociation <strong>of</strong> NaCl in H2 at 1.0 mn for Various Input Powers<br />
t - -<br />
4 *O I<br />
0.240 0 280<br />
I<br />
I<br />
F'igure 2 shows the effect <strong>of</strong> carrier gas pressure at constant<br />
power. A marimum in metal production occurred at about 1.5 mm pressure.<br />
For H2 this pressure coincides closely with the highest concentration <strong>of</strong><br />
H atoms, and is probably related to the concentration <strong>of</strong> electrons having<br />
sufficient energy to cause dissociation. The reaction rates appeared to<br />
be substantially independent <strong>of</strong> the nature <strong>of</strong> the carrier gas,<br />
The fact that considerable quantities <strong>of</strong> highly reactive metals<br />
are deposited in atmospheres <strong>of</strong> even more highly reactive gases, namely<br />
halogen atoms, suggests that these halogen species may be in the form <strong>of</strong><br />
negatively charged ions.<br />
Such ions, having a complete outer shell <strong>of</strong><br />
eight electrons, would be non-reactive <strong>chemical</strong>ly, and if neutralization<br />
<strong>of</strong> their charges is delayed until they are swept clear <strong>of</strong> the metal<br />
deposit, an explanation <strong>of</strong> the apparent absence <strong>of</strong> appreciable back<br />
reaction would be afforded. Mass spectroscopic studies are being made<br />
on the nature <strong>of</strong> the species in these <strong>discharges</strong> to elucidate this matter.<br />
The &OUR I1 compounds. The majority <strong>of</strong> the halides <strong>of</strong> Be, ldg, Ca, Sr<br />
and Ba were investigated. Rates <strong>of</strong> dissociation did not appear to vary<br />
as widely as those <strong>of</strong> Group I. As optimum conditior.8 were approached<br />
for each compound, all the halide vapourized was dissociated and deposits<br />
were formed on the W d h <strong>of</strong> the reriction tube in which metals and dihalides<br />
were founO in equi-olar proportions. This points strongly to the<br />
formation <strong>of</strong> unstable monohalide molecules in the discharge, which dis-<br />
'proportionate to yield the observed products. Again the dissociations<br />
were not dependent on the carrier gas employed. BeC12, one <strong>of</strong> the more<br />
volatile halides in this group, end one <strong>of</strong> the most easily dissociated <strong>of</strong><br />
all the compounds studied, may be broken down in the lower frequency<br />
apparatus mentioned previously-<br />
Metals could be separated from the dihalidss in the deposits by<br />
means <strong>of</strong> suitable solvents for the letter, or in some cases by vacuupl<br />
sublimation <strong>of</strong> the dihalide.
,<br />
”..’<br />
3or<br />
25 -<br />
5 20 -<br />
s<br />
3<br />
w<br />
Y 15-<br />
<<br />
z’<br />
1<br />
f 10-<br />
I<br />
0 8.0<br />
SODIUM CHLORIDE SUBLIMATION RATE (MMOLE/HR) AT Po0 W
160<br />
Group I11 compounds. No in estigations have been made on the compounds<br />
<strong>of</strong> boron. Ywkovskii t& &.' reported in 1958 that BC1 was reduced to<br />
elemental boron by H atoms, and the present work on alum?nium halides<br />
supports this claim.<br />
When inert csrrier gases were used there was a very limited<br />
deposition <strong>of</strong> metal from Al and Sc halides amounting to onu a few<br />
per cent <strong>of</strong> the vapour passed through the discharge, but when hydmgen<br />
wee employed much higher yields <strong>of</strong> metals were obtained. It would<br />
appear, therefore, that at least one step in the decomposition involves<br />
a H atom reaction, and the process may well be, for AlCl :- initial<br />
dissociation <strong>of</strong> UC~ to uc13 : H atom reduction <strong>of</strong> UC~, to UCI :<br />
disproportionation 03 fic1 to U and fic13. A maas spectromet+c<br />
examination <strong>of</strong> the species present in the discharge revealed AN1 In<br />
readily detectable quantities but no AlCl was observed. Yields <strong>of</strong> up<br />
to 55% Al metal have been obtained from Ab1 in a single pees through<br />
3<br />
a 2450 mC plasma, and if a second discharge was produced downstreem<br />
more Al deposited. The rate at which AlCl decomposes, with<br />
comparatively low power, and with ita intenae blue plama makes this<br />
reaction one <strong>of</strong> the most spectacular <strong>of</strong> those studied. The relatively<br />
involatile AlF3 also dissociated quite readily to glve the metal,<br />
although it is not easily handled in the experimental 8pparatUe described.<br />
The chloride and fluoride <strong>of</strong> ecandiun behaved in a similar manner to the<br />
corresponding aluminium compounds.<br />
NCl also dissociated readily in the lower frequency apparatus.<br />
It should be Lntioned here that the chief difference between the wavesuide<br />
and the coil-coupled apparatus appear8 to be that in the formar<br />
the electrons are accelerated to higher energlea since the electrostatic<br />
field is concentrated between the resonator walls where a high potential<br />
gradient exists in a direction axial to the discharge tube. By<br />
comparison the E field in a coil is much more randomly distributed.<br />
Rare Earth Comounda. We have not as yet had an opportunity <strong>of</strong> studying<br />
these halides systematically, but preliminary erperiments have shorn<br />
that they behave in a menner similar to AlCl That is, there is only<br />
limited dissociation to metal in an inert c&ier gas, but with hydroem<br />
satlafactory yields <strong>of</strong> metals are obtained. Thae Ce and Le result<br />
from &F3, CeC13, LaB and LaC13. If, for example, chlorides contsirr<br />
ing appreciable quautjties <strong>of</strong> oxychloride are used for the dirsooiation<br />
this inpurity is left in the sample boat and a highly pure metal is<br />
deposited. Such reactions may therefore prove <strong>of</strong> use in the preparation<br />
<strong>of</strong> certain metalm.<br />
2. Markovakii, L. V., Lvova, V. I., Kondrashev, Y. D.,<br />
PO Bora i Ego Sveedin, 36 (1958).<br />
Ber. Tr. KO&.<br />
1<br />
i
I<br />
161<br />
THE GLOW DISCHARGE DEPOSITION OF BORON<br />
A. E Hultquist and M. E. Sibert<br />
Materials Sciences <strong>Laboratory</strong><br />
Lockheed Palo Alto Research <strong>Laboratory</strong><br />
Palo Alto, California<br />
INTRODUCTION<br />
Many ncw materials concepts are evolving out <strong>of</strong> aerospace materials technology. One <strong>of</strong><br />
' the most promising <strong>of</strong> these is that <strong>of</strong> boron filament-reinforced composite structures.<br />
Incorporation <strong>of</strong> continuous boron filament in a suitable matrix could yield a structure with<br />
' the strength <strong>of</strong> high-strength steel, the rigidity <strong>of</strong> beryllium, and the density <strong>of</strong> magnesium.<br />
A large-scale development effort is currently concerned with fabrication <strong>of</strong> such structures.<br />
, Once this has been achieved, large amounts <strong>of</strong> filament must be made available at reason-<br />
able cost to make large-scale usage practical. Present techniques for filament production<br />
' are based on scaled-up laboratory <strong>chemical</strong> vapor deposition procedures, and major tech-<br />
nological development is required to result in large-scale production. As a result, many<br />
i other new and novel techniques for filament preparation have been investigated with the<br />
, intent <strong>of</strong> developing alternate approaches.<br />
This study is concerned with investigation <strong>of</strong> one such approach, that <strong>of</strong> deposition in an<br />
electrodeless glow discharge. Basically a high-voltage, low-amperage, high-frequency rf<br />
currcnl is imposed across a boron-containing gas, resulting in a high degree <strong>of</strong> ionization/<br />
' activation <strong>of</strong> the ions present. Boron then deposits out in elemental form on all surfaces<br />
within the glow discharge area. Use <strong>of</strong> proper deposition conditions confines the glow<br />
largcly to the filament substrate. By passage <strong>of</strong> the filamentary substrate continuously<br />
' through the discharge, the approach is potentially capable <strong>of</strong> high rate filament formation<br />
with excellent uniformity and reliability. Variation <strong>of</strong> current input can result in deposi-<br />
L. tion at any point from room temperature up to 800- 1000°C.<br />
Both metallic and non-<br />
, metallic substrates can be employed. Deposition is achieved at low pressures <strong>of</strong> the order<br />
<strong>of</strong> a few mm.<br />
reaction <strong>of</strong> boron trichloride and hydrogen in a glow discharge. However, efforts to dupli-<br />
cate this work indicate that the conditione employed lead to effects more like an arc than<br />
a glow discharge. Related, although distinctly thermally rather than electrically initiated<br />
I
162<br />
EXPERIMENTAL WORK<br />
Apparatus. The basic apparatus utilized for parametric studies employing a fixed substrate<br />
is shown in Fig. 1. The reaction vessel is a simple pyrex tube with the substrate filament<br />
suspended down the central axis and suitably sealed at each end. The reactant gases are<br />
admitted at one end <strong>of</strong> the tube and exit at the opposite end. Electrodes are simple con-<br />
centric copper strips which may be moved along the tube for optimum positioning. The<br />
glow discharge then forms between these electrodes. Both horizontal and vertical reactor<br />
configurations have been employed; the vertical system is preferred since dubstrate align-<br />
ment is better controlled. The vertical arrangement is also more amenable ,to moving<br />
filament systems for continuous filament preparation. Both air and water cooled reaction<br />
vessels have been evaluated. Water cooling was originally instituted to minimize sidewall<br />
deposition; later work demonstrated that this could be accomplished by regulation <strong>of</strong> gas<br />
composition and current input so as to confine the glow area to the central area <strong>of</strong> the<br />
reaction tube. A three compartment chamber was used so that these experiments could be<br />
performed before the system was opened for examination.<br />
The balance <strong>of</strong> the system is largely a gas flow system. Hydrogen is passed through a<br />
catalytic cartridge and a liquid nitrogen trap for removal <strong>of</strong> oxygen and water impurities.<br />
Boron trichloride is fed into the hydrogen stream. Both lines have appropriate flowmeter<br />
devices. The boron trichloride is controlled at the cylinder valve; satisfactory control<br />
with respect to potential condensation is obtained by bleeding the gas into a section <strong>of</strong> tubing<br />
at operating pressures and heated to 35°C. Hydrogen flow is regulated by a stainless steel<br />
steel needle valve just downstream from the flowmeter. The combined hydrogen-boron ,<br />
trichloride stream then passes into the reaction chamber. Exit gases from the reactor<br />
pass successively through a trap for condensation <strong>of</strong> unused boron trichloride, past a<br />
manometer, through a Cartesian manostat, and thence to a mechanical vacuum pump.<br />
The power supply is a Lockheed-designed 3 Mc oscillator capable <strong>of</strong> about 30 W output;<br />
however, only a portion <strong>of</strong> this output is available as rf power, about 20 W at 1400-1500 V.<br />
The first 20 turns <strong>of</strong> the large coil (Fig. l), and the capacitance connected across them,<br />
act as the tank. The tank is a resonance combination wherein the energy is alternately<br />
stored in the condensers and in the field <strong>of</strong> the coil, with interchange occurring at a fre-<br />
quency determined by the coil inductance and capacitance <strong>of</strong> the condensers. A variable<br />
condenser included in the tank allows its frequency to be matched to that <strong>of</strong> the secondary.<br />
When such matching occurs, the coils are efficiently coupled, and rf power is drawn from<br />
the secondary. This power is fed into the primary by a 6146A transmitter tube activated<br />
at proper frequency by feedback from the tank. This is done through a condenser and a<br />
resistor wound with a coil, serving as a low-Q resonance circuit to suppress high frequency<br />
parasitic oscillation. A 47K resistor allows the grid to be self-biased. An rf choke pre-<br />
vents loss <strong>of</strong> power into the backup power supply. TWO 1OK resistors and a 7500 fl resis-<br />
tors serve as a voltage divider for the screen grid.<br />
Materials. The boron trichloride used was electronic grade supplied by American Potash<br />
and Chemical Co. Hydrogen was a high purity laboratory grade (BB-H-886) supplied by<br />
Air Reduction Co. Quartz mon<strong>of</strong>ilament utilized as a substrate for most <strong>of</strong> the work w88<br />
obtained from the General Electric Co. Glass filaments were drawn from pyrex glass rods<br />
by the LMSC glass shop.<br />
General Procedure. Experiments were started by evacuating the assembled apparatus to<br />
1 mm Hg pressure. The leak rate was determined by turning <strong>of</strong>f the pump and noting the<br />
increase - in Dressure over extended time periods. A leak rate <strong>of</strong> less than 0.05 mm Hg/min<br />
is necessary to avoid boron oxide formation. When the system was determined to be essentially<br />
leak free, a flow <strong>of</strong> hydrogen was introduced and allowed to sweep through the apparatus<br />
for 15 to 30 min. The desired gas pressures and flow rates <strong>of</strong> hydrogen and boron<br />
trichloride were established, the power supply for the oscillator turned on, and the flow Was I initiated by adjusting the variable capacitor. At the end <strong>of</strong> the experiment, the flow discharge<br />
and the boron trichloride flow were turned <strong>of</strong>f and hydrogen allowed to sweep through the
Fig. 1 Process Apparatus<br />
-__l.ll__ I _I_ -<br />
____<br />
Fig. 2 Boron on Glass, Longitudinal Section (3000~)<br />
Boron<br />
B2°3<br />
oxygen<br />
Enriched<br />
or<br />
Oriented<br />
Material
164<br />
apparatus for 10 min to remove unreacted boron trichloride. The pump was turned <strong>of</strong>f and<br />
the apparatus returned to ambient pressure using hydrogen. The apparatus was then dis-<br />
mounted and the sample removed.<br />
RESULTS<br />
Preliminary Experiments. Initial experiments were made to observe operating character-<br />
istics while cxciting hydrogen gas in a glow discharge. Glow <strong>discharges</strong> were initiated in<br />
hydrogen at pressures <strong>of</strong> 6, 15, 36, 40, 60, and 81 mm Hg and at flow rates <strong>of</strong> about 40 to<br />
100 cc/min. Electrode separations <strong>of</strong> 0.75, 1.0, 2.0, 3.5, and 4.5 cm were used. The<br />
glow was initiated by tuning the capacitor at maximum voltage to obtain an input current <strong>of</strong><br />
105 mA. The effect <strong>of</strong> power input to the oscillator on the glow discharge was determined<br />
by varying the voltage. The change in glow is reflected in the current drawn by the oscil-<br />
lator at any particular voltage and capacity setting. The following characteristics were<br />
observed:<br />
rn Increasing gas-flow rates reduces the glow intensity.<br />
0 Increasing pressure reduces th'e glow intensity.<br />
0 In general, glow intensity is reduced as the electrode distance is increased<br />
Glows were maintained in the presence <strong>of</strong> a dielectric and a conductor filament to determine<br />
their effect on glow characteristics. The presence <strong>of</strong> a strand <strong>of</strong> quartz, QFY-150 to 204<br />
end, 1/0, with oil starch finish, d = 0.004 in. , did not affect the operating characteristics<br />
<strong>of</strong> the glow. However, the presence <strong>of</strong> a 5-mil tungsten wire resulted in considerable<br />
changes in the glow. The tungsten wire acted as a ground connection, and the ground lead<br />
from the oscillator could be disconnected without affecting the glow. The glow was con-<br />
centrated at and spread out along the wire. This system used much higher currents and,<br />
as a result, considerable heat was generated. A mirror was deposited on the reaction tube<br />
walls.<br />
A series <strong>of</strong> experiments were performed using glass and quartz substrates to determine<br />
approximate conditions necessary for the deposition <strong>of</strong> boron. A qualitative description <strong>of</strong><br />
the process was obtained from these observations. The data shown in Table I describes<br />
the experimental conditions along with pertinent observations. The results have been<br />
summarized in the following qualitative statements:<br />
Wall deposition is favored by high hydrogen and low BCl3 flow rates.<br />
rn Deposition on the filament substrate is more pronounced at higher flow rates <strong>of</strong><br />
BCl3.<br />
0 Use <strong>of</strong> a water cooled reactor lowers wall deposition.<br />
0 Small leaks <strong>of</strong> air result in the formation <strong>of</strong> the oxide rather than the metal (see<br />
experiment 07-18A and Fig. 2).<br />
0 Under certain conditions <strong>of</strong> power input and gas flow, a brown deposit is formed<br />
that hydrolyzes on exposure to air and evolves a gas (see experiment 07-20A and<br />
07-2lA).<br />
0 Thick deposits <strong>of</strong> boron can be produced in reasonable times and conditions (see<br />
Fig. 3).<br />
Following these preliminary experiments, flowmeters were calibrated and the quartz mono- '<br />
filament substrate was characterized in terms <strong>of</strong> physical and mechanical properties. A<br />
study was then initiated on effects <strong>of</strong> the various process parameters.<br />
Paramctric Study. A study <strong>of</strong> the effect <strong>of</strong> process parameters on the deposition <strong>of</strong> boron<br />
using the static filament apparatus was conducted. The parameters studied include system<br />
pressure, reactant gas flows and ratios, electrode separation and configurations, and field<br />
strengths. The evaluation <strong>of</strong> certain parameters has been quantitative and waa baaed on<br />
the weight <strong>of</strong> boron deposited or on the thickness <strong>of</strong> the deposit at the midpoint <strong>of</strong> the fila-<br />
ment. Other parameters were evaluated by a qualitative deecription <strong>of</strong> the deposit.
v)<br />
t-<br />
at- InIo Io0 t-Q) loo lcua O 0) O ua I<br />
0 0<br />
3 2<br />
oo o<br />
m a
166<br />
(a) Filament Emerging From Broken End (220~)<br />
- W<br />
-' - - ---<br />
(b) Surface <strong>of</strong> Deposit at Broken End (220X)<br />
Fig. 3 Boron on Quartz /<br />
--<br />
I
This study is only an approximation <strong>of</strong> the moving filament system. The movement <strong>of</strong> the<br />
filament in a continuous system averages the effects <strong>of</strong> all the parameters so that the fila-<br />
ment sample is prepared in a uniform manner. The edge effects noted on the filament<br />
situated under the electrodes would not be evident in a continuous system.<br />
Material efficiencies are based on the amount <strong>of</strong> boron produced from the total amount <strong>of</strong><br />
boron passed through the reactor. The electrical efficiency is based on the amount <strong>of</strong><br />
boron produced divided by the theoretical amount that could be produced by the total charge<br />
passed wing the reduction equation B+3 - BO + 3 ~ The . current was measured by 811 rf<br />
ammeter placed in the electrical circuit.<br />
(1) Electrode Configuration<br />
The experiments performed previously have used electrodes made from copper foil. The<br />
electrodes are shown in Fig. 4. It was the purpose <strong>of</strong> one <strong>of</strong> the series <strong>of</strong> experiments<br />
conducted to determine if other electrode configurations would be desirable. The configu-<br />
rations shown in Figs. 4b, 4c, 4d, and 4e were used. The following qualitative observa-<br />
tions were sufficient for determining the best design at this time:<br />
The plate electrodes parallel to the gas flow shown in Fig. 4 produce a glow but<br />
no boron deposition with gas flows and reactant concentrations known to deposit<br />
boron when ring electrodes are used.<br />
0 The large sheet electrodes shown in Fig. 4d encourage the deposition <strong>of</strong> boron on<br />
the side walls between the electrodes.<br />
0 Electrodes, using No. 16 copper wire, hooked up as in Fig. 4a produce a minimal<br />
amount <strong>of</strong> sidewall deposition, and influence <strong>of</strong> the electrodes on the deposit under-<br />
neath is also reduced.<br />
0 Electrodes, using No. 16 copper wire, hooked up as in Figs. 4b and 4c produce<br />
uneven glows and deposits. The glow will be intense between two <strong>of</strong> the electrodes<br />
while the glow between the other electrodes is very faint. The conductivity <strong>of</strong> the<br />
gas phases, electrode separation, and limitation <strong>of</strong> the current output <strong>of</strong> the oacil-<br />
lator are probably important factors.<br />
On the basis <strong>of</strong> the results observed, copper wire electrodes wound around the reactor tube<br />
as in Fig. 4a have been used in the process study.<br />
(2) Electrode Separation<br />
The effect <strong>of</strong> electrode separation on the deposition <strong>of</strong> boron is show in Table II. The gm<br />
flow, reactor pressure, and electrical parameters were the same in all cases. The data<br />
show a rise in efficiency as the electrode separation is increased to 2.54 cm.<br />
Electrode Deposit Deposit<br />
distance length weight<br />
(cm) Icm) (Ccgm/cm)<br />
0.62 0.85 , 35.3<br />
2.22 2.15 23.4<br />
3.16 2.22 25.3<br />
2.64(a) 2.15 46.6<br />
Table II<br />
EFFECT OF ELECTRODE SEPARATION<br />
.<br />
Total Materld<br />
thickness efficiency emciency<br />
1.8 0.34 I 0.76<br />
1.67 0.69 I 1.62<br />
1.72 0.66 I 1.41<br />
2.22 1.16 I 2.66
la<br />
lb H&i GASFLOW<br />
GROUND<br />
IC ‘-0 GAS FLOW<br />
GROUND<br />
GROUND<br />
Id 0 GAS FLOW<br />
le<br />
GROUND<br />
0 GAS FLOW<br />
Fig. 4 Electrode Configuration<br />
0<br />
/<br />
c
Mole Reaction Material<br />
ratio time efficiency<br />
BC13/H2 (min) (96 per pass)<br />
0.22 7.80<br />
0.19 5.0<br />
0. 12 10.0<br />
0.08 20.0<br />
Electrical<br />
efficiency<br />
(% per pms)<br />
0.046 1.2<br />
0.044 1. 1<br />
0. 06 1. 1<br />
0. 14 1.2<br />
As the mole ratio <strong>of</strong> BCl3 to hydrogen is increased from 0.22 to 0.66 (near 8toichiometric)<br />
and by reducing the hydrogen gas fl~w, the appearance <strong>of</strong> the glow and the character <strong>of</strong> the<br />
deposit are changed significantly. The hydrogen flow rate is an estimated value because<br />
the calibration curve does not extend to these low flowmeter readings. However by cutting<br />
the hydrogen flow rate in half, making the flow rate near stoichiometric for the reaction,<br />
the glow is intensified around the filament substrate and will migrate along the subatrate<br />
beyond the rf electrodes. The deposit character produced by this procedure ia shown in<br />
Fig. 40. These pictures show the types <strong>of</strong> deposita that are formed at different points on<br />
the substrate.<br />
The deposition rate ig significantly affected by the flow ratio a8 shown in Table W.<br />
Table IV<br />
EFFECT OF FLOW RATIO ON DEPOSITION RATE<br />
Mole flow ratio<br />
(miWmin)<br />
0.66 (est. )<br />
-- _. . . -_ __ _. - -
1 6<br />
170<br />
Fig. 5 Filaments Produced at Near Stoichiometric Flow<br />
Ratios (220 X)<br />
1<br />
4<br />
I<br />
\,
\<br />
i<br />
(4) Effect <strong>of</strong> System Pressure<br />
The effect <strong>of</strong> system pressure on deposit characteristics is shown in Table V.<br />
Pressure<br />
(mm Hg)<br />
5<br />
20<br />
30<br />
(6) Interaction <strong>of</strong> Parameters<br />
171<br />
Table V<br />
EFFECTS OF SYSTEM PRESSURE I<br />
Type <strong>of</strong> deposit<br />
Needles, trees, and dendrites.<br />
Exploded from substrate if run was long<br />
Thick, fairly uniform, some craters or knobs<br />
Thick, small mounds, no craters<br />
The gas phase being ionized is an integral part <strong>of</strong> the electrical circuit. Thus as gas com-<br />
position, pressure, or electrode spacing is varied, the impedance <strong>of</strong> the gas phaee is ala0<br />
changed. The necessary voltage and rf currents are thus changed. These changes are<br />
reflected in the type <strong>of</strong> deposit obtained.<br />
DISCUSSION<br />
General. The results <strong>of</strong> these experiments show that boron can be deposited on a dielectric<br />
substrate in a glow discharge. There is no limit on the coating thickness inherent in the<br />
process. Complete control <strong>of</strong> the process parameters has not been achieved, but there is<br />
no reason to believe that control cannot be achieved. The results obtained so far indicate<br />
that gas purity, gas composition, and rf power input are among the most important factors.<br />
Gas Purity. The deposit obtained can be considered to be in <strong>chemical</strong> equilibrium with the<br />
gas composition. The introduction <strong>of</strong> certain impurities such as oxygen or water will result<br />
in certain amounts <strong>of</strong> oxygen in the deposit. The solid solubility <strong>of</strong> oxygen in boron is prob-<br />
ably not great, and its effect in the mechanical properties <strong>of</strong> the deposit is unknown. How-<br />
ever, if a certain concentration <strong>of</strong> oxygen or water is present, the equilibrium solid phase<br />
resulting from the processing will be the oxide and not the element. Experimental experi-<br />
ence has shown that this concentration value is probably not very great, and a tight process<br />
system must be used with the reduced pressure process.<br />
Gas Composition. The effect <strong>of</strong> gas composition, i. e. , the ratio <strong>of</strong> hydrogen to boron tri-<br />
chloride, will influence the type <strong>of</strong> reaction that occurs and the type <strong>of</strong> product formed. It<br />
has been observed that the amount <strong>of</strong> (1) sidewall deposition; and (2) fine particles that fall<br />
out <strong>of</strong> the gm downstream <strong>of</strong> the glow, increase with increasing hydrogen concentrations.<br />
This indicates that redlrction in the gas phase increases with the higher hydrogen to boron<br />
trichloride ratios. With increasing hydrogen ratios, the glow is more uniform over the<br />
space and the formation <strong>of</strong> fine particles is encouraged.<br />
Increasing the ratio <strong>of</strong> boron trichloride to hydrogen causes the glow to concentrate toward<br />
I<br />
'<br />
the center mound the substrate. This concentration <strong>of</strong> the glow toward the centerdiscourages
172<br />
the formation <strong>of</strong> small particles by the boron atoms and encourages the growth <strong>of</strong> the coat-<br />
ing by deposition <strong>of</strong> the boron atoms directly on the substrate. As the flow ratio approaches<br />
the stoichiometric value <strong>of</strong> 1.5 moles <strong>of</strong> H2 per mole BCl3, the glow concentrates very<br />
close to the substrate and is observed to migrate along the filament beyond the electrode<br />
rings. At the same time the rate <strong>of</strong> deposition increases about fourfold or more.<br />
The resistance <strong>of</strong> the filament decreases as the boron coating is formed, which allows more<br />
current to flow. This increases the temperature <strong>of</strong> the filament, which encourages more<br />
rapid deposition <strong>of</strong> boron. Under these conditions, the reaction goes out <strong>of</strong> control and the<br />
filament heats to a general red-orange glow with brighter spots and finally breaks.<br />
Careful adjustment <strong>of</strong> the reactant gas concentration and the rf power input to the coil will<br />
minimize the gas-phase reaction, sidewall deposition, and fine-particle formation.<br />
The gas composition also obviously influences the nature <strong>of</strong> the material deposited. The<br />
glow discharge <strong>of</strong> boron trichloride has been studied by several workers (Refs. 1, 5, and<br />
8 to 16) in the field, and their results should indicate the deposit to be expected at high<br />
ratios <strong>of</strong> boron trichloride to hydrogen. Holzmann and Morris (Ref. 1) analyzed the light<br />
from glows <strong>of</strong> boron trichloride at 1 mm <strong>of</strong> Hg pressure. The glow <strong>discharges</strong> were<br />
caused by a 2.4 to 2.5 x lo9 Hz (2,400 to 2,500 Mc). They observed bands due to boron<br />
monochloride and lines due to atomic boron and atomic chlorine. Frazer and Holzmann<br />
(Ref. 9) have used this technique to prepare laboratory quantities <strong>of</strong> B2Cl4. which is the<br />
primary product under these conditions.<br />
The compound B2Cl4 can be decomposed under a variety <strong>of</strong> conditions. Rosenberg (Ref. 8)<br />
used 60-Hz 10-kV <strong>discharges</strong> in B2Cl4 and obtained BC13(g) and (BCl),, which hedescribed<br />
ns a light brown or yellow film which is hydrolyzed by water to form B2O3, hydrochloric<br />
acid, and hydrogen. From the rather incomplete data on this system and from other<br />
authors, it appears that a (BC1)n phase can be obtained at compositions down to (BC10. g)n,<br />
and a material which analyzes as BC10.6 has been reported. At the present time the (BC1)n<br />
material is considered to be a polymer, and a series <strong>of</strong> higher unit structures other than<br />
B2Cl4, (such as B4Cl4) can also be obtained.<br />
In addition to these considerations, experiments have shown that, by operating at reduced<br />
rf currents, a deposit possessing the reported properties <strong>of</strong> (BC1)n can be obtained with<br />
gas compositions that would normally deposit elemental boron.<br />
In summarizing the results and literature survey, it is possible by glow-discharge tech-<br />
niqucs to obtain a variety <strong>of</strong> products from boron trichloride and hydrogen gas streams.<br />
These products can range from a polymer-type (BCl)n, a liquid B2Cl4, or a solid BC10.6<br />
to the boranes. In between these gas compositions that will give the foregoing products is<br />
the gas-composition range that will give baron as a product. In addition to the gas com-<br />
position, the amount <strong>of</strong> rf power used will also determine the product. It is fortunate that<br />
the gas composition obtainable with our experimental apparatus is in the range for boron<br />
deposition, otherwise selection <strong>of</strong> gas composition from present information would be<br />
impossible.<br />
Power Input. The power input to the glow is <strong>of</strong> great importance in determining the product<br />
and character <strong>of</strong> the product. The rf voltage at any gas composition and electrode geometry<br />
must be a value greater than the breakdown potential <strong>of</strong> the gases at that pressure. The rf<br />
current is a direct measurement <strong>of</strong> the ionization <strong>of</strong> the gases to produce electrons and<br />
positive ions. In electro<strong>chemical</strong> processing, these terms are or can be considered analogs<br />
<strong>of</strong> the redox potentials and current density, respectively. At higher currents, a fastercoat-<br />
ing rate will be attained, but changes in deposit characteristics will occur as a result <strong>of</strong><br />
such changes in plating rate. Perhaps part <strong>of</strong> the nonuniformity <strong>of</strong> the deposit can be<br />
attributed to a nonuniform plating rate. The uniformity should be improved by moving the<br />
filament substrate through the glow.<br />
/<br />
i
173<br />
Pressure. Since pressure is a concentration function, it may be expected to influence the<br />
type <strong>of</strong> deposit and the rate <strong>of</strong> deposition. At low pressures, needle-like deposits are<br />
observed. As the pressure is increased, the type <strong>of</strong> deposit changes and a coating or continuous<br />
layer is formed. Increasing the pressure further provides a smoother deposit.<br />
However as the pressure is increased the voltage necessary to maintain a glow discharge<br />
is increased.<br />
I<br />
Post-Deposition Treatment. One sample prepared at 30 mm Hg pressure, and at flowratios<br />
and electrical conditions which normally provide a typical coating, was exposed to a hydro-<br />
gen glow discharge before removal from the system. This was done to explore possible<br />
methods <strong>of</strong> reducing the brown, glassy material observed near the end <strong>of</strong> filaments. The<br />
treatment was done in 30 mm Hg H2 pressure and at low flow rates - 25 cc/min. The rf<br />
current and dc voltage were 250 mA and 480 V respectively.<br />
The glow was normal in every respect except that just outside the electrode on the down-<br />
stream side a faint green fluorescence or glow was observed. The end areas <strong>of</strong> the sample<br />
did show some evidence <strong>of</strong> reduction. The most interesting part <strong>of</strong> the sainple is shown in<br />
Fig. 6. This is a longitudinal cross section <strong>of</strong> a fracture. Averythin (lessthan 0.187-mil)<br />
layer is much darker than the bulk <strong>of</strong> the deposit. This has not been observed in any sam-<br />
ples prepared during this process study.<br />
FILAMENT EVALUATION<br />
Density. The density <strong>of</strong> filaments produced by this process has been measured by two tech-<br />
niques. A direct measurement was made by dropping a boron-coated pyrex rod in adensity<br />
gradient tube. The average density was 2.24 gm/cc, which agrees satisfactorily with the<br />
value estimated from the density <strong>of</strong> boron, 2.35 gm/cc, and pyrex, 2.24 gm/cc.<br />
An alternate indirect measurement has been performed during the process study by com-<br />
paring the observed thickness <strong>of</strong> samples with that calculated-from-weight measurements.<br />
It was anticipated that the optically-measured value would be higher than the value calcu-<br />
lated from weight measurements.which is an average value,since it is measured near the<br />
center <strong>of</strong> the filament.<br />
This’is observed in the data for samples 07-42 26A and,B. The total thickness measured<br />
by optical methods are 2.2 and 2.4 mils, and calculated by weight the thicknesses are 1.45<br />
and 1.72 mils. The results illustrate the nonuniformity <strong>of</strong> the static filament samples.<br />
However, the data is sufficiently in agreement to indicate that the filaments produced<br />
possess a density in accordance with the relative amounts <strong>of</strong> boron deposit and quartz<br />
substrate.<br />
X-Ray Identification. A typical x-ray analysis is shown in Table VI. The diffuse lines<br />
observed in the x-ray pattern correspond to the alpha rhombohedral structure. This is<br />
the low-temperature form <strong>of</strong> boron. However, the structure is not well developed as<br />
indicated by the broad lines. The boron may have been in the process <strong>of</strong> changing from<br />
an amorphous structure to a crystalline structure at the time the experiment was termi-<br />
nated, or the crystalline boron was mixed with a matrix <strong>of</strong> amorphous boron.<br />
Other samples <strong>of</strong> boron-coated 1.0-mil quartz filament show only two broad diffuse halos.<br />
No lines indicating B2O3 were seen. No positive identification <strong>of</strong> the material was possi-<br />
ble from the data.<br />
Tensile Strengths. Several sample filaments <strong>of</strong> boron-coated 1-mil quartz substrate have<br />
been tested for ultimate strengths. No attempts have been made to obtain an elastic modulus<br />
since the samples prepared in the static apparatus are not <strong>of</strong> sufficient length. These<br />
data are presented in Table VII. The coating cracked and in some cases completely exfoliated.<br />
Thus, the values reported here basically represent breaking strengths <strong>of</strong> quartz<br />
rather than composite filament <strong>of</strong> boron-coated quartz mon<strong>of</strong>ilaments.
Fig. 6 Longitudinal Cross Section <strong>of</strong> Deposit After<br />
Exposure to H2 Glow Discharge<br />
$ 4 -<br />
I<br />
! I-<br />
@)<br />
Fig. 7 (a) Longitudinal and (b) Cross Section <strong>of</strong> Boron-Coated<br />
Quartz Mon<strong>of</strong>ilament (220 x)<br />
-k<br />
I
175<br />
Table VI<br />
X-RAY DATA OF DEPOSIT<br />
Unknown intensity<br />
(a) Alpha rhombohedral form.<br />
Table WI<br />
BREAKING WEIGHT OF SELECTED SAMPLES<br />
Sample Sample condition Breaking Filament<br />
Number prior to test weight (lb) diameter (mile)<br />
07-40-24 Not observed 0.07 1.5<br />
07-42-26 Not observed 0.07 1.5<br />
07-52-36 Longitudinal cracks 0. 11 2.0<br />
07-53-36 Coating missing near tab 0.033 2.0<br />
Coating Evaluation. A typical sample cross section prepared during the process study is<br />
shown in Fig. 7, demonstrating that acontinuous coating can be successfully deposited. In<br />
the longitudinal cross section two mounds are fortuitously shown and it can be seen that<br />
the growth extends to the substrate. This indicates that the cause <strong>of</strong> this different growth<br />
originates at the substrate surface, possibly as a microcrack or particle <strong>of</strong> dust. The<br />
problem <strong>of</strong> handling the filament and its contamination would be minimized with use <strong>of</strong> a<br />
continuous system.<br />
The bond between the coating and the substrate is essentially mechanical in nature. It<br />
would be very desirable to improve this by effecting some sort <strong>of</strong> reaction between the two<br />
materials to improve the overall properties <strong>of</strong> the filament. It would perhaps be undesir-<br />
able to clean the organic or silicone finish from the substrate as has been done,<br />
Microscopic examination <strong>of</strong> one <strong>of</strong> the filaments,has revealed a possible explanation <strong>of</strong> the<br />
reduced strength <strong>of</strong> boron-coated quartz filaments. The pictures in Fig. 8 are typical <strong>of</strong><br />
what is occasionally seen on samples. The chip and hole in the top and middle picture<br />
were not measured, but the rather deep cut made indicates that the quartz mon<strong>of</strong>ilament<br />
possesses weak portions that may separate before the bond between the boron and the<br />
quartz ruptures. These weak volumes contribute to the spread <strong>of</strong> the strength values<br />
observed earlier since they are probably randomly distributed. The hole and ball shown<br />
in the bottom figure have been measured during the microscopic examination. The ball<br />
W- &at 1.0 mil in diameter, the hole WBB observed to 0.8 mil, while the filament cram<br />
eection averaged 1.8 mils. The coating thickness can be only 0.4 mil, so that the hole<br />
must have been formed by the loss <strong>of</strong> 0.4 mil <strong>of</strong> the quartz substrate.
4<br />
176<br />
Fig. 8 Boron-Coated Quartz Mon<strong>of</strong>ilament (x 340)<br />
Top - Hollow in Boron-Coated Quartz Mon<strong>of</strong>ilament<br />
Middle - Material From Hole<br />
Bottom - Ball and Hollow in Thick Coating on Quartz<br />
Mon<strong>of</strong>ilament<br />
1<br />
i
177<br />
Another source <strong>of</strong> weakness is the cracking and peeling. Many <strong>of</strong> these cracks and breaks<br />
are the result <strong>of</strong> handling during removal from the static apparatus. Use <strong>of</strong> a continuous<br />
filament moving through the apparatus and improvement in the adhesion <strong>of</strong> the Coating to<br />
the substrate would possibly eliminate this problem.<br />
CONCLUSIONS<br />
This paper basically represents a feasibility study <strong>of</strong> the deposition <strong>of</strong> d o n from a glow<br />
discharge system produced in a boron trichloride-hydrogen medium. Feasibility <strong>of</strong> the<br />
basic concept has been conclusively demonstrated for dielectric or insulating substrates.<br />
It is probable that conditions can be adjusted to provide for deposition on metallic or conductive<br />
substrates as well. The following secondary conclusions can also be drawn from<br />
this work:<br />
0<br />
0<br />
'0<br />
0<br />
e<br />
0<br />
0<br />
0<br />
0<br />
Glow <strong>discharges</strong> can be initiated a d sustained in hydrogen-boron trichloride mix-<br />
tures at pressures <strong>of</strong> 50-100 mm.<br />
There is no apparent limit on coating thickness which can be deposited.<br />
The deposited boron is amorphous with a partial alpha rhombohedral crystalline<br />
character.<br />
Deposition rate is <strong>of</strong> the order <strong>of</strong> 5 x 10-5 gm/sec.<br />
Filament resistance increases with thickness; temperature in turn increases<br />
resulting in a higher deposition rate; this also results in increased bodning to the<br />
substrate by <strong>chemical</strong> or diffusive action.<br />
The process is quite sensitive to small amounts <strong>of</strong> impurities in the gas stream,<br />
particularly oxygen and moisture.<br />
The ratio <strong>of</strong> hydrogen to boron trichloride has a major effect on nature <strong>of</strong> the<br />
deposit; both sidewall deposition and downstream particle fallout increase with<br />
hydrogen concentration. However, the glow is more uniform at high hydrogen<br />
values, so these factors must be balanced for optimum performance.<br />
An increase in the boron trichloride to hydrogen ratio tends to concentrate the<br />
glow around the substrate, promoting efficiency and minimizing extraneous<br />
deposition.<br />
High hydrogen to boron trichloride ratios tend to produce boranes; operation at<br />
reduced rf currents tends to yield (BCl), polymers.<br />
Current investigations are concerned with further development <strong>of</strong> the glow discharge deposi-<br />
tion procedure to operate on a continuous basis utilizing moving filament techniques. Indica-<br />
tions are that the technique may be used to prepare a high-quality boron filament at<br />
reasonable processing rates.<br />
ACKNOWLEDGMENT<br />
This work was sponsored by the U.S. Air Force under Contract AF 33(615)-2130. Acknowl-<br />
edgement is given to J. G. Bjeletich, W. C. Coons, J. Robinson, B. A. Traina, R. N.<br />
Varney, and R. D. Wales for assistance on photographic and experimental portions <strong>of</strong> the<br />
program. All are members <strong>of</strong> the Lockheed Palo Alto Research <strong>Laboratory</strong>. Further<br />
acknowledgment is made to R. M. Neff <strong>of</strong> the Air Force Materials <strong>Laboratory</strong> for con-<br />
tinuing consultation on progress <strong>of</strong> the program.<br />
1.<br />
2.<br />
3.<br />
4.<br />
REFERENCES<br />
R. T. Holzmann and W. F. Morris, J. Chem. Phys. , Vol. 29, 1958, p. 677<br />
W. V. Kotlensky and R. Schaeffer, J. Am. Chem. Soc., Vol. 80, 1958, p. 4517<br />
R. M. Rosenberg. Univ. Micr<strong>of</strong>ilms (Ann Arbor, Mich. ) L. C. Card No. MIL-59-2911.<br />
160 pp: Dissertation Abst., Vol. 20, 1959, p. 526<br />
L. Ya. Markovskii, V. I. L'viva, and Yu D. Kondrashev, "Obtaining Elementary<br />
Boron in a Glow Discharge." Bor. Trudy Konf. Khim, Bora; Ego Soedineii, pp. 36-45<br />
(1958); cf. FTD-MT-64-427, Foregin Technol. Division, AF System Command, 1985
5. F. Weintraub, Trans. Am. Electrochem. Soc., Vol. 21, 1909, pp. 165-184 ,<br />
6. W. Kroll, 2. Anorg. Allgem. Chem., Vol. 101, 1918, p. 1<br />
7. "Report <strong>of</strong> the International Symposium on Electrical Discharges in Gases," Delft,<br />
Netherlands, April 1955; The Hague, 1955<br />
8. R. M. Rosenberg, PhD Thesis, Pennsylvania State University, 1959<br />
9. J. W. Frazer and R. T. Holzmann, J. Am. Chem. Soc., Vol. 80, 1998, pp. 2907-2908<br />
10. T. Wartik, R. Moore, and H. I. Schlesinger, J. Am. Chem. Soc., Vol. 71, 1849,<br />
p. 3265<br />
11. G. Urry-, T. Wartfk, and H. I. Schlesinger, J. Am. Chem. SOC., Vol. 76, 1954,<br />
p. 4223<br />
12. A. Stock, A. Brandt, and H. Fischer, Ber. Deut. Chem. Ges., Vol. 58B, 1925, p. 653<br />
13. H. I. Schlesinger and A. 3. Bing, J. Am. Chem. Soc., Vol. 53, 1931, p. 4321<br />
14. A. Stock, H. Martin, and W. Sutterlin, Ber. Deut. Chem.. Gee., Vol. 67, 1934, p. 396<br />
15. F. F. Apple, PhD Thesis, Pennsylvania State University, 1955<br />
16. H. I. Schlesinger, H. C. Brown, BI Abraham, N. Davidson, A. E. Finholt, R. A. Lad,<br />
J. Knight, and A. M. Schwartz, J. Am. Chem. Soc., Vol. 75, 1953, p. 191<br />
L
179<br />
PLATING IN A CORONA DISCHARGE<br />
R. D. Wales<br />
Materials Sciences <strong>Laboratory</strong><br />
Lockheed Palo Alto Research <strong>Laboratory</strong><br />
Palo Alto, California<br />
ABSTRACT<br />
It has been demonstrated that boron can be deposited on a suitable<br />
substrate in a corona discharge. Application <strong>of</strong> high-voltage electri-<br />
cal energy across a gaseous hydrogen-boron halide mixture forms<br />
ionized/activated states, resulting in the deposition <strong>of</strong> metallic boron.<br />
The deposition process is electro<strong>chemical</strong> in nature, the boron being<br />
deposited cathodically.<br />
INTRODUCTION<br />
Literature on electric <strong>discharges</strong> is quite extensive. However, most <strong>of</strong> this iiterature concerns<br />
the nature and properties <strong>of</strong> <strong>discharges</strong> taking place in stable as systems. Dctailed<br />
discussions <strong>of</strong> electrical <strong>discharges</strong> are presented in several books(f* 2* 39 4). Relatively<br />
little information is available concerning <strong>chemical</strong> reactions initiated and sustained by<br />
clectrical <strong>discharges</strong>. Arc reactions are basically thermally initiated reactions deriving<br />
their thermal energy from the arc and thus are not applicable in the present sense. Glow<br />
<strong>discharges</strong> have been utilized for the polymerization <strong>of</strong> organic materials and the deposition<br />
<strong>of</strong> oxide films. The formation <strong>of</strong> pure boron powded5) and the decomposition <strong>of</strong> diboratd)<br />
and boron tri~hloride(~) have been studied in a glow discharge. No references have becn<br />
located concerning <strong>chemical</strong> reactions initiated and sustained by a corona discharge.<br />
Those references(*$ 99 lo) which refer to the utilization <strong>of</strong> a corona discharge to catalyze,<br />
or cause, a <strong>chemical</strong> reaction do not in fact utilize a corona discharge. The discharge<br />
utilized in these references has an odd resemblance to a glow discharge, but is actually a<br />
suppressed spark. Thus, the discharge goes through the phenomena <strong>of</strong> spark buildup without,<br />
however, generating a spark. Direct current cannot be used in these systems since<br />
the buildup <strong>of</strong> charge at the insulated electrodes quenches the discharge, and a reverse<br />
cycle is necessary to remove this charge and reinitiate the discharge.<br />
A corona discharge is, essentially, intermediate between a glow and an arc discharge<br />
being, however, a low current phenomenon. Glow <strong>discharges</strong> are generally generated at<br />
low pressures while an arc is generally a high-pressure phenomenon. The gas is ionized<br />
more or less uniformly throughout the system in a glow discharge. An arc is obtained<br />
when a narrow path <strong>of</strong> ionized particles is generated between two electrodes, resulting in a<br />
low resistance path which further aggravates the situation until extremely energetic particles<br />
exist in the narrow path. A corona discharge is not as pressure-dependent and is obtained<br />
at a small electrode which is opposed by a much larger electrode. There is a very large
160<br />
change in field through the corona, but very little change over the remaining distance to the<br />
opposing electrode. Most studies <strong>of</strong> corona <strong>discharges</strong> have been in inert gas systems, and<br />
have been generated at a point electrode.<br />
This study has been directed toivard the tletcrmination <strong>of</strong> the feasibility for depositing :1<br />
coating, c.g., boron, on a suitablc substrate in :I corona discharge. A further object has<br />
becn to determine some <strong>of</strong> the critical pnramctcrs and a possible indication <strong>of</strong> the mechanism<br />
and characteristics <strong>of</strong> this technique for coating the substrate material. ;<br />
EXPERIMENTAL WORK<br />
A schematic diagram <strong>of</strong> the system used in this study is indicated in Fig. 1. The system<br />
pressure was controlled with a Cartesian Manostat GA (Manostat Corporation) and a Welch<br />
mechanical pump Model 1402 (W. M. Welch Manufacturing Company). The discharge was<br />
initiated and sustained with a 7,000/12,000 V Jefferson luminous tube outdoor-type trans-<br />
former (Jefferson Electric Company) connected to GO-cps 110-V power source through a<br />
Variac variable transformer.<br />
To dcterrnine the effect <strong>of</strong> alternating and direct current, an RCA CR 212 half-wave recti-<br />
fier \vas added to the circuit. At the same time a resistance bridge, a current measuring<br />
rcsistor, and an ammeter were added to the circuit. A schematic <strong>of</strong> the circuit is presented<br />
in Fig. 2. The resistance bridge consisted <strong>of</strong> six 2-W 2-megohm resistors (R2) and one<br />
2-\V 1,000-ohm resistor .(R1), all in series; the total voltage being 11,775 times the voltage<br />
across the 1,000-ohm resistor. The current measuring resistor ,was a 2-W 10.075-ohm<br />
resistor. The voltage measurements were made with a Hewlett-Packard Model 130B<br />
oscilloscope, and alternating current measurements were made with a Simpson Model 378<br />
milliammeter.<br />
The reactants utilized in this study were hydrogen, helium, or argon, and boron tribromide.<br />
The hydrogen was passed through a ffDe-oxo'7 unit (Engelhard), through a drying column <strong>of</strong><br />
magnesium perchlorate, and then through a flowmeter. After the flowmeter, the hydrogen<br />
was split into two streams, one <strong>of</strong> which was bubbled through the boron tribromide, and<br />
then recombined into one stream before entering the reaction chamber or cell.<br />
The temperature <strong>of</strong> the bubbler, and <strong>of</strong> the cell, was controlled by wrapping each (and the<br />
associated gas lines) with heating tape and controlling the power input with a Variac variable<br />
transformer.<br />
The system was maintained at or below atmospheric pressure for this study, with most <strong>of</strong><br />
the data being obtained at 2 to 3 in. <strong>of</strong> mercury below atmospheric pressure.<br />
Tungsten wire was chosen as the substrate material, This wire was cleaned by passing it<br />
successively through a train consisting <strong>of</strong> concentrated nitric acid, a distilled water rinse,<br />
and an acetone rinse.<br />
Using the hydrogen-boron tribromide reactant system and 60-cps alternating current to<br />
develop n corona, approximately a dozen cell designs were tried. The cell design which 1<br />
appears to be the most satisfactory is pictured in Fig. 3. This cell consists <strong>of</strong> a 2-in.long<br />
pyrex glass tube with a neoprene O-ring inside each end and wrapped with eight turns<br />
<strong>of</strong> 3-mil tungsten wire along the tube length such that the wire is parallel and concentric<br />
to the axis <strong>of</strong> the tube. The wire is 0.9 cm from the axis. This assembly is then inserted<br />
in a 5-in.-long pyrex glass tube 1.5-in. in diameter and supported such that the tubes are<br />
concentric. A tungsten wire lead is then extended out <strong>of</strong> the tube, and neoprene rubber<br />
stoppers inserted in each end. Each stopper has a capillary tube through its center such<br />
that there are approximately 3 in. between the capillaries, and the filament enters and<br />
leaves the cell through these capillaries. The reactants also enter (and leave) the cell<br />
i!<br />
through the stoppers.<br />
I<br />
4<br />
1<br />
,<br />
I
\<br />
3<br />
,<br />
,\<br />
110 v<br />
60 CPS<br />
MANOMETER .<br />
Fig. 1 Schematic <strong>of</strong> System Utilized for Plating in a Corona Discharge<br />
7 300 TO 15 000 V<br />
TRANSFORMER<br />
VARIAC<br />
MILLIAMMETER<br />
Fig. 2 Schematic <strong>of</strong> Power Input and Measuring Circuit Used With the Corona<br />
I' Discharge Plating Apparatus<br />
i<br />
I<br />
I
Deposition Utilizing Altcrnating Current<br />
1s2<br />
PRIAIXRT \'XRI;\DI,E COXSIDERATIONS<br />
Irydrogcn and boron trihronude vapors iverc used to obt:iin a co:iting <strong>of</strong> boron on 1-niil<br />
tungsten irirc in a corona dcvelopctl ivith (X)-c.ps alternating currcnt. It \vw determined th;it,<br />
at about 70 and 350 mA/cni2, no coating is ol,t:lined irhen the cell (and reactants) are at room<br />
tcniperature, \rhilc a co:iting <strong>of</strong> boron is obtnincd if the temperature is raiscd to approximately<br />
100" C. At highcr currcnt densities, the coating is concentrated in nodules. If enough power<br />
is supplied to heat the filament to a dull red glow, :I good coating is obtained in the hot zone,<br />
At lower current densities, a more uniform coating is obtained, depending upon the reactant<br />
ratios and flow rates. Thus, hydrogen was bubbled through boron tribromide at a rate <strong>of</strong><br />
approximately 400 ml/min, and the system \viis maintained at approximately atmospheric<br />
pressure. When the boron tribromide \vas maintained at about room temperuture and at a<br />
total hydrogen flow rate <strong>of</strong> about, 1.5 liter/min, thc corona was discontinuous along the wire<br />
and boron deposited in the area <strong>of</strong> the coronas to give discrete coated and uncoated areas.<br />
With a hydrogen flow rate <strong>of</strong> about 400 ml/min and a plating time <strong>of</strong> about 10 min, a much<br />
smoother coating was obtained. There was some tendency for the corona to break into<br />
discontinuous sections, which resulted in a nonuniform buildup <strong>of</strong> boron.<br />
When the boron tribromide was heated to about 30" C and the hydrogen flow rate was main-<br />
tained at 400 ml/min, the corona was somewhat more continuous and the coating nearly<br />
uniform. However, the coating was not nearly as smooth as in the previous example.<br />
There was also a tendency for the formation <strong>of</strong> corona points on the opposing electrode<br />
system with a resulting deposition <strong>of</strong> boron under the corona.<br />
Deposition Utilizing Direct Current<br />
Using hydrogen and boron tribromide as reactants to obtain a deposit <strong>of</strong> boron on 1-mil<br />
tungsten wire in a corona developed with half-wave rectified 60-cps alternating current,<br />
no deposit was obtained when the 1-mil wire was the anode. However, a coating was<br />
obtnincd when the 1-mil wire was the cathode. Good, continuous corona and boron deposits<br />
were obtained when the corona was initiated in a large excess <strong>of</strong> hydrogen (the hydrogen<br />
flow was then decreased to the desired amount after about 5 min).<br />
The coating is quite uniform and has an orange peel appearance at 2,OOOX magnification.<br />
Some <strong>of</strong> the morphology <strong>of</strong> the substrate is apparent at high magnification (e.g., die marks).<br />
The voltage requirements for a particular current increased as the hydrogen flow decreased,<br />
increasing from more than 1,000 V peak to about 7,000 V peak as the excess hydrogen was<br />
decreased in these runs.<br />
Attempts to utilize pure de power were unsuccessful. The power supplies available were<br />
designed to deliver several hundred milliamperes and had relatively coarse controls. Thus,<br />
as the corona was initiated, the power supplies did not allow sufficient control, resulting in<br />
"runaway" or burning in two <strong>of</strong> the substrate wire.<br />
Deposition Utilizing Argon and Helium<br />
Using helium instead <strong>of</strong> hydrogen, a coating was obtained, but arcing was much easier and<br />
thus lower current (and lower flow rates) were necessary. Peak voltages <strong>of</strong> 7,000 to 8,000 v<br />
were used.<br />
Using argon instead <strong>of</strong> hydrogen required higher voltage (with lower currents) and resulted in<br />
the formation <strong>of</strong> corona points under which boron was deposited to give whiskers. Peak<br />
voltages <strong>of</strong> about 9,000 V were used. Whiskers greater than 6 mils long and about 0.5 mil<br />
in diameter were obtained before discontinuing these experiments.<br />
I<br />
.1<br />
I!
I<br />
i<br />
, \<br />
I<br />
i<br />
& a -<br />
P<br />
Fig. 3 Reaction Cell Used for Plating in a Corona Discharge<br />
Fig. 4 Boron Deposit on 1-mil Tungsten Wire, Run 28-1,<br />
Tungsten Substrate Material at Top (340x)<br />
Fig. 5 Boron Deposit on 0.5-mil Tungsten Wire,<br />
RLUI 35-1 (450X)
184<br />
Bromine was a product when helium or argon was used while hydrogen caused the formation<br />
<strong>of</strong> hydrogen bromide.<br />
Discussion<br />
jl) Cell Design. The reaction cell should be designed so that the concentration <strong>of</strong> reactants<br />
and products is essentially constant throughout the cell to give a more uniform corona and<br />
deposit. The electrode geometry and separation are to some extent depended upon the power<br />
requirements, which are in turn dependent upon the resistance heating effects in the filament<br />
and upon the arcing probability. Thus, in practice, the current in the filament should not be<br />
great enough to heat the filament excessively. With this current limitation, the electrode<br />
separation is dependent upon the arcing probability and should be great enough to'level out<br />
any small variations in electrode separation which could cause the current to concentrate in<br />
a small region. Furthermore, to obtain a unfirom corona around the filament, the opposing<br />
electrode should be concentric around the filament. If the power input is not pure direct<br />
current, a "motoring" effect results if the opposing electrode is a wire coiled around the<br />
filament. That is, a vibration or circular oscillation will be induced on the filament. There-<br />
fore, the opposing electrode has been made up <strong>of</strong> a group <strong>of</strong> connected electrodes arranged<br />
concentrically around the filament and parallel to it in order to minimize the "motoring"<br />
effect. The opposing electrode could also have been a metal cylinder or screen, but in order<br />
both to observe the filament and be able to operate at reasonable electrode separations<br />
(reasonable voltages), interconnected parallel wire electrodes were selected.<br />
12) Mechanism <strong>of</strong> Deposition. The results obtained indicate that the mechanism <strong>of</strong> boron<br />
deposition in a corona discharge is cathodic. In this type system, electrons are generated<br />
and repelled from the cathode while positive ions are drawn to it. At the anode, positive ions<br />
are generated or electrons are drawn to it. Thus, the boron radical is positively charged and<br />
drawn to the cathode where it is deposited as elemental boron.<br />
Helium and argon were used to determine if the boron tribromide was ionized directly, or if<br />
its ionization was a secondary reaction. Table I indicates some <strong>of</strong> the ionization levels for<br />
these gases. Helium in a corona discharge has only one or two potential (or ionization) levels,<br />
and these are quite high. However, helium is ionized at lower applied voltages than most<br />
gases because the electrons generated at the cathode have only elastic collisions with helium<br />
atoms until they acquire enough kinetic energy from the field to ionize the helium. Argon has<br />
a potential (or ionization) level which is relatively low (about half that <strong>of</strong> helium) but higher<br />
than the ionization levels for hydrogen. In helium the system was very susceptible to arcing,<br />
while in argon higher voltages were required with some susceptibility to arcing. In hydrogen<br />
arcing was not such a problem because <strong>of</strong> the many types <strong>of</strong> inelastic collisions possible and<br />
the consequent tendency to decrease the electron energy and concentration. Boron was<br />
deposited in all three systems. However, in hydrogen, hydrogen bromide was obtained while<br />
bromine was obtained in both argon and helium. Deposition was slightly easier and there<br />
was a slightly increased rate <strong>of</strong> deposition <strong>of</strong> boron with an increased tendency to arcing in<br />
helium and argon. The difference, however, was not sufficient to be due to direct reaction<br />
with the free electron, and indicates that the boron tribromide is ionized by collision with<br />
ionized particles rather than electrons or the field. Thus, the mechanism includes: (a) the<br />
ionization (or activation) <strong>of</strong> the hydrogen (helium or argon), @) the collision or exchange <strong>of</strong><br />
energy <strong>of</strong> the ionized particle with boron tribromide to result in (c) the formation <strong>of</strong> a<br />
positively charged boron radical and hydrogen bromide (or bromine), (d) the transport <strong>of</strong><br />
the charged boron radical to the cathode in the field, and (e) the electro<strong>chemical</strong> deposition<br />
<strong>of</strong> boron on the cathode.<br />
It is apparently necessary to add a small amount <strong>of</strong> kinetic (thermal) energy to aid the<br />
deposition in hydrogen. However, this reactant system <strong>of</strong>fers certain advantages Over<br />
helium or argon systems. That is, with hydrogen, there are apparently more ionized or<br />
activated particles with fewer electrons and consequently less likelihood <strong>of</strong> creating<br />
conditions conducive to arcing.
\<br />
\'<br />
C<br />
0<br />
.d *<br />
d<br />
.-<br />
C<br />
U<br />
d<br />
0)<br />
c(<br />
1)<br />
L-<br />
. . . .<br />
I n w I n c u<br />
O d d 0<br />
N d d d<br />
e 8 m
125 I<br />
I The C'orol;:~. In tiic Iiigii-iicld rcfiioii ni':~~' the' sul,..;trnte wire. positive ions can nttain<br />
high encrgics :ind ttic sccond:ir!- clc~ctron cmission process is quite clficient. 'rhese<br />
secont1:iry electrons \\-ill start electron avalanctics. \[.hick \vi11 iti' turn produce rna~iy:positive<br />
I<br />
I<br />
I<br />
ions, \vhich protlucc niorc :cv:il:inchcs, ctc. ;\t lo\\ ~)rcssur~'s, tlilf~ision ol thc electron<br />
avatanctics sprcati..; thc glo\v anti tiistril)utcs !lie iiositive sixice cti;irge so that ihc discharge<br />
is not yucnchcti. 1)ifiusion docs not talic p1:icc at higher pressure, and ;I localized dense<br />
!<br />
sixicc charge estinguishcs 'thc corona. 'l'he extinction lnsts until the positivc space ch;!rge<br />
tiiffuscs to ttic clcctrode and the last ions rcinitiiite ttic ~ischargc, resulting in ;t periodic<br />
coron:i \vith a frequency dependent upon the ficltl ~uid the velocity <strong>of</strong> thc positiveions.<br />
1<br />
\\'itti the ncgativc wirc (cathode), the ionization increases first with distance from the wire,<br />
rcaches ;I ~iculr, then drops sharply. ?'hat is, the electrons move out'from thc wire and<br />
produce relatively stationary positive ions I)ct\vecn thcniselves and the wire, thus weakening ,<br />
the field at greater distances. The ninsimum ioniz:ition occurs ;it several ionizing; free paths .<br />
from the \vim, antl there is ;I dark space Iict\veen thc wire and the luminous glow. Furtherniorc,<br />
as mentioned, at low pressures the lxtcrnl diffusion <strong>of</strong> electron avalances spreads the<br />
coron;~ over the wirc surfncc. Uecausc the vnluc <strong>of</strong> the coefficient <strong>of</strong> electron epission is<br />
iniport:int. antl because the glow will not be uniform if the coefficient <strong>of</strong> electron emission<br />
varics over the surface <strong>of</strong> the wire, the glow may settle in p?tches <strong>of</strong> greater or lesser , '.<br />
luminosity and at high pressures will appear :IS a single small area <strong>of</strong>.corona. The corona<br />
niny cshibit a marked flickering near threshold because the positive ion bombardment :may<br />
changc the effective coefficient <strong>of</strong> electron emission by denuding the surface <strong>of</strong> its gas film..<br />
The discharge thus decreases or ceases in that rcgion, resuming again when the surface Ins<br />
recovered. At low pressures and voltages well above the threshold value, the corona is fairly,<br />
steady.<br />
In this work it was necessary to clean the substrate wire so that there were no variations in<br />
thc,coefficient <strong>of</strong> electron emission along the wire, and thus.a localization <strong>of</strong> the corona into'<br />
points. During deposition the corona was distributed over the substrate wire in a uniform<br />
manner. However, the discharge was periodic as indicated in the above discussion. That is,<br />
the shcnth (<strong>of</strong> light) dong the wire was not <strong>of</strong> a uniform brightness, but vnsisted <strong>of</strong> brighter<br />
nncl darker areas which seemed to move along the wire in a somewhat random manner,ibut<br />
which appeared periodic in nature at a given point on the wire.<br />
I.:s pe r i men t a1 \V or k<br />
SECONDARY VARIABLE CONSIDERATIONS<br />
The deposition system was that previously described. The tungsten wire was cleaned as<br />
before, thcn placed in the cell. The corona was then initiated in excess hydrogen and mintained<br />
for about 5 min, or until the corona was continuous or nearly continuous along the<br />
substrate electrode, before the conditions were changed to give boron deposition. This<br />
initial "cleaningt1 process was effected with hydrogen flowing at about 16Occ/min through<br />
the boron tribromide at about 26" C and with hydrogen flowing through the bypass line at<br />
about 270cc/min. The electrical input during this time was about 17 mA peak and about .<br />
4,700 V peak for 1-mil tungsten wire and about 5,600 V peak and 4.5 and 11.0 mA peak<br />
for 0.5-mil tungsten wire.<br />
Results<br />
11) Deposition on 1-mil Tungsten Wire. Some <strong>of</strong> the results obtained are presented in<br />
TaMc 2. The corona was maintained continuously in all run.s. A picture <strong>of</strong> the fihment<br />
olitained in Run 28-1 is shown in Fig. 4. In Run 28-1 the current fell from an approximate<br />
initial value <strong>of</strong> 12.3 mA peak to about 3.. 5 mA peak in about 9 min, after which it was<br />
nearly constant. The peak voltage changed from 6,900 to 13,190 V. The filamedC obtained<br />
in this run is apparently the desired type, although not <strong>of</strong> optimum thickness.<br />
,<br />
. '<br />
'<br />
1<br />
4<br />
.J<br />
6.<br />
. , i<br />
I
I<br />
i<br />
I .<br />
Q)<br />
I,<br />
s<br />
w<br />
0<br />
v)<br />
Y<br />
0<br />
a<br />
w<br />
W<br />
a<br />
!<br />
rn<br />
cu<br />
I<br />
0<br />
W<br />
rl<br />
187<br />
c) In I SI SI<br />
0 0 * 00 00 00<br />
*In .ma
188<br />
obtained in the experiments presented, and in other similar experiments, indicate<br />
the voltage requirements increase as the boron tribromide to hydrogen ratio<br />
increases.<br />
The voltage requirements increase as the system pressure increases.<br />
The substrate is etched with little or no boron deposited as the boron tri-<br />
bromide to hydrogen ratio decreases.<br />
The corona tends to break up into points at high flow rates, resulting in nonuniform<br />
boron deposits.<br />
Good coatings were obtained at high boron tribromide to hydrogen ratios and relatively low<br />
currents.<br />
12) Deposition on 0.5-mil Tungsten Wire. Some <strong>of</strong> the results obtained are given in Table 3.<br />
The corona was maintained continuously in Runs 30-1 and 30-2. The power input was dis-<br />
continued in all remaining runs until the boron tribromide and the cell has attained the desired<br />
temperature. The currents and voltages indicated for all runs below 35 are approximate<br />
since no adjustments were made once the run had begun.<br />
The effects <strong>of</strong> operating conditions are similar to those observed for deposition on 1.0-mil<br />
tungsten wire. However, the decreased heating effects <strong>of</strong> the 0.5-mil wire necessitate heating<br />
the cell to about 200°C to prevent condensation <strong>of</strong> the boron tribromide.<br />
In Run 31-1, an apparently desirable coating was obtained at the ends <strong>of</strong> the reaction zone<br />
while the central portion <strong>of</strong> the filament was quite noddar. In Run 31-1 the peak current was<br />
6.2 mA, falling to about 1.8 mA at the end <strong>of</strong> the run. During this time the peak voltage<br />
changed from 12,480 V to about 15,780 V.<br />
Runs 35-1 and 35-2 yielded filaments having uniform coatings <strong>of</strong> boron. The coating, as<br />
shown in Fig. 5, had ridges along its surface resembling die marks on wire. Figure 6 shows<br />
a cross section <strong>of</strong> filament from Run 35-1 in normal and in polarized light. The higher magni-<br />
fications indicate that the markings on the surface are reflections <strong>of</strong> the markings on the sub-<br />
strate in almost a one-to-one ratio. Furthermore, there is no indication <strong>of</strong> boride formation.<br />
However, polarized light indicates some anisotropic properties similar to pyrolytic graphite.<br />
The material seems s<strong>of</strong>ter and much darker than that grown by gas plating techniques. Although<br />
there are some unidentified lines, x-ray data indicate that the coating is amorphous boron with<br />
no borides in the filament.<br />
Cursory examination <strong>of</strong> the surface for a few <strong>of</strong> the substrates has indicated surface finishes<br />
varying from the ridged (die marks) through smooth to an orange-peel-type finish, while the<br />
filaments obtained have had surface finishes ranging from ridged through smooth to nodular.<br />
Therefore, the substrate surface is a critical factor in determining the appearance and sur-<br />
face <strong>of</strong> the final filament.<br />
13) Effect <strong>of</strong> Morphology <strong>of</strong> the Substrate. The 0.5-mil tungsten substrate wire has been<br />
examined after various stages in the process. Some <strong>of</strong> the results are presented in Table 4.<br />
It has been determined that the liquid cleaning train cleans and slightly etches the wire,<br />
and the high hydrogen corona etches the wire as indicated in Table 4. At low peak currents<br />
the die marks were etched away, but the surface has a very rough orange peel finish. Using<br />
both high and low currents consecutively, a much smoother orange peel finish is obtained.<br />
Boron deposited on wire etched to an orange peel finish gives a coating similar to that<br />
pictured in Fig. 7. The morphology <strong>of</strong> boron deposited on variously etched wire verifies the<br />
hypothesis previously presented in that the coating morphology and growth structure are<br />
directly dependent upon the morphology <strong>of</strong> the substrate.<br />
1
I<br />
(b) Polarized Light<br />
Fig. 6 Cross Section <strong>of</strong> Filament Obtained by Plating in a Corona<br />
Discharge, cf. Fig. 5 (3,000~)<br />
I
0<br />
5 .d<br />
3<br />
v)<br />
*<br />
.d<br />
3<br />
0<br />
u<br />
a 6<br />
.d E *g<br />
e-<br />
3%<br />
SP)<br />
0 a-<br />
><br />
0 00<br />
e ' (0<br />
I<br />
*<br />
W<br />
Q) d<br />
d<br />
rl<br />
rl<br />
t-<br />
CD<br />
I<br />
In<br />
E-<br />
In 0<br />
m w<br />
In<br />
r(<br />
d N<br />
m<br />
In<br />
O E-<br />
c) . h l<br />
0 d<br />
N<br />
03 m<br />
9 . (D<br />
0<br />
N<br />
N<br />
N<br />
0 0 m 0 0<br />
dl rl d m rl<br />
0<br />
u)<br />
0<br />
v)<br />
0<br />
v)<br />
0<br />
In<br />
0<br />
In<br />
I0 (0 E- In<br />
c-<br />
01 c) ea N<br />
N<br />
I<br />
1
i<br />
m<br />
v)<br />
Y<br />
C<br />
i!<br />
u<br />
o)<br />
23<br />
a a-<br />
+<br />
191<br />
m m 0 v)<br />
* * * P)<br />
v)<br />
0<br />
N<br />
~<br />
N<br />
m<br />
0<br />
@a<br />
*<br />
0<br />
01<br />
m<br />
W<br />
m<br />
N<br />
*<br />
v)<br />
v)<br />
@a<br />
@a<br />
@a<br />
v)<br />
OD 0 *<br />
@a<br />
@a<br />
0 0 9<br />
hl 4 OD<br />
0<br />
m<br />
r(<br />
I<br />
m<br />
0<br />
m<br />
0<br />
m<br />
0<br />
N<br />
0 In
192<br />
(a) 604x<br />
Fig. 7 Boron Deposit on 0.5-mil Tungsten Wire, Run 52-1,<br />
cf. Table 4
i,<br />
d<br />
C<br />
.d<br />
c c<br />
W Q ) 0 0<br />
L1Y<br />
0 000 00 00 ov) 0<br />
t- t- P-t- t-t- t-e t-m<br />
8 8 .z .z N N NN NN NN N
N<br />
LI Y<br />
0)<br />
ucd +<br />
194<br />
mol 0<br />
rl<br />
7 s I I<br />
4<br />
(D 1<br />
m<br />
(D<br />
i<br />
I?<br />
1
i<br />
e 4 0 wcr<br />
-4<br />
4<br />
al<br />
0<br />
0 0<br />
c)<br />
cr 13:- I<br />
m<br />
‘ E<br />
100<br />
Q)<br />
uao<br />
Q)v)<br />
I<br />
0<br />
P-<br />
cu<br />
0<br />
(0<br />
l-l<br />
195<br />
w e N N m (D<br />
rl rl<br />
0<br />
a<br />
0<br />
C<br />
. w<br />
x<br />
E<br />
._<br />
E:<br />
0<br />
c Y<br />
a,<br />
4<br />
k<br />
al<br />
5<br />
o e o<br />
Ncua<br />
CJ<br />
cu l-l d<br />
I<br />
I<br />
d<br />
m<br />
P-<br />
P-<br />
l?<br />
CJ 00<br />
cd dc;<br />
mu3<br />
4P)<br />
Y<br />
cd
(-1) \'ariation <strong>of</strong> 1'l;iting Conditions. Comliining thc iniorm:ition obtained in the previous tests,<br />
several runs irere macle using.-vnri,:itions <strong>of</strong> the- pl;itinfi conditions.<br />
ohtainctl are prcscnte!l in Table 5 and in Fig. 5.<br />
Some <strong>of</strong> thc, results<br />
The current-tinic :intl.voltage-time chnr:ictkkistics for t\ro runs :ire indicated in Fig. 8.'<br />
TIIC fil:imcnt oI1t:iincd in Ilun 73- 1 (T:il)Ie 5) \vas cvenly and uniformly coated similar to<br />
thc filamcnt picturcd.in Fig. 5. Except for oiic defect, the filnmcnt obtained from Run<br />
74-1 (Tablc 5) \v:is evenly and uniformly coated similar to the filament pictured in Fig. 7.<br />
As indicated in Fig. 8, the defect probably occurred after an elapsed time <strong>of</strong> ?bout 21 min<br />
at a cell voltage <strong>of</strong> about 23,600 V.<br />
Thc results obtained.indicate that the best process conditions and plating procedure for<br />
boron deposition on a stationary 0.5-mil'tungsten !\ire, are as indicated in Fig. 8.<br />
Discussion<br />
Thc morphology <strong>of</strong> the deposit obtainecl reflects, in general, the morphology <strong>of</strong> the sub-<br />
strate. 'I'hc coating is essentially amorphous in nature although there is indication <strong>of</strong><br />
anisotropy.<br />
One set <strong>of</strong> operating conditions giving ;I .satisfactory coating <strong>of</strong> boron on tungsten is presented<br />
graphically in 13g. 8. In general, good coatings have been obtained at high boron tribromideto-hydrogen<br />
ratios and relatively low currents. Furthermore, if the flowrate is too great,<br />
the corona tends to break up into points, resulting in nonuniform deposits.<br />
Other effects <strong>of</strong> varying operation conditions which suggest limitations for deposition, and<br />
which also tend to verify the mechanism for deposition .. .. presented previously, include:<br />
. .<br />
(a) The voltage requirements increase as the boron tribromide to hydrogen ratio<br />
increases.. ._<br />
(b) The voltage requirements increase as the system pressure increases.<br />
(c) The substrate is etched with.little or no boron deposited as the boron tribromide-<br />
to-hydrogen ,ratio decreases. '..<br />
CONTINUOUS. BORON DEPOSITION ON A MOVING FILAMENT<br />
,.<br />
Experimental Work<br />
.. . . ._<br />
A system was designed, assembled, and tested for. continuous boron deposition on a moving<br />
filament. Figure 9 is a schematic <strong>of</strong> the apparatus. The design resembles that for the<br />
batch plating process (Fig. 1);. ,The filament is pulled through the system with a Graham<br />
constant speed motor. The spool is mounted ,on a shaft and suspended in;such a manner as<br />
to <strong>of</strong>fer minimum friction.. The seal and,electrical contact between cells is a unique arrangement<br />
such that the filament moves through the seal in a vertical direction without breaking<br />
.. . . ., I'<br />
the seal.<br />
: I .I. . j ..<br />
The power for the ceils has been obtained'in a manner similar to that used for the batch plating<br />
process (big. 2). There has been a, considerable problem with 60-cycle pickup in the measurement<br />
system, Shielding;-ahd grounding, at various critical positions were necessary to elimin-<br />
. I<br />
ate this problem. 2. .<br />
IIydrogen-boron trichloride was 'chosen ab the'reactant 'system'. The reactant mixture has<br />
bccn obtained by bubbling hydrogen through boron trichloride maintained at about -2O'C.<br />
Thc reaction cells havemot been heate$ externally, and the vapor flow was split between all<br />
three cells. .,. ... .<br />
The etching cell was n& Zil as'such;'arid Was eventually converted to use for cleaning )<br />
the substrate filament by the hot-wire technique. Thus,, the <strong>chemical</strong> cleaning train was<br />
eliminated. Hydrogen only was passed through the cleaning cell and the substrate heated to I<br />
, . . _'. I, .. "3:. :_<br />
~ ,,. _. , . ..<br />
t<br />
i<br />
6
\<br />
E<br />
‘ 1<br />
TIME FOR RUN<br />
13-1 14-1<br />
- 1<br />
0 0<br />
a 1<br />
b 2<br />
c 3<br />
d 4<br />
e 5<br />
H2 FLOW<br />
BUBBLER<br />
(M L/MIN)<br />
160<br />
160<br />
95<br />
50<br />
50<br />
e<br />
H2 FLOW BBr3<br />
BYPASS TEMPERATURE<br />
LML/MW (* C)<br />
270 -22<br />
270 Htrs. on<br />
. 95 -35<br />
0 -50<br />
0 -72<br />
Shut down<br />
I I 1 I I I 1 I<br />
4 8 12 16 20 24 28 32<br />
PROCESSING TIME (MIN)<br />
28<br />
24<br />
S<br />
2o 3<br />
B<br />
16 0”<br />
Fig. 8 Electrical Characteristics During Deposition <strong>of</strong> Boron on<br />
0.5-mil Tungsten Wire in a Corona Discharge<br />
12<br />
E<br />
4<br />
w’<br />
0<br />
4<br />
s<br />
cl<br />
w<br />
u
H2 --1<br />
DE-OXO DRYING<br />
TOWER<br />
TAKE-UP REEL<br />
CHEMICAL<br />
CLEANING TRAIX<br />
COLD TRAPS<br />
Fig. 9 Schematic <strong>of</strong> Continuous System for Plating in a Corona Discharge<br />
Fig. 10 Boron Deposit on 0.15-mil Tungsten Wire,<br />
Continuous System (600~)<br />
i
199<br />
a red heat in this cell using dc power. Tests performed in this system have utilized both<br />
0.5-mil and 0.15-mil tungsten substrate.<br />
Results<br />
11) AC Pickup. The use <strong>of</strong> shielded leads and wire cages around the transformers has,<br />
with the use <strong>of</strong> 1,000 ohm current measuring resistors, virtually eliminatyd the ac pickup.<br />
j2) Dcposition on 0.5-mil Tungsten IVire. Satisfactory results have been obtained at a<br />
hydrogen flow rate through the bubbler <strong>of</strong> about 100 cc/min and additional hydrogen at a<br />
flow rate <strong>of</strong> about 350 cc/min. Hydrogen is flowing through the cleaning cell at about 140 cc/<br />
min and the wire is cleaned by resistance heating at a dc voltage <strong>of</strong> about 70 V and a DC<br />
current <strong>of</strong> about 320 milliamperes. Some <strong>of</strong> the results obtained are indicated in Table 6.<br />
Using only one cell, at a filament speed <strong>of</strong> about 3.6 in. /min a coating about 0.25-mil<br />
thick was obtained; at a filament speed <strong>of</strong> about 14.4 in./min, a coating ab-out 0.05-mil<br />
thick was obtained.<br />
Using two cells, there was some problem in maintaining the corona in the second cell. At<br />
a filament speed <strong>of</strong> about 3,6 in. /min a coating about 0.7-mil thick was obtained in the<br />
areas where a good corona was maintained. At a filament speed <strong>of</strong> about 14.4 in./min and<br />
a current input to the second cell <strong>of</strong> about half that to the first cell, a coating about 0.07-mil<br />
thick was obtained.<br />
13) Deposition on 0.15-mil Tungsten Wire. The 0.15-mil tungsten substrate wire was cleaned<br />
'<br />
by the hot-wire technique at hydrogen flow rate <strong>of</strong> about 140 cc/min and at a dc power input<br />
<strong>of</strong> about 208 V and 90 mA. While the input vapors were split between the three plating cells,<br />
only one cell was utilized for plating studies. Some <strong>of</strong> the results are indicated in Table 7.<br />
The best results were obtained at a filament speed <strong>of</strong> about 5 in./min with a hydrogen flow<br />
rate through the bubbler <strong>of</strong> about 70 cc/min, and additional hydrogen at a flow rate <strong>of</strong> about<br />
350 cc/min. To maintain a uniform corona, it was necessary to switch the power on and<br />
<strong>of</strong>f relatively slowly during the run. Under these conditions a coating thickness <strong>of</strong> about<br />
0.17 mil was obtained.<br />
There is apparently some difference in either the morphology <strong>of</strong> the 0.15-mil substrate<br />
as compared to the 0.5-mil material, or less effect by this substrate upon the morphology<br />
<strong>of</strong> the deposit. Although the morphology <strong>of</strong> the material deposited on 0.5-mil tungsten<br />
appeared similar to that indicated in Fig. 5, the morphology <strong>of</strong> the deposit on the 0.15-mil<br />
tungsten appeared more like that indicated in Fig. 4. Figure 10 indicates the morphology<br />
usually obtained 0.15-mil tungsten substrate. The total diameter <strong>of</strong> this sample is 0.4 mil.<br />
Discussion<br />
An apparatus has been presented, and tests performed, indicating the feasibility <strong>of</strong> depositing<br />
boron on a continuously moving substrate in a corona discharge.<br />
Although no tests were made for verification, the results obtained with the 0.15-mil sub-<br />
strate indicate that the substrate diameter also effects the operating conditions, probably<br />
though buildup <strong>of</strong> the positive ion sheath to such an extent that the corona cannot recover<br />
after being quenched. This effect is possibly related to the similar effect <strong>of</strong> high flowrates<br />
on larger substrates noted in the previous section. The effects might be explained by postu-<br />
lating a higher concentration <strong>of</strong> boron radicals around the smaller substrate simply through<br />
volume considerations, and around the larger substrate through an increased concentration<br />
resulting from increased availability due to higher flowrates. The charged species would<br />
tend to remain in the volume near the substrate because <strong>of</strong> the greater effects <strong>of</strong> the elec-<br />
trical field.
.-<br />
c<br />
0<br />
c<br />
m<br />
A<br />
' r n<br />
rn<br />
C<br />
.d 44<br />
C<br />
0<br />
V<br />
3 .d<br />
2<br />
u3<br />
M<br />
.d<br />
x<br />
:<br />
c<br />
0<br />
v)<br />
4<br />
0<br />
0) c<br />
w<br />
200<br />
1 , " I I<br />
-r m<br />
W r-<br />
4<br />
I<br />
0<br />
O<br />
N<br />
I 1<br />
0<br />
N c-<br />
0<br />
W<br />
c)<br />
c- W (9<br />
0 0<br />
0 0<br />
0 0<br />
0 0<br />
0 0<br />
w o<br />
0 0<br />
0<br />
0<br />
0<br />
0<br />
c - t - w w W (9<br />
0 0 0<br />
m u 3 In<br />
m m m<br />
0 0<br />
0 0<br />
3 4<br />
0<br />
0 4<br />
.<br />
0<br />
m<br />
0<br />
0<br />
rl<br />
0 0 0 0<br />
e z 5 F l 2<br />
hl<br />
I<br />
E-<br />
OD<br />
I<br />
B
\<br />
,<br />
t<br />
j<br />
\<br />
I,<br />
v)<br />
<br />
2<br />
Y .<br />
C<br />
0<br />
u<br />
m<br />
.I C<br />
Y<br />
C<br />
z<br />
d<br />
cu<br />
X<br />
p:<br />
&<br />
a<br />
I<br />
I -<br />
a 0<br />
n 2<br />
2<br />
m<br />
201<br />
( D o<br />
I 2 2 2<br />
0 0 0 0<br />
0 0 0 0<br />
N 4 N - F<br />
v) P - t - P -<br />
0 0 0 0<br />
m o a m a<br />
a0 a J O D O D<br />
0 0 0 0<br />
N N N N<br />
0 0 0 0<br />
v) m m v )<br />
m m m m<br />
I 7<br />
v) c w m<br />
m a m 0<br />
m Q D m m
202<br />
CONCLUSIONS<br />
Chemical reacticns can be initiated and sustained by a corona discharge. Boron has been<br />
deposited on a tungsten wire substrate in a corona discharge at relatively low temperatures.<br />
The deposition process is electro<strong>chemical</strong> in nature, the boron being deposited cathodically,<br />
and the mechanism for the reaction is indicated to be <strong>of</strong> the form:<br />
(a) Emission <strong>of</strong> electrons from the cathode<br />
(b) Ionization (or activation) <strong>of</strong> hydrogen, argon, or helium, and production <strong>of</strong><br />
secondary electrons which in turn ionize more hydrogen, etc.<br />
(c) Collision <strong>of</strong> ionized hydrogen, argon, or helium with boron tribromide and<br />
exchange <strong>of</strong> energy<br />
(d) Formation <strong>of</strong> positive boron radicals and hydrogen bromide or bromine<br />
(e) Transfer <strong>of</strong> the positive boron radical to the cathode<br />
(f) Electro<strong>chemical</strong> reaction <strong>of</strong> the boron radical to form an essentially amorphous<br />
deposit <strong>of</strong> boron<br />
The morphology <strong>of</strong> the deposit is essentially the same as the morphology <strong>of</strong> the substrate.<br />
There is no interaction <strong>of</strong> the boron with the tungsten and there is apparently some anisotropy<br />
<strong>of</strong> the deposit.<br />
The corona discharge during deposition is not essentially different from a corona discharge<br />
developed in an inert gas system, and the same criteria and properties are extant. Like a<br />
corona discharge in an inert gas, the coefficient <strong>of</strong> electron emission and the ability <strong>of</strong> the<br />
electrons to diffuse along the wire are most important in obtaining a uniform corona, and<br />
thus a uniform deposit, on the substrate. Furthermore, if the sheath <strong>of</strong> positively charged<br />
boron radicals becomes too dense. the corona is quenched and will not recover fast enough<br />
to prevent the breakup <strong>of</strong> the corona into points.<br />
ACKNOWLEDGEMENT<br />
This work was supported by the U. S. Air Force Contract AF 33(615)-2130, and was based<br />
upon an idea presented by J. B. Story. The filament cross-sections were prepared by<br />
W. C. Coons and the continuous system was operated by Barbara Traina. Grateful acknow-<br />
ledgement is also made to R. N. Varney, M. E. Sibert, and A. E. Hultquist for many help-<br />
ful discussions <strong>of</strong> gas <strong>discharges</strong> in general and <strong>of</strong> corona <strong>discharges</strong> in particular.<br />
1.<br />
2.<br />
3.<br />
4.<br />
5.<br />
6.<br />
7.<br />
8.<br />
9.<br />
10.<br />
11.<br />
REFERENCES<br />
N. A. Kapzow, IIElelstrische Vorgange in Gasen und in Vakuum, Veb Deutscher<br />
Verlog der Wissenschaften, Berlin, 1955<br />
S. C. Brown, "Basic Data <strong>of</strong> Plasma Physics," The Technology Press <strong>of</strong> MIT and<br />
John Wiley and Sons, Inc., New York, 1959<br />
E. J. Helland, The Plasma State, Reinhold Publishing Corporation, New York, 1961<br />
L. B. Loeb, Electrical Coronas, Univ. <strong>of</strong> Calif. Press, 1965<br />
L. Ya. Markovskii, V. I. L'vova, Yu. D. Kondrashev, Bor. Trudy Konf. Khim, Bora;<br />
Ego Soedineii. 36-45 (1958) cf. FTD-MT-64-427, Foreign Technology Division,<br />
A. F. Systems Command, 1965<br />
W. V. Kotlensky andR. Schaeffer, J. Am. Chem. SOC. 80, 4517 (1958)<br />
R. T. Holzmann and W. F. Morris, J. Chem. Phys. 29, 677 (1958)<br />
N. R. Dibelius, J. C. Fraser, M. Kawahata, and C. D. Doyle, Chem. Engr.<br />
Progress, 60, No. 6, 41-44 (June 1964)<br />
S. J. Lawrence, Chem. Engr. Progress, 60, No. 6, 45-51 (June, 1964)<br />
J. A. C<strong>of</strong>fman and W. R. Browne, Scientific American, 212, No. 6, 90-98 (June 1965)<br />
N. A. Lange, Editor, Handbook <strong>of</strong> Chemistry, 10th Ed., 111, McCraw-Hill Book .<br />
Company, Inc., N. Y. 1961
203<br />
VAPOR PHASE FORMATION OF NONCRYSTALLINE FILMS<br />
BY A MICROWAVE DISCHARGE TECHNIQUE<br />
D. R. Secrist and J. D. Mackenzie<br />
International Business Machines Corporation, Poughkeepsie, 1 New York<br />
Rensselaer Polytechnic Institute, Troy, New York<br />
ABSTRACT<br />
Numerous variations <strong>of</strong> the microwave discharge method have been employed to<br />
prepare thin films. In this study, non-crystalline films were deposited at low<br />
temperatures and pressures by the vapor phase reaction <strong>of</strong> suitable compounds<br />
with the energetic gaseous species generated in a discharge operated at 2,450<br />
megacycles/sec. The major part <strong>of</strong> the investigation concerned the decomposition<br />
<strong>of</strong> metal-organic compounds <strong>of</strong> the type (R),M or (RO),M, where R may<br />
be C2H5, C3H7. etc. Amorphous oxide films <strong>of</strong> silicon, germanium, boron,<br />
tin, and titanium were readily formed when an oxygen plasma was maintained.<br />
Silica films were also grown via an argon plasma. These films were deposited<br />
on metallic and non-metallic substrates positioned outside the discharge region.<br />
Conversely. the oxidation <strong>of</strong> silicon tetrafluoride to yield a fluorsiloxane polymer<br />
<strong>of</strong> composition SOl. SF was found to occur only within the confines <strong>of</strong> the<br />
plasma. It is shown that the films are free from decomposition products when<br />
deposited above a certain critical temperature, which is dependent on the reactant<br />
flow rate. The effects <strong>of</strong> temperature, pressure, flow rate, .and hygroscopic<br />
nature bf the substrate on the deposition rate are examined. Optical<br />
properties are presented. Therutilization <strong>of</strong> a nitrogen or nitrogen oxide<br />
plasma to generate nitride or oxynitride films is discussed.<br />
INTRODUCTION<br />
Extensive studies have been conducted with crystalline films in regard to film<br />
structure, epitaxy, and property variations as a function <strong>of</strong> the history <strong>of</strong> a<br />
particular deposition process. The nature <strong>of</strong> glassy films, however. has not<br />
been investigated with the same thoroughness. This is due, in part, to the<br />
inability <strong>of</strong> most materials to form in the glassy state. In general, the approach<br />
to film formation has been from the vapor phase. Some methods from the vapor<br />
include evaporation, vapor phase hydrolysis, thermal decomposition, and<br />
"sputtering". All <strong>of</strong> these techniques will yield non-crystalline films with the<br />
proper conditions. However, with the exception <strong>of</strong> the latter method, relatively<br />
high temperatures are required. As a result, most films invariably<br />
crystallize immediately after being deposited. The non-cryetalline films prepared<br />
by these techniques are usually materials which readily form a glaea<br />
by the conventional cooling <strong>of</strong> a melt; e. 8.. SiOz. The method <strong>of</strong> sputtering,<br />
on the other hand, occasionally leads to the formation <strong>of</strong> uncommon non-<br />
crystalline films when the substrates are maintained at low temperatures<br />
In this paper. the low temperature preparation <strong>of</strong> a wide variety <strong>of</strong> non-crystalline<br />
films is discussed utilizing a microwave discharge method as an alternative<br />
to sputtering. With this technique, the rate <strong>of</strong> film deposition can be,<br />
carefully controlled by regulation <strong>of</strong> the reactant flow rates. The quality <strong>of</strong><br />
any particular film is shown to be a sensitive function <strong>of</strong> (1) the concentration<br />
(1) .
and type <strong>of</strong> reaction products, (2) the <strong>chemical</strong> nature <strong>of</strong> the substrate, and<br />
(3) the reactor (substrate) .temperature.<br />
204<br />
APPARATUS<br />
A schematic drawing <strong>of</strong> the main flow system employed in this work is shown<br />
in Figure 1. A gaseous plasma, in this instance oxygen, could be maintained in<br />
a 1" 0. D. pyrex tube when the system pressure was less than 800 microns.<br />
The electrodeless discharge was produced with a Raytheon PGM-10 Microwave<br />
Generator. This unit could supply 100 watts <strong>of</strong> microwave energy at a fixed<br />
frequency <strong>of</strong> 2, 450 mc/sec. The energized species were allowed to flow into<br />
the bell jar through a 0. 5" dia. pyrex tube. The metal-organic compounds<br />
selected for this work were liquids with a vapor pressure <strong>of</strong> about 0. 5 mm.<br />
near room temperature and were contained in a graduate cylinder immersed<br />
in a constant temperature bath. The metal-organic evaporation rate (flow rate)at<br />
any temperature could be altered by adjusting a 1 mm. dia. Teflon stopcock.<br />
The ultimate vacuum achieved in the system was 240 microns pressure with a<br />
gas flow rate <strong>of</strong> 1.76 cm<br />
3<br />
/minute. The majority <strong>of</strong> the studies were performed<br />
inside an aluminum furnace 2 114" in dia. x 4" long. A fused silica cylinder<br />
1 3/8" in dia. x 2 I/Z" long served as a second furnace core. The constant<br />
temperature zones for these furnaces extended one cm. above and below the<br />
gas inlet port with a maximum radial temperature variation <strong>of</strong> t 5OC for points<br />
one cm. from the cylinder axes. A fused silica balance havinga sensitivity<br />
<strong>of</strong> i cm/mg. was employed to determine the rate <strong>of</strong> film deposition. Details<br />
<strong>of</strong> this apparatus have been previously reported(2). In order to provide a<br />
secondary working geometry, the basic apparatus was modified as illustrated<br />
in Figure 2. This adaption was used to study the oxidation <strong>of</strong> silicon tetrafluoride<br />
FILM DEPOSITION AND EVALUATION<br />
The substrates were positioned in the furnace reaction vessel with their wide<br />
dimension perpendicular to the gas inlet tube. The general cleaning procedure<br />
consisted <strong>of</strong> dipping the specimens into a 48% solution <strong>of</strong> hydr<strong>of</strong>luoric acid,<br />
followed by a rinse in distilled water. During the period when the furnace temperature<br />
was increasing, the system was purged with the gas selected to form<br />
the plasma. In all work, the microwave generator was operated at 90% <strong>of</strong> its<br />
rated power output.<br />
The films were deposited onto NaCl or KBr discs to facilitate examination by<br />
infrared transmission. Substrates <strong>of</strong> platinum, aluminum, A12O3, and fused<br />
Si02 approximately one cm. in diameter were prepared for the rate studies.<br />
The index <strong>of</strong> refraction and isotropic character <strong>of</strong> film specimens obtained<br />
from the substrates and/or gas inlet tube were determined with a petrographic<br />
microscope. Debye -Scherrer x-ray diffraction powder patterns were made to<br />
establish whether the films were amorphous or crystalline.
BELL JAR<br />
STANC<br />
TO VACUUM<br />
SYSTEM<br />
U T 2 HELIX BALAWE<br />
0 TELESCOR J<br />
AWTE CYLINDER<br />
TEMPERATURE WTH<br />
BAY PLATE<br />
OXYGEN ENERGlZlffi CHAMBER<br />
TO CAPILLARY<br />
LEAK ASSEMaY<br />
Figure 1 : Schematic Drawing <strong>of</strong> Microwave Discharge Apparatus.<br />
+<br />
si F4<br />
I, INLET<br />
I<br />
'h<br />
1)<br />
\I,<br />
P<br />
\<br />
1<br />
J<br />
'\<br />
I\<br />
MICROWAVE ANTENNA<br />
1<br />
I OXYGEN<br />
I<br />
I 4- INLET<br />
I<br />
I 4<br />
SUBSTRATE 1" 0. D.<br />
PYREX TUBE<br />
Figure 2: Modified Microwave Discharge System.
206<br />
RESULTS AND DISCUSSION<br />
Preliminary studies indicated that extensive water was formed as a reaction product<br />
when the metal-organic compounds were decomposed via an oxygen discharge.<br />
This water could be <strong>chemical</strong>ly incorporated into the oxide films when the substrates<br />
were held below a certain temperature which depended on the organic<br />
evaporation rate and the hygroscopic nature <strong>of</strong> the substrate. For'instance, with<br />
NaCl substrates, the silica films were found to be free <strong>of</strong> water above 18OoC<br />
with a tetraethoxysilane evaporation rate <strong>of</strong> 0. 015 cm<br />
3<br />
/hour. (3) The presence<br />
<strong>of</strong> silicic acid was detected below 18OoC by infrared transmission studies. Conversely,<br />
with a KBr substrate and similar conditions <strong>of</strong> temperature and evaporation<br />
rate, extensive water was incorporated into the silica film.<br />
The,majority <strong>of</strong> the films prepared in this study were formed with low deposi-<br />
tion rates <strong>of</strong> about 20 Elminute on NaCl substrates heated to a temperature<br />
<strong>of</strong> 200°C. The optical properties <strong>of</strong> some <strong>of</strong> the films are presented in Table 1.<br />
TABLE 1<br />
COMPARISON OF THE OPTICAL PROPERTIES OF<br />
VAPOR-FORMED FILMS AND THEIR RESPECTIVE<br />
GLASSY OR CRYSTALLINE OXIDES<br />
Principal Infrared Index <strong>of</strong><br />
Mate rial Frequency (CM-l) Refraction<br />
Si02: vapor-formed film 1, 045;800 1.458) . 002<br />
GeOZ :<br />
fusion-formed glass<br />
vapor -formed film<br />
1, 080;800<br />
85 0<br />
1.458: . 002<br />
1. 5827 . 002<br />
B203 :<br />
fusion -formed glass<br />
vapor -formed film<br />
860<br />
1,350<br />
1. 534-1. 607<br />
1.470i . 002<br />
fusion-forme d glass 1,370 1.464p)<br />
TixOy: ' vapor-formed film 950-700 b1.7<br />
Ti02: anatase 1,200-500 >l. 7<br />
Sn Oy:<br />
SnljT:<br />
vapor-formed film<br />
cassiterite<br />
1,425<br />
85 0 -5 00<br />
1.536t - . 002<br />
31.7<br />
All <strong>of</strong> the films were isotropic when examined with polarized light and were<br />
amorphous byx-ray diffraction. The silica films were formed by (1) decomposing<br />
tetraethoxysilane, (C H 0)4 Si, in either an oxygen or argon plasma or (2)<br />
2 5<br />
decomposing tetraethysilane in an oxygen plasma. The germania and boron<br />
oxide films were prepared by decomposing tetraethoxygermane and triethylborate<br />
via an oxygen discharge. The infrared absorption frequencies and refractive<br />
indices <strong>of</strong> the latter three vapor-formed films are similar to those<br />
<strong>of</strong> the respective fusion-formed glasses, which suggests that a random network<br />
structure can be achieved by methods other than the conventional cooling<br />
<strong>of</strong> the melt. The spread in the index <strong>of</strong> refraction for germania glass was<br />
realized by slowing cooling one specimen and quenching another from 1 loO°C;<br />
the slow-cooled specimen had the larger refractive index. With the exception
207<br />
<strong>of</strong> the boron oxide film, the films listed in Table 1 adhered well to the NaCl substrates.<br />
The B 0 film was immediately coated with mineral oil after removal<br />
from the glow dfsciarge apparatus because <strong>of</strong> its extreme moisture sensitivity<br />
and was only partially adherent.<br />
I<br />
The non-crystalline titanium oxide films were prepared from titanium tetra-<br />
isopropylate while maintaining an oxygen discharge. Since titanium oxide is<br />
not a common glass-forming system, one may inquire as to whether or not the<br />
viscosity <strong>of</strong> such a film would be similar to that <strong>of</strong> a glass near its transition<br />
region, e. i. 1013-1014 poises. For many undercooled sy t ms, the growth<br />
rate can be described in terms <strong>of</strong> the reciprocal viscosit$eas:<br />
U = AHf (Tf-T)<br />
3TfTf ,A L % N<br />
where u is the rate <strong>of</strong> growth (cmlsec), T is the undercooled temperature,<br />
v\ is the viscosity (poises), Tf is the fusion temperature, A is the mean jump<br />
distance (cm), I-$ is the latcnt heat <strong>of</strong> fusion (ergs), and N is Avogadro's number.<br />
If this relationship is valid for a system, then an approximate estimation<br />
<strong>of</strong> the viscosity at the crystallization temperature can be made if the rate<br />
<strong>of</strong> growth (crystallization) is known. In this study, the titanium oxide film exhibited<br />
birefringence (crystallized) after 45 minutes at 325OC. An approximate<br />
rate <strong>of</strong> crystallization can be calculated for the film if it is assumed that the<br />
particles grew from some arbitrary value, for example, 10 8 to 110 before<br />
crystallinity was detected. The jump distance A was taken as 2 2 units. Although<br />
Ti02 dissociates, the heat <strong>of</strong> fusion has been reported as 15. 5 kcal/mole<br />
at a melting point <strong>of</strong> 184Ood6). The growth rate determined from the rate <strong>of</strong><br />
crystallization is about 3. 7 x 10-l' cmlsec. The corresponding viscosity<br />
at 5980K is 5. 5 x 10" poises, which would classify the film as a high1 viscous<br />
supercooled liquid since the viscosity value is slightly below 1013-10'4 poises.<br />
Since viscosity is approximately an exponential function <strong>of</strong> temperature, it is<br />
reasonable to conclude that at some slightly lower tem erature the viscosity<br />
<strong>of</strong> the film is characteristic <strong>of</strong> a solid glass, e. i. 101f-1014 poises.<br />
An amorphous tin oxide film was formed by decomposing dibutyltin-diacetate<br />
via an oxygen plasma. Preliminary measurements indicated that the electrical<br />
resistivity <strong>of</strong> the film near room temperature was greater than lo7 ohm-cm.<br />
Comparing th I. R. spectra <strong>of</strong> the film and SnOZ, it is seen in Table 1 that the<br />
main absorption mode for the amorphous film occurs at a much higher frequency<br />
than that <strong>of</strong> the crystalline modification. This shift can probably be attributed<br />
to a large structural variation such as a change in coordination number.<br />
The basic apparatus shown in Figure 1 was modified as illustrated in Figure 2 in<br />
order to study the oxidation <strong>of</strong> SiF4 via an oxygen discharge. Research grade<br />
SiF4 gas was metered into the pyrex chamber at a rate <strong>of</strong> 1.8 cm3/minute.<br />
Simultaneously, dry oxygen was admitted at the rate <strong>of</strong> 5 cm3/minute. The<br />
electrodeless discharge was maintained at 800 u total pressure. Under these<br />
conditions, a thick non-adherent film was deposited onto the walls <strong>of</strong> the pyrex
chamber within the boundaries <strong>of</strong> the plasma in about one hour. The wall temperature<br />
in this region was approximately 100°C. The film can be described as<br />
a translucent blue-white gel which was amorphous by x-ray diffraction and isotropic<br />
when observed with polarized light. The index <strong>of</strong> refraction <strong>of</strong> the gel<br />
was estimated to be considerably less than 1.400; infrared absorption bands<br />
were evidenced at 1080, 920, and 750 cm-’. A micro<strong>chemical</strong> dnalysis <strong>of</strong> the<br />
film indicated that its composition could be represented as (SiOl. 5F)n. It is<br />
interesting to note that the film formed only within the confines <strong>of</strong> the plasma,<br />
which suggests that thermal and/or microwave energy are required for the<br />
oxidativn and/or polymerization processes.<br />
Effect <strong>of</strong> Pressure, Temperature, and Substrate - An extensive study was made<br />
with t e silica films with respect to the mechanism and kinetics <strong>of</strong> film forma-<br />
tion(2’. It is reasonable to suspect that the general conclusions formulated in<br />
this work with respect to pressure, temperature, and nature <strong>of</strong> the substrate<br />
are applicable to most <strong>of</strong> the other oxide films discussed in this manuscript.<br />
Support for this viewpoint is also documented in regards to the incorporation<br />
<strong>of</strong> water-into the films(3).<br />
Basically, the effect <strong>of</strong> minor pressure fluctuations on the rate <strong>of</strong> deposition <strong>of</strong><br />
silica films was found to be negligible. However, above 500 microns pressure,<br />
the rate <strong>of</strong> deposition was rapidly decreased due to (1) an increased atomic<br />
oxygen recombination and (2) poisoning effects from the reaction products.<br />
The films discussed in the present work were deposited near 240 u pressure.<br />
Typical rates <strong>of</strong> deposition for the silica films ranged from 10 to 80 A/min.<br />
by variation <strong>of</strong> (1) the organic evaporation rate or (2) the substrate temperature.<br />
The rate <strong>of</strong> deposition was constant with time. The relationship between<br />
logarithm <strong>of</strong> the deposition rate and reciprocal temperature is depicted<br />
in Figure 3 for various substrates. The data have been spread to show the<br />
temperature dependence <strong>of</strong> deposition with each substrate. With a tetraethoxy-<br />
- silane evaporation rate <strong>of</strong> 0. 15 cm3/hour, the silica films were free <strong>of</strong> water<br />
and/or organic inclusions (by infrared transmission studies) above 29OoC.<br />
At higher temperatures, the rate <strong>of</strong> deposition decreased with increasing<br />
temperature and was ascribed to a process <strong>of</strong> physical adsorption. The apparent<br />
heat <strong>of</strong> adsorption <strong>of</strong> silica glass on the fused silica, NaC1, and platinum<br />
substrates was calculated to be 12. 7 t 0. 2 kcal/mole. For the aluminum<br />
or A120 substrates, the apparent heat <strong>of</strong> adsorption <strong>of</strong> silica was calculated<br />
to be 9. 3 t 0. 3 kcal/mole. It was concluded that the hygroscopic nature <strong>of</strong> the<br />
A1 0 surface was responsible for the lower heat <strong>of</strong> adsorption observed.<br />
It<br />
2 3<br />
was postulated that a hydrated surface tends to incorporate further hydroxyl<br />
groups into the film structure. With this reasoning, one might expect that the<br />
hygroscopic nature <strong>of</strong> the KBr substrate discussed earlier would also lead to<br />
a decreased heat <strong>of</strong> adsorption. In conclusion, it is postulated that the rate <strong>of</strong><br />
deposition <strong>of</strong> an oxide film with a moisture sensitivity similar to that <strong>of</strong> silica<br />
is most likely affected in the same manner, e. i. physical adsorption controlled<br />
below 410OC. The apparent heat <strong>of</strong> adsorption <strong>of</strong> the oxide films on the various<br />
substrates, is <strong>of</strong> course, dependent on the molecular structure <strong>of</strong> the films and<br />
the hygroscnpic nature <strong>of</strong> the substrate.
i<br />
I,<br />
4<br />
I.(<br />
Y I-<br />
* 0<br />
i<br />
f<br />
i<br />
3 0.t<br />
I<br />
I I<br />
I I .6!3 1.85<br />
&”~’<br />
Figure, 3: Relationship <strong>of</strong> Logarithm Silica Deposition Rate and Reciprocal<br />
Temperature for Deposition on Various Substrates.<br />
Other Systems - The microwave discharge technique can be employed to deposit<br />
a variety <strong>of</strong> thin films at low temperatures. One obvious variation <strong>of</strong> the technique<br />
utilizes gaseous plasmas other than oxygen. For instance, in the work<br />
with silica films it was found that (C2H5O)qSi contained sufficient oxygen to<br />
facilitate the formation <strong>of</strong> Si02 when the c mpound was decomposed with energetic<br />
argon species. Sterling and S~ann‘~’ have recently deposited amorphous<br />
Si3N4 films by the reaction <strong>of</strong> silane and anhydrous ammonia in an RF discharge.<br />
Non-crystalline silicon nitride films have also been prepared at Rensselaer<br />
Polytechnic Institute by the decomposition <strong>of</strong> ( C2H5)4Si via a nitrogen discharge.<br />
The use <strong>of</strong> a N20 plasma to form Si20N2 poses another equally interesting<br />
possibility. Success to date with the latter reactions has been complicated by<br />
the ease <strong>of</strong> formation <strong>of</strong> silica. Since metal-organic compounds are now available<br />
for a large number <strong>of</strong> metals, it would seem that the microwave discharge<br />
technique could lead to many new and unusual films by a simple variation <strong>of</strong><br />
the reactant and/or plasma compositions.<br />
i<br />
4
21 0<br />
References<br />
1. R. A. Mickelson, "Electrical and Optical Properties <strong>of</strong> Amorphous Zinc<br />
Oxide Films", Sc. D. Thesis, M. I. T, 1963.<br />
2. D. R. Secrist and J. D. Mackenzie, "Deposition <strong>of</strong> Silica Films by the Glow<br />
Discharge Technique", J. Electrochem. SOC. 113 (9), 914-920 (1966).<br />
3. D. R. Secrist and J. D. Mackenzie, Incorporation <strong>of</strong> Water Into Vapor<br />
Deposited Oxide Films", Solid State Electronics, 9, 180 (1966).<br />
4. G. W. Morey, "The Properties <strong>of</strong> Glass", 2nd Edition, Chap. XVI, p. 370,<br />
Reinhdd Publishing Corp., New York, 1954.<br />
5. David Turnbull, p. 225 in Solid State Physics, Vol. 3, edited by F. Seitz<br />
& D. Turnbull, Academic Press, .Inc., N. Y. 1956.<br />
6. Q. Kubachewski and E. Evans, Metallurgical Thermodynamics, p. 306,<br />
Pergamon Press, N. Y. 1958.<br />
7. H. F. Sterling and R. C. G. Swann, "Chemical Vapor Deposition Promoted<br />
by R. F. Discharge", Solid State Electronics 8, (8) p. 653-54 (1965).<br />
Acknowledgements<br />
The authors are grateful to the Pittsburgh Plate Glass Company for the support<br />
<strong>of</strong> a fellowship. (D. R. S.).<br />
I<br />
A<br />
I
211<br />
The Reacticn 3f Oxygen with Carbonaceous Compounds<br />
in the Electrodeless Ring Discharge<br />
b Chester E, Gleit<br />
Department <strong>of</strong> Chemistry, North Carolina State University, Raleigh, North Carolina, 27607<br />
D Abstract<br />
The electrodeless ring discharge has recently found use in <strong>chemical</strong> analysis as<br />
a means <strong>of</strong> removing carbon from graphitic and organic compounds preceding elemental<br />
analysis and determination <strong>of</strong> microstructure. To determine appropriate reaction conditions,<br />
the relationship between oxidation rate and sample temperature was studied.<br />
Samples were immersed in a plasma produced by passing molecular oqgen through a<br />
radi<strong>of</strong>requency field. An inductively coupled, 13.56-We generator producing 95 watts<br />
was utilized. Gas pressure was 1.2 torr, and flow 150 cc/min. The apparent activation<br />
energy <strong>of</strong> pure graphite was found to be 6.5 Kcal/mol in the temperature range<br />
'' 100 to 3OO0C, and zero at higher temperatures. The activation energy <strong>of</strong> both impure<br />
graphite and sucrose were slightly lower. These values are consistent with those <strong>of</strong><br />
atomic oqgen. The reaction <strong>of</strong> graphite with C02 did not occur below 1X)OC and<br />
yielded an apparent activation energy <strong>of</strong> 3.2 Kcal/mol in the temperature range 150<br />
to 300OC. Oxidation rate was observed to depend on electrical field configuration.<br />
Increasing the temperature <strong>of</strong> the reaction tube's walls decreased the rate <strong>of</strong> carbon<br />
oxidation. Trace ions, such as Fe(I1) , Se(IV), Os(1V) , and iodide, were converted<br />
to higher oxidation states. Neither oxidation nor volatility was observed to be<br />
strongly temperature dependent.<br />
0 Introduction<br />
In 1962 a method was reported for the decomposition <strong>of</strong> organic substances based<br />
on reaction with an oxygen plasma, produced by passing molecular gas through a radi<strong>of</strong>requency<br />
electrodeless discharge (5). This method has found use in a variety <strong>of</strong><br />
<strong>chemical</strong> studies. Loss <strong>of</strong> trace elements through volatilization and diffusion is less<br />
than that in conventiczd dry ashing. Destruction <strong>of</strong> mineral structure is reduced.<br />
\As a small quantity <strong>of</strong> purified oxygen is the only reagent, the possibility <strong>of</strong> <strong>chemical</strong><br />
contamination is diminished. Applications <strong>of</strong> this method include: microincineration<br />
<strong>of</strong> biological specimens (l3), the ashing <strong>of</strong> coal (6) and filter paper (4), and the<br />
recovery <strong>of</strong> mineral fibers from tissue (1). Several reviews <strong>of</strong> analytical applications<br />
! have been published (9) (14) .<br />
In conventional ashing both reaction rate and retention <strong>of</strong> volatile components<br />
lare strongly temperature dependent. However, disagreement exists as to the effect <strong>of</strong><br />
temperature in plasm ashing. In part, this is due to difficdty in applying conven-<br />
1 tional temperature measuring techniques to solids immersed in a strong radi<strong>of</strong>requency<br />
"field. The question is further complicated by the fact that a single temperature<br />
\,cannot be assigned to the system. Electrons in the low pressure plasma are not in<br />
, thermal equilibrium with the ions and neutral species. Furthermore, the temperature<br />
4 <strong>of</strong> the specimen and the surrounding vessel may differ considerably. In this study the<br />
)\ effects <strong>of</strong> temperature on oxidation rate, recombination <strong>of</strong> active gaseous species,<br />
a d volatility <strong>of</strong> inorganic reaction products are considered.<br />
I<br />
I<br />
Experimental<br />
The reaction system employed in these studies (figure 1) consisted <strong>of</strong> a 100-cm<br />
long, 3.5-cm I .D., borosilicate cylinder (Pyrex No. 7740). Specimens were placed on<br />
i<br />
I
212<br />
the principal axis 43 cm from the gas inlet. A 0.6-cm tube, C, passed diametrically<br />
through the cylinder and held the spechen on a central projection. Sample temperature<br />
was regulated, in part, by circulating liquids between this tube and a constant<br />
temperature bath. Radi<strong>of</strong>requency excitation was supplied from a 250-watt, crystal<br />
controlled 13.56-MHz generator, described previously (3). Except as otherwise noted,<br />
an output <strong>of</strong> 115 watts was employed. Power was transferred to the gas by means <strong>of</strong><br />
impedance matching network terminating in a lo-turn coil <strong>of</strong> $-inch O.D. copper tubing<br />
tapped 2 turns from ground.<br />
The coil, B, which had an inside diameter <strong>of</strong> 4.5 cm and<br />
was 12.5 cm long, was coaxial with the reaction tube and placed 10 hm from the gas<br />
inlet.<br />
After passage through a rotometer, molecular gas entered the reaction system<br />
through a capillary orifice, A. Pressure was monitored by a McCloud gauge attached<br />
to side ann E, located 50 cm beyond the gas inlet. Pressure was maintained at 1.2<br />
torr; and flow rates at 150 cc per minute, S.T.P. U.S.P. grade oxygen and instrument<br />
grade carbon dioxide were used.<br />
The temperature <strong>of</strong> solid specimens in the plasma was determined by means <strong>of</strong> an<br />
infrared radiation thermometer (Infrascope Model 3-lC00, Huggins Laboratories Inc.,<br />
Sunnyvale, Calif.) attached to a chart recorder. This device employs a lead sulfide<br />
detector and suitable filters to permit remote measurement <strong>of</strong> 1.2 to 2.5~ radiation<br />
emitted by the specimen. To compensate for variations in emissitivity and inhomogeneity<br />
in the optical field, empirical calibration curves were constructed.<br />
Themcouples were employed to determine cylinder-wall temperatures. To eliminate<br />
interaction with the radi<strong>of</strong>requency field, the transmitter was inactivated during<br />
the latter measurements.<br />
High purity graphite rods (<strong>National</strong> Carbon Co., Grade AGKSP) were employed as<br />
standard specimens. These rods were 0.61 cm in diameter and had a cavity machined<br />
into their base to affix them to the cooling tube. Pellets <strong>of</strong> sucrose and carbon<br />
containing small quantities <strong>of</strong> cupric acetate were also epployed. The latter were<br />
produced by heating and then pressing a slurry <strong>of</strong> the salt solution and m-mesh<br />
graphite powder.<br />
Results and Discussion<br />
Sample Temerature: The temperature variation <strong>of</strong> oxidation rate <strong>of</strong> graphite<br />
exposed to the oxygen plasma is shown in figure 2. Over tpe range 120 to 300OC, the<br />
Arrhenius equation, X = Ce'Ea/RT, fits the data well and yields a value <strong>of</strong> 6.5<br />
Kcal/mol for the apparent activation energy, Ea. Between 300 and 450°C, the highest<br />
temerature studied, oxidation rate is not dependent on temperature. The oxidation<br />
<strong>of</strong> graphite by molecular oxygen at high tempgrature is strongly dependent on the purity<br />
<strong>of</strong> the graphite. To determine if a similar effecb occurs in plasma oddation, pellets<br />
composed <strong>of</strong> pure graphite powder and inorganic salts were utilized. The results <strong>of</strong><br />
the addition <strong>of</strong> 0.01 _M cupric acetate are shown in figure 2. At low temperature the<br />
change in oxidation rate <strong>of</strong> pure and impure graphite with temperature is similar.<br />
However, the rate <strong>of</strong> oxidation <strong>of</strong> the impure graphite becomes independent <strong>of</strong> taprature<br />
at a lower temperature. Measurements performed with specimens contabhg 1-r ,<br />
concentrations <strong>of</strong> cupric acetate led to results which fell between the illustrated<br />
curves.<br />
The gas in the law pressure electrodeless discharge is <strong>chemical</strong>ly similar to that k<br />
in the positive column <strong>of</strong> a low pressure arc. Wlecule-molecule and ion-molecule<br />
collisions are frequent. The ions and neutral species are, therefore, nearly in ther- 1<br />
mal quildbrium.<br />
Elastic collisions between these species and electrons are less<br />
' frequent. At low pressure electron temperatures are quite high. In the oxygen die-<br />
Charge atodc oxygen (%) is believed to be the most abundant active species. Higher<br />
energy states <strong>of</strong> atomic oxygen, positive and negative ions, and electronically<br />
1<br />
,,
2<br />
E,<br />
W<br />
t-<br />
a<br />
Z<br />
9<br />
I-<br />
a<br />
2<br />
X<br />
0<br />
I50<br />
too<br />
50<br />
30<br />
20<br />
A A A A<br />
1<br />
K<br />
SOURCE<br />
~ ~~ ~<br />
Fig. 1. Experimental apparatus : capillary inlet, A;<br />
power coil, B; specimen mounting tube, C; sidearm to<br />
manometer, D; outlet to vacuum pump, E.<br />
1<br />
IO<br />
1.5 2-0 2.5<br />
SURFACE TEMPERATURE )/r x<br />
Fig. 2. Rate <strong>of</strong> oxidation vs. surface temperature for<br />
graphite rods,@, and graphite pellets containing 0.01 ,M<br />
cupric acetate,A.<br />
I
214<br />
states <strong>of</strong> molecular oqgen are also present.<br />
It is instructive to compare these results with measurements <strong>of</strong> the activation<br />
energy <strong>of</strong> graphite with atomic oxygen removed from the luminous discharge. In severearly<br />
studies a vdue <strong>of</strong> zero was reported (2) (16). Hennig (8) observed no dependence<br />
on temperature at a pressure <strong>of</strong> 1.0 torr and a value <strong>of</strong> 7 Kcal/mol at 10 torr. This<br />
difference was attributed to ozone formation. In a recent detailed study <strong>of</strong> the<br />
atomic oxygen-graphite reaction, Marsh et al. (U) reported an activation energy <strong>of</strong><br />
10 Kcal/mol between U and 2OO0C, which approached zero at 350°C. 'Although the rate<br />
<strong>of</strong> oxidation in these studies was considerably lower than that obtained in the plasm,<br />
their values are consistent with the belief that atomic oqygen is a major reactant<br />
within the discharge.<br />
Of a variety <strong>of</strong> specimens, only graphite exhibited an abrupt change in activation<br />
energy near 3Oo0C. For example, the apparent activation energy in the decomposition<br />
<strong>of</strong> sucrose is approximately 4 Xcal/mol over the entire measurement range. The independence<br />
<strong>of</strong> oxidation rate for graphite at high temperatures is ascribed to the formation<br />
<strong>of</strong> surface oxides. Numerous studies <strong>of</strong> graphite combustion have shown that such surface<br />
compounds control rate at elevated temperatures in excess oxygen. As anticipated,<br />
increasing radi<strong>of</strong>reqiiency power to the discharge does not measurablv increase the rate<br />
<strong>of</strong> graphite oxidation at high sample temperature.<br />
The exhaust gas <strong>of</strong> the ovgen-graphite reaction contains both carbon monoxide a d<br />
carbon dioxide, with the latter predominating. Tn the lumi~ous discharge region these<br />
gases react with graphite. The results <strong>of</strong> measuremepts in which carbon dioxide was<br />
passed through the radi<strong>of</strong>requency field and then exposed to graphite rods activated<br />
are shown in figure 3. Between 150 and 4OOOC the Arhennius equation fits well, yielding<br />
an apparent activation energy <strong>of</strong> 3.2 Kcal,/mol. Below 120°C less than one mg per<br />
hour <strong>of</strong> carbon was removed. This small weight loss is attributed to ion and electron<br />
bombardment.<br />
W& Conditions: In a number <strong>of</strong> experiments, increasing power beyond an optimum<br />
value led to the anomolous result <strong>of</strong> reducing ashing rate. Earlier it was noted that<br />
placing a dry ice-acetone trap in the exhaust stream extended the length <strong>of</strong> the luminous<br />
discharge (5). To study this effect, wall temperatures were varied while the<br />
carbon rod was maintained at 200°C. The results <strong>of</strong> a series <strong>of</strong> measurements are shown<br />
in figure 4.<br />
Assuming that the decrease in oxidation rate at increased wall tempera-<br />
ture ip due to recombination <strong>of</strong> active species at the wall, the overall activation<br />
energy at low temperatures for this process is approximately 2 Kcal/ml.<br />
The major sources leading to loss <strong>of</strong> active oxygen species are recombination <strong>of</strong><br />
atomic oxygen and ions at the'walls <strong>of</strong> the vessel. The recombination <strong>of</strong> discharged<br />
oxygen on glass surfaces has been investigated. Linnett and Marsden (10) reported<br />
that the recombination coefficient <strong>of</strong> atomic oxygen on clean borosilicate glass<br />
(Pyrex) was independent <strong>of</strong> temperature. However, positive values were observed on<br />
contaminated surfaces. In a later series <strong>of</strong> papers Greaves and Linnett (7) studied<br />
a number <strong>of</strong> oxide surfaces, in all cases the recombination coefficient was temperature<br />
'<br />
dependent. For silica apparent activation energies were 1 to 13 Kcal/mol over the<br />
temperature range 15 to 3OOOC. The exceptional nature <strong>of</strong> Pyrex was noted.<br />
' The second major source <strong>of</strong> loss <strong>of</strong> active species results from electron-ion<br />
recombination at the chamber walls. The electric field restricts the drift <strong>of</strong> ions<br />
to the chamber walls. Rate <strong>of</strong>-recombination is controlled by ambipolar diffusion,<br />
but is limited by the presence <strong>of</strong> negatively charged oqgen ions in the discharge (15).<br />
Although diffusion is not dependent on wall temperature, a distortion <strong>of</strong> the<br />
symmetric electrical field will enhance wall recombination, producing an inarease in<br />
This effect is noted in t&le 1.<br />
The presence <strong>of</strong> a 6-cm long metallic ring on the exterior wall <strong>of</strong> the<br />
reduced oddation rate by more than ten per cent. Grounding the ring in comn with<br />
. wall temperature and a reduction <strong>of</strong> oxidation rate.<br />
<<br />
/<br />
I<br />
1<br />
I
'i'<br />
100<br />
50<br />
IC<br />
21 5<br />
2.0 2.5<br />
SURFACE TEMPERATURE, I/T i103<br />
Fig. 3. Rate <strong>of</strong> gassification vs. temperature for graphite<br />
rods exposed to carbon dioxide discharge.<br />
5c<br />
4c<br />
3c<br />
20<br />
IC<br />
1.5 2 .o 2.5 3.0 3.5<br />
WALL TEMPERATURE, x lo3<br />
Fig. 4. Effect <strong>of</strong> wall temperature on the oxidation<br />
rate <strong>of</strong> graphite.
.<br />
216<br />
the output coil further reduced the oxidation rate. This is a local effect, in that<br />
oxidation rate beyond the ring was not greatly reduced. Distortion <strong>of</strong> the field can<br />
also be introduced by changing the angle between the end <strong>of</strong> the output coil and the<br />
reactSon tube.<br />
le&h <strong>of</strong> the luminous discharge. Similar effects have been observed in electrodeless<br />
<strong>discharges</strong> at higher pressure (12).<br />
Variations from axial symmetry led to reduced rates and decreased the ,<br />
Table I I<br />
Effect <strong>of</strong> Field Distortion on Carbon Oxidation Rate<br />
Cbcidation Rate<br />
Angle w/hr<br />
Pyrex tube 00 79<br />
Pyrex tube and ungrounded<br />
copper ring<br />
Pyrex tube and grounded<br />
copper ring<br />
00 70<br />
00 62<br />
mrex tube 1.0 71<br />
Pyrex tube 3." 63 '<br />
Effect on Nineral Constituents: It has been reported that there is no appreciable ,,<br />
loss <strong>of</strong> a number <strong>of</strong> metal ions in plasma oxidation (4,s). Nonvolatile species include:<br />
Na(I), Cs(II), Cu(II), Zn(II), Mn(II), Pb(II), Cd(II), Co(II), Ho(III), Er(III), Fe(III),<br />
Cr(IIL), As(III), Sb(SII), and Mo(V1). The effect <strong>of</strong> ashing temperature, the reason<br />
for the surprisingly low volatility, and the final oxidation dtate <strong>of</strong> the product were<br />
not previously explored. As part <strong>of</strong> the present study, 20 to 100 mg <strong>of</strong> compounds containing<br />
radioactive tracers were deposited on Whatman cellulose filters. After<br />
tupxure to ashing for a sufficient period to remove the filter paper, generally 30<br />
dnutes, the activity <strong>of</strong> the ash was measured and the oddation state <strong>of</strong> the element<br />
determined. Specimen temperature was adjusted during ash- by altering input power.<br />
These measurements are summarized in table 11. In general, temperature has little<br />
effect on retention. These results and earuer observations Indicating complete retention<br />
<strong>of</strong> metals in compounds such as arseneous chloride and metalloporphyrFns (5) are<br />
believed to be due to competition between volatilization and oxidation to leas volatile<br />
compounds.<br />
Unlike the other elements studied, the highest valence oxide <strong>of</strong> o d u ,<br />
is the most volatile. Therefore, this element is volatilized In the plasmrr<br />
on process.<br />
The volatility <strong>of</strong> iodide, shown in figure 5, is also consistent wlth the hypothe- {<br />
sis <strong>of</strong> competition between volatilization and oxidation. During these measurements,<br />
ashing was stopped at 5 minute intervals. It is Been that loss <strong>of</strong> 1131 closely foumS<br />
ttie curve for filter paper g ssification. No loss <strong>of</strong> 1131 occurs after the filtar<br />
paper is remved. Loss <strong>of</strong> I f 3l varied between 15 and 35 per cent. However, none Of<br />
the residual 1131 could be preci itated with silver nitrate, indicating that the iodide<br />
had been oxidized. When the 113 E tracer was converted to NdO3 before ashing, all Of<br />
the iodine activitywas retained.<br />
I-<br />
z<br />
W<br />
0<br />
w<br />
a<br />
a<br />
100<br />
80<br />
60<br />
40<br />
20<br />
215<br />
0<br />
0 IO 20 30<br />
TIME (MINI<br />
Fig. 5. Loss <strong>of</strong> sample weight,n; and loss <strong>of</strong> Il3’<br />
tracer,O, during ashing <strong>of</strong> filter paper in an owgen<br />
plasma.
- Tracer<br />
Fe59<br />
Fe59<br />
' ~e75<br />
se75<br />
,8875<br />
se75<br />
218<br />
Table I1<br />
Recovery <strong>of</strong> Trace Elements from Ashed Filter Paper<br />
~. Highest Percent in<br />
Max. sample Percent Oxidation Highest<br />
Compound Temperature Recovered State Oxidation State<br />
I<br />
123<br />
I11<br />
95<br />
120<br />
I11<br />
100<br />
110<br />
250<br />
ll0<br />
250<br />
ll0<br />
250<br />
120,<br />
150<br />
* Recovered after rinsing tube with HF-HN03<br />
V<br />
V<br />
V<br />
V<br />
I<br />
I<br />
VI11<br />
.VI11<br />
The behavior <strong>of</strong> silver ion represents a different type <strong>of</strong> ashing loss. As<br />
opposed to the other metals in this study, only 75 to 90 per cent <strong>of</strong> the Agum could<br />
be recovered from the borosilicate sample holder by rinsing with dilute nitric acid.<br />
To recover the remainder <strong>of</strong> the Ague? the glassware had to be repeatedly rinsed with<br />
a warn mixture <strong>of</strong> nitric and hydr<strong>of</strong>luoric acids. gY maintaining the system at low<br />
temperature this difficulty was ameliorated, but not eliminated.<br />
Conclusion: A number <strong>of</strong> active species are known to be present in the luminous<br />
electrodeless discharge. Although a sikilarity between reactions in the plasma and<br />
those <strong>of</strong> discharged oxygen is,noted, it cannot be concluded that within the plasma<br />
onlythe reactions <strong>of</strong> atomic owgen are significant. The present study indicates<br />
appropriate conditions for the application <strong>of</strong> the electrodeless discharge to decompose<br />
carbonaceous materials prior to elemental analysis. Specifically, increasing sample<br />
temperature to 200-300OC leads to an improvement in ash- rate without appreciably<br />
increasing volatility losses.<br />
91<br />
93<br />
100<br />
100<br />
--<br />
c<br />
-<br />
-<br />
--<br />
l<br />
Acknowledgement //<br />
, The electrical apparatus used in this study was constructed by James Breitmsier.<br />
The assistance <strong>of</strong> Juan Wong and Daniel Silvers is appreciated. Walter Holland, Harvey<br />
Beaudry and Robert Reinhart assisted in preliminary studies performed under a contract<br />
between Union Carbide Chemical Corporation and the Laboratorg for Electronics.<br />
stw was supported by grants from the North Carolina Engineering Foundation and North<br />
C a r o b State University Pr<strong>of</strong>essional Development Fund.<br />
r<br />
I<br />
1<br />
'<br />
d<br />
'<br />
I<br />
t
s.,<br />
Literature Cited<br />
Berkley, C. , Churg, J., Selik<strong>of</strong>f, I. J. , and Smith, W. E., e.<br />
132, 48 (1965).<br />
New York Acad.<br />
Blackwood, J. D., and McTaggart, F. K., -. J. w., 12, 114 (1959).<br />
Gleit, C. E. , Microchem. J. , lo, 7 (1966).<br />
Gleit, C. E., Benson, P. A. , Holland, W. D. , and Russell, I. J., &&. m., 36,<br />
2067 (1964).<br />
Gleit, C. E. , and Holland, W. D., And.. Chem., 2, 1454 (1962).<br />
Gluskoter, H. J., m, 44, 285 (1965).<br />
Greaves, J. C., and Linnett, J. W., Trans. Farad. u., a, 1355 (1958).<br />
Hennig, G. R. , Dienes, G. J., and Kosiba, W., Proc. Second Intern. Conf. Peaceful<br />
Uses At. Energy, Geneva, 1, 301 (1958). -<br />
--<br />
HOUahan, J. R., J. Chem. FdUC., &J A@1, U97J 392 (1966).<br />
Linnett, J. W., and Marsden, D. G. H., m. &. w.<br />
(1956).<br />
I<br />
489<br />
(Londod E. A, a,<br />
Marsh, H., O'Hair, T. E., and Wynne-Jones, W.F.K., Trans. Farad. &., 61, 274<br />
(1965).<br />
Mitin, R. V., and Pryadkin, K. K., 2. =. m., z? 1205 (1965).<br />
Thomas, R. S., J. Cell Biol. , 2, 113 (1964).<br />
Thomas, R. S., "Techniques <strong>of</strong> Electron Microscopy: Microincineration for the Fine<br />
Localization <strong>of</strong> Mineral Constituents".in fISubcellular Pathology" ed. by Steiner,<br />
J. W., Morat, H. Z., and Ritchie, A. C., Harper and Row, New York (in press).<br />
Thompson, J. B., E. m. =. (London), 22, 818 (1959).<br />
Wrkevich, J., and Streznewski, T., Revue L'Inst. Fran. du Petrole, 3, 686 (1958).
220<br />
THE EFFECT OF CORONA ON THE REACTION OF CARBON MONOXIDE AND STEAM<br />
T.C. Ruppel, P.F. Mossbauer, and D. Bienstock<br />
U.S. Department <strong>of</strong> the Interior, Bureau <strong>of</strong> Mines,<br />
Pittsburgh Coal Research Center, Pittsburgh, Pa.<br />
SUMMARY I<br />
The water-gas shift reaction was chosen for study in a corona discharge with<br />
the aim <strong>of</strong> establishing the important variables associated with the production <strong>of</strong><br />
hydrogen.<br />
The reaction was carried out in a quartz Siemen's type ozonizer having an<br />
electrode length <strong>of</strong> 12 inches and an electrode separation <strong>of</strong> 8 nun. The electrodes<br />
consisted <strong>of</strong> fired-silver paint--one electrode coating the inside <strong>of</strong> the inner (high<br />
voltage) tube, and the other coating the outside <strong>of</strong> the outer (ground) tube.<br />
The reaction was studied by means <strong>of</strong> a four-factor, two-level, factorially<br />
designed set <strong>of</strong> experiments. This consisted <strong>of</strong> input power levels <strong>of</strong> 60 and 90<br />
watts,. pressures <strong>of</strong> .Si and 1 atmosphere, hourly space velocities <strong>of</strong> 200 and 800, and<br />
reactor temperatures <strong>of</strong> 127 and 527OC (400 and 800'K). Although voltage was not a<br />
factor, the voltage ranged from 3100 to 11,000 rms volts.<br />
The prediction equation resulting from the statistical analysis <strong>of</strong> the data<br />
indicates that more hydrogen is produced at higher pressures, temperatures, and<br />
power inputs, and lower space velocities. Practically no hydrogen is produced in<br />
the absence <strong>of</strong> a discharge, regardless <strong>of</strong> the values <strong>of</strong> space velocity, temperature,<br />
pressure, or power dissipated.<br />
The highest yield <strong>of</strong> hydrogen within the factorial region studied was 4.5 per-<br />
cent. An extra-factorial region, which was indicated by the prediction equation to<br />
be fruitful, was explored and 11.5 percent hydrogen was obtained.<br />
It was found that the water-gas shift reaction in a corona discharge is<br />
kinetically, rather than thermodynamically controlled.<br />
INTRODUCTION<br />
Novel techniques are being investigated at the Bureau <strong>of</strong> Mines to introduce<br />
energy into coal and coal products in an effort to find new uses for coal. One<br />
technique under study involves <strong>chemical</strong> reactions in a corona discharge. Initially<br />
the gas-phase reaction <strong>of</strong> carbon monoxide and steam to produce carbon dioxide and<br />
hydrogen will be investigated.<br />
The immediate aim <strong>of</strong> this studv is to establish the imoortant variables associated<br />
with the production <strong>of</strong> hydrogen in the water-gas shift reaction in the presence<br />
<strong>of</strong> a corona discharge. A more general goal is to determine the dependence <strong>of</strong> product<br />
yield on the important variables in a corona discharge for several gas-phase reactions'<br />
As knowledge is gained on the effect <strong>of</strong> corona discharge in <strong>chemical</strong> reactions, mre<br />
complex reactions involving coal and its products will be studied. 4<br />
<<br />
The water-gas shift reaction, which is usually carried out industrially over<br />
an Fefl3-Cr203 catalyst at 300-500°C and 100-300 psig, has been discussed at great<br />
length in the literature. Summary articles are a~ailable.l-~ It is almost certain-<br />
/<br />
'<br />
,'<br />
ly a surface reaction5 as opposed to the apparent homogeneous nature <strong>of</strong> the reaction ;<br />
in the corona discharge. Experiments with the quartz Siemen's type ozonizer to be<br />
described have shown that no reaction occurs between water and carbon monoxide at ,p<br />
i
7 h<br />
\ F<br />
221<br />
600°C and 1 atmosphere pressure in the absence <strong>of</strong> a corona. This suggests that the<br />
reaction is homogeneous when it does occur with a corona. That is, the reactor walls<br />
probably do not enter into the reaction by acting as a catalyst or as a third body.<br />
The <strong>physics</strong>6-8 and chemistry9-" <strong>of</strong> corona discharge have been surveyed by<br />
several authors. A bibliography <strong>of</strong> <strong>chemical</strong> reactio s in electrical <strong>discharges</strong> over<br />
a thirty-year period, 1920-1950, has been compiled. 19<br />
PROCEDURE<br />
The corona discharge unit is shown in figure 1. The operational scheme can be<br />
seen in the flow diagram, figure 2. Carbon monoxide from a compressed gas cylinder<br />
(1) is passed through an activated carbon adsorption trap (5). The metered flow (7)<br />
passes through a flow control valve (9); a second stream <strong>of</strong> gas may be blended here<br />
if desired. The carbon monoxide at the pressure <strong>of</strong> the system passes through a<br />
steam generator (11). The thermocouple immediately above the reflux condenser (17)<br />
controls the steam flow rate via a time-proportionating temperature controller (not<br />
shown). The carbon monoxide and water flow rates may be individually varied between<br />
0.2 and 3 ft.3fhr. by adjusting the flow controller (9) and temperature controller<br />
settings. The carbon monoxide-steam mixture then enters the Siemen's-type reactor<br />
through preheaters (18) and (21). The reactor is enclosed in a tube furnace (23).<br />
The product gases pass through a series <strong>of</strong> three cold traps (25, one shown) at<br />
-8OOC. Bypass cold traps (26) are available if required. The non-condensable gases<br />
pass through a sample train (28,29) and then through a flow meter (30) which is at<br />
system pressure. On leaving the flow meter the non-condensable gases pass through<br />
the system-pressure regulator (32), vacuum pump (34), and finally a wet test meter<br />
(35). The system is designed to operate from 0.5 to 2.0 atmospheres.<br />
The Siemen's type reactor, shown in figures 3 and 4, consists <strong>of</strong> two concentric<br />
quartz tubes, 45 and 33 mm. OD, having a 4 mm. annular space. The electrode length<br />
is 12 inches and there is an 8 mm. electrode-to-electrode separation. The electrodes<br />
consist <strong>of</strong> fired-silver paint--one electrode coating the inside <strong>of</strong> the inner (high<br />
voltage) quartz tube, and the other coating the outside <strong>of</strong> the outer (ground) tube.<br />
Tungsten (or optionally copper) wires are attached to the outside and inside <strong>of</strong> the<br />
reactor with two or three coatings <strong>of</strong> fired-silver paint. The capacitance <strong>of</strong> the<br />
reactor was measured as 86-89 pfad using an impedance bridge.<br />
The average wall temperature was used as a measure <strong>of</strong> the temperature <strong>of</strong> the<br />
system. Three thermocouples were equally spaced vertically along the inside wall <strong>of</strong><br />
the reactor (fig. 2) and three along the outside wall. The inside wall thermocouples<br />
were at the corona potential and thus were isolated from ground. A millivoltmeter<br />
was connected in series to each <strong>of</strong> the inside wall couples, and thus was<br />
similarly "floated". To simplify the electrically hot thermocouple circuits, no<br />
compensating ice junction was included; the room temperature correction was added to<br />
each millivoltmeter reading. The corona potential had a positive effect on the<br />
millivoltmeter readings in the hot circuit. Therefore the millivoltmeters were read<br />
before the corona potential was impressed on the system.<br />
taken as the average <strong>of</strong> all six thermocouple measurements.<br />
The system temperature was<br />
The study was made according to a factorially designed set <strong>of</strong> experiments to<br />
determine the effect <strong>of</strong> pressure, space velocity, temperature, and input electrical<br />
power on the yield <strong>of</strong> hydrogen.<br />
This was a four factor, two level set <strong>of</strong> experiments as shown in Table 1.<br />
I
222<br />
Figure 1.- Unit for studying <strong>chemical</strong> reactions in a corona,.discharge.<br />
1<br />
1
223<br />
e<br />
I'
224<br />
Figure 3.- Siemen's type reactor.<br />
i<br />
I
R<br />
L<br />
'<br />
-1<br />
9mm O.D. 5mm I.D.<br />
45mm 0.D 41mm I.D.<br />
2mm wall with a tolerance -.2mm<br />
,33 mm O.D. 29 mm I.D.<br />
/<br />
9mm O.D.<br />
5mm I.D.<br />
4mm annular space<br />
Figure 4. Siernen's t:ne reactor.<br />
!
J<br />
4<br />
226 (<br />
1<br />
Table 1.- Factorial design <strong>of</strong> the experiments<br />
2 level, 4 factor, full factorial<br />
Z4 = 16 experiments 4<br />
1:1 molar mixture, H2:CO<br />
Leve 1<br />
Factor LOW High<br />
Pressure, atm. 0.5 1.0<br />
Space velocity, hr.-' 200 800<br />
Temperature, "K 400 800<br />
Input power, watts 60 90<br />
I It was found that no hydrogen was produced in the absence <strong>of</strong> a discharge, no<br />
matter what the potential impressed on the high voltage electrode. Thus Paschen's<br />
law curves, discharge-initiating potential VS. the product <strong>of</strong> pressure and electrode<br />
separation, were developed for the reactor. These are shown in figure 5. A second<br />
reactor with 12 m. electrode separation was also used to obtain these curves.<br />
Initially it was intended to factorialize secondary voltage, rather than input<br />
power. However, it was not possible to factorialize voltage (set a high and low<br />
level) without losing the discharge at low temperatures or drawing too high a current<br />
at high temperatures. At high currents the arc would puncture the quartz reactor.<br />
Maintaining a discharge in some cases and not others would introduce a very signif-<br />
icant unfactorialized (uncontrolled) qualitative variable, discharge vs. no discharge,<br />
into the design. While it might be possible to factorialize discharge VS. no dis-<br />
charge, it was known that no hydrogen would be produced with no discharge, regardless<br />
<strong>of</strong> the power level.<br />
Input power was factorialized since the voltage and amperage required for a<br />
chosen power level are determined by the discharge and therefore need not meet set<br />
requirements <strong>of</strong> a factorial design.<br />
Although the voltage was not a factor, the voltage ranged from 3100 to 11,000<br />
rms volts.<br />
The above factors were chosen intuitively. Other factors, such as AC frequency,<br />
distance between reactor electrodes, and electrical current, may also have an effect.<br />
Future experiments using a frequency <strong>of</strong> 10,000 cps are planned to detect any fre-<br />
quency effect on hydrogen production.<br />
The factorial design form <strong>of</strong> experbentation has the advantages <strong>of</strong> 1) obtaining<br />
an empirical regression equation with minimum effort, 2) isolating the magnitudes <strong>of</strong><br />
the effects <strong>of</strong> each variable and its interaction with other variables (synergism) on<br />
the dependent variable, 3) allowing optimization <strong>of</strong> the dependent variable within<br />
the levels <strong>of</strong> the factors studied, and 4) suggesting the direction to be taken out-<br />
side the region studied for further optimization.<br />
After the desired carbon monoxide and steam flow rates were obtained and stabilized<br />
at .?; or 1 atmosphere, the discharge was initiated. It was monitored with an<br />
oscilloscope (Tektronix Model RM 35A), typical oscillographs <strong>of</strong> which are shown in<br />
figure 6.<br />
The waveforms on the left show the voltage trace above the current trace.<br />
The area enclosed by the Lissajous figure on the right is a measure <strong>of</strong> the true Power<br />
used by the discharge.<br />
I<br />
I<br />
i<br />
I<br />
4<br />
1
Figure 5.- Paschen's law curves.<br />
2-15-67
228<br />
W<br />
h<br />
3<br />
tn<br />
.r(<br />
w<br />
rn<br />
5<br />
w<br />
W<br />
><br />
3<br />
U<br />
91<br />
M<br />
I4<br />
G<br />
v)<br />
.rl<br />
a<br />
W<br />
0<br />
rl<br />
m<br />
0<br />
.rl<br />
E<br />
1<br />
W
\<br />
'?<br />
I<br />
The water-free product gases were analyzed by gas chromatography. However the<br />
hydrogen yields are reported with water present.<br />
RESULTS AND DISCUSSION<br />
The data were analyzed according to the linear hypothesis statistical model<br />
resulting in the following empirical prediction equation for the percent hydrogen<br />
(water included):<br />
I<br />
Pct. H2 = -1.88e.762 - (1.946+0.499)P + (0.00402+0.00042)SV<br />
+(0.00486+0.00121)T - (O.O106K).O0830)W<br />
+(0.0047~.000791)(P*T) - (0.0000103+0.0000006)(T~SV)<br />
+(0.000028&H).0000132)(T-W)<br />
where P = pressure, atmospheres<br />
SV = space velocity, hr.-1<br />
T = temperature, "K<br />
W = input power, watts.<br />
An inspection <strong>of</strong> the terms in the equation, after numerical values are inserted,<br />
shows that tempzrature has the largest effect on the production <strong>of</strong> hydrogen, with<br />
space velocity being the least important.<br />
The coefficient with the largest error associated with it is in the input power<br />
term. Since the factorial levels were maintained quite closely, the inference which<br />
can be drawn is that the yield <strong>of</strong> hydrogen tends to be least correlatable with input<br />
power. The reverse is also true, that hydrogen production tends to correlate best<br />
with the interaction term T-SV.<br />
The coefficients were determined with an IEN 7090 digital computer available at<br />
the University <strong>of</strong> Pittsburgh, using a program for a regression equation previously<br />
developed by Bureau personnel.<br />
According to the equation, maximum production <strong>of</strong> hydrogen, within the range<br />
studied, was found to be at 1 atmosphere, 200 hourly space velocity, 800%, and 90<br />
watts input. At these levels the percentage <strong>of</strong> hydrogen was 4.5, compared with the<br />
figure 4.1 calculated by the equation. The extra-factorial region <strong>of</strong> interest suggested<br />
by the equation would be higher pressures, temperatures, and inputs, and<br />
lower space velocities.<br />
Within the limitations <strong>of</strong> our equipment, and operating on a<br />
flow basis, this was P=2, SV=lOO, T=800, and W=lOO. The hydrogen percentage in-<br />
creased to 8.6. By stopping the gas flow, 11.6%H2 was obtained in the product.<br />
Extrapolating an empirical equation is always dangerous. There is no guarantee<br />
that an indicated extra-factorial region will yield good results. In the present<br />
case, the extra-factorial region proved to be more fruitful than predicted. The<br />
advantage <strong>of</strong> an empirical equation is the ease in which it is obtained while representing<br />
the data well in a compact form within the region in which the data were<br />
taken.<br />
If one could define the reaction temperature in a corona discharge, it would be<br />
possible to discuss the thermodynamics <strong>of</strong> the system. However it is difficult, if<br />
not impossible, to define an equilibrium with respect to the corona reaction. While<br />
there are methods available for obtaining measurements <strong>of</strong> the average (translational)<br />
temperature <strong>of</strong> the gas or plasma, there is a valid question as to the "local tmperature"<br />
at the reaction site <strong>of</strong> the activated species. However it was attempted in<br />
this study to correlate the average wall temperature <strong>of</strong> the system with hydrogen<br />
production and thus to define the system sufficiently for design purposes. me<br />
prediction equation strongly indicates that this approach is satisfactory.
230<br />
The decrease in equilibrium constant <strong>of</strong> the water-gas-shift reaction with I<br />
temperature is shown in figure 7. As the prediction equation indicates a positive<br />
temperature effect on the production <strong>of</strong> hydrogen, the water-gas-shift reaction in<br />
an electrical discharge must be kinetically rather than thermodynamically con-<br />
I<br />
trolled. I<br />
The yields <strong>of</strong> hydrogen shown here are less than the yields achieved industrially13<br />
by catalytic means. What is <strong>of</strong> more interest to us is! the empirical<br />
determination, for several reactions, <strong>of</strong> the functional dependence <strong>of</strong> the yield <strong>of</strong><br />
a given product on the several independent variables in a corona discharge. This<br />
information is a prelude to future experiments where powdered coal or coal volatiles<br />
will be treated with certain gases in a corona discharge.<br />
CONCLUSIONS<br />
The conclusions that can be drawn from this study are:<br />
(1) No hydrogen is produced in the absence <strong>of</strong> a discharge, regardless <strong>of</strong> the values<br />
<strong>of</strong> space velocity, pressure, temperature, or power dissipated.<br />
(2) The prediction equation arising from the statistical analysis <strong>of</strong> the data in-<br />
dicates that more hydrogen is produced at higher pressures, temperatures, and power<br />
inputs, and lower space velocities.<br />
(3) Within the range <strong>of</strong> variables studied, temperature has the largest relative<br />
effect on the production <strong>of</strong> hydrogen, while space velocity has the least relative<br />
effect.<br />
(4) Of the four factors chosen, input power is the least correlatable with the<br />
production <strong>of</strong> hydrogen, while the first order interaction:<br />
velocity) is the most correlatable.<br />
(temperature) X (space 4<br />
(5) The water-gas-shift reaction in a corona discharge is kinetically, rather than<br />
thermodynamically controlled.<br />
REFERENCES I<br />
1. Morgan, J.J. Water Gas. Ch. in Chemistry <strong>of</strong> Coal Utilization. Vol. 2, H.H.<br />
Lowry, ed., John Wiley & Sons, Inc., New York, N.Y., 1945. i<br />
2. Fredersdorff, C.G. von, and M.A. Elliott. Coal Gasification. Ch. in Chemistry<br />
<strong>of</strong> Coal Utilization, Supplementary Volume, H.H. Lowry, ed., John Wiley & Sons,<br />
Inc., New York, N.Y., 1963.<br />
3. Blackwood, J.D. Kinetics <strong>of</strong> Carbon Gasification. Ind. Chem. 36, 55-60, 129-133,<br />
171-175 (1960).<br />
4. Sherwood, P.W. Water Gas Conversion.<br />
Petroleum (London) 24, 338-340 (1961).<br />
1<br />
5. Reference 1, p. 1704. ,<br />
'<br />
6. Loeb, L.B. Electrical Coronas, Their Basic Physical Mechanisms, Univ. <strong>of</strong> Calif.<br />
Press, Berkeley & Los Angeles, 1965, 694 pp. I<br />
7. Brown, S.C. Basic Data <strong>of</strong> Plasma Physics, Mass. Inst. <strong>of</strong> Tech. and John WileY<br />
& Sons, Inc., New York, N.Y., 1959, pp. 250-273. i<br />
8. Meek, J.M., and J.D. Craggs.<br />
1953, pp. 148-176.<br />
Electrical Breakdown in Gases. Oxford Univ. Press, f<br />
4<br />
i<br />
4<br />
4<br />
f<br />
I<br />
,<br />
;<br />
I<br />
J<br />
t
N<br />
I<br />
+<br />
0"<br />
ON<br />
i<br />
I<br />
+<br />
0<br />
u<br />
/<br />
0<br />
0<br />
I<br />
0<br />
cu<br />
-<br />
g<br />
-<br />
0<br />
0<br />
4<br />
-<br />
0<br />
0<br />
cn<br />
Y<br />
0<br />
g:<br />
"2<br />
a:<br />
a<br />
O Q<br />
[L<br />
W<br />
I-<br />
w<br />
F<br />
0 3<br />
01<br />
(Do<br />
ul<br />
m<br />
a<br />
0<br />
s:<br />
0<br />
0<br />
t<br />
0<br />
0<br />
m
232<br />
9. Glockler, G., and S.C. Lind. The Electrochemistry <strong>of</strong> Gases and Other Di- I<br />
electrics, John Wiley & Sons, Inc., New York, N.Y., 1939.<br />
10. Thomas, C.L., G. Egl<strong>of</strong>f, and J.C. Morrell. Reactions <strong>of</strong> Hydrocarbons in Elec-<br />
trical Discharges. Chem. Rev. 28, No. 1, 1-70 (1940).<br />
11. Jolly, W.L. The Use <strong>of</strong> Electrical Discharges in Chemical Syntheses. Ch. in<br />
Technique <strong>of</strong> Inorganic Chemistry, ed. by H.B. Jonassen and A. Weissberger, v. 1,<br />
Interscience Div. <strong>of</strong> John Wiley & Sons, Inc., New York, N.Y., 1963, pp. 179-208.<br />
12. Akerl<strong>of</strong>, G.C., and E. Wills. Bibliography <strong>of</strong> Chemical Reactions in Electric<br />
Discharges, Aug. 1951, Office <strong>of</strong> Technical Services, Dept. <strong>of</strong> Commerce,<br />
Washington , D . C . I<br />
13. Christain, D.C., and P,B. Boyd, Jr. What to Look for in CO Conversion Catalysts.<br />
Chem. Eng. 56, No. 5, 148-150 (1949).<br />
.<br />
/<br />
I<br />
<<br />
,<br />
d
-<br />
233<br />
SOME PROBLEMS OF THE KINETICS OF DISCHARGE REACTIONS<br />
Robert Lunt<br />
Department <strong>of</strong> Physics<br />
University College, London<br />
This essay is concerned with the kinetics <strong>of</strong> reactions in electrical<br />
<strong>discharges</strong> through gases that are either effected by, or initiated by,<br />
the charged particles present in the gas.<br />
The principal experimental investigations on the kinetics <strong>of</strong> such reactions<br />
which have led to the development <strong>of</strong> the statistical theory <strong>of</strong> discharge<br />
I reaction are recalled briefly before stating spme elaborations <strong>of</strong> the formal<br />
analysis that are now seen to be pertinent and which have not been summarized<br />
hitherto.<br />
The present state <strong>of</strong> the theory is then illuminated by outlining some examples<br />
<strong>of</strong>:<br />
(1) the achievements <strong>of</strong> the theory in providing a detailed and<br />
quantitative Interpretation <strong>of</strong> the values found from experiment<br />
for the empirical reaction rate coefficients considered as<br />
functions <strong>of</strong> the discharge parameters,<br />
(2)<br />
the apparent inadequacy <strong>of</strong> the theory to account for the results<br />
<strong>of</strong> experiment in two gases, and<br />
(3) the lacunae in the experimental data hitherto available that<br />
impede the interpretation <strong>of</strong> the data in term8 <strong>of</strong> statistical<br />
theory: the recognition <strong>of</strong> these lacunae may pave the way to<br />
the better design <strong>of</strong> experiment and to a =re precise uader-<br />
standing <strong>of</strong> the mechanism <strong>of</strong> discharge reaction and <strong>of</strong> the<br />
usefulness <strong>of</strong> electrical <strong>discharges</strong> in <strong>chemical</strong> and in<br />
electrical engineering.<br />
I
234<br />
The Plasma Induced Reaction <strong>of</strong> Hydrogen Sulfide with Hydrocarbons<br />
F. J. Vastola and W. 0. Stacy<br />
Department <strong>of</strong> Fuel Science<br />
The Pennsylvania State University, University Park, Pa.<br />
Introduction I<br />
The interaction <strong>of</strong> sulfur with hydrocarbons under a range <strong>of</strong> experimental<br />
conditions has been investigated by various workers. Knight and co-workers1 re-<br />
acted excited sulfur atoms, produced by 'in situ' photolysis <strong>of</strong> COS at 25OoC, with<br />
a range <strong>of</strong> paraffinic hydrocarbons and found as primary products only the corre-<br />
ponding mercaptans. Bryce and Hinsnelwood2 studied the reaction <strong>of</strong> sulfur vapor<br />
with parafinnic hydrocarbons in the temperature range 320°C to 349°C and observed<br />
that the primary products were hydrogen sulfide and unsaturated hydrocarbons.<br />
Study <strong>of</strong> the reaction <strong>of</strong> sulfur and methane 3-7 over a catalyst at 500°C to 7OO0C<br />
has shown the products to be carbon disulfide and hydrogen sulfide according to<br />
the reaction<br />
CH4 + 2S2 -+ CS 2 + 2H2S. (1)<br />
Thomas and Strickland-Constable8 studied the interaction <strong>of</strong> sulfur and hydrocarbons<br />
at temperatures 1200.OK to 1500'K and in the absence <strong>of</strong> a catalyst observed no carbon<br />
disulfide formation. With a catalyst, however, the reaction proceeded yielding<br />
carbon disulfide, hydrogen sulfide, and hydrogen; reactions similar to (1) for<br />
methane, above 1200°K, being increasingly superseded by those similar to (2):<br />
CH4 + S + CS2 + 2H2*<br />
2<br />
Reported now are some results <strong>of</strong> a study <strong>of</strong> the reactions <strong>of</strong> excited atomic<br />
species, generated by the dissociation <strong>of</strong> hydrogen sulfide in a plasma jet, with<br />
methane and neopentane.<br />
9<br />
This study was suggested by an earlier investigation <strong>of</strong> Vastola and co-workers<br />
into the reaction between carbon and the products <strong>of</strong> water vapor microwave discharge,<br />
where it was observed that the presence <strong>of</strong> carbon downstream inhibited the recom-<br />
bination, to either oxygen or water, <strong>of</strong> the active atomic species. Instead, the<br />
active oxygen reacted preferentially with carbon.<br />
Experimental Procedure<br />
The plasma reactor is shown in Figure 1. The reactor consists <strong>of</strong> a 10 x 0.75<br />
inch fused quartz tube. The gas to be brought to the plasma state is introduced<br />
tangentially upstream from the output coil <strong>of</strong> a 1.2 KW, 120 MHz RF induction heater.<br />
Ihe spiraling motion imparted by the tangential input increases the dwell time <strong>of</strong><br />
the gas in the plasma zone. The hydrocarbon gases to be reacted with the plasma<br />
dissociation products are introduced down stream from the plasma flame.<br />
By moving<br />
the hydrocarbon feed tube the distance between the plasma flame and the point <strong>of</strong><br />
hydrocarbon introduction can be varied.<br />
TO initiate the plasma discharge argon is passed through the reactor (0.5 CU<br />
ft/hr, 1 Atm) and a graphite rod is placed in the induction field. After the heated<br />
graphite rod starts the discharge it is removed and the gas feed is switched to the<br />
desired H2S-Ar mixture. Mass spectrometric gas analyses were made before and after<br />
the reaction.<br />
1<br />
i
.. .<br />
:. ,. .I , .<br />
i ? ..<br />
. ,<br />
... .<br />
. .<br />
,235 . . . . . . 1<br />
. .. . I >.<br />
, . . .<br />
. (<br />
. a<br />
.*. . .<br />
'LI<br />
0 .<br />
H
236<br />
Results and Discussion<br />
When hydrogen sulfide is introduced into an argon plasma condensing elemental<br />
sulfur produces a dense fog dovnstream from the plasma flame. As the amount <strong>of</strong><br />
nydrocarbon that is introduced into this region is increased the elemental sulfur<br />
production decreases to a point where it can no longer be observed. With large<br />
excesses <strong>of</strong> hydrocarbon the color <strong>of</strong> the plasma flame changes from blue to orange<br />
due to back diffusion <strong>of</strong> the hydrocarbon.<br />
Table 1 gives the reactant and product concentrations for reactions <strong>of</strong> dis-<br />
sociated liydrogen sulfide with methane and neopentane. With the exception <strong>of</strong> run<br />
number 3 the only gaseous products detected were hydrogen sulfide, unreacted hydro-<br />
carbon, carbon disulfide, and hydrogen.<br />
Table 1<br />
Reactant and Product Analyses<br />
Reactant Mixture Products<br />
Composition Mole % Composition Mole %<br />
RUXI<br />
No. Reactants HC H2S hK HC H2S H2 CS2 C2H2 AK<br />
1 H2S, CH4 6.4 17.6 76.5 2.2 4.5 18.5 3.8 0 70.1<br />
2 I, 6.4 17.6 76.5 4.3 6.7 13.4 1.6 0 71.1<br />
3 11 25.5 9.9 64.7 13.0 1.3 21.9 3.5 3.0 57.8<br />
4 H2S, C5Hl2 15.3 14.9 70.2 11.0 0.7 18.1 3.9 0 69.7<br />
5 11 15.3 14.9 70.2 13.4 4.8 12.9 2.8 0 70.0<br />
The following stoichiometry would be expected for the reaction <strong>of</strong> methane:<br />
2H2S + CH4 -+ CS2 + 4H2. (3)<br />
In run number 3 the acetylene was produced by the back diffusion <strong>of</strong> methane into the<br />
plasma region.<br />
The stoichiometry €or the neopentane reaction would be<br />
lOH2S + C5H12 -f 5cs2 + l6HzS. (4)<br />
The reactions <strong>of</strong> neopentane were performed with a large excess <strong>of</strong> hydrocarbon with<br />
the hope <strong>of</strong> detecting sulfur insertion compounds OK reaction intermediates. How-<br />
ever, hydrogen and carbon disulfide were the only gaseous species produced. This<br />
indicates that once the neopentane reacts with one sulfur atom its resistance to<br />
further reaction is lowered.<br />
Table 2 shows the variation <strong>of</strong> reactant decomposition with changes <strong>of</strong> hydrogen<br />
sulfide to hydrocarbon flow ratio and plasma-hydrocarbon injection distance. From<br />
Table 2 it can be seen that the extent <strong>of</strong> hydrogen sulfide decomposition is a<br />
function <strong>of</strong> the amount <strong>of</strong> hydrocarbon present downstream from the plasma flame and<br />
the distance from the flame that the hydrocarbon is introduced. the highest - decom-<br />
Position, 95 per cent, occurring in run number 4 with an approximately ten fold excess ’<br />
Of neopentane introduced 1/4 inch from the flame. The figures for the decomposition ’<br />
Of hydrogen sulfide tend to be misleading, the hydrocarbon addition does not change 1<br />
the degree <strong>of</strong> dissociation <strong>of</strong> the hydrogen sulfide but rather it reacts with the<br />
atomic sulfur minimizing formation <strong>of</strong> hydrogen sulfide by the recombination <strong>of</strong> the I<br />
i
237<br />
Table 2<br />
Experimental Variables and Xeaction Conversion Efficiencies<br />
Flow<br />
HC inlet<br />
distance Conversion <strong>of</strong> decomposed<br />
Run<br />
- NO. Reactants<br />
rate<br />
(total)<br />
*<br />
c.f.h.<br />
HZS/HC<br />
from<br />
plasma<br />
inches<br />
Reactant reactants to:<br />
decomposition CS?<br />
H? -<br />
H2S HC H2S IHC &S + HC<br />
% % -%- x %<br />
1 H2S, CH4 0.14 2.75 114 72 63 65 *lo0 %lo0<br />
2 I, 0.14 2.75 1 59 28 33 1.100 %lo0<br />
3 ,I 0.33 0.39 114 85 43 93 36 81<br />
4 HzS, CgH12 0.25 1.03 1/4 95 28 55. 19 46<br />
5 I, 0.25 1.03 1 68 12 56 30 61<br />
* cu ft/hr.<br />
atomic hydrogen and sulfur. The data on extent <strong>of</strong> conversion to hydrogen and<br />
carbon disulfide in Table 2 show that in runs 1 and 2 essentially all <strong>of</strong> the<br />
carbon from the decomposed methane is converted into carbon disulfide and the<br />
hydrogen into molecular hydrogen. In run number 3 the acetylene produced accounts<br />
for the missing carbon and hydrogen. In runs number 4 and 5 a large fraction <strong>of</strong><br />
the neopentane that was decomposed was converted into a solid product.<br />
Table 2 also shows the percentage <strong>of</strong> the sulfur, resulting from the decom-<br />
position <strong>of</strong> the hydrogen sulfide, that is converted into carbon disulfide; the<br />
remainder <strong>of</strong> the sulfur appears as elemental sulfur (runs 1-3) or can be incor-<br />
porated with the carbon-hydrogen polymeric solid that is produced in the runs<br />
us ing neopentane .<br />
These results indicate that hydrogen sulfide can be,completely dissociated by<br />
an electric discharge and the excited atomic hydrogen and sulfur generated can be<br />
reacted with hydrocarbons to produce molecular hydrogen and carbon disulfide.<br />
Under conditions which minimize back diffusion <strong>of</strong> the hydrocarbon into the electric<br />
discharge substantially complete conversion <strong>of</strong> the hydrogen sulfide and hydrocarbon<br />
to hydrogen and carbon disulfide can be effected.<br />
1.<br />
2.<br />
3.<br />
4.<br />
5.<br />
6.<br />
7.<br />
8.<br />
9.<br />
References<br />
Knight, A. R., Strausz, 0. P. and Gunning, H. E.,J. Am. Chem. Soc. 85, 2349,<br />
(1963).<br />
Bryce, W. A. and Hinshelwood, C., J. Chem. Soc. 4, 3379, (1949).<br />
Nabor, G. W. and Smith, J. M., Ind. Eng. Chem. 45, 1272, (1953).<br />
Thacker, C. M. and Miller, E., Ind. Fng. Chem. 36, 182, (1944).<br />
Folkins, H. O., Miller, E. and Hennig, H., Ind. Eng. Chem. 2, 2201, (1950).<br />
Fisher, R. A. and Smith, J. M., Ind. Eng. Chem. 42, 704, (1950).<br />
Forney, R. C. and Smith, J. H., Ind. Eng. Chem. 43, 1841, (1951).<br />
Thomas, W. J. and Strickland-Constable, R. F., Trans. Farad. Soc. 53, 972, (1957)<br />
Vastola, F. J., Walker, P. L., Jr. and Wightman, J. T., Carbon 1, 11, (1963).
238<br />
The extensive literature <strong>of</strong> "active" nitrogen chemistry does not appear to<br />
I<br />
record any study <strong>of</strong> competition between organic substrates. This paper reports an<br />
investigation <strong>of</strong> consumption <strong>of</strong> hydrocarbon in the competition between an olefin,<br />
ethylene, and a paraffin, propane. Reaction probably went to completion under most,<br />
if not all, experimental conditions.* Consumption <strong>of</strong> hydrocarbon as a result <strong>of</strong><br />
both primary attack by one or more components <strong>of</strong> "active" nitrogen and <strong>of</strong> secondary<br />
attack by reactive intermediates must be considered possible.<br />
Expcrimental<br />
Apparatus and metnods are described more completely elsewhere, 3'4. Active<br />
nitrogen was generated by electrodeless glow discharge supported by 2450MHz<br />
microwaves at a total pressure <strong>of</strong> 321 Torr. Transport rates were 230cm. sec.-l,<br />
150p mole set.-' and 1.3t0.2~ mole set.-' for linear flow, N2 flow and N(4S) flow,<br />
respectively. !he latter was measured by nitric oxide "emission" titration.'<br />
Hydrocarbon substrate was introduced countercurrent3 into the active nitrogen stream<br />
at autogenous temperature (approx. 5G°C) acm. (.G87 sec.) downstream from the glow<br />
discharge. The gas stream psssed through liquid pitrogen traps 5Gcm. (.22,sec.)<br />
downstream from the substrate inlet. Amounts <strong>of</strong> recovered reactants were determined<br />
by gas chromato raphy on silica &el.4 Relative activities <strong>of</strong> 14Clabeled compounds<br />
were determined g by proportional counting <strong>of</strong> CO2 obtained by combustion <strong>of</strong> the gas<br />
chromatographically purified compounds in 02 over CuO at 450°. Propane and labeled<br />
ethylene were Matheson products. Cold ethylene was Phillips Research Grade.<br />
Data<br />
Table I smarizzs 2ercep.t consumption <strong>of</strong> ethylene consequent upon reaction <strong>of</strong><br />
the pure substrate and three <strong>of</strong> its mixtures with propane with six different ratios<br />
<strong>of</strong> N(4S). Table I1 sunmarizes analogous data for propane. The data formixtures<br />
given in'the two Tables are derived from the same sets <strong>of</strong> experiments. Table I11<br />
summarizes apparent relative specific rates <strong>of</strong> consumption, IlkCpH4/kC3H8tt. lhese<br />
ratios were calculated by means <strong>of</strong> e . 1, the expression which would be appropriate<br />
c ~ ~ ~ / ~ c logC(C2H4)J ~ H ~ I ~ (C,H4)tIq10gC(C,H8)o/(C~H8)tI ,(I)<br />
if the relative rates <strong>of</strong> consmption depended entirely on the bimolecular reaction<br />
<strong>of</strong> each <strong>of</strong> the substrates with the same reagent. Table IV summarizes molar<br />
radioactivities <strong>of</strong> recovered reactants or product ethane relative to that <strong>of</strong> reactant<br />
ethylene for the reaction <strong>of</strong> equimolar mixtures <strong>of</strong> 14C-labeled ethylene and ordinary<br />
propane.
239<br />
Zhbie I<br />
Eerccnt Consumption <strong>of</strong> Ztcylene<br />
(Total Hydrocarbon) Average Percent Consumption<br />
(N)a Pure (C&4),/ (c3H8) e<br />
C2H4 nb 3.0 nb 110 n6 -0.33 nb<br />
4 19 3 13 3 25' 10 -<br />
2 24' 3 27 3 37+ 1c 51 6<br />
1 3?+ 4 45 4 47X 16 56 9<br />
21 3 45 2 59 1 - 730 2<br />
112 7PX 3 87 3 93 10 90 7<br />
, 116 9;o 2 - 93 7 84 3<br />
a.<br />
b.<br />
a.<br />
b.<br />
Flow rate <strong>of</strong> N(4S) is l.3*O.& mole sec.-l throughout.<br />
Number <strong>of</strong> independent determinations averaged to give the tabulated figure.<br />
Table I1<br />
Percent Consumption <strong>of</strong> Propane<br />
41" 2 39 3 39 10 37 7<br />
480 2 - 4 5 7 4 4 3<br />
now rate <strong>of</strong> N(4S) is l.3?S10.2p mole set.-' throughout.<br />
Number <strong>of</strong> independent determinations averaged to give the tabulated<br />
figure.<br />
-<br />
I
( rot-1 -!y-roccrbon)<br />
240<br />
xprr2nc <t1\. ., 3czctlvltlcs<br />
"kC _.<br />
/kc Ii I'<br />
(14 2'14 3 8<br />
1<br />
Sz>craco'<br />
(CPfi4) J( C3II.S 10 I<br />
3.c 1.c 0.35<br />
- o.:? 1.1 -<br />
2 (0.30) 0.33 1.2 2.9<br />
1 (1.2) 1.5 1.7 2.3<br />
2/3 - 2.0 - 3.5<br />
1/2 (2.9) 4.1 5.4 5.2<br />
1/5<br />
(4.0) - 4.5 3.1<br />
a. Numbers in this column are calculated from the data for pure substrates.<br />
(Total Hydrocarbon) is t,d.cn arbitrarily as the concentration <strong>of</strong> one <strong>of</strong> the<br />
pure hydrocarbons. Values are based on data for identical concentrations<br />
<strong>of</strong> pure hydrocarbons.<br />
,<br />
bot& ii drocarbon<br />
Ilelative 1,iolar Activities <strong>of</strong> Recovered<br />
Reactants and Product Etiannea<br />
2 0.9s 7 - 0.01c 1<br />
1 0.98 4 - - /<br />
- 0.8 1 0.0lC 1<br />
0.3') 2 0.5 1 O.OlC 1<br />
a. Conpared to unrcacted ethylene; hydrocarbon reactmts equbols.<br />
b. '&e number or^ indepcndcnt ex?erimcnts.<br />
c.<br />
d.<br />
Indistinguishable from the value found for,unreacted propane.<br />
Flow rate <strong>of</strong> N(4S) was 1.z20.2p mole set.-' throughout.<br />
P<br />
I<br />
I
241<br />
Discussion<br />
The systematic variation <strong>of</strong> the ratio IlkC a /k 'I over the range <strong>of</strong> concentration<br />
parameters summarized in Table I11 demonstrateg thaE3r8lative consumption <strong>of</strong><br />
competing hydrocarbons is not determined simply by the relative rates <strong>of</strong> attack <strong>of</strong><br />
N(4S) (or any set <strong>of</strong> reactive species) on ethylene and propane sinc? such determination<br />
would lead to constancy <strong>of</strong> the ratio. A similar conclusion can be drawn from the<br />
data <strong>of</strong> Tables I and I1 by comparison <strong>of</strong> the consumption <strong>of</strong> a given substrate at a<br />
fixed concentration upon reaction witln a fixed proportion <strong>of</strong> N(4S) in the presence and<br />
absence <strong>of</strong> the competing substrate. Such matched points are designated in Tables I<br />
and I1 by identicd superscripts. If the simple model <strong>of</strong> the competition from which<br />
eq. 1 is derived were correct, ccnd Leeping in mind the similar degrees <strong>of</strong> consumption<br />
<strong>of</strong> the two Axtrates, -.ddition <strong>of</strong> the second substrate should always substantially<br />
suppress the consumption <strong>of</strong> the first and should have the largest effect when substrate<br />
is in excess over reagent. In fact, with (reactant hy&ocarbon)/(N)>/l such<br />
suppression is absent or negligible. It becomes significant only when N(4S) is in<br />
excess. The complexity <strong>of</strong> the reaction is also indicated by the shall.ow dependence<br />
<strong>of</strong> percent consumption <strong>of</strong> pure hydrocarbon on the ratio, (pure hydrocarbon)/(N),<br />
particularly with propane for which a twelvefold decrease in the raeo is associated<br />
with increase in percent consumption by a factor <strong>of</strong> only 1.65. Such behaviour is<br />
consistent with destruction <strong>of</strong> the substrate by both attacking reagent and reactive<br />
+itexmediates arising from the primary attack if, as the proportion <strong>of</strong> N(*S) is raised,<br />
/these intermediates are increasingly consumed by the reagent before they can attack<br />
the substrate. This feature is also present with ethylene but to a smaller extent.<br />
,<br />
Except with the largest excess <strong>of</strong> N(*S), the value <strong>of</strong> 1'kC,Hd/kC2HiI (Table 111)<br />
increases with decrease in the ratio (CpH.+)o/(CjH~)o at constit -val;es <strong>of</strong> (total<br />
hydrocarbon)/ (N). 'Ihis systematic change is consistent with occurrence <strong>of</strong> competition<br />
'between the two substrates with respect to their destruction by reactive intermediates<br />
arising from primary attack <strong>of</strong> the reagent. More specifically, (cf. Tables I and 11)<br />
.ethylene appears to be more sensitive to consumption by intermediates arising from<br />
propane than vice versa. Presumably, the effect <strong>of</strong> attack by reactive intermediates<br />
can be reduced or eliminated by using sufficiently large excesses <strong>of</strong> N(*S) that the<br />
intemediates react with the latter rather than with hydrocarbon. 'Ihis analysis<br />
hggests that the values <strong>of</strong> 1'kC,H,/kC3He11 obtained with the largest excess <strong>of</strong> N(4S)<br />
-most closely approximate the &e valie-<strong>of</strong> this ratio for primary attack and indicates<br />
that this true value is at least 5. (See further discussion <strong>of</strong> the data at (hydrocarbon)<br />
/(N) = 116 below.)<br />
3<br />
9<br />
'Be above discussion tacitly asrmmes that N(4S) is the only component <strong>of</strong> "active"<br />
\!nitrogen which is significantly involved. Ilbat this is BO for ethylene is widely<br />
'<br />
accepted. The work <strong>of</strong> Jones and bluer6 suggests that it is erobabl also the case<br />
%or propane. Ilhe latter study provides a value <strong>of</strong> 1.0 x lo6 M 'set.-' for the specific<br />
\ate <strong>of</strong> reaction <strong>of</strong> CSH~ at 50°C. Since nitrogen atom cancedtrations were determined<br />
from HCN yields produced by reaction with ethylene, this constant should be adjusted<br />
Uownward 30 to pk, e.g. to 7 x lo5 M-'sec.-'. Since the constant was apparently<br />
evaluated from experiments in which propane was in molar excess over propane it may also<br />
be significantly high because it does not correct for the consequences <strong>of</strong> attack by<br />
,,'reactive, intermediates arising from initial attack by N(*S). Heerron has estimated from<br />
his elegant mass spectrometric study7 <strong>of</strong> the reaction <strong>of</strong> N(*S) with ethylene that the<br />
bate <strong>of</strong> primary attack in the (virtually temperature independent) reaction <strong>of</strong> N(4S)<br />
(with ethylene is equal to or less than 7 x lo6 M-'=ec.-'. A retboe OS lthe ratio <strong>of</strong><br />
dspecific rates <strong>of</strong> primary attack by N(4S) on ethglene and prbpae, respectively, in<br />
the vicihity <strong>of</strong> 10 is thus indicated by earlier work. Agroemmt with the present<br />
t<br />
I<br />
\
242<br />
result is well within the range <strong>of</strong> the mutual uncertainties. Herronls work further .<br />
establishes that hydrogen atoms produced in the reaction <strong>of</strong> N(4S> with ethylene<br />
compete with the reagent for the substrate and, in fact, provides the model upon which<br />
interpretation <strong>of</strong> the oresent data is based.<br />
!he data <strong>of</strong> Table IV establish that even with a sixfold excess <strong>of</strong> N(4S),<br />
synthesis <strong>of</strong> propane4 in the reaction mixture at least in part from fragments<br />
originating in ethylene is not detectable. Production <strong>of</strong> ethylene ejther by degradation<br />
<strong>of</strong> propane or synthesis at least in part from fragments originating in<br />
propane occurs to H detectable extent with a sixfold excess <strong>of</strong> N(4S) and equal initid<br />
concentrations <strong>of</strong> the hydrocarbons. Presumably such processes are more important<br />
with propane in excess over ethylene. !he values <strong>of</strong> 1~c3n8'1 obtained with a<br />
sixfold excess <strong>of</strong> N(4S) are, accordingly, reduced by such replacement <strong>of</strong> consumed<br />
ethylene, the reduction being greatest with excess propane. !his effect results in<br />
reversal <strong>of</strong> the trend to higher values <strong>of</strong> tlkC2H4/+jHst1 with increasing excess <strong>of</strong><br />
N(4S) which is apparent in the data <strong>of</strong> Table 111. It can also account for the<br />
unusual sequence <strong>of</strong> values <strong>of</strong> this ratio with increasing proportion <strong>of</strong> propane<br />
observed with (hydrocarbon)/(N) = 6 and to a lesser extent when this ratio equals 2.<br />
!he data <strong>of</strong> Table IV also establish that ethane is produced from both ethylene and<br />
propane.<br />
1.<br />
2.<br />
3.<br />
4.<br />
5.<br />
6.<br />
7.<br />
Footnotes<br />
Paper VI in the series, "Reactions <strong>of</strong> Active Nitrogen with Organic Substrates."<br />
lhis statement is based on a study <strong>of</strong> the effect <strong>of</strong> trapping times on droduct<br />
yields in the reaction <strong>of</strong> "active" nitrogen with propylene by Dr. Y. IPitani<br />
in these laboratories. Cf. N. N. Lichtin, Int. J. Radiation Biol., in press.<br />
Y. Shinocaki, R. Shaw and N. N. Lichtin, J. Am. Chem. Soc., 5 $1 (1964).<br />
P. T. Hinde, Y. Titani and N. N. Lichtin, z., in press.<br />
P. Harteck, G. G. Mannella and R. R. Reeves, J. Chem. Phys., 3 608 (1958).<br />
Y. E. Jones and C. A. Winkler, Can. J. Chem., 42<br />
&?<br />
J. T. Herron, J. Phys. Chem., 69 2736 (1965).<br />
d<br />
1948 (1964).
I<br />
\<br />
1<br />
243<br />
FORMATION OF HYDROCARBONS FROM H2 + CO IN<br />
MICROWAVE-GENERATED ELECTRODELESS DISCHARGES<br />
Bernard D. Blaustein and Yuan C. Fu<br />
U. S. Bureau <strong>of</strong> Mines, Pittsburgh Coal Research Center,<br />
4800 Forbes Avenue, Pittsburgh, Pennsylvania 15213<br />
I<br />
Formation <strong>of</strong> hydrocarbons from H2 + CO over metal catalysts is <strong>of</strong> considerable<br />
interest and has been studied at length. Hydrocarbons can also be formed in<br />
electrical <strong>discharges</strong>.3, 5-9. l1-I7/ and this <strong>of</strong>fers an interesting alternative for<br />
producing hydrocarbons from H2 + CO.<br />
Fischer and Peters?/ worked with a flow system<br />
where the gases at 10 torr pressure circulated through a discharge (50 Hz) between<br />
metal electrodes, then through a mercury vapor pump, a liquid air-cooled trap, and<br />
back to the discharge. Conversion <strong>of</strong> CO to hydrocarbons was very low for one pass<br />
through the discharge, but by recirculating the gases for 100 minutes, practically<br />
all <strong>of</strong> the CO would react.<br />
Unt?’ and Epple and Ap&1 worked with electrodeless radi<strong>of</strong>requency (2-110 MHz)<br />
<strong>discharges</strong> in static reactors at pressures up to 300 torr. Conversions <strong>of</strong> H2 + CO<br />
and H2 + C02 to C q were quite high f?f/reaction times <strong>of</strong> several minutes; no other<br />
hydrocarbons were formed. McTaggart,- and Vastola et a1.161 working with<br />
electrodeless microwave (2450 MHz) <strong>discharges</strong> in flow systems at pressures <strong>of</strong> a few<br />
torr formed only traces <strong>of</strong> hydrocarbons from H2 + CO. Here, the gas passed through<br />
the discharge only once, and the residence time was a fraction <strong>of</strong> a second. However,<br />
work in our laboratory has shown that in a static reactor, and with reaction times <strong>of</strong><br />
the order <strong>of</strong> a minute, CO can be converted in high yields to hydrocarbons in an<br />
electrodeless microwave discharge in H2 + CO. under conditions where essentially no<br />
polymers are formed.<br />
EXPERIMENTAL PROCEDURES<br />
Reactions were carried out in 11-nun ID cylindrical Vycor reactors placed in a coaxial<br />
cavity (Ophthos Instruments, similar to type 2A described by Pehsenfeld et a1.21)<br />
connected by a coaxial cable to a Raytheon KV-lWA CMD-10 2450 MEz generator.<br />
For a run, the reactors were evacuated to < 3 p and filled with either 5:l Hz-CO<br />
or various H2-CO-Ar (typically 5.1:1.0:0.4) mixtures prepared from tank gases and<br />
stored in the vacuum system. Product analyses were made on a CEC 21-1OX mass<br />
spectrometer. Net power into the discharge, measured with a Microwave Devices<br />
model 725.3 meter, was approximately 36 watts, although lower power levels could be<br />
used. Air was blown through the cavity to cool the reactor somewhat, but the ,<br />
estimated wall temperature in the discharge was still several hundred degrees C.<br />
Runs with argon gave the same results as in the absence <strong>of</strong> Ar, and the ratio <strong>of</strong><br />
total-carbon-to-argon in the product was usually a few percent lower in the product,<br />
but did not vary by more than + 10 percent from its value in the original mixture,<br />
indicating that only negligible amounts <strong>of</strong> polymers were formed in the reaction.<br />
For H2 + CO mixtures without Ar it was assumed that polymer formation was negligible,<br />
so long as reaction conditions were similar.<br />
As a precaution against the gradual accumlation <strong>of</strong> small amounts <strong>of</strong> polymer in the<br />
reactors, they were cleaned before each run by maintaining a discharge in O2 at<br />
about 10 torr for 3 minutes to oxidize any carbonaceous material present. This was<br />
repeated twice, with fresh samples <strong>of</strong> 02. A discharge in H2 (about 10 torr) was<br />
then maintained for 3 minutes in the reactor. This was also repeated twice, with<br />
fresh samples <strong>of</strong> H2. Since the reactions to be studied were to be carried out in<br />
a reducing environment, this treatment with H2 was felt to be desirable after the<br />
O2 discharge.
244<br />
RESULTS<br />
Figure 1 shows the results <strong>of</strong> experiments made at initial gas pressure <strong>of</strong> 12 + 1<br />
torr in 36-cm long by 11-mm I D reactors. The percent <strong>of</strong> carbon present in the<br />
product as C2Hp and as CH4 + C2H2 is plotted vs. reaction time. Figure 2 shows<br />
the percent <strong>of</strong> carbon present in the product as CH4 for the same runs. Water, C02.<br />
and occasionally slight traces <strong>of</strong> other hydrocarbons, were also present in the<br />
product.<br />
times <strong>of</strong> 30-120 seconds. Figure 3 shows the results <strong>of</strong> experimentslmade at initial<br />
gas pressures <strong>of</strong> 50 t 3 torr in 20-cm long by 11-mm ID reactors, Here, the percent<br />
<strong>of</strong> carbon present as CH4 + C2H2 is at a m3ximum <strong>of</strong> 24-25); for reaction times <strong>of</strong><br />
3-4 minutes. There is considerable scatter In the data; the dashed curves are<br />
intended to show only the general trend.<br />
The conversion <strong>of</strong> CO to CHq + C2H2 reaches a maximum <strong>of</strong> 17-18% for reaction<br />
These data show that conversion <strong>of</strong> CO to hydrocarbons in the discharge under these<br />
conditions is Limited. The composition <strong>of</strong> the gases in the reactor approaches a<br />
stationary state, for reaction times <strong>of</strong> the order <strong>of</strong> 0.5 to 3 minutes, depending<br />
upon the pressure. Because <strong>of</strong> the geometry <strong>of</strong> the reactors, and the volume <strong>of</strong> a<br />
reactor relative to the volume occupied by the discharge, the time required to<br />
reach the stationary state appears to be longer than is actually so, due to the<br />
relatively long times required for diffusion <strong>of</strong> gases into and out <strong>of</strong> the discharge.<br />
(The 36-cm long reactors were used for the runs made at 12 torr; for the runs mede<br />
at 50 torr, the shorter 20-cm long reactors were used.<br />
Preliminary experiments<br />
showed that the conversion <strong>of</strong> CO to CH4 + C2H2 at 50 torr in 36-cm long reactors<br />
was essentially the same as for the 20-cm long reactors, but took 1-2 minutes<br />
longer to achieve mximm conversions.)<br />
High yields <strong>of</strong> gaseous hydrocarbons from H2 + CO have now been shown to occur in<br />
low-frequency (50 Hzll and 60 H Z / ) <strong>discharges</strong>, radi<strong>of</strong>requency (2-110 MHz)?/ and<br />
microwave (2450 MHz) <strong>discharges</strong>. However, in all <strong>of</strong> these cases, the reaction does<br />
not appear to be an extremely rapid one, such as is the case for dissociation <strong>of</strong><br />
diatomic molecules in a discharge, for instance.<br />
For reaction times longer than those needed to reach maximum conversions, the<br />
conversions appear to decrease. This is probably due to some polymer fomtion,<br />
but not enough to show up as a 10% decrease in the analytically determined C/Ar<br />
ratio. Several runs at 12 torr (not plotted) <strong>of</strong> 5 minutes duration, and some<br />
longer runs, including one at 50 torr for 10 minutes, gave values <strong>of</strong> the C/Ar<br />
ratio more than 10% below the initial value <strong>of</strong> the ratio, and this was considered<br />
evidence <strong>of</strong> polymer formation. Here, also, the yields <strong>of</strong> CH4 and C2H2 were lover.<br />
Conversions <strong>of</strong> CO could be increased by removing reaction products ae they forrmd,<br />
by having a cold trap surround the bottom 5 cm <strong>of</strong> the reactor before and during<br />
the time that the discharge was maintained at a distance <strong>of</strong> 10 cm from the bottom.<br />
The discharge is localized and extends over a distance <strong>of</strong> about 2 cm. As shown in<br />
table 1, the conversion <strong>of</strong> CO increased markedly and the product distribution<br />
changed. Values for the C/Ar ratio indicated that polymers were not formed. Also,<br />
there is no indication that the product distribution is any different in the presence<br />
<strong>of</strong> Ar than in its absence. The first three columns in the table are for the 3-<br />
minute runs shovn in figures 1 and 2.<br />
When the discharge was maintained for 3 minutes’<br />
or longer, with Dry Ice (-78O) surrounding the end <strong>of</strong> the reactor, the percent Of<br />
carbon present in the final product as CHq is 51%; C2H2, 17%; CzRg, la. At this<br />
temperature, the only reaction product frozen out is H20. Apparently, the removal<br />
<strong>of</strong> this is sufficient to increase the convereion <strong>of</strong> CO to hydrocarbons to approxi-<br />
mately 78%.<br />
I<br />
I<br />
/J
h<br />
z<br />
-<br />
a<br />
o m<br />
a<br />
20<br />
T IME, minutes .<br />
Fiqure 1.- Formation <strong>of</strong> hydrocarbons from H2+ CO in<br />
microwave- generated discharge: acetylene 0,<br />
acetylene + methane 0. Initial gas pressure =<br />
1251 torr.<br />
11-30-66 L-9694
20<br />
15<br />
IO<br />
5<br />
0<br />
0<br />
0<br />
L 0<br />
0<br />
I 2 3 4<br />
TIME, minutes<br />
Figure2.-Formation <strong>of</strong> CH, from H,t CO in<br />
microwave -generated discharge. Initial<br />
gas pressure= 12 t I torr.<br />
11-30-66 L- 9695<br />
0<br />
0<br />
5<br />
r<br />
i
I 1 I I I I<br />
0 I 2 3 4 5<br />
TIME, minutes<br />
Figure3.-For mation <strong>of</strong> hydrocarbons from He + CO in<br />
microwave- generated discharge: methane CY,<br />
acetylene A, acetylene + methane 0. Initial<br />
gas pressure=50+ 3 torr.<br />
11-30 -66 L- 9696
d<br />
r-<br />
0<br />
r-<br />
,-I<br />
d<br />
N<br />
2<br />
N<br />
2<br />
co<br />
Ln<br />
N<br />
0<br />
m<br />
m<br />
N<br />
m<br />
d<br />
d<br />
0<br />
d<br />
4<br />
m<br />
d<br />
N<br />
4 0<br />
m<br />
N<br />
d<br />
co<br />
m<br />
d<br />
h<br />
2<br />
d<br />
h<br />
N<br />
4<br />
E)<br />
d<br />
N<br />
0<br />
N<br />
QI<br />
m<br />
N<br />
0<br />
4<br />
N<br />
m<br />
N<br />
0<br />
d<br />
.s<br />
0%<br />
N<br />
e<br />
..<br />
c<br />
a<br />
d<br />
00<br />
co 4<br />
U<br />
4<br />
a<br />
c<br />
0<br />
U<br />
W * ?N.g<br />
W *
The next five coluws in table 1 show that with liquid N2 cooling (-196'), the<br />
percent <strong>of</strong> carbon in the product as CH4 in only 4%; C2H2 + C2H6 (with the latter<br />
predominating) 81%; C3 + C4 hydrocarbons, 69.; more H20 and C02 are also formed.<br />
The mass spectrometric analyses indicated that traces <strong>of</strong> oxygenates were also<br />
present in the products <strong>of</strong> these runs, but in such small amunts that no identi-<br />
fications could be attempted. For some unknown reason, C2H4 is not formed under<br />
these conditions. (Several runs were analyzed by gas chromatography For C2H4 and<br />
none was found.) At a temperature <strong>of</strong> -196O, the only non-condensables present in<br />
the mixture are H2, CO, and CH4; all the other products are frozen but. One can<br />
speculate that the high yield <strong>of</strong> C2H6 is due either to a recombination <strong>of</strong> CH3<br />
radicals at the cold surface, or hydrogenation <strong>of</strong> C2H2, but the mechanism <strong>of</strong><br />
formation <strong>of</strong> any <strong>of</strong> the hydrocarbon products is not known.<br />
The next-to-last pair <strong>of</strong> columns in table 1 show that very high conversions <strong>of</strong> CO<br />
to C2- and higher hydrocarbons can be obtained using a 2.2:l H + CO mixture.<br />
Here, also, the C02 production is higher, and the CH4 lower, tian compared with<br />
the more hydrogen-rich reactant mixture. The last pair <strong>of</strong> columns, giving data<br />
for runs made at 50 torr, indicate that C2H2 was, by far, the predominant product<br />
in these runs.<br />
The almost complete conversion <strong>of</strong> CO to hydrocarbonqH20, and C02, obtained by<br />
cooling the bottom <strong>of</strong> the reactor, is reversible. Several additional experiments<br />
(at 12 torr), where the gases were reacted for 3 minutes while the reactor was<br />
cooled, the bottom <strong>of</strong> the reactor then warmed to room temperature in a few seconds<br />
with a water bath, and the gases reacted for 2 more minutes, gave the same product<br />
compositions as for the runs shown in figures 1 and 2.<br />
Table 2 gives the results <strong>of</strong> some preliminary experiments where water vapor was<br />
added to the H2 + CO + A r mixture before reaction. The water vapor was added to<br />
the previously evacuated reactor and a portion <strong>of</strong> the vacuum system connected to<br />
a Pace Engineering Co. Model P7 pressure transducer containing a + 1 psi diaphragm.<br />
The output <strong>of</strong> this was indicated on a Pace Model CD25 transducer indicator. After<br />
the water vapor partial pressure was measured, the stopcock to the reactor was<br />
closed and the water frozen at,the bottom <strong>of</strong> the reactor. The reactor was then<br />
filled with the H2-CO-Ar mixture, the bottom <strong>of</strong> the reactor warmed slightly, and<br />
3 minutes allowed for mixing <strong>of</strong> the gases before the discharge was initiated.<br />
Even from these few experiments, it can be seen that adding water vapor to the<br />
initial reactant mixture has a strong inhibitory effect on the production <strong>of</strong><br />
hydrocarbons.<br />
Comparison <strong>of</strong> the second and third runs listed in the table shows that 3 minutes<br />
reaction time increases the yield <strong>of</strong> hydrocarbons only slightly as compared to'one<br />
minute. The last two runs in the table show that when the water vapor partial<br />
pressure is equal to or greater than the CO partial pressure in the reactant gas<br />
mixture, no hydrocarbons are produced at all.<br />
(The C02 yield is increased due to the reaction H20 + CO +C02 + H2.)<br />
The pronounced inhibitory effect <strong>of</strong> H20 vapor on hydrocarbon formation in this<br />
reaction may explain some <strong>of</strong> the scatter <strong>of</strong> the data shown in figures 1, 2, and 3.<br />
It is possible that small (and variable) amounts <strong>of</strong> H20 vapor were adsorbed on the<br />
walls <strong>of</strong> the reactor tubes, and that this decreased the hydrocarbon yield in (some<br />
<strong>of</strong>) the runs by a varying amount which shows up as scatter in the data. Further<br />
experiments would have to be done to shed more light on this point. However, there<br />
are other, as yet unknown, sources <strong>of</strong> variability in the experimental conditions<br />
which also undoubtedly contribute to the scatter in the data.
250<br />
TABLE 2. - Effect <strong>of</strong> adding water to the H7-CO-Ar mixture.<br />
Bepression <strong>of</strong> hydrocarbon formation<br />
Run 11153<br />
Initial H2-CO-Ar<br />
pressure torr<br />
(5.1 : 1.0:0.4) 10.8<br />
Initial H20 pressure<br />
added, torr<br />
.35<br />
Time <strong>of</strong> discharge, min. 1<br />
Percent <strong>of</strong> carbon present<br />
in product as<br />
CH4<br />
4.0<br />
C2H2 4.2<br />
co2 3.3<br />
Hydrocarbons 8.2<br />
DISCUSSION<br />
11152 11154 11152<br />
12.6 13.9 11.8<br />
.85 .95 1.83<br />
1 3 1<br />
1.9 1.7 0.0<br />
1.8 2.6 0.0<br />
4.6 4.5 8.8<br />
3.7 4.3 0<br />
11013<br />
12.7<br />
1 6<br />
1<br />
0.0<br />
0.0<br />
23.1<br />
0 .i<br />
I<br />
the reaction in I 1<br />
a stationary i<br />
The different conversions <strong>of</strong> CO can be explained by assuming that<br />
the discharge. without cooling the bottom <strong>of</strong> the reactor, reaches<br />
state where-the production <strong>of</strong>-CH4 and C2W2 is limited by-the back reaction <strong>of</strong> these<br />
hydrocarbons with H20 and/or C02 to form H2 + CO. The data in figure 3, by comparison<br />
with the runs in figures 1 and 2, indicate that conversion <strong>of</strong> CO to hydrocarbons is<br />
increased at higher initial pressures, as would be expected for a reaction where the<br />
volume <strong>of</strong> the products is less than the reactants. When one or more <strong>of</strong> the reaction<br />
products are removed from the discharge zone by being frozen out, the Stationary<br />
state shifts and more CO reacts to form hydrocarbons. When the frozen-out hydrocarbons<br />
are re-introduced into the discharge, they react very readily to re-form<br />
the initial stationary state composition. If, on the other hand, water vapor i0<br />
added to the initial reactant gas mixture, the conversion <strong>of</strong> CO to hydrocarbons is<br />
repressed.<br />
These experimental observations can be sunmed up by discussing the reaction<br />
H + CO + C q + H20 in terms <strong>of</strong> a stationary state in the discharge which can<br />
shft in the direction that would be predicted by applying Le Chatelier's principle.<br />
Actually, in this respect, the system behaves as if it were in <strong>chemical</strong> equilibrium.<br />
Qualitatively, this interpretation o the data is very reasonable. Fischer and<br />
Peters,?/ Wendt and Evans,El Lunt,?j and Epple and ApS/ have all discurrcd the<br />
production <strong>of</strong> hydrocarbons from H2 + CO in terms <strong>of</strong> equilibria in the diocharge.<br />
However, simple arguments shov that any discussion <strong>of</strong> equilibria in <strong>discharges</strong> is<br />
not a straightforward one because <strong>of</strong> the absence <strong>of</strong> temperature equilibri<br />
the various species - electrons, ions, and molecules - in the discharge.- i$-*<br />
Manes/ has pointed out that in the statistical mechanical derivation <strong>of</strong> the<br />
equilibrium constant in terms <strong>of</strong> the part,ition functions, the functional form Of<br />
the equilibrium constant expression follows from the conservation <strong>of</strong> atom in the<br />
system. This leads to the likelihood that the H2-CO-CH4-B20CO2 rysta vlll eoafom<br />
to some equilibrium "constant" in an electrical discharge system, although th<br />
magnitude <strong>of</strong> this constant vi11 not be derivable from equilibrium thermodynndc<br />
properties.<br />
1<br />
1<br />
,<br />
1<br />
)<br />
j<br />
I<br />
' 1
\<br />
.\<br />
t<br />
251<br />
Given the existence <strong>of</strong> this equilibrium "constant" for (some) reactions occurring<br />
in a discharge, we would then expect Le Chatelier's principle to apply. In fact,<br />
the "constant" need be only approximately constant, as conditions change over a<br />
certain range, for the system to exhibit qualitative changes in accordance with<br />
Le Chatelier's principle.<br />
For a preliminary inspection <strong>of</strong> the quantitative aspects <strong>of</strong> these equilibria<br />
"constants," from the data in table 2 and the tWo one-minute runs in figures 1 and<br />
2, values were calculated for I<br />
and<br />
P X P<br />
CO H20<br />
K1 = P XP<br />
co2 H2<br />
P X P<br />
CH4 H20<br />
K2 = ---<br />
P x P3<br />
GO n2<br />
These are given in table 3. If these equilibria had been established in an ordinary<br />
thermal system, the values for K1 and K2 would depend only on temperature. However,<br />
these "equilibria" were established in a reactor system where part <strong>of</strong> the gas was in<br />
an electrical discharge. Since we cannot define one temperature for the discharge,<br />
the experimental values obtained for K1 and K2 depend on the parameters which<br />
characterize the discharge, such as rate <strong>of</strong> energy input, geometry <strong>of</strong> the reactor,<br />
the volume <strong>of</strong> gas in the discharge relative to the volume <strong>of</strong> the reactor, and the<br />
electrical variables, such as field strength, etc.<br />
The values obtained for K1 and K2 can be used to define a "<strong>chemical</strong>ly equivalent<br />
temperature" for the gases in the discharge. Thus, the average value for K1<br />
corresponds to a "temperature" <strong>of</strong> 1300'K. This is very approximate, due to the<br />
small change <strong>of</strong> K with temperature for this reaction near 130O0K. The average<br />
value for K2 corresponds to a "temperature" <strong>of</strong> approximately 800'K. Pursuing<br />
much the same line <strong>of</strong> thought, Epple and Apgl calculated "equivalent temperatures"<br />
<strong>of</strong> about 750'K from values <strong>of</strong> K1 and 850'K from values <strong>of</strong> K2 obtained from the data<br />
on the H2-CO-CQ-H20-C02 system in their static radi<strong>of</strong>requency reactor experiments.<br />
The significance, if any, <strong>of</strong> these "temperatures" calculated from the discharge<br />
data, is unknown.<br />
TABLE 3. - Values <strong>of</strong> equilibrium "constants" calculated<br />
from the data in table 2<br />
"Temperature, I'<br />
Run 11153 11151 11154 11152 11013 2154 4272 OK<br />
K1<br />
1.4 1.8 1.9 2.0 1.9 1.1 1.9 1300<br />
K2<br />
K3 x Id<br />
21<br />
7.4<br />
12<br />
17<br />
9<br />
37<br />
- 1/<br />
-<br />
K4<br />
3.2 2.5 3.2 -<br />
11 -<br />
11 -<br />
- 11 22 33 800<br />
11 4.5 6.3 1300<br />
11 2.1 6.9<br />
11 me to the large concentration <strong>of</strong> H20 vapor, no hydrocarbons were produced<br />
in these runs.
252<br />
One unusual finding <strong>of</strong> Epple and Apt is that CH4 was the sole hydrocarbon produced<br />
under their coaditions. Even though CH4 was present in the discharge for minutes,<br />
no C2H2 was produced. We have repeated their experiments (using a 400 mz<br />
generator) and can confirm this finding, as well as their values for the amount <strong>of</strong><br />
CO converted to CH4 in a radi<strong>of</strong>requency discharge. On the other hand, C2H2 is<br />
always produced in the reaction in a microwave discharge.<br />
this difference in products in the two cases, perhaps some significance can be found<br />
in the approximate values <strong>of</strong> 1300°K calculated from K1 for our microwave discharge<br />
runs as contrasted with 750'K from K1 calculated by Epple and Apt. The difference<br />
in these "temperatures" may be another indication <strong>of</strong> what we feel intuitively -<br />
namely, that the microwave discharge is "hotter" than the radi<strong>of</strong>requency discharge,<br />
because <strong>of</strong> the higher value <strong>of</strong> energy input per mole <strong>of</strong> gas in the discharge.<br />
Values were also calculated for<br />
and<br />
Kg =<br />
K4 =<br />
P x P3<br />
CZH2 "2<br />
P2<br />
CH4<br />
2<br />
P XP<br />
C2H2 H20<br />
P2 x P3<br />
CO HZ<br />
In attempting to explain<br />
and are given in table 3. From the average value for K3, one can calculate a<br />
"temperatu e" for this reaction <strong>of</strong> approximately 130OOK. The values for<br />
K4 (K4 - K: x K ) are mrch higher than there is any reason to expect, since for<br />
this reaction, jog K = -4.6 at 300°K, and decreases with increasing temperature.<br />
The values calculated for K4 indicate that the reaction<br />
is not even remotely near any sort <strong>of</strong> equilibrium in the discharge; the amount <strong>of</strong><br />
CZHZ formed is far greater than can be accounted for at equilibrium.<br />
In summary, it is helpful to discuss qualitatively at least some reactions in<br />
<strong>discharges</strong> from the point <strong>of</strong> view <strong>of</strong> stationary states or equilibria, which can<br />
19 are also attempting to discuss<br />
shift according to Le Chatelier's princip . Admittedly, this approach te<br />
speculative at the moment. Other workersdischarge<br />
reactions in terms <strong>of</strong> equilibria, and it will be <strong>of</strong> interest to see how<br />
useful these ideas prove to be in interpreting <strong>chemical</strong> reactione in <strong>discharges</strong>.<br />
The authors wish to thank Gus Pantages, Waldo A. Steiner, and Paul Golden for their<br />
technical assistance; A. G. Sharkey, Jr. and Janet L. Shultr for the mass-<br />
spectrometric analyses; and Drs. Irving Wender and Frederick Kaufman for their<br />
valuable discussions. /<br />
,<br />
REFERENCES<br />
1. Arzimovich, L. A. Elementary Plasma Physics. Blaisdell Pub. Co., New York,<br />
N. Y., 1965, pp. 5-6.<br />
Eck, R. V., E. R. Lippincott, M. 0. Dayh<strong>of</strong>f, and Y. T. Pratt.<br />
August 5, 1966, pp. 628-633.<br />
2. Scbcnce, 153,<br />
i<br />
1<br />
I<br />
/<br />
I<br />
(<br />
,i
I<br />
L<br />
253<br />
Epple, R. P. and C. M. Apt. The Fonnation <strong>of</strong> Methane from Synthesis Gas by<br />
High-Frequency Radiation. Gas Operations Research Project FT-27(ext.), Am.<br />
Gas Assoc. Catalog No. 59/OR, Am. Gas ASSOC., Inc., July 1962, 47 pp.<br />
4. Fehsenfeld, F. C., K. M. Evenson, and H. P. Broida. Rev. Sci. Instruments,<br />
- 36, March 1965, pp. 294-298.<br />
5. Fischer, F. and K. Peters. Btennst<strong>of</strong>f-Chem., 2, No. 14, 1931, pp. 268-273.<br />
6. Lefebvre, H. and M. van Overbeke. Chimie et Industrie, Special No., April<br />
7.<br />
1934, pp. 338-342.<br />
Lefebvre, H. and M. van Overbeke. Compt. rend., 198, 1934, pp'. 736-738.<br />
8.<br />
9.<br />
10.<br />
11.<br />
12.<br />
Losanitsch and Jovitschitsch in Ch. IV <strong>of</strong> "The Conversion <strong>of</strong> Coal into Oils,"<br />
by P. Fischer, E. Benn, Led., London, 1925<br />
Lunt, R. W. Proc. Roy. SOC. (London), m, 1925, pp. 172-186.<br />
Manes, M. Private Co-nication.<br />
McTaggart, F. K. Austral. J. Chem., l7, 19.64, pp. 1182-1187.<br />
Sahasrabudhey, R. H. and S. M. Deshpande. hoc. Indian Acad. Sci., =,<br />
1950. pp. 317-324.<br />
13. I Sahasrabudhey, R. H. and S. M. Deshpande.<br />
pp. 361-368.<br />
J. Indian Chem. SOC., 27, 1950,<br />
14.<br />
15.<br />
Sahasrabudhey, R. H. and S. M. Deshpande.<br />
pp. 377-382.<br />
J. Indian Chem. SOC., 28, 1951,<br />
16.<br />
1948, pp. 366-374.<br />
Vastola, F. J., P. L. Walker, Jr., and J. P. Wightman.<br />
1963, pp. 11-16.<br />
Carbon, 1, No. 1,<br />
17. Wendt, C. L. and G. M. Evans. J. Am. Chem. SOC., 50, 1928, pp. 2610-2621.<br />
Sahasrabudhey, R. H. and A. Kalyanasundaram. Proc. Indian Acad. Sci., z,
254<br />
RADIOFREQUENCY ELECTRODELESS SYNTHESIS OF POLYMERS:<br />
REACTION OF CO, N2 AND H2<br />
John R. Hollahan<br />
Richard P. McKeever<br />
Research and Development Department<br />
TRACERLAB, A Division <strong>of</strong> LFE<br />
2030 Wright Avenue<br />
Richmond, California 94804<br />
Introduction<br />
A certain amount <strong>of</strong> the large literature concerning the synthesis <strong>of</strong> compounds<br />
in electrical <strong>discharges</strong> relates to the synthesis <strong>of</strong> polymeric materials, and many<br />
industrial patents exist in this category. Most <strong>of</strong> these syntheses have employed<br />
1) hydrocarbon reactant( s) or 2) discharge apparatuses involving internal electrode<br />
configurations as the source <strong>of</strong> excitation, and the glow discharge or "cold plasma"<br />
was initiated by a. c. or d. c. fields <strong>of</strong> several hundred volts between the internal<br />
electrodes (1).<br />
We wish to report an electrodeless discharge synthesis <strong>of</strong> a polymeric material<br />
involving the simple inorganic gases CO, N2 and H2. The advantages <strong>of</strong> the electrodeless<br />
technique are principally avoidance <strong>of</strong> electrode erosion or kinetic interaction<br />
<strong>of</strong> the electrodes with the reacting gas mixtures, the capability <strong>of</strong> filling uniformly<br />
a plasma reactor with a volume <strong>of</strong> excited gas, and engineering advantages<br />
<strong>of</strong> radi<strong>of</strong>requency excitation.<br />
Run Procedure<br />
Experimental<br />
An apparatus for precision metering <strong>of</strong> several gases into plasma reactors was<br />
designed and constructed for this work. Figure 1 shows the system design. Up to<br />
four different gases may be metered individually at known flowmeter pressures.<br />
Since the gases were non-corrosive, mercury manometers adeq ately served as the<br />
flowmeter pressure measurement device. Flows <strong>of</strong> gases in cm' min-' NTP were<br />
calculated from the gas viscosity at the operating temperature, the gas density at<br />
the operating pressure and temperature, and the flowmeter scale reading. The flowmeter<br />
operating pressure P was calculated from P = AP t P where Psys<br />
is the system pressure measured<br />
FM<br />
at Point A and AP i%%e different%lspressure<br />
across the manometer.<br />
The accuracy <strong>of</strong> flow rates are taken to be f 2% to f 10% depending on flow meter<br />
condition and flow rate. In nearly every case, the f 2'7'0 accuracy was maintained.<br />
The reagent gases, sources, and statement <strong>of</strong> purity are as follows:<br />
-<br />
Gas Grade Source Purity<br />
co c. P. Mathe s on 99. 570<br />
N2<br />
H<br />
Extra Dry<br />
Pre- Purified<br />
II<br />
,I<br />
99.7%<br />
99. 95%<br />
Coleman<br />
11<br />
99. 99%<br />
After calculations <strong>of</strong> the desired flow rates and consequent required flow conditions,<br />
the entire gas line is purged by alternate pumping and filling with the desired<br />
gases. The metering valve V(2) and the flow meter pressure regulating valve V(3)<br />
are adjusted to provide the conditions <strong>of</strong> flow meter pressure P and flow meter<br />
indication necessary to produce with accuracy the desired flow-Mes. Leak checks
1 '<br />
Fig. 1 Schematic <strong>of</strong> flow discharge apparatus and gas monitoring system.<br />
I<br />
, .\\<br />
\<br />
255
256<br />
with a helium mass spectrometer leak detector are made before and after the experi-<br />
mental run. Vrlve V(1) is a gas flow on-<strong>of</strong>f valve.<br />
With the flow rates accurately adjusted, the RF Generator/RFG-600 (see below)<br />
connected to the capacitive exciter is energized and the activator tuned while bringing<br />
the generator to maximum power, - 300 watts. Trap T(l) which is filled with 3<br />
mm diameter glass beads and some pyrex wool to assure efficient trapping is chill,&<br />
with liquid nitrogen. Traps T(2) and T(3) are chilled respectively with a dry ice/2propanol<br />
slurrey and liquid nitrogen. T(3) contains 5 mm glass beqds again to assure<br />
efficient trapping and to assure non-interference from vapors originating from the<br />
pump. Next, the second RF generatorlRFG-600,connected to the inductive exciter<br />
is energized and the activator tuned while bringing the generator to maximum power,<br />
-- 300 watts.<br />
With the gases and flows selected for the experiment, the reaction proceeds and<br />
the polymer is allowed to accumulate along with the other products. After running a<br />
length <strong>of</strong> time, the generators and gas flows are turned <strong>of</strong>f and the traps allowed to<br />
warm up. The pump is then valved <strong>of</strong>f and the system let up to atmosphere with Argon.<br />
The polymer <strong>of</strong> interest is removed from the inner surface <strong>of</strong> the center tube<br />
<strong>of</strong> T(2) and retained for analysis.<br />
Radi<strong>of</strong>requency Equipment<br />
The two plasma reactors were each activated either capacitively or inductively<br />
with RF generators (RFG-600, Tracerlab, Inc. ) with deliverable power from mini-<br />
mum up to 300 watts. The generators consist <strong>of</strong> an RF section composed <strong>of</strong> a two<br />
stage system utilizing a crystal controlled oscillator, and a Class C power amplifier<br />
stage. The second part is a power supply producing plate, screen, bias, and fila-<br />
ment power. The details <strong>of</strong> the electronic requirements for generation and trans-<br />
mission <strong>of</strong> RF energy have been described elsewhere ( 2 ).<br />
Energy from the generator is transmitted to the exciter plates R(1), or coil,<br />
R(2) via a plasma activator, the circuitry <strong>of</strong> which allows maximization <strong>of</strong> the RF<br />
energf to the gas load and minimization <strong>of</strong> the reflected power back to the generator.<br />
A great experimental advantage <strong>of</strong> this means <strong>of</strong> power delivery and measurement is<br />
the ability the operator realizes in being able to reproduce his discharge conditions<br />
for the different experiments or from run to run.<br />
The net power delivered by the<br />
generator is a function <strong>of</strong> gas type and concentrations, which in turn are related to<br />
system pressures and flow rates. Each time an excited gas parameter is changed,<br />
there is a concomitant change in the intrinsic impedance <strong>of</strong> the gas load, which must<br />
be rematched to the impedance <strong>of</strong> the secondary activator circuits. Thus,<br />
the convenience <strong>of</strong> simply being able to achieve this matching by the power meter on<br />
the generator, which reads forward and reflected power, is a decided advantage.<br />
-<br />
All experiments were carried out at a radi<strong>of</strong>requency <strong>of</strong> 13.56 MHz*487 KHz, an FCC<br />
approved non-interference band. The choice <strong>of</strong> inductive vs.capacitive exciters for<br />
R(1) and R(2) was rather arbitrary, both were employed totest any significant differences<br />
in the reactions induced. The conclusion is that the chemistry is independent<br />
<strong>of</strong> the mode <strong>of</strong> excitation. There are, however, definite advantages either configuration<br />
<strong>of</strong>fers in certain systems, such as gas-solid reactions.<br />
cribed in Ref. (2).<br />
Observations and Discussion<br />
This has been des-<br />
I<br />
;<br />
Synthesis<br />
The polymers to be described below can be produced either <strong>of</strong> two ways: (1) the<br />
reaction directly <strong>of</strong> CO, H2, and N2; or (2) the reaction first <strong>of</strong> C02 and H2 to produce<br />
CO, via the water gas reaction, and then subsequent reaction <strong>of</strong> the produced<br />
1<br />
i<br />
,)
5'<br />
.<br />
'<br />
I 1<br />
257<br />
co, with excess H2, and N admitted either downstream <strong>of</strong> T(1), as indicated in<br />
2<br />
Fig. 1, or in the manifold system. The latter experiment was believed to be a purer<br />
The liquid nitrogen.<br />
Source Of CO and was employed for most <strong>of</strong> the preparations.<br />
temperature <strong>of</strong> T( 1) and its packing trapped all significant water produced in the<br />
water-gas reaction, as well as remaining COz. It became apparent that if T(l) does<br />
not trap all the significant water, that no polymer forms at T(2), due either to a reaction<br />
<strong>of</strong> H20 or discharge products therefrom (3) with polymer precursors, or to<br />
the interference <strong>of</strong> water to the deposition <strong>of</strong> the polymer as a film on the cold inner<br />
tube <strong>of</strong> T(2). I<br />
The polymers at low N flows are characterized by being transparent, yellow,<br />
tightly adherent films. At fiigher N2 flows, the transparency decreases, and film<br />
strength appears to decrease, indicating perhaps lower molecular weight materials<br />
being formed.<br />
The production <strong>of</strong> the polymer under the flow conditions listed in Tabie I appears<br />
critically dependent on the system pressure. In the experiments, the flow8 bf CO or<br />
CO and H were maintained at nearly constant values while the nitrogen flow rate<br />
waH varied The very low flow rates <strong>of</strong> N resulted from permeation through a very<br />
short segment <strong>of</strong> Tygon tubing initially connecting<br />
2<br />
R(l) to the manifold outlet. These<br />
rates <strong>of</strong> permeation were measured by observing rates <strong>of</strong> system pressure increases<br />
with time, and checked by He permeation with the mass spectrometer at the same<br />
point. In Run 861, the uncertainty is probably an order <strong>of</strong> magnitude; in Runs 631<br />
and 291, the uncertainties are half that. In any case, the low flows <strong>of</strong> N2 in the initial<br />
experiments were much less than measured flow rates in subsequent experiments,<br />
which indicate (Table I) that less and less polymer is produced other ases fixed) as<br />
ca. 107 cm' min-', no polymer<br />
was o%served to form at all.<br />
1 the N flow rate increases. At a very high N2 flow, -<br />
1<br />
).<br />
TABLE 1. Flow Conditions for Several Sample Preparations<br />
I<br />
Run<br />
I Flow Rates , cm3 min-l<br />
Power, Watts Run<br />
~<br />
h '<br />
b<br />
co2 co<br />
N2 H2<br />
861 1. 18 --- 1-3. 1~10-~ 3. 65<br />
63 1 1.43 --- 3. 9x10-3 3. 95<br />
I 291 ---- 2. 18 -42. 5x10-' 4.43<br />
bl 931 1.25 --- 1. 26 3. 60<br />
6 890 1.36 --- 107 3. 74<br />
i<br />
I 991 1.36 --- 9. 88 3. 83<br />
psYs, Time;<br />
torr R(1) R(2) Mins.<br />
.310 330 1, 355 680<br />
.320 --- 280 414<br />
.36 300 --- 720<br />
.45 320 350 463<br />
.75 320 410 460<br />
2.47 320 360 273<br />
1 Table I1 indicates elemental analyses for C, H, N, and 0 for several runs. The per<br />
cent nitrogen is found to increase as the flow <strong>of</strong> N increases. This suggests, up to<br />
a certain point, that one can produce in this experiment 2 tailored polymers containing<br />
a controllable amount <strong>of</strong> a given constituent by controlling its flow rate. The dependence<br />
<strong>of</strong> per cent nitrogen found in the polymer on flow rate is given in Figure 2.<br />
Why no polymer forms at high system pressures, ca. 1.0 mm Hg pressure, is<br />
perhaps due to competing gas phase processes (4). ThFsame types <strong>of</strong> polymers no<br />
doubt may be arrived at by substitution <strong>of</strong> ammonia for the N - H2 mixture, since<br />
one <strong>of</strong>ten achieves many similar results irrespective <strong>of</strong> whetker one uses N<br />
or NH3. Little molecular or atomic oxygen, as such, should be produced in 2 these +H2<br />
.
Spectroscopy<br />
Run 70 Element Found<br />
C H 0 N<br />
861 80.51 10.31 8. 06 0. 2<br />
63 1 75. 0 9. 5 12. 7 2. 8<br />
291 68. 19 9. 12 13.23 8.84<br />
93 1 55.90 7.77 18.40 17.64<br />
very little formed<br />
no polymer formed<br />
Total<br />
I<br />
99. 1<br />
100. 0<br />
99.38<br />
39.71<br />
The transmission infrared (Fig. 3) and internal reflectance, ATR (Fig. 4) spectra<br />
for samples 291 and 633, respectively, show very nearly identical features for the<br />
principal bands.<br />
Structural moieties such as ’<br />
0<br />
-z-NH2, -CH2-, -CH3, C-N, C=N, and -OH<br />
in organic compounds give the best comparative infrared spectral features to those<br />
observed for the synthesized polymer. Spectra <strong>of</strong> polyacrylamides, polyacryloni-<br />
triles and proteins (5) <strong>of</strong> some types, give quite similar spectra. Carbon-nitrogen<br />
double or single bonds may exist in the synthesized pyymer, but there is no evidence<br />
for the strong -CEN frequency at ca. 2240 - 2260 cm- (5). Nicholls and Krishna-<br />
machari (6 ) found in microwave eTFctrodeless discharees at 2450 MHz emission<br />
u<br />
bands <strong>of</strong> NCO formed at cryogenic temperatures from the reaction<br />
I<br />
N(2P) tC0 (X Ct)+NCO<br />
at the cold surface. Thus an NCO species may enter into the polymer as an important<br />
structural entity in the present case.<br />
Conclusions<br />
Under the flow discharge conditions <strong>of</strong> this study, the RF electrodeless method<br />
appears to <strong>of</strong>fer interesting synthetic possibilities with the simple inorganic gases.<br />
One might better understand the mechanism <strong>of</strong> origin <strong>of</strong> some polymers as a function<br />
<strong>of</strong> simple excited molecular, atomic or free radical precursors, generated in<br />
the discharge. The composition <strong>of</strong> the gas phase products formed in various reactions<br />
would be worth monitoring. Some <strong>of</strong> the gas products in the present study are<br />
being analyzed.<br />
t<br />
<<br />
/<br />
4
1 1 1<br />
moo 1400 1300<br />
c V'<br />
I<br />
IPOO<br />
I<br />
IIUO<br />
I<br />
1000<br />
I<br />
eo0<br />
I<br />
800<br />
Fig. 3 1nfrared.transmission spectrum <strong>of</strong> polymer, Run 291.<br />
I 4000 3500 SO00 2500 2000 1002 I600 1400 I200 1000 800 600<br />
1 CY<br />
!, Fig. 4 Infrared ATR spectrum <strong>of</strong> polymer, Run 633.<br />
c<br />
O<br />
100<br />
so<br />
80<br />
10<br />
60<br />
60<br />
40<br />
30<br />
20<br />
10<br />
0
Acknowledgement<br />
260<br />
The authcrs are pleased to acknowledge the analytical assistance <strong>of</strong> Dr. Robert 1<br />
Rinehart <strong>of</strong> Huffman Laboratories, <strong>of</strong> Wheatridge, Colorado. Mr. Jim Beaudry en- ,<br />
gineered all the RF generator and activator circuitry, and for whose assistance we<br />
are deeply indebted. Finally, technical discussions with Richard Bersin were most ~<br />
helpful,<br />
Literature Cited I<br />
1. J. Goodman, J. Polymer. -- Sci. 44, 551 (1960); pertinent references to past works<br />
are located here.<br />
2. J. R. Hollahan, --- J. Chem. Ed., - 43, A401, A497 (1966).<br />
3. N. Hata and P. A. Giguere, Can.' J. Chem. , 44, 869 (1966); F. Kaufman, Ann.<br />
de Gkophys., 20 106 (1964). - --<br />
P<br />
-<br />
4. B. D. Blaustein, U. S. Bureau <strong>of</strong> Mines, Pittsburgh, Pa. , personal communica-<br />
tion, has studied a system similar to ours at higher pressure, but has observed<br />
no polymer formation.<br />
5. J. Schurz, H. Bayzer, and H. Sturchen, Makromol. Chem. 23, 152 (1957); I,<br />
P. A. Wilks and M. R. Iszard, "Identification <strong>of</strong> FibersdTabrics by Internal J<br />
Reflection Spectroscopy", paper presented at Fifteenth Mid-America Spectroscopy '<br />
Symposium, Chicago, Illinois, June 2-5, 1964.<br />
6. R. W. Nicholls and S. L. N. G. Krishnmachari, Can. J. Chem., 38 1952 (1960). 1 ---<br />
1<br />
I
i<br />
25i<br />
The Dlssocfatior <strong>of</strong> ToIuere VaFor in a Radi<strong>of</strong>requency Discharge<br />
Frank J. Dinan<br />
Departrent <strong>of</strong> Cher.istry, Carisius Collem , Buffalo, Nev Tork<br />
The emission spctra resulting from the excitation <strong>of</strong> toluene wpcr in electrode<br />
<strong>discharges</strong> have teen investiqatec' by Schuler as a part <strong>of</strong> his extenelre work<br />
dealine dth the behavior <strong>of</strong> Folecules in discharps.(l) He found that two distinctly<br />
diffennt spectra worn emitted. One, the normal toluene emission rpectrum,<br />
WE centered in the 2600-3000 A' naion snd was related to the forbidden benzenoid<br />
abeorption band <strong>of</strong> toluene in the crude minor Image relationehip vhich characbriz%s<br />
fluorescence emission. This spectrum, as it was obtained In the premnt study,<br />
le shown In figure 1. In addition to this ultraviolet eniselon, Schuler also<br />
observed a vfsible erission spectrum from tolusne vhich appeared In the 4300 to<br />
5000 Ao reqlon, the details <strong>of</strong> vhich ven not published. This spctrum was<br />
designated the "blue spectrum" and was aesipned to the benzyl radial vithout stronk,<br />
supportine evidence.<br />
In a subeequert study <strong>of</strong> the react1 n<br />
<strong>of</strong> toluere vapor in a high voltage elec-<br />
trode discharce, Kraaijveld and Watenran92f investigated the fornation <strong>of</strong> the<br />
bibenzylmolecule fmr toluene and found that, under optimum conditions in a 2400 v<br />
arc, LO$ <strong>of</strong> the toluene vapor could te converted to tiberzyl. The other products<br />
formed under these conditione vere not lnveetigabd. This result, ae vel1 as those<br />
obtained by Schuler, imply the existence <strong>of</strong> benzyl radicals as the major species<br />
present in electrode ivduced tolwne <strong>discharges</strong>.<br />
Yore recently, Stmitwieser and Ward conducted the first comprehensive study<br />
vhich vas concerned vith the ranee <strong>of</strong> r ducts formed from the electrodelees<br />
rnicrovave excitation <strong>of</strong> tolwne vapor.p3? In this study, flowina tolmne vapor in a<br />
helium csrrier rrae stream was excjted by a 3 RMc lricrovave peneratar, The epsctrm<br />
<strong>of</strong> the lfaht enitted vas not investigated, hovever the products were carefully<br />
detemined. The composition <strong>of</strong> the mixture <strong>of</strong> producte obtained is sbovn in figure 2.<br />
The mv minor amounts <strong>of</strong> diger biaryla which mm formed in this discharge<br />
sueepsted that radical intenredfated ven not <strong>of</strong> dominant Importance. The lack <strong>of</strong><br />
fornation <strong>of</strong> the xylene isorera vas also interpreted as eupportlng the absence <strong>of</strong><br />
methyl radicals in the plasm.<br />
Two other possibilities, the fiolecular cation and anion vere coneidemd as<br />
likely intermediates in the rnicrovave discharge. Results obtained from labeling<br />
experiments ruled out consideration <strong>of</strong> the cation, and it was tentatively concluded<br />
that anion irtenrediates remained as the moat likely peesibility.<br />
This result contrasted intereetingly vith the conclusione which had been<br />
previously dravn regarc!inu the douinance <strong>of</strong> radjcals in toluene <strong>discharges</strong> and =st<br />
some doubt on these conclueions. Hovever, work vhicb had been done in W. D. Cooke's<br />
laboratory at Cornel1 University siivested that very significant differences might<br />
be anticiptcd 'ietveen the products resulting from the excitation <strong>of</strong> organic vapors<br />
in a microvave povend discharge ard the excitation <strong>of</strong> these vaprs vlth a radlo-<br />
frequency source. It vas, therefore, decided to investigate both the spectre and the<br />
prodrrets obtained vhen toltrepe vapor 1.88 excfted in a 28 Mc. mdi<strong>of</strong>requency dis-<br />
charge.<br />
The apparatus used in this study in ehom in block diagram form in figure 3.<br />
?loving tol-ve rapr vas pemd throuqh a discharge povered by an R.P. transmitter<br />
opemtinp at 28 mgcycles and 100 watts output. The pressure <strong>of</strong> the vapor uas<br />
maintajned constant at 0.10 to 0.15 mm. Materials fomed in the plam wm<br />
collecbd in traps maintained at Qo and 80° and vere aubseqwntly investigated by<br />
chromatoer%phic and smctroscopfc techniques. The liqht emitted from the discharge<br />
VB- focumd into a scannine Fonochroaator and detected by 3 IP-28 photomultiplier<br />
tube. The a~pllfierl output <strong>of</strong> the photomltiplier vas recorded electronically.<br />
Dnder the conditions used in these experiments, tolucne vae converted<br />
to products jn 12 to 1s yield vfth 75 to 8% <strong>of</strong> the toluene being recovered unchaneed.
2152<br />
TOLUENE ULTRAVIOLET EMISSION<br />
PRODUCTS FROM TOLUENE VAPOR<br />
IN A 3 KMc* POWERED DISCHARGE<br />
BENZENE 49<br />
ETHYLBENZENE 30<br />
PHENYLACETYLENE 8<br />
STYRENE 3<br />
XYLENE ISOMERS TRACE<br />
BIBENZYL -0.5<br />
DIPHENYLMETHANE - 0.2<br />
81 PHENYL<br />
Figure 2<br />
"lo 1<br />
i<br />
i<br />
- 0. I<br />
I<br />
4<br />
1<br />
(<br />
j<br />
J
L<br />
!<br />
,<br />
c<br />
PRODUCTS FROM A 28 MC.<br />
DISCHARGE IN TOLUENE<br />
PRODUCT<br />
- YO<br />
BENZENE 35<br />
ETHYLBENZENE 41<br />
6 I BE N Z'YL 16<br />
01 PHENYL METHANE 7<br />
BIPHENYL 2<br />
XYLENE ISOMERS
254<br />
A summary <strong>of</strong> the products formed in the R. F. powered dfscharge is presented<br />
in figure L. It should Fe noted that these results am normalized to exclude<br />
recovered tolmne, non-oondensatle cases and polymeric materials which in<br />
combination accounted for 3 to 6 <strong>of</strong> the oripinal toluene vapor.<br />
The formation <strong>of</strong> Substantial amounts <strong>of</strong> dimer biaryls; bibenzyl, diphenylmethane<br />
and biphenyl is <strong>of</strong> prticulqr signifiance since, as noted above, the formation<br />
<strong>of</strong> only trace amounts <strong>of</strong> dlmer bisryls vas observed in the microwsve<br />
discharqe. Additional experiments -re condmted in which a helium carrier gas was<br />
uaed, and no substantial difference in the nature and amounts <strong>of</strong> products formed was<br />
noted. The chanp in product ratios, therefore, appears to reflect a change In<br />
mechanism vhich results frm the use <strong>of</strong> an R. F. povered discharee.<br />
The products formed jn this study, in particular the large amounts <strong>of</strong> biaqls,<br />
indicate that free radicals am the major intennediatee leadlne to the formation <strong>of</strong><br />
products. The compounds fonned can be readily explained by the series <strong>of</strong> radical<br />
combination and hydrown sbstraotion reactions involving benzyl, phenyl and methyl<br />
radicals which are outlined in fieurn 5.<br />
It should be noted that minor, but definite amounts <strong>of</strong> the three xJrlene isomers<br />
-re fomed In the radi<strong>of</strong>requancy discharee. The trace amounts <strong>of</strong> these isomers<br />
which were formed from the dlssociation <strong>of</strong> toluene raper in a microwave powered discharge<br />
hss previously been used aq an a ament against the presence <strong>of</strong> methyl<br />
radlcals in the mlcrowave discharp. (3f As is shovn in figure 6, however, it has<br />
been demonstrated that methyl radlcals mact vith toluene wp r with 3 hundred fold<br />
preference for the side chafn rathe- than the rinc positions.~4) It is, therefore,<br />
entirely co~slstent with the presence <strong>of</strong> methyl radicals In the R. F. disoharee that<br />
msctfon should take place predominantly vith the toluene slde chain to fonn<br />
ethylbenzene rather than with the rlng positiona to yield xylene isomers, although<br />
the formation <strong>of</strong> small amounts <strong>of</strong> qlene should be anticipated.<br />
Experiments in which speoifically labeled deuteriotoluene was passed through<br />
the R.F. disoharee afforded additional experimental data which supported the<br />
importance <strong>of</strong> radioal intermediates. The producte formed from the labeled toluene<br />
were collected, separated by chromatoyraphio techniques, and the distribution <strong>of</strong> the<br />
deuterium label determined by infrared and nuclear magnetic resonance epctroscopy<br />
and mass spctmmetry.<br />
The slde chain <strong>of</strong> the recovered ethylbenzene was found to be heavily deuterated.<br />
The partisl pass spectrum <strong>of</strong> the recovered ethylbenzene compared to the mass<br />
speotrm <strong>of</strong> un-deuterated raterfal is shevn in figure 7. The parent peak <strong>of</strong> the<br />
recovemd material is five mass units higher than the corresponding peak in the<br />
un-deuterated material, and deronstrates the incluslon <strong>of</strong> five deuterium atoms in<br />
the molecule. The bene pak <strong>of</strong> the unlabeled ethylbenzene molecule resulte from a<br />
P-15 cleawae correspondinp to the lam <strong>of</strong> a methyl group. In the spectrum <strong>of</strong> the<br />
reoovemd saterial, the base peak results from P-18 cleavaae and clearly<br />
demonotrataa the rethyl group <strong>of</strong> the side chain to be fully deuterated. This<br />
molecule preswmbly forms from the combination <strong>of</strong> C7H5D2 radical vith a CD3 radical.<br />
The distribution <strong>of</strong> the deuterium label in the benzyl frapent can be demonstrated<br />
by considerlnc the WR epectrm <strong>of</strong> the recovered bibenzyl. In figure 8, the<br />
100 VC. proton rapetic ma30nance spectrum <strong>of</strong> un-deutered bibenzyl and that <strong>of</strong> the<br />
mcovered material ere compared. Ria important to note that the 5:2 ratio <strong>of</strong><br />
aromatic to sethylene protons observed in the non-deutereted material vould also be<br />
observed with the prtjally deutemted nolecule if the deuterium label vere uniformly<br />
distrituted. The ematly reduced methylene proton peak in the spectrum <strong>of</strong> the<br />
remwred material, however, clearly demonetrates that the label remains localiced<br />
fn the e!de chain <strong>of</strong> the hnayl fragment, and is not distributed throughout the<br />
ring.<br />
This result is entfrely consistent vlth the presence <strong>of</strong> fme radical interlpsdiatee<br />
in the discharge slnce, as io show in fim 9, it has been demonstrated<br />
that the un=ioniced benzyl radical does not undergo any type <strong>of</strong> rearrangement vhkb<br />
t in the randomlzatlon <strong>of</strong> a label, such as the fornation <strong>of</strong> the tropyliw<br />
z%a?eg By contrast, the benzyl cation doee maergo an immediate arranqement to<br />
1ltm lon, which vould result in uniform distribution <strong>of</strong> the deuterium<br />
labl. Since this is not observed, the benzyl catien can be ruled out as a<br />
tr?8
'\<br />
J<br />
?. , .. . :<br />
PRODUCT FORMATION REACTIONS<br />
Figure 5<br />
REACTION OF METHYL RADtCALS<br />
WITH TRITIATED TOLUENE<br />
RELATIVE RATE<br />
CONSTANTS AT 85'<br />
6- 0.76<br />
m- 0.26<br />
P-<br />
c Ha-<br />
I .oo<br />
156<br />
X.V. BEREZIN, et.a\., ZHUR. OBSCHE\ KH\W.><br />
3p,4093 (1960). C.A., a, 27153 b.<br />
Figure 6
1.<br />
266<br />
PARTIAL MASS SPECTRA OF mn BENZWB<br />
c?-18) -100<br />
PRODUCT<br />
Pigurt 7<br />
PARTIAL NMR SPECTRA OF BIBENZYL<br />
*ZcntQ<br />
I I I<br />
PRODUCT<br />
(1 )<br />
I<br />
-100<br />
4<br />
4<br />
,
'9<br />
/<br />
I<br />
'\<br />
"1<br />
BENZYL CATION AND RADICAL BEHAVIOR<br />
Figure 9<br />
PROOUCTS FROM IODINE AND<br />
TOLUENE VAPOR IN A2Sk DISCHARGE<br />
PRODUCT<br />
BLNLYL 10010E L)7<br />
IO DOBEN ZEN€ IY<br />
ETHYLBENZENE It<br />
BENtEdE 13<br />
METHYL IODIDE 6<br />
UNIDENTIFIED 9<br />
Figure EO
significant reaction int-ermedia te.<br />
Additional experiments were conducted in which toluene and iodine vapors were<br />
passe6 through the R.F. discharge together. Figure 10 shows the composition <strong>of</strong> the<br />
products Eormed in this experiment. In .:iew <strong>of</strong> the known affinity <strong>of</strong> iodine €or free<br />
radicals, (7) the formation <strong>of</strong> benzyl iodide, iodobenzene and methyl iodide together<br />
with greatly reduced amounts <strong>of</strong> the previously observed products argues for the<br />
presence <strong>of</strong> benzyl, phenyl and methyl radicals i'n the discharge. ;<br />
It should also be noted that the visible emission spectrum which is shown in<br />
figure 11 was observed vith the toluene discharge: This spectrum is similar to that 4<br />
vhich Schuler had previously observed from electrode toluene <strong>discharges</strong> and assigned<br />
to the benzyl radical.<br />
In sumrary, the diCferent products and intermediates formed in this work as<br />
contrasted to Streitwieser and Ward's study indicates that significant differences<br />
are to be anticipated betveen R;F. and microwave powered <strong>discharges</strong>, and<br />
unequivocal ly establish the importance <strong>of</strong> radical intermediates in the toluene R.F.<br />
discharge. Addit!onally, the hazards involved in the Tenera1 ization <strong>of</strong> data<br />
obtained in a specific type <strong>of</strong> discharge are readily apparent from these observations.<br />
Literature Cited<br />
1. H. Schuler and A. Michel, A. Naturforch., li)a. 495 (1955)<br />
2. H.J. Kraaijveld and H.I. Waterman, Brenst<strong>of</strong>f-Chem., 43, 33 (1962),<br />
C.A. x, 10627f<br />
3. A. Strcftuicser and H.P.Ward, J. 4m. Cliem. Soc., 2, 539.. (1963).<br />
,I<br />
\<br />
4.<br />
5.<br />
6.<br />
I.V. Herezin, N.F. Kazankava and K. Martinek, Zhur. Obschei<br />
Khim.. 2. 4093 (1960). C.A., 2, 27153b.<br />
R.F. Pottie and F.F. Lossing, J. Am. Chem. SOC., 2. 2634 (1361)<br />
P.N. Rylander, s. Meyerson and H.E. Grubb, J. Am. Chem. SOC. 2.<br />
842 (1957).<br />
' '<br />
4<br />
7. J.F. Carst and R.F. Cole, Tetrahedron Letters. w, 679 (1963).
a
270<br />
R .ACTION OF 'bI.4LEI.C J.NI?Y?'lRITE WITH ARCFl;\TTC FT'rllRCC.4IIRDNS<br />
UKDER TIi5 INFZUXNCE OF SILENT ZL'XTRIC ?ISC?!.RGX<br />
A.T ATWSPHTRIC F'RZSSVRRE.<br />
Stylianos Sifniades, David Jerolamcn, Robert Fuhrmam'<br />
Allied Chemical Ccrporation, Central Research <strong>Laboratory</strong>, Box 309,<br />
Yorristown, N. 3.<br />
I<br />
Maleic anhydride is lmoyn to form two olasses <strong>of</strong> adducts with<br />
alkyl benzenes, depending on the conditions <strong>of</strong> excitation. A t reflux<br />
. t-peratures and in the presence <strong>of</strong>. catalytic amounts <strong>of</strong> peroxides<br />
adducts <strong>of</strong> type'I 2re formed (1,2). A free radical chain reaction is<br />
assumed involving abstraction <strong>of</strong> a benzylic hydrogen. The chain length,<br />
based on the ratio <strong>of</strong> product to' added peroxide, is 20 to 100.<br />
R<br />
I (R,Rf = II or Me; R"= Me,Et,i-Pr) I1 (R,Rf= H,Me,t-Ru,Cl)<br />
4t FGOIU or somewhat higher temperatures and in the presence <strong>of</strong><br />
ultra-violet radiation aPducts <strong>of</strong> type I1 are formed (3 to 8). Excited<br />
aromatic molecules or excited charge-transfer complexes <strong>of</strong> maleic<br />
anhydride with an aromatic molecule are claimed tc be the reaction<br />
intermediates. The addition is sensitized by benzophenone, but the<br />
adduct can be formed in the absence <strong>of</strong> a sensitizer, although at a<br />
much lower rate. It appears that the presence <strong>of</strong> benzophenone is. iridis-<br />
pensable if sun light is used as the source <strong>of</strong> exciting radiation (9).<br />
It is interesting to note that in this case only benzene forms an<br />
adhct IT with maleic anhydride, while alkylbenzenes are only partly<br />
Incorporated in poly-anhydride chains which are formed.<br />
Recently formation <strong>of</strong> the adduct I1 <strong>of</strong> benzene and maleic anhydride<br />
'under the influence <strong>of</strong> gamma radiation was reported (10). The adduct<br />
is only a minor product ,<strong>of</strong> the reaction, corresponding to about 4% <strong>of</strong><br />
the maleic anhydride spent. The main product is a mixture <strong>of</strong> polyanhydrides.<br />
These can be considered to arise through a free radical<br />
chain similar to the one yielding adducts <strong>of</strong> type I.<br />
It is then clear that in the system maleic anhydride-benzene (6r<br />
alkylbenzene) two types <strong>of</strong> reactions are prevalent, one by free radical<br />
chain yielding adduct T or poly-anhydrides, the other by means <strong>of</strong><br />
excited molecules or charge-transfer complexes yielding adduct II. It<br />
was thought <strong>of</strong> interest to investigate the influence <strong>of</strong> silent or corona<br />
discharge on this system. This type <strong>of</strong> discharge was chosen because it<br />
is easy to maintain at atmospheric pressure.<br />
-iharge apparatus was essentially a modif led Siemens ozonizer<br />
Vertically mounted. It was made <strong>of</strong> Pyrex glass. A solution <strong>of</strong> sodium<br />
chloride in glycerol circulating in the jacket constituted the ground<br />
electrode, while a silver coating In the interior surface <strong>of</strong> the central
271<br />
tube serve? as the high voltare electrode. The action mixture was<br />
circulated at a hi-h rate from top Lo bottom through the reactor by<br />
means <strong>of</strong> a custom-made membrane pump constructed <strong>of</strong> Halon (registered<br />
mark <strong>of</strong> Allied Chemical Corooration). $n inert gas, nitrogen or helium,<br />
was introdwed at the top <strong>of</strong> the reactor and vented at the bottom<br />
through a condenser. The temwerature <strong>of</strong> the reaction mixture was measured<br />
at the exit <strong>of</strong> the discharpe. Temaerature control was ensured by<br />
regulating the temperature <strong>of</strong> the circulating glycerol solution.<br />
I<br />
Current at the desired frequencies was produced by an audi<strong>of</strong>requency<br />
generator (Heathkit ?lode1 17-71?) cou2led with a 1000 VA<br />
custom-madeamdifier. It was raised to high voltage by means <strong>of</strong> a 25<br />
KV transformer. Lower voltages could be obtained by acting on the outgut<br />
<strong>of</strong> the avlifier. The power input to the system was monitorcd by<br />
nekns <strong>of</strong> a watt-meter (Westinphouse Type PYb, specially compensated<br />
for high frequency work) at the primary circuit <strong>of</strong> the transformer.<br />
Eastman W.iite Label <strong>chemical</strong>s were user! I ithout purification. In<br />
a tjpicpl experiment 29.L~ grams nf maleic anhydride in 416 ml <strong>of</strong> cumene<br />
were kirculated for two hours in the appmatus while the discharge was<br />
mainth'ined at a power level <strong>of</strong> 400 watts. Nitrogen was suppli?d at the<br />
rate o'f'50 al/min. The teqperatiire <strong>of</strong> the reclction mixture was 127O C.<br />
Tne renction mixture iuas ccole.' to 20° C. and the resiilting crystals<br />
were filtered and washed with 50 ml <strong>of</strong> cold benzene. Additional crystals<br />
here obtained after the mother liquor had been condensed to one third<br />
<strong>of</strong> its initbal volume by flash evaporation. T!ie combined product was<br />
drier! at 50 '-C. in a vacuum oven. It weigned 5.18 grams and had melting<br />
point 255-7' C. Elementary analysis and neutralization equivalent were<br />
consistent with formula I1 (R = H, R' = i-Pr). The infra-red spectrum<br />
showed absence <strong>of</strong> aromatic cnaracter. Flash evaporation <strong>of</strong> the unreacted<br />
cumene and maleic anhydride left 23.0 grams <strong>of</strong> a viscous residue which<br />
had neutralization equivalent corresponding to a mixture <strong>of</strong> a 1:l and<br />
2:l adduct <strong>of</strong> maleic anhydride and cumene. It was assumed that it containLd<br />
adduct <strong>of</strong> formula I (R = R' = Me) together with products <strong>of</strong><br />
further condensation with maleic anhydride. Infra-red and NMR spectra<br />
were consistent with this assumption.<br />
Similar results were obtainPd ~laing bencene, toluene, and ethylbenzene<br />
as substrates. Chlorobenzene and nitrobenzene failed to yield<br />
a crystalline product. "he exwrimental conditions and the results<br />
are summarized in table I.<br />
Dis cu s s ion.<br />
It is apparent from the present results that maleic anhydride<br />
forms an adduct <strong>of</strong> type IT with benzene and alkylbenzenes under the<br />
influence <strong>of</strong> silent discharge. The generally accepted path <strong>of</strong> formation<br />
<strong>of</strong> IT in photo<strong>chemical</strong> (3) or gamma-radiation induced (10) reaction is<br />
% I
~ tation<br />
272<br />
It is reasonable to expect that the same path is followed also in 1<br />
the present case <strong>of</strong> discharge excitation. Since the rate <strong>of</strong> formation<br />
cf adduct 11 is independent <strong>of</strong> the concentration <strong>of</strong> maleic anhydride<br />
(experiments 2 and 4) it is concluded that the formation <strong>of</strong> the intermediate<br />
monoadduct is the rate-determining step. The same conclusion<br />
has been reached in the photo<strong>chemical</strong> fcrmation <strong>of</strong> adduct I1 (3,4).<br />
Adduct I1 is not, however, the only product formed in the reaction;<br />
a considerably larger amount oi resinous material (tabulated as adduct<br />
"TyDe I") is also formed in all cases. This material arlses from a free<br />
radical reaction similar to the one giving. rise to adduct I in peroxide- '<br />
initiated reactions. The chain length <strong>of</strong> these reactions is about 20 to ;<br />
100, as mentioned earlier. On the other hand it has been shown that the<br />
quantum yield <strong>of</strong> adduct TI formed in photo<strong>chemical</strong> reaction is about<br />
0.1 (4). Since in the present experiments adduct IT is formed in amounts 4<br />
about one quarter <strong>of</strong> the RInoUnt <strong>of</strong> "type I" adduct although about ten<br />
excited molecules, or charge-transfer complexes, are necessary to produce<br />
one molecule <strong>of</strong> TI, while one free radical will produce 20 to 100 !<br />
molecules <strong>of</strong> T, it must be concluded that the great majority <strong>of</strong> active<br />
species in the liquid phase in contact with the discharge are excited<br />
molecules and not free radicals. Tn fact it can be estimated that 98%<br />
to 99% <strong>of</strong> the active species are excited molecules. The absence <strong>of</strong> free<br />
radicals as important reaction intermediates has also been deduced by<br />
product analysis in the microwave glow discharge <strong>of</strong> aromatics in helium,<br />
(11)<br />
In the photo<strong>chemical</strong> reaction benzene forms an adduct I1 at a higher<br />
rate than the alkylbenzenes, which in turn react in relative rates indi-<br />
cative <strong>of</strong> steric hindrance. No such effect was observed in the present 1<br />
work, as shorn by the power yields in the table. These yields are also 4<br />
measures <strong>of</strong> the rate <strong>of</strong> formation <strong>of</strong> adduct I1 since roughly equal power<br />
levels <strong>of</strong> discharge were maintained in all cases.<br />
Comparison <strong>of</strong> runs 1 and 2 shows that formation <strong>of</strong> adduct I1 is<br />
fevored at fhe lower frequency with a correspondingly higher potential<br />
difference applied across the electrodes. Although the electric field<br />
strength between the dielectric surfaces is not necessarily proportional 4<br />
to the externally measured potential difference (12), it appears that<br />
electrons <strong>of</strong> higher average energy favor the production <strong>of</strong> adduct 11,<br />
as evidenced by the fact that the rate <strong>of</strong> formation <strong>of</strong> I1 is higher in<br />
a helium than in a nitrogen atmosphere (runs 4 and 5). It is known that<br />
the average electron energy for equal field strength is quite larger in<br />
the former gas (13). Of cource the presence <strong>of</strong> organic vapors modifies<br />
the electron eneru distribution but it is reasonable to expect that 4<br />
some difference still exists.<br />
"he mechanism <strong>of</strong> energy transfer cannot be deduced with certiihty<br />
f'rom the present data. ComDarison <strong>of</strong> yields at two temperatures (runs<br />
2 and 3) shows higher yield at the higher temperature, a fact which may'<br />
be interpreted as meaning that benzene vapors in the dircharqe are excited<br />
by collision with electrons: at the higher temperature the conced<br />
tration <strong>of</strong> benzene in the gas phare is higher. While this conclusion<br />
may be correct it is not unambiguous because <strong>of</strong> mechanical difficultie~<br />
at the lower temperature, arising from the fact that adduct 11 was<br />
sparingly soluble in benzene at this temperature and quickly coated the<br />
dielectric surfaces where it was partly decomposed. Another way <strong>of</strong> excf<<br />
would involve bombardment <strong>of</strong> the liquid phase with electrons<br />
generated in the discharge. This method <strong>of</strong> excitation has been shown to '<br />
prevail in the discharge-Induced oxidation <strong>of</strong> acidified water and ferrou)<br />
4<br />
d<br />
I<br />
'
273<br />
ion (1L.). Zxcitei-ion by collision with excited nitrcpr. or helium cannot<br />
be an inportant 3rocess, cmsi+eri~~ tiiet the first excited state <strong>of</strong><br />
'neliixn contoins 19.81 EV <strong>of</strong> enert7y (15). Tollision with siich a species<br />
~0~~1.' result in cxicnsivf f'rapentation <strong>of</strong> pn or,-anic rnoleclrlo, vrhereas<br />
the preseyt results point to the fact that free radical initiation is<br />
;.cry li lite6 it: this sgsten.<br />
Table I. Cordcnseticn <strong>of</strong> maleic anhydride with ben;-ene nnd slkylben7enes<br />
under tne influence <strong>of</strong> silent t lectric discharie.<br />
Clar e, Trequ. Potent.Tgmn.Adduct, rms Yie d <strong>of</strong> I1 g.p.<br />
.briber fas Kc/s* c KV C. Type 1 mmo:e/kw-hr C.<br />
1 250 TII benz.<br />
l?.O<br />
26.5;<br />
a) 15.6<br />
i2 .s: MA, N2 3.6 17.0 74 14.5 2.77 26.2<br />
5 380 ml benz.<br />
17 F TU, I!€ 3.6 13.0 75 15.1 11.16 33.3 355-57<br />
6 390 ml tolu.<br />
13 g XA, ?e 3.6 13.0 '32 30.0 11.79 30.4 245-60<br />
7 168 ml -tbz.<br />
29 . w, N;2 3.6 13.0 121 25.1 7.35 36.0 260-61<br />
3 LlC ml cme.<br />
7-<br />
29 g X', 12 3 .6 13.0 1?7 23.0 5.18 29.8 255-57<br />
T ~ product F was brown.<br />
i.bbreviations: HA, benz, tolu, etbz, cume denote correspondingly maleic<br />
R~I hy c? r i de , b en zene , t olu en e, ethyl h en z ene and cumsne.<br />
References.<br />
1. b4.G. Bickford, ".S. Fisher, Y.:!. Dcllear an? C-.E. Swift, J.Am.011<br />
. Chern.Soc. w, 251<br />
2. 3.Y. Beavers to Rohm and. IIaas Coy 1J.S. Patent 2,692,270 (1954)<br />
3. T). Pryce-Smith and, b.. Gilbert, J.Chem.Soc. 1965, 918<br />
4. G.S. 3amond and l,'.:!. !lardham, ?roc.Shexn.Son963, 63<br />
5. D. Bryce-Snith 2nd J.Y. Lod.ye, J.Chcm.Soc. xm67.5<br />
6. ' 2. Grovehstein, Jr, q.V. Rao and J.W. Taylor, J.Am.Chem.Soc. a,<br />
1705 (196.1)<br />
7. O.n. Schenck and R. Steinmetz, Tetr.Letters,.lo 21, 1 (1960)<br />
8. H.J.F. Ants and 9. Bryce-smith, J.Chem.Soc. 4791<br />
9. D. ?ryce-Smith, 1. 'lilbert- an?< €3. Vickery, Chznd - 1962, 2060;<br />
British Patent 986,348 (1965)<br />
10. Z. Raciszevski, Chem.In?. s, 418<br />
11. A. Streitwieser, Jr, an& H.R. ':ard, J.Am.Chem.Soc. 539 (1963)<br />
12. il. Rartnikas and J.Y.E. Levi, Rev.Sci.Instr. 2, 12%*(1966)<br />
13. L.E. Loeb, "3asic 9rocesses <strong>of</strong> "rase.ous Electronics", University<br />
<strong>of</strong> California Press (1961)<br />
1L. .I. Yokohatp an? S. Tsuda, Sull.Chem'.Scc.Japan, 3, 46, 1640 (1966)<br />
15. L.B. Loeb, "Xectric:,l Coronas", Tlniversity <strong>of</strong> California Press<br />
(1965)
274<br />
The Polymerization <strong>of</strong> Benzene in a Radio-Frequency Discharge<br />
David D. Neiswender<br />
Mobil Oil Corporation, Princeton, New Jersey<br />
INTRODUCTION<br />
The reactions <strong>of</strong> benzene in various types <strong>of</strong> electrical<br />
<strong>discharges</strong> have been studied by a number <strong>of</strong> researchers over a<br />
span <strong>of</strong> nearly 70 years. Reported results vary widely. Several<br />
early workers (1-3), using ozonizer tubes, obtained a gummy,<br />
wax-like substance, along with hydrogen, acetylene and other light<br />
hydrocarbon gases. One <strong>of</strong> these researchers later obtained a liquid<br />
and a solid, both analyzing as C24H25 compounds (4). In 1930 Austin<br />
and Black (5) found diphenyl and a solid which they suspected to be<br />
a polyphenylene containing phenol groups. Harkins and Gans (6).<br />
using an electrodeless discharge, got complete conversion to a red-<br />
. brown insoluble solid analyzing for (CH)x. Davis (7). on the other<br />
hand, obtained diphenyl, pterphenyl, a resin (CgH4)x, hydrogen,<br />
acetylene, ethylene and light paraffins. A 1935 U.S. patent (8)<br />
describes a discharge apparatus for preparing diphenyl from benzene.<br />
More recently, Streitwieser and Ward (9.10) obtained a 5% conversion<br />
<strong>of</strong> benzene in a microwave discharge, the products being low molecular<br />
weight gases, toluene, ethylbenzene and phenylacetylene. Stille and<br />
co-workers (ll), using a radiefrequency discharge, got a 10% conver-<br />
sion to poly(ppheny1enes) , diphenyl, fulvene, acetylene, allene,<br />
and methylacetylene. Vastola and Wightman (12) obtained a solid film<br />
and concluded from the infrared spectrum <strong>of</strong> the film that no aromati-<br />
city remained in the polymer. Jesch et a1 (13), on the other hand,<br />
also obtained a solid film, but interpreted its infrared spectrum as<br />
suggesting the presence <strong>of</strong> aromatic groups as well as olefinic and<br />
acetylenic unsaturation. The wide disparity <strong>of</strong> the results certainly<br />
indicates there is still much to be learned about the chemistry <strong>of</strong><br />
benzene in electrical <strong>discharges</strong>. This disparity is probably due<br />
largely to widely varying reaction conditions. Especially important<br />
are considerations such as the pwer dissipated in the discharge, the<br />
pressure, etc.<br />
The work described here is an attempt to systematize the study<br />
<strong>of</strong> benzene reactions in radio-frequency <strong>discharges</strong>. The only products<br />
isolated in these reactions were diphenyl, a liquid polymer and a<br />
solid polymer. The polymers appear to be polystyrenes.
Apparatus and Procedure<br />
275<br />
EXPERIMENTAL<br />
The apparatus consisted <strong>of</strong> a 3.69 MHz radio-frequency generator<br />
capacitively coupled to a cylindrical pyrex flow reactor by means <strong>of</strong><br />
two external copper electrodes. An inductively coupled tank circuit<br />
was used to match the impedances <strong>of</strong> the generator and the reactor.<br />
An approximate measure <strong>of</strong> the pwer dissipated in the dipcharge was<br />
determined by measuring the voltage and current during the experiments<br />
and making a phase correction for the capacitive component <strong>of</strong> the<br />
reactor current. A simple schematic <strong>of</strong> the apparatus is shown in<br />
Figure 1.<br />
The hydrocarbon or helium-hydrocarbon mixture was metered into<br />
the reactor at various rates. Reactor pressures from 1-20 torr were<br />
employed. In most cases the discharge established itself as soon as<br />
the r.f. signal was applied. If it did not, it was triggered with a<br />
Tesla coil. Products were collected in a series <strong>of</strong> traps, one at room<br />
temperature, one at -78OC and one at -195OC.<br />
Chemicals<br />
Reagent grade, thiophene free benzene (Baker) and vacuum<br />
distilled styrene (Matheson, Coleman and Bell) were used in the<br />
discharge experiments. The helium (Matheson) had a reported minimum<br />
purity <strong>of</strong> 99.995%.<br />
RESULTS<br />
Depending on the conditions employed, the reaction <strong>of</strong> benzene<br />
in the r.f. discharge resulted in either a complete conversion to a<br />
solid polymer or a lower conversion to a liquid polymer and diphenyl.<br />
The solid tends to form under conditions <strong>of</strong> high power dissipation<br />
and/or low partial pressures <strong>of</strong> benzene in the reactor. Conversely,<br />
the liquid polymer and diphenyl result under low power dissipation<br />
and/or high partial pressures <strong>of</strong> benzene.<br />
The solid polymer, which can sometimes be observed leaving<br />
the discharge zone as a fine smoke, deposits throughout the trap<br />
system, but principally in the dry ice trap. When collected, it is<br />
a very light, fluffy, nearly white powder which picks up a considerable<br />
static charge upon handling. Some <strong>of</strong> the substance's physical and<br />
<strong>chemical</strong> properties are as follows:<br />
1. It is completely insoluble in water and in all organic<br />
solvents which were tested.<br />
2. It does not melt up to 435OC.<br />
3. Thermogravimetric analysis shows that the polymer<br />
undergoes a stegwise loss in weight, with the loss<br />
being complete at 580OC.<br />
4. X-ray diffraction studies show it t o be completely<br />
amorphous.<br />
5. The density <strong>of</strong> a pressed pellet is 1.10 g/cc.
276<br />
Figure 1<br />
czb<br />
Raoio Frequency Discharge Reactor<br />
500 watt<br />
radio frequency<br />
genera tor<br />
tuning<br />
circuit<br />
J. reactants<br />
products
.<br />
277<br />
6. A freshly prepared sam le was found to have an electron<br />
spin density <strong>of</strong> 2 x lop7 spins/cc.<br />
7. Its surface area ,#(nitrogen adsorption) is 42 m2/g, a<br />
rather high value for an organic substance.<br />
8. It is neither thermosetting nor thermoplastic.<br />
9. It chemisorbs oxygen from the air at room temperature,<br />
the adsorption continuing for extended peribds <strong>of</strong> time.<br />
10. Its carbon-hydrogen ratio is 1:l.<br />
In those experiments which did not yield the solid polymer,<br />
the conversion <strong>of</strong> the benzene was <strong>of</strong> the order <strong>of</strong> 30%. About 5% <strong>of</strong><br />
the benzene was converted to diphenyl: the other 25% was converted<br />
to a liquid polymer with an average molecular weight <strong>of</strong> 617. This<br />
polymer, a viscous amber liquid, was not characterized as completely<br />
as the solid, but infrared spectral data indicate it to be very<br />
similar structurally to the solid.<br />
Polvmer Structure<br />
DISCUSSION<br />
The physical properties <strong>of</strong> the solid suggest that it is a high<br />
molecular weight, highly cross-linked polymer with an irregular<br />
structure. The extreme insolubility precludes spectral studies<br />
requiring solutions. It was possible to obtain an infrared spectrum<br />
by preparing a KBr pellet containing 2% <strong>of</strong> the solid. The liquid<br />
polymer, which was soluble in organic solvents had an infrared spectrum<br />
very similar to the solid.<br />
-<br />
Of several structural possibilities considered, the one which<br />
agrees best with the infrared data and seems the most likely from a<br />
<strong>chemical</strong> viewpoint is a polystyrene type structure. In Figure 2, the<br />
infrared spectrum <strong>of</strong> a reference polystyrene film (A) is compared with<br />
the spectra <strong>of</strong> the solid (B), the liquid (C) and a solid polymer<br />
obtained when a styrene-helium mixture was passed through the discharge<br />
(D). The spectra are nearly identical, the only significant differences<br />
being the OH absorptions in the 3300-3400 cm-l range and the carbonyl<br />
absorptions at about 1700 cm-l. These bands in all three <strong>of</strong> the<br />
spectra from the discharge-derived polymers are due to rapid oxidation<br />
by molecular oxygen. These bands become quite pronounced if the<br />
polymers are allowed to stand in air for a few hours.<br />
It is believed that the principal difference between the solid<br />
and liquid products is that the liquid is essentially a linear polymer,<br />
while the solid is highly cross-linked. Such a highly cross-linked<br />
polystyrene would be expected to have a lower ratio <strong>of</strong> aromatic C-H<br />
to aliphatic C-H bonds than would a linear polymer. Comparison <strong>of</strong><br />
spectrum B or D with C in Figure 2 shows that the intensity <strong>of</strong> the<br />
C-X stretching vibrations agrees with this expectation.<br />
Because the solid and liquid seem to be structurally similar,<br />
the NMR spectrum <strong>of</strong> the latter was examined and compared with that <strong>of</strong><br />
an authentic polystyrene sample. As is generally true with polymeric<br />
materials, the resolution was quite poor and only broad bands were
h<br />
B<br />
C<br />
D<br />
2-'?<br />
Figure 2. Infrared Spectra<br />
-7<br />
/I/ ' !I '!<br />
i<br />
T<br />
A = Polystyrene film<br />
B = Solid polymer from benzene<br />
C = Liquid polymer from benzene<br />
D = Solid polymer from styrene
observed. The polystyrene (10% solution in ccl4) spectrum simply<br />
showed two broad peaks - one centered at 6 = 1.50 due to aliphatic<br />
protons and one at 6 = 7.08 (with a small companion peak at 6 = 6.58)<br />
due to aromatic protons. The spectrum <strong>of</strong> the liquid polymer (lo”/.<br />
solution in cc14) was quite similar with the peaks appearing at<br />
6 = 1.60 and 7.04 (no peak at 6 = 6.58). In addition, a very small<br />
peak at 6 = 5.70 was observed. This is probably due to protons on<br />
olefinic double bonds, and suggests either that some unspturation is<br />
present in the polymer backbone, or that some unpolymerized vinyl<br />
groups are present. An attempt was made to increase the resolution<br />
<strong>of</strong> the NMR spectra by using a time averaging computer on very dilute<br />
solutions <strong>of</strong> the polymers, but the resolution was unchanged. Though<br />
<strong>of</strong> limited value, the NMR data do support a polystyrene type Structure.<br />
Coupled with the infrared data, it seems quite likely that the pOlyITIerS<br />
are both polystyrenes.<br />
Mode <strong>of</strong> Formation <strong>of</strong> Polymer<br />
The most likely route from benzene to polystyrene involves<br />
several steps, the first <strong>of</strong> which is the establishment <strong>of</strong> an<br />
equilibrium between benzene and acetylene:<br />
This interconversion has been observed in many high energy systems<br />
including electrical <strong>discharges</strong>. In the next step, the benzene and<br />
acetylene, one or both <strong>of</strong> which may be in a reactive state, combine<br />
to give styrene which then polymerizes:<br />
The simple linear polymer thus formed, limited to small chains such<br />
as pentamers, hexamers, etc., would explain the liquid polymer<br />
product.<br />
The formation <strong>of</strong> the highly cross-linked solid polymer can<br />
be explained by postulating the formation <strong>of</strong> polyvinylbenzenes which,<br />
when polymerized, would yield an extensive, irregular structurer<br />
4.<br />
Highly cross-linked polystyrene
280<br />
Both ionic and free radical mechanisms can be written to<br />
explain the foregoing reactions in more detail, but these would be<br />
strictly speculative, since no definitive experimental evidence has<br />
been obtained. Some sort <strong>of</strong> benzene ions must form as a result <strong>of</strong><br />
inelastic collisions between the electrons and benzene molecules in<br />
the plasma, but whether these ions or some derivative species are<br />
the reactive intermediates is not known. Recently, Potter et a1 (14)<br />
have shwn that styrene, when perfectly dry, does polymerize via an<br />
ionic mechanism when irradiated with gamma rays. One cah picture a<br />
similar mechanism occurring in the electrical discharge.<br />
On the other hand, the fact that the polymer has a high<br />
electron spin density suggests free radical involvement. However,<br />
it can be questioned whether the unpaired electrons arose during or<br />
after the polymerization reaction. An attempt was made to induce a<br />
spin signal in a finely divided polystyrene sample by passing the<br />
solid through a helium discharge. No signal was detected after this<br />
treatment.<br />
I<br />
The question <strong>of</strong> mechanism must await the results <strong>of</strong> more<br />
fundamental studies <strong>of</strong> the phenomena occurring within the discharge.<br />
Effect <strong>of</strong> Reaction Variables<br />
In an attempt to find a correlation between the reaction<br />
conditions and the type <strong>of</strong> polymer obtained, about 50 experiments<br />
were examined. The dependence <strong>of</strong> the nature <strong>of</strong> the product and the<br />
benzene conversion on r.f. power dissipated per mole <strong>of</strong> benzene was<br />
tested first. Those experiments which gave a complete conversion to<br />
solid polymer had an average dose <strong>of</strong> 1.9 x LO7 watt-sec/mole 8H.<br />
Those which gave a low conversion to liquid polymer plus diphenyl had<br />
an average dose <strong>of</strong> only 7.0 x 106 watt-sec/mole 8H. This correlation<br />
with dose is reasonable since more energy would be required to<br />
furnish the additional acetylene required for cross-linking and to<br />
gain the complete conversion.<br />
A number <strong>of</strong> experiments within the group examined were conducted<br />
with varying proportions <strong>of</strong> helium, neon and argon mixed with the<br />
benzene. The presence <strong>of</strong> these gases did not alter the correlation<br />
with dose provided that the r.f. paver dissipated was considered as<br />
deposited in the benzene alone. This is a reasonable presumption<br />
because the ionization potential <strong>of</strong> these gases is well above that<br />
<strong>of</strong> benzene.<br />
Within the group <strong>of</strong> experiments, some produced solid polymer<br />
at doses as low as 4.9 x 106 watt-sec/mole 8H, but with reduced<br />
conversions. This suggests that while the dose does correlate with<br />
the conversion obtained, it alone does not determine the product.<br />
It is evident that this system will require additional investigation<br />
to understand how the products form.
i<br />
i (12)<br />
c<br />
(11)<br />
(14)<br />
I 281<br />
LITERATURE CITED<br />
Losanitsch, S. M. and Jovitschitsch, M. Z., Ber. '2, 135 (1897)<br />
DeHemptinne, A., Bull. sci. acad. roy. Belg. (3) 34, 269 (1897)-<br />
DeHemptinne, A,, 2. physik. Chem. 22, 360 (1897)i 25, 284 (1898).<br />
Losanitsch, S. M., Ber. 41, 2683 (1908).<br />
Austin, J. B. and Black, I. A., J. Am. Chem. SOC. 52, 4552<br />
(1930).<br />
Harkins, W. D. and Gans, D. M. ibid. 52, 5165 (1930).<br />
Davis, A. P., J. Phys. Chem. 35, 3330 (1931).<br />
Kleinschniidt, R. V., U.S. 2,023,637 (1930).<br />
Streitwieser, A. and Ward, H. R., J. Am, Chem. SOC. 84, 1066<br />
(1962).<br />
Streitwieser, A. and Ward, H. R., ibid. 85, 539 (1963).<br />
-<br />
Stille, J. K., Sung, R. L. and Vander Kooi, J., J. Org. Chem.<br />
30, 3116 (1965).<br />
Vastola, F. J. and Wightman, J. P., J. Appl. Chem. 2, 69<br />
(1964) .<br />
Jesch, K., Bloor, J. E. and Kronick, P. L., J. Polymer Sci.<br />
- 4, 1487 (1966).<br />
Potter, R. C., Bretton, R. H., Metz, D. J., ibid. 4, 2295<br />
(1966)
INTRODUCTION<br />
Chemistrv <strong>of</strong> Electrical %?scharge Polymerizations<br />
Dr. Peter M. Ray<br />
Central Research Laboratories, J. P. Stevens c Co.<br />
Garfield, N. J. 1<br />
The conversion <strong>of</strong> volatile organic compounds into liquid and solid prod- I<br />
ucts by the action <strong>of</strong> a high-voltage gas discharge has been investigated<br />
by many people over the past 100 years and more (1). Recently some<br />
companies have been reported to be working on the application <strong>of</strong> this<br />
principle to the coating <strong>of</strong> containers (2). steel strip (3) or fabric<br />
(4). These references all have indicated that work was to be under<br />
low-pressure conditions where the only gases would be the volatile<br />
monomer. In this paper I will describe some results obtained under<br />
atmospheric-pressure conditions, with the electrical discharge taking<br />
place in a mixture <strong>of</strong> nitrogen and volatile organic compounds.<br />
EXPERIMENTAL METHODS<br />
All polymerizations were carried out at room temperature in a mixture<br />
<strong>of</strong> organic vapors and nitrogen at a total pressure <strong>of</strong> one atmosphere.<br />
The gases were delivered to the reaction zone through the system shown<br />
schematically in Figure 1. Nitrogen passed through liquid monomer in<br />
one bubble tube and through additive in another tube. The nitrogen<br />
and entrained vapors entered the enclosed reaction chamber at one<br />
corner and exited to the atmosphere at the opposite corner. The ratio<br />
<strong>of</strong> monomer to additive was determined by weighing the tubes before and<br />
after the experiment.<br />
Details <strong>of</strong> the reaction chamber are shown in Figure 2. Polymeriza-<br />
tion was initiated by corona discharge between two cylindrical,<br />
parallel, insulated electrodes (A) made by lining the inner surfaces<br />
<strong>of</strong> Pyrex glass tubing with aluminum foil. The glass tubing had a wall<br />
thickness <strong>of</strong> 2.5 mm. and an outside diameter <strong>of</strong> 63 mm. The two glass<br />
surfaces were separated by a gap <strong>of</strong> 4 nun. The alternating current<br />
high voltage was supplied by a Tesla generator, manufactured by Lepel<br />
High Frequency Laboratories, Inc. (Model HFSG-2). The peak voltage,<br />
as estimated by spark length in air, was about 20,000 volts.<br />
A moving strip <strong>of</strong> flexible substrate, (B) was positioned in the elec-<br />
trode gap. As shown in Figure 2, this substrate, which in most cases<br />
was 2-mil (50 microns) poly(ethy1ene terephthalate) film, was formed<br />
into a closed loop 47 cm. long and 5 an. wide and was moved by the<br />
rotating roller, C. Vapors entered through D and exited through E.<br />
Polymer which formed in the corona zone deposited both on the glass<br />
electrode coverings and on the moving substrates. The substrate strip<br />
was dried and weighed before and after the polymerization. The coated<br />
strips were also baked in a circulating air oven and reweighed. The<br />
final weight gain was taken as the yield. In some cases polymer was<br />
!
FLOW-<br />
METER<br />
I<br />
ADDITIVE<br />
SUPPLY<br />
* ,283<br />
VACUUM<br />
PUMP<br />
1<br />
7<br />
REACTION CHAMBER<br />
VAPOR POLYMERIZATION - SCHEMATIC LAYOUT<br />
Figure 1<br />
PRESSURE<br />
GAUGE<br />
VAPOR POLYMERIZATION ON MOVING SUBSTRATE<br />
Figure 2<br />
TO HIGH<br />
VOLTAGE
254<br />
removed from the glass surfaces by solvent and recovered for infrared<br />
or <strong>chemical</strong> analysis.<br />
RESULTS<br />
Survey <strong>of</strong> Polymerizable compounds<br />
A number <strong>of</strong> volatile orqanic compounds were subjected to corona discharge<br />
with a wide variety <strong>of</strong> results. In almost all cases a deposit<br />
<strong>of</strong> oily or solid brown material formed on the substrate strip.<br />
weight gain resulting from 15 minutes <strong>of</strong> .corona polymerization ranged<br />
from tenths <strong>of</strong> a milligram to over 30 milligrams. When the coated<br />
strips were heated at 15OoC for 10 minutes,, part <strong>of</strong> the added weight<br />
was lost. Some'<strong>of</strong> this is thought to be unreacted monomer which was<br />
absorbed by the substrate and coating. Another part <strong>of</strong> it could be<br />
very-low-molecular-weight products <strong>of</strong> the corona reaction. A third<br />
possibility is thermal decomposition <strong>of</strong> the coating.<br />
The compounds which gave the heaviest coatings after heating, (taking<br />
into account the amount <strong>of</strong> monomer volatilized) included triallylamine,<br />
acrylonitrile, toluene and styrene. As the list in Table 1 shows it<br />
is not necessary for the monomer to be a vinyl compound in the strjct<br />
sense <strong>of</strong> the word in order for a non-volatile polymer product to form<br />
in corona discharge. Toluene, benzene, benzotrifluoride and even<br />
acetone gave measurable yields. There seems to be no pattern'<strong>of</strong><br />
relationship between the structure <strong>of</strong> a monomer and its yield in<br />
corona polymerization.<br />
Triallylamine<br />
Acrylonitrile<br />
To 1 ue n e<br />
Styrene<br />
Acrylic Acid<br />
Benzene<br />
I<br />
TABLE 1<br />
v<br />
Benzotrifluoride<br />
4-Vinyl cyclohexene<br />
Ethyl acrylate<br />
1-Octene<br />
Allyl amine<br />
Vinyl acetate<br />
Acetone<br />
The
285<br />
TABLE 2<br />
Effect <strong>of</strong> Haloqenated Additives on Corona Polymerization <strong>of</strong> Styrene<br />
Yield in Milliqrams per Gram <strong>of</strong> Styrene<br />
Additive Percent Added Yield Additive Percent Added Yield<br />
Chlor<strong>of</strong>orm<br />
Bromo f o m<br />
0.5<br />
0.5<br />
22.4<br />
11.6<br />
Chlorine<br />
Bromine<br />
7<br />
1<br />
1<br />
5.6<br />
19.0<br />
Brom<strong>of</strong>orm 1.0 14.2 Bromine 5 32.0<br />
Bromo form 2.5 19.7 Bromine 10 16.7<br />
Bromo form 5.0 19.2 Iodine 5 9.7<br />
Brom<strong>of</strong>orm<br />
Iod<strong>of</strong>orm .<br />
10.0<br />
5.0<br />
12.0<br />
21.2<br />
None<br />
-<br />
0<br />
-<br />
10.7<br />
-<br />
Other additives that enhanced the yields <strong>of</strong> styrene polymer were<br />
carbon tetrachloride, 1,2-dibromo-1,1,2,2-tetrafluoroethane, l-bromo-<br />
butane and 2-bromobutane. Yields from monomers other than styrene<br />
were not all increased by halogenated additives: some even were de-<br />
creased. It was impossible to develop any rational relationship<br />
between monomer structure and susceptibility <strong>of</strong> the monomer to yield<br />
enhancement by halogenated additive.<br />
The results given above were all derived from experiments in which the<br />
additive and the monomer were mixed and volatilized from a single<br />
bubble tube. Thus, the exact composition <strong>of</strong> the vapor was not known.<br />
A new set <strong>of</strong> experiments was carried out using separate bubble tubes<br />
for_monomer and additive. The weight changes <strong>of</strong> the tubes during a<br />
run were used to calculate mole ratios <strong>of</strong> additive to styrene. The<br />
results <strong>of</strong> experiments with four additives are shown in Figure 3.<br />
The conversion to polymer <strong>of</strong> styrene without additives was 0.75 to<br />
0.9 percent. As increasing amounts <strong>of</strong> brom<strong>of</strong>orm, 1-bromobutane or<br />
2-bromobutane were added, the conversion increased and then fell <strong>of</strong>f<br />
again. The pattern <strong>of</strong> points in the case <strong>of</strong> 1-bromobutane was widely<br />
scattered but most <strong>of</strong> the points lay well above the level for styrene<br />
itself. With 2-bromo-2-methylpropane as an additive there was no<br />
significant increase in conversion. The weight gain from the additives<br />
alone, with no styrene, were all low compared to styrene.<br />
In considering <strong>chemical</strong> explanations for corona polymerization, both<br />
free radical and ionic intermediates are possibilities. Experiments<br />
were run with various additives to styrene that might be expected to<br />
inhibit each kind <strong>of</strong> reaction through combination with the active<br />
intermediate but no clear-cut reduction in yield was observed. Benzo-<br />
quinone at l and 2 mole percent gave normal yields. Water and butyl<br />
amine were extensively studied but, as Figure 4 demonstrates, the<br />
tendency for these supposed cation scavengers to depress the conversion<br />
is slight and not clear-cut. Butyl amine alone gave a surprisingly<br />
high yield. Ammonia, triethylamine, acetone and carbon dioxide as<br />
additives had no substantial effect on yield.
t-<br />
z<br />
W<br />
0<br />
LL<br />
W<br />
4 2<br />
LL<br />
w<br />
1.0<br />
O.! 5<br />
-<br />
A o<br />
1.5- d<br />
z O//<br />
A<br />
- 92 3 '<br />
@ STYRENE WITH I- BROMOBUTANE<br />
A STYRENE WITH 2-BROMOBUTANE<br />
0 STYRENE WITH 2-BR-2-ME-PROPANE<br />
rn STYRENE WITH BROMOFORM<br />
v)<br />
LL<br />
W<br />
0 0<br />
2 0.5- 0<br />
0<br />
0<br />
k<br />
z<br />
W<br />
0<br />
a<br />
W<br />
-<br />
a<br />
a<br />
W<br />
2<br />
3<br />
0<br />
a<br />
0<br />
I-<br />
z<br />
9<br />
v)<br />
a<br />
w<br />
><br />
z<br />
0<br />
0<br />
I<br />
I<br />
0.1 0.2 0.3 0.4 0.5<br />
MOLE FRACTION OF ADDITIVE<br />
A.<br />
A * A<br />
A<br />
Figure 3<br />
* STYRENE WITH WATER<br />
A STYRENE WITH BUTYL AMINE<br />
1<br />
0.1 0.2 0.3 0.4 0.5 0.6 0.7<br />
MOLE FRACTION OF ADDITIVE<br />
Figure 4<br />
b<br />
A<br />
A<br />
/.<br />
\<br />
f !<br />
\
ANALYTICAL RESULTS<br />
Some evidence was collected on the <strong>chemical</strong> nature <strong>of</strong> some <strong>of</strong> the<br />
products. The products deposited on the moving film and on the glass<br />
electrode covers were always easily dissolved in common solvents like<br />
acetone, chlor<strong>of</strong>orm and benzene, indicating low molecular weight and<br />
that there was little cross-linking.<br />
Infrared spectra were obtained <strong>of</strong> polymers made from styiene, benzene<br />
and toluene by dissolving the deposit from the glass electrode cover<br />
with chlor<strong>of</strong>orm and evaporating the solution on a salt plate. The<br />
spectra were all similar and closely resembled conventional poly-<br />
styrene. As Figure 5 shows, there were additional absorptions at<br />
1050, 1220, 1720 and 3400 wave numbers indicating oxygenated products<br />
<strong>of</strong> various kinds including hydroxyl or amino, ester and probably<br />
ether. These spectra are very similar to those published by Jesch,<br />
Bloor and Kronick (5).<br />
The infrared evidence <strong>of</strong> oxygen-containing groups was obtained on a<br />
styrene product which was prepared and transferred in a nitrogen<br />
atmosphere. The only contact with air was during the time the spec-<br />
trum had been run.<br />
The sample was exposed to the atmosphere for 24<br />
hours and it changed only slightly. As Figure 6 shows there was little<br />
further increase <strong>of</strong> bands attributed to oxygenated groups. The only<br />
noticeable change in the spectrum was in the relative peak heights at<br />
700 and 760 wave numbers. The spectrum <strong>of</strong> a product made with l-bromo-<br />
butane mixed with the styrene was almost identical to the unmodified<br />
styrene product with a stronger absorption ascribed to ketone carbonyl<br />
at I720 wave numbers.<br />
The final evidence <strong>of</strong> <strong>chemical</strong> composition was elemental analysis.<br />
The styrene product had considerably less carbon than styrene itself,<br />
as seen in Table 3. It also contained a significant amount <strong>of</strong> nitrogen<br />
and, as determined by difference, a large amount <strong>of</strong> oxygen. Heating<br />
the polymer in air did not change the composition significantly. Sty-<br />
rene polymer made in the presence <strong>of</strong> 25 to 30 weight-percent l-bromo-<br />
butane had almost the same analysis plus a significant bromine content.<br />
TABLE 3<br />
Elemental Analysis <strong>of</strong> Corona Polymers<br />
Monomer - C - H - N 0 (by difference)<br />
Styrene, unheated product 72.23 6.06 3.2 - 17.71<br />
Styrene. oven-heated 73.46 6.60 3.39 - 16.47<br />
Styrene plus bromobutane<br />
unheated product 73.74 6.95 3.59 6.00 9.64<br />
oven-heated product 73.29 6.69 3.56 6.97 9.49<br />
Styrene, calculated 92.26 7.74 - - -<br />
Bromobutane, calculated 35.06 6.62 - 50.32 -
'603 ~---~ _-_- T_--r.-<br />
Figure 5<br />
1000 900 800 700 650<br />
E - TOLUENE<br />
F. BE NZENE<br />
G -SlVRENE<br />
i<br />
\
DISCUSSION<br />
289<br />
The salient features <strong>of</strong> the information presented above might be<br />
summarized as follows. In a mixture <strong>of</strong> organic vapor and nitrogen<br />
subjected to high-frequency, high-voltage, electrodeless discharge,<br />
the nitrogen, the organic compound and trace amounts <strong>of</strong> oxygen and<br />
water are activated and combine <strong>chemical</strong>ly to yield products <strong>of</strong> higher<br />
molecular weight than the starting materials. These proqucts condense<br />
on any solid surface available and may even undergo further <strong>chemical</strong><br />
reaction within themselves and with more monomeric material which is<br />
not electrically activated.<br />
The <strong>chemical</strong> mechanism <strong>of</strong> this series <strong>of</strong> reactions must be complex,<br />
possibly produced by simple ionization through collision with a<br />
rapidly-moving electron followed by expulsion <strong>of</strong> two secondary<br />
electrons:<br />
N2 + e-+N2+ + 2e- or<br />
+<br />
RH + e---+RH + 2e-<br />
I' These are the products ordinarily found in mass spectrometry and on<br />
exposure to gamma or beta radiation. These activated cationic products<br />
may activate vinyl polymerization or undergo secondary reactions,<br />
u' yielding neutral free radicals or even anions, either <strong>of</strong> similar<br />
structure or in the form <strong>of</strong> fragments and combination or rearrange-<br />
9 ment products. In the present case there exists the further possibility<br />
that reaction products become reactivated, since they are formed in<br />
the presence <strong>of</strong> the high-voltage field, and undergo further reaction.<br />
1 The simplest mechanism to consider would be polymerization <strong>of</strong> the<br />
I vinyl monomers to long-chain products after initiation by some active<br />
c<br />
,, species. The fact that non-vinyl compounds gave good yields may be<br />
' explained through a mechanism involving some fragmentation <strong>of</strong> every<br />
1 monomer molecule and combination <strong>of</strong> the fragments to products <strong>of</strong> higher<br />
molecular weight. The vinyl monomers could be reacting through this<br />
P*<br />
i<br />
non-vinyl mechanism also.<br />
The action <strong>of</strong> halogenated additives suggests that the yield <strong>of</strong><br />
reactive intermediates is increased by some <strong>of</strong> them and that these<br />
intermediates increase the initiation <strong>of</strong> vinyl polymerization. There<br />
is not enough data to .establish this firmly. It may also be consid-<br />
ered that, by changing the dielectric constant <strong>of</strong> the gas mixture, the<br />
additive can increase the efficiency <strong>of</strong> energy transfer from the elec-<br />
tric field to the monomer.<br />
The other additives had little or no effect, indicating that the<br />
situation is not very similar to bulk or solution polymerizations by<br />
5 free radical or ionic catalysis such as are now being actively studied<br />
by several laboratories (6,7). Certainly we do not have a simple<br />
vinyl polymerization. Otherwise some <strong>of</strong> the potential inhibitors<br />
would have shown an effect.
230 . .<br />
One <strong>of</strong> the unexpected results was t e fixation <strong>of</strong> nitrogen. This<br />
can be seen in retrospect to be related to work with discharge-acti-<br />
vated nitrogen which has been reported from time-to-time (8,9,10).<br />
No complete mechanistic explanation will be presented. More experi-<br />
ments are in process but it is not expected that a system which is<br />
as potentially complex as this can soon be completely explained.<br />
ACKNOWLEDGEMENTS<br />
The able assistance <strong>of</strong> Messrs. Roger Kolsky and Walter Miner and<br />
the helpful analytical interprelations <strong>of</strong> Mr. Elliot Baum are grate-<br />
fully acknowledged.<br />
REFERENCES<br />
1.<br />
2.<br />
3.<br />
4.<br />
5.<br />
6.<br />
7.<br />
a.<br />
9.<br />
10.<br />
A. Charlesby, “Atomic Radiation and Polymers, “ Pergamon Press,<br />
Oxford, 1960, p.1.<br />
R. M. Brick and J. R. Knox, Modern Packaging, January 1965,<br />
pp. 123-128.<br />
T. Williams, J. Oil and Colour Chemist’s Association 2, 936-951<br />
(1965) .<br />
Manmade Textiles, July, 1965, pp. 58-9.<br />
K. Jesch, J. E. Bloor and P. L. Kronick, J. Polymer Sci.,<br />
A-1, 4, 1487-97 (1966).<br />
R. C. Potter, R. H. Bretton, D. J. Metz, J. Polymer Sci. A-1,<br />
2295-2306 (1966).<br />
K. Ueno, K. Hayashi, S . Okamura, J. Polymer Sci. BA, 363-8,<br />
(1965).<br />
L. B. Howard and G. E. Hilbert, J. Am. Chem. SOC. 60, 1918<br />
(1938).<br />
T. Hanafusa and N. N. Lichtin, J. Am. Chem. SOC. 82, 3798-9,<br />
(1960) .<br />
G. D. Steinman and H. A. Lillevik, Arch. Biochem. Biophys.<br />
- 105, 303 (1964): CA 61, 4198 (1964).<br />
I
? ~<br />
MENBERSHIP<br />
IN THE DIVISION OF FUEL CHEMISTRY<br />
I The Fuel Chemistry Division <strong>of</strong> the American Chemical Society is an internationally<br />
!recognized fol'um for scientists, engineers and technical economists concerned with<br />
I<br />
the conversion <strong>of</strong> fuels to energy, <strong>chemical</strong>s, or other forms <strong>of</strong> fuel. Its interests<br />
center on the <strong>chemical</strong> problems, but definitely include the engineering and economic<br />
aspects as wen.<br />
Any chemist, <strong>chemical</strong> engineer, geologist, technical economist,,or other scientists<br />
1 concerned with either the conventional fossil fuels, or the new high-energy fuels--<br />
' whether he be in government, industry or independent pr<strong>of</strong>essional organizations-would<br />
benefit greatly from participation in the progress <strong>of</strong> the Fuel Chemistry<br />
I<br />
Division.<br />
The Fuel Chemistry Division <strong>of</strong>fers at least two annual programs <strong>of</strong> symposia and general<br />
papers, extending over several days, usually at <strong>National</strong> Meetings <strong>of</strong> the American<br />
! Chemical Society. These include the results <strong>of</strong> research, developnent, and analysis in<br />
t the many fields relating to fuels wbich are so vital in today's energy-dependent<br />
economy. Members <strong>of</strong> the Division have the opportunity to present papers <strong>of</strong> their own,<br />
or participate in discussions with experts in their field. Most important, the Fuel<br />
Chemistry Division provides a permanent record <strong>of</strong> all <strong>of</strong> this material in the form <strong>of</strong><br />
' complete preprints.<br />
\\<br />
I<br />
Stding in September 1959, the biennisl Fuel Cell Symposia <strong>of</strong> the Division have been<br />
the most important technical meetings for chemists and <strong>chemical</strong> engineers active in<br />
,,this field. These symposia have all been published in book form. The recent land-<br />
,mark symposium on Advanced Propellant Chemistry has been published in book form also.<br />
J '<br />
I Further, the Division is strengthening its coverage <strong>of</strong> areas <strong>of</strong> air and water pollu-<br />
4 tion, gasification, and related areas.<br />
In addition to receiving several volumes <strong>of</strong> preprints each year, as weU as regular<br />
news <strong>of</strong> Division activities, benefits <strong>of</strong> membership include: (1) Reduced subscrip-<br />
;" tion rates for '~uel" and "Combustion and Flame," (2) Reduced rates for volumes in<br />
the "Advances in Chemistry Series" based on Division symposia, and (3) The receipt<br />
card sent in acknowledgment <strong>of</strong> Division dues is good for $1.00 toward a ccmplete set<br />
,',<strong>of</strong> abstracts <strong>of</strong> all papers presented at each <strong>of</strong> the <strong>National</strong> Meetings.<br />
,I<br />
To join the Fuel Chemistry Division as a regular member, one must also be or becme a<br />
member <strong>of</strong> the American Chemical Society. Those not eligible for ACS membership<br />
, because they are not practicing scientists, engineers or technical econdsts in areas<br />
related to chemistry, can become Division Affiliates. They receive all benefits <strong>of</strong>'a<br />
$)regular member except that they cannot vote, hold <strong>of</strong>fice or present other than invited<br />
papers. Affiliate membership is <strong>of</strong> particular value to those in the informational. and<br />
library sciences who must maintain awareness <strong>of</strong> the fuel area. Non-ACS scientists<br />
'active in the fuel area and living outside <strong>of</strong> the United States are invited also to<br />
\pecome Division Affiliates.<br />
Membership Fn the Fuel Chemistry Division costs only $4.00 per year, or $pL.oO for<br />
in addition to ACS membership. The cost for a Division Affiliate, with-<br />
ACS, is $10.00 per year. For further information, write to:<br />
Dr. Frank Rusinko, Jr.<br />
Secretary-Treasurer<br />
ACS Division <strong>of</strong> Fuel Chemistry<br />
c/o Speer Carbon Ccunpany<br />
St. ~ y s Pennsylvania ,<br />
15857<br />
Telephone: 814 - 834-2801
i<br />
.;<br />
Chemical Engineering Aspects <strong>of</strong> %?&mica1 Synthesis<br />
in Electrical Discharges.<br />
J.D. THORNTON.<br />
Department <strong>of</strong> Chemical Engineering, The University,<br />
Newcastle upon Tyne, England.<br />
I 1.0.Introduction.<br />
Although numerous investigations have been published dealing with the effects<br />
<strong>of</strong> electrical <strong>discharges</strong> upon <strong>chemical</strong> reactions, most <strong>of</strong> the work is fragmentary<br />
1<br />
,I<br />
'<br />
r/.<br />
in the sense that even now no clear picture has emerged <strong>of</strong> the relative importance<br />
<strong>of</strong> discharge characteristics and reactor geometry upon the reaction yield. This<br />
is largely because no systematic studies have been undertaken with a fixed<br />
reaction System covering a reasonably wide range <strong>of</strong> discharge conditions and<br />
reactor configurations. Furthermore most <strong>of</strong> the published work has been concerned<br />
with laboratory-scale investigations so that, with the possible exception <strong>of</strong> ozone<br />
synthesis, little or no attention has been paid to the <strong>chemical</strong> engineering problems<br />
involved'in gas phase electrosynthesis. This is no doubt consequent upon the low<br />
yields obtained in many laboratory studies and is unfortunate in that a proper<br />
application <strong>of</strong> the engineering factors affecting the reactor efficiency could, in<br />
many Cases,. lead to considerably improved reaction yields.<br />
Whereas physical chemistry is usually concerned with the individual kinetic<br />
/ steps contributing to a particular reaction scheme, <strong>chemical</strong> engineering is<br />
concerned with the translation <strong>of</strong> the laboratory Teaction to a full scale contin-<br />
! uous process wherein the desired reaction product is produced at the lowest capital<br />
' and operating costs. In the case <strong>of</strong> gas discharge synthesis, these financial<br />
< criteria imply that the reactor should have three highly desirable characteristics:<br />
(a) The energy yield (Gms. product per Kw.Hr.1 should be high.<br />
(b) The con\.ersion per pass should be high.<br />
d<br />
/<br />
(c) The reactor should be selective for the desired product, i.e. side<br />
reactions with their consequent wastage <strong>of</strong> material and energy should be<br />
minimised.<br />
In practical terms, it is not necessary to have a detailed step by step know-<br />
,p'ledge <strong>of</strong> the reaction kinetics in order to design the reactor, so long as an<br />
{ I<br />
empirically determined rate equation is available which adequately describes the<br />
influence <strong>of</strong> the operating variables upon the net rate <strong>of</strong> reaction. Clearly a more<br />
informed approach to the design problem is possible when fundamental data are<br />
' available but this is seldom the case at the design stage.<br />
,d Assuming that the electrons in a discharge are able to excite the reactant<br />
molecules, it is possible that many <strong>of</strong> the low product yields hithertoreported may<br />
E be attributed to one or more <strong>of</strong> the following factors:<br />
(a) The use <strong>of</strong> unsuitable reactor geometries in which a sizeable fraction<br />
J' <strong>of</strong> the reactant by-passes the discharge zone.<br />
II<br />
'Y<br />
\<br />
?<br />
\<br />
,<br />
(b) The use <strong>of</strong> non-optimum discharge conditions in which the average<br />
activation cross-section for the desired reaction is low.<br />
(c),The use <strong>of</strong> inappropriate residence times or residence time distributions<br />
<strong>of</strong> the reactants and products.<br />
\/<br />
1<br />
(d) The presence <strong>of</strong> parallel reactions which compete for the reactant and<br />
the occurrence <strong>of</strong> rapid reverse and degradation (or consecutive) reactions which<br />
destroy the primary product.<br />
j<br />
It is with these aspects.<strong>of</strong> gaseous electrosynthesis that the present paper<br />
is concerned, since it is possible that many <strong>of</strong> these effects can be minimised by<br />
', proper consideration at the reactor design Stage.<br />
T<br />
, 2.0 Reactor Classificatlon.<br />
I It is useful to conslder initially the basic reactor geometries that can be<br />
,, employed in a contlnuous-flow reaction system. Although no formal reactor classlf-<br />
/ lcation has hlthert0 been proposed, the electrode configuration and direction <strong>of</strong><br />
I<br />
3
292<br />
flow <strong>of</strong> the reactant gas stream can be made the basis <strong>of</strong> a convenient system <strong>of</strong> /<br />
classification. Thus the electrodes may consist <strong>of</strong> parallel plates, co-axial<br />
cylinders or a pair <strong>of</strong> points whilst the gas flow may be parallel or at right<br />
angles to the plane <strong>of</strong> the discharge. Figure 1 shows in diagrammatic form the<br />
4 \.arious electrode anti flow arrangements on the basis <strong>of</strong> this classification.<br />
Parallel flow in parallel plate and coaxial electrode reactors necessitates the use<br />
<strong>of</strong> porous electrodes through which the gas phase flowsas shown in Figures 1.1 and<br />
1.2. On the other hand, the commoner cross-flow arrangements shown in Figures 1.4,<br />
1.5 and l.G usually employ impervious metal electrodes, there being :no necessity<br />
for the reactants to flow through the electrodes themselves. The usual parallel/ 3<br />
point anti cross-flow/point reactors are depicted in Figures 1.3 and 1.7 respectively.<br />
The distinction between parallel-flow and cross-flow is not an academic one<br />
by reason <strong>of</strong> the non-uniform nature <strong>of</strong> the electric field in many<strong>of</strong> these reactor<br />
arrangements. In most <strong>of</strong> the laboratory investigations reported in the literature,<br />
the areas <strong>of</strong> the electrodes are small and little or no attempt has been made to<br />
minimise c.{lgr effects by the use <strong>of</strong> suitable electrode pr<strong>of</strong>iles. The field<br />
intensity is therefore a function <strong>of</strong> position in the inter-electrode gap. Furthermore<br />
the iise <strong>of</strong> coaxial geometries where the respective electrode radii are very<br />
different also gives rise to highly non-uniform fields in the vicinity <strong>of</strong> the<br />
central electrode. It follows therefore that the or.erall activation effects<br />
might well be different in the parallel/parallel and cross-flow/parallel systems<br />
in Figures .1.1, 1.4 and 1.5. In the former case, molecules traversing a centreline<br />
path will encounter electrons <strong>of</strong> different energies to molecules following a<br />
peripheral path whilst, in a cross-flow reactor, all the molecules encounter<br />
electrons with a wide spectrum <strong>of</strong> energies. In coaxial reactors, the position is<br />
further complicated by virtue <strong>of</strong> the non-uniform field associated with the central<br />
electrode. Thus in Figures 1.2 and 1.6 not only is there a transverse field<br />
variation but there is also an axial variation as the molecules approach the<br />
bottom and top planes <strong>of</strong> the outer,plectrode.<br />
Such considerations are purely speculative at the present time because no<br />
experimental data are available which would enable a direct comparison <strong>of</strong> reactant<br />
conversions for parallel and cross-flow systems to be made for the same reactants<br />
under similar conditions. Nevertheless these factors must be born in mind when<br />
interpreting the influence <strong>of</strong> reactor geometry on a quantitative basis.<br />
Coaxial electrode systems in which the reactant flows through the annular<br />
space between the two electrodes have an advantage in that all the reactlnt<br />
molecules must pass through the discharge. There is therefore no by-passing and<br />
subject to suitable electron energies, there will be a high level <strong>of</strong> activation<br />
in the gas phase. Such ccnditions are difficult to achieve in parallel plate<br />
arrangements unless the reactant is introduced at the centre <strong>of</strong> one <strong>of</strong> the electrodes<br />
as in Figure 1.4. Much work has been reported with point electrode Systems<br />
such as those illustrated in Figures 1.3 and 1.7 and here it is difficult to see<br />
how by-passing <strong>of</strong> the discharge can be avoided. Furthermore the flow pattern<br />
within the reactor is also dependent upon the thr.?ulence level so that changes<br />
in the gas flowrate will almost certainly be accompanied by corresponding changes<br />
in the fraction <strong>of</strong> the gas stream by-passing the discharge zone.<br />
All the reactor systems discussed so far can be described as homogeneous in<br />
the sense that only a single, gaseous, phase is present in the reactor. In many<br />
instances, however, it is advantageous to remove one or more products <strong>of</strong> reaction<br />
as rapidly as possible' in order to minimise subsequent reactions which would<br />
adversely affect the primary rehction yield. A convenient method <strong>of</strong> achieving<br />
this involves the introduction <strong>of</strong> a second.phase into the discharge zone in the<br />
form <strong>of</strong> a liquid absorbent (6) or fluidised solid adsorbent (\&I. In principle<br />
a second phase can be introduced between any <strong>of</strong> the electrode arrangements shown<br />
in Figure 1. This modification gives rise to a second series <strong>of</strong> reactor-systems<br />
which will be referred to subsequently as heterogeneous reactors.<br />
3.0 Nature <strong>of</strong> the Electrical Discharge.<br />
For optimum reactor performance it is necessary to ensure that the reactants<br />
\<br />
I<br />
i
ELECTRODE ARRANGEMENT<br />
PLRALLEL CO-AXIAL I POINT<br />
I- Fig-1 ~<br />
Ihsir:<br />
rcaclor p?ornrrtrics.<br />
-<br />
i t
294<br />
have suitable residence times in that part <strong>of</strong> the discharge zone where activation<br />
is most favoured. In this connection it will be recalled that the relative rate '<br />
<strong>of</strong> ammonia synthesis from its elements has been shown to be greatest in the region<br />
<strong>of</strong> the cathode potential drop (3,) whilst in the case <strong>of</strong> hydrazine synthesis from<br />
ammonia, the significant fraction <strong>of</strong> the discharge is the positive column region<br />
(k). In theory, it is desirable to i<br />
be able to predict the discharge conditions<br />
necessary for optimum conversion in any particular reaction; in practice however,<br />
this is not yet possible. This is-because in the past it has been customary to (<br />
describe <strong>discharges</strong> in general terms such as glow, arc or silent digcharges without<br />
regard to the local or point properties existing in different parts <strong>of</strong> the discharge;<br />
zone. Since there is a gradual transition between our regime and the next, qualit-<br />
ative descriptions <strong>of</strong> this type are not <strong>of</strong> great value in interpreting reaction<br />
rate data. Any fundamental correlation <strong>of</strong> rate data with discharge characteristics<br />
would involve not only a knowledge <strong>of</strong> the concentrations and energy distributions<br />
<strong>of</strong> the various particle -species present in the discharge space)including electrons<br />
but also an understanding <strong>of</strong> the way in which the probability <strong>of</strong> excitation varied 'k<br />
with electron energy . A typical example <strong>of</strong> the way in which the degree <strong>of</strong><br />
excitation is dependent,upon electron energy is shown in Figure 3 which depicts .<br />
total cross-section curves for argon and neon ( 5). The cross-section for an mn -)<br />
type transition by electron collision is zero when the electron energy is less than<br />
the energy required to raise a valency electron <strong>of</strong> an atom from the mth to the \<br />
nth level,-but increases with electron energy to a maximum after,which any further<br />
increase in electron energy is accompanied by a decrease in the value <strong>of</strong> the<br />
total cross-section. There is therefore an optimum value <strong>of</strong> the electron energy<br />
corresponding to the maximum cross section for the process under consideration. 1<br />
It is the lack <strong>of</strong> data <strong>of</strong> this type together with a knowledge <strong>of</strong> the reactive<br />
species present in the discharge that makes it difficult to correlate reaction \<br />
rates with discharge parameters for systems <strong>of</strong> engineering interest. I<br />
Nevertheless despite the absence <strong>of</strong> fundamental information relating to the<br />
\<br />
.activation characteristics <strong>of</strong> <strong>discharges</strong>, one or two general observation can be<br />
made with regard to the nature'<strong>of</strong> the <strong>discharges</strong> commonly employed. All the<br />
reactor geometries discussed so far, and illustrated diagrammatically in Figure 1,<br />
employ the same type <strong>of</strong> discharge in the sense that both electrodes are located<br />
within the reactor and are therefore in contact with the reactant gas. This class<br />
<strong>of</strong> discharge in which electrons pass freely between the two electrodes is represeh<br />
schematically in Figure 2a. An alternative type <strong>of</strong> discharge which has been used<br />
I<br />
frequently is the so called barrier discharge shown diagrammatically in Figure 2C.<br />
Here one <strong>of</strong> the electrodes is separated from the gas phase by means <strong>of</strong> a dielectric)<br />
barrier such as quartz so that the reactor may be looked upon as a capacitative '<br />
and resistive load in series. In practice a dielectric barrier may be employed \<br />
in conjunction with any <strong>of</strong> the reactor geometries shown in Figure 1. This<br />
arrangement has the advantage <strong>of</strong> providing a more uniform type <strong>of</strong> discharge which<br />
is current limited so that the build up <strong>of</strong> spark or arc type <strong>discharges</strong> is avoided.\<br />
If this principle is carried to its logical conclusion, both electrodes may be<br />
isolated from the reactant gas and molecular excitation set up by applying a high<br />
frequency potential.to the external electrodes or by induction from an external \<br />
conductor carrying a high frequency current.(Figure 2d.) This type <strong>of</strong> "electrodeless:<br />
reactor has not been mentioned in the classification discussed above but is Of<br />
interest since in the absence <strong>of</strong> metal electrodes. in the gas phase, the degree Of<br />
dissociation <strong>of</strong> the gas and therefore the concentration <strong>of</strong> atoms would be expected<br />
to be high (LO). The high concentration <strong>of</strong> atomic species in such <strong>discharges</strong> is<br />
evidenced by the afterglow phenomena frequently encountered in electrodeless<br />
systems.<br />
One type <strong>of</strong> electrodeless sytem which might well prove <strong>of</strong> interest in eleCtrO-;<br />
synthesis is' the microwave discharge. Here the degree <strong>of</strong> activation can be highcq)<br />
and it has been reported that the atomic yields in hydrogen, nitrogen and oxygen 1<br />
are approximately ten times greater than the yields <strong>of</strong> atoms and radicals in low<br />
frequency and direct current <strong>discharges</strong> at the same field strength and pressures.<br />
From the bngifieering point <strong>of</strong> view, microwave systems have the added advantage<br />
1<br />
,I<br />
-I<br />
t<br />
'<br />
i'<br />
c<br />
S
295<br />
'' that they can be sustained at higher pressures but on the other hand the detailed<br />
,t economics still require'evaluation.<br />
The crossed discharge arrangement shown in Figure 2b comprises a low frequency<br />
discharge at right angles to one <strong>of</strong> high frequency. The discharge zone itself is<br />
said to be characterised by a low energy density and to result in unexpectedly<br />
high activation <strong>of</strong> the reactants and relatively high yields <strong>of</strong> the reaction products<br />
(3 ). These claims ha$-e not however been confirmed by recent work in these<br />
j<br />
laboratories in which methane was subjected to a crossed discharge at 31 mm.Hg<br />
pressure. The reactor consisted <strong>of</strong> a 15 mm. internal diameter glass: tube fitted<br />
i/ with two pairs <strong>of</strong> 3 mm.diameter stainless steel electrodes arranged at right angles.<br />
The percentage decomposition <strong>of</strong> methane was observed using a 0.91 Mc/s high<br />
frequency discharge, a 50 c/s low frequency discharge and combinations <strong>of</strong> high<br />
1. and low frequency <strong>discharges</strong> simultaneously. Figure 4 shows some typical data<br />
plotted in the form'$ methane decomposed versus $ H.F. power for a series <strong>of</strong> runs<br />
at constant total power. The methane flowrate was constant throughout at 93 cc/<br />
'<br />
minute,at N.T.P. These results confirm the general trend whereby an increase in<br />
disch,arge power is accompanied by an increase in the percentage decomposition <strong>of</strong><br />
the methane but on the other hand do not substantiate the claim made for the higher<br />
activating effect <strong>of</strong> the crossed discharge. Had this been the case, the curves<br />
shown in Figure 4 would have exhibited maxima whereas, in fact, the observed<br />
methane decomposition at constant total power increased steadily with increasing<br />
percentage pf H.F. power. In this particular case, there is therefore no advantage<br />
in the use <strong>of</strong> crossed <strong>discharges</strong> and the characteristics claimed by Cotton still<br />
lemain to be confirmed . -<br />
4.0 Product Selectivity and Residence Time Considerations.<br />
In the simple plug flow reactor, the conversion (x) obtained is related to the<br />
residence time (f) by the expression:<br />
. .<br />
"where U, and r are the molal volume <strong>of</strong> the reacting mixture and the reaction rate<br />
respectively. If the reaction is one which can only take place under the influence<br />
i <strong>of</strong> an electrical discharge, then the residence time refers to the time spent by<br />
1 the reactants in the discharge zone itself. If however subsequent reactions involv-<br />
,;ng the primary reaction products are possible outside the discharge zone, these<br />
I<br />
$1. will usually proceed at a different rate and must be taken into account separately<br />
in analysing the overall performance <strong>of</strong> the reactor system.<br />
\ If the reaction rate (r) could be written in terms <strong>of</strong> the concentractions <strong>of</strong><br />
the reactants and a rate constant (k) which in turn was dependent upon the discharge<br />
characteristics, then in principle it would be possible,to integrate the above<br />
'I expression for any set <strong>of</strong> discharge conditions and compute the residence time for<br />
! any desired con--ersion. In practice however this is seldom possible because the<br />
functional relationship between the reaction rate and the discharge characteristics<br />
'~<br />
Dishcarge reactions are frequently complicated by reason <strong>of</strong> the interactions<br />
) between the atomic, molecular, ionic and free radical species present. Moreover<br />
because <strong>of</strong> the wide range <strong>of</strong> species present, reactions are rarely simple in the<br />
sense that only one primary reaction product is produced. Competing parallel,<br />
consecutive and degradation reactions are <strong>of</strong>ten present, the net result <strong>of</strong> which<br />
is to produce a wide spectrum <strong>of</strong> reaction products. One <strong>of</strong> the key problems at<br />
the present time therefore is how to design reactors in which unwanted side<br />
reactions are reduced to a minimum. There is no clear-cut anawer to this p'roblem<br />
as yet, but an examination <strong>of</strong> some <strong>of</strong> the more elementary types <strong>of</strong> reactions such<br />
as those listed in Table I indicates a promising method,<strong>of</strong> approach.
0<br />
E<br />
<<br />
h(<br />
E<br />
l!<br />
d K<br />
c 0<br />
.-<br />
Y<br />
0<br />
t<br />
u<br />
t<br />
loo Po 300 100<br />
Elrctron Energy, V<br />
Fig. 3. Typical Cross-Scction versus Electron Biergy Curves.<br />
High Frequency Power z.<br />
Fie.4. Effect <strong>of</strong> a Crossed Discharge on % Metha*$ l’)nro-ipnnition.
\<br />
,<br />
,<br />
~ ~~ -<br />
Consecutlve Reactlons A + B A C<br />
A+C-D<br />
Polymerisation Reactlons A + A h B<br />
A + B--.C<br />
A + C-D<br />
Parallel Reactlons<br />
TABLE I<br />
297<br />
Examples <strong>of</strong> Elementary Reactions.<br />
A + Bgg<br />
Reversible Reactions A + B S C + D<br />
I<br />
Degradative Reactions A + B 4 C d D - E<br />
In all the react,ions listed in Table I it is assumed that C is the<br />
preferred product. Intermediate steps involving excited species are omitted so<br />
that the equations merely represented the overall stoichiometry <strong>of</strong> the various<br />
situations. If therefore product C is being produced from reactants A and B<br />
and a second (consecutive) reaction is present which destroys C , then the rate<br />
<strong>of</strong> the second reaction can be reduced by maintaining the concentration <strong>of</strong> C at<br />
a low value.<br />
Since the primary reaction responsible for the formation <strong>of</strong> C<br />
should, on economic grounds, proceed as rapidly as possible, the only way <strong>of</strong><br />
maintaining a low product concentration is to remove C from the discharge zone<br />
as rapidly as it is formed. The methods by which this can be achieved will'be<br />
discussed further below.<br />
The same principles apply to the other reactions in Table I. Thus in the<br />
case. <strong>of</strong> polymerisation reactions, the molecular weight <strong>of</strong> the product can be<br />
controlled by selective removal <strong>of</strong> certainconstituents from the discharge. If<br />
product B is preferred, then further polymerisation can be minimised by rapid<br />
removal <strong>of</strong> B. If on the other hand, the higher molecular weight product C is<br />
required, B is allowed to remain in the discharge and C is selectively removed.<br />
It frequently happens that the product yield is limited by reverse and parallel<br />
reactions. In the latter case, C can be produced in preference to D by<br />
selective removal <strong>of</strong> C. In the former instance, any yield limitations imposed<br />
by equilibrium. considerations can be overcome by rapid removal <strong>of</strong> C so that the<br />
equilibrium is displaced almost entirely in favour <strong>of</strong> products.<br />
Similar reasoning shows that in the case <strong>of</strong> degradative reactions, product<br />
C can be produced in preference to D or E provided that C is removed rapidly<br />
enough from the activating influence <strong>of</strong> the discharge.<br />
A number <strong>of</strong> methods are available for the rapid removal <strong>of</strong> a particular<br />
component from the discharge zone, namely:<br />
(1) Quenching the reaction at low temperatures and freezing out the product (x)<br />
(2) Absorbing the desired product selectively in an inert liquid absorbant (6)<br />
(13 1<br />
(3) Adsorbing the desired product in a fluidised bed <strong>of</strong> solid adsorbent (I&).<br />
(4)'By using a low residence time in the discharge zone. This may be achieved<br />
either by high gas flowrates or by using a pulsed discharge technique. These<br />
are all devices which reduce both the produce concentration and -residence time in<br />
the gas phase and so minimise product losses through subsequent reactions.<br />
Methods (1) - (3) are means <strong>of</strong> controlling selectively the residence time and<br />
concentrations <strong>of</strong> certain specific products in the discharge and a choice can only<br />
be made between them in the light <strong>of</strong> the physical properties <strong>of</strong> the various<br />
constituents <strong>of</strong> the gas phase. The fourth method, which does not discriminate<br />
between the concentrations or residence times <strong>of</strong> reactants and products, can only<br />
be evaluated when the relative rates <strong>of</strong> the competing reactions are known. Of the<br />
various techniques available, simultaneous reaction and absorption in a heterogeneous<br />
reactor (method 2) opens up interesting possibilities for producing economic<br />
I
298<br />
yields <strong>of</strong> many <strong>chemical</strong> products where hitherto the overall yield has been<br />
restricted by decomposition reactions. The theoretical analysis <strong>of</strong> such systems<br />
is howe!.er extremely complex and even if the kinetics <strong>of</strong> the process were fully<br />
understood, it is doubtful if the absorption rate into the liquid phase could be \<br />
predicted with any degree <strong>of</strong> certainty because <strong>of</strong> complications arising from<br />
dielectrophoretic stirring <strong>of</strong> the liquid. If the liquid phase is polarizable<br />
the electrical forces in a non-homogeneous field will cause polar molecules to<br />
move towards regions <strong>of</strong> maximum non-homogeneity. Any turbulence thereby set up I<br />
within the liquid phase will enhance the rate <strong>of</strong> gas absorption but at the same<br />
time render the prediction <strong>of</strong>. mass transfer coefficients extremel-J uncertain.<br />
From a practical point <strong>of</strong> view, such effects are desirable ins<strong>of</strong>ar as it is<br />
important that the rate <strong>of</strong> absorption <strong>of</strong> the product should be high compared with<br />
its rate <strong>of</strong> production by synthesis.<br />
Some illustrations <strong>of</strong> the factors -d'iscussed are <strong>of</strong> interest at this stage and<br />
Figs.5 and 6 show how the molecular weight <strong>of</strong> the product is influenced by<br />
residence time in the.case <strong>of</strong> consecutive type ractions involving methane as the a<br />
reactant. Figure 5 shows how the higher molecular weight product, in this case<br />
propane, is favoured by longer residence times in the discharge (11 ). These data<br />
were obtained at 80 mm Hg pressure in a cross-flow/coaxial reactor with a 1.60 mm. \<br />
thick quartz barrier adjacent to the inside surface <strong>of</strong> the outer electrode. The<br />
relevant electrode diameters were\-l mm and 3 mm respectively and the power \<br />
density was0 .O&w/cc throughout. Figure 6 shows the effect <strong>of</strong> longer residence<br />
times on the higher molecular weight fraction from methane at a pressure <strong>of</strong><br />
10 mm Hg and again the molecular weight <strong>of</strong> the product is strongly dependent on<br />
residence time (7 ).<br />
In the discharge synthesis <strong>of</strong> hydrazine from ammonia, it has been suggested<br />
( l.+ )( I ) that the hydrazine can be subsequently destroyed by further electron \<br />
excitation or by reaction with atomic hydrogen produced in the primary electron I<br />
bombardment <strong>of</strong> ammonia. If therefore th' concentration and residence time <strong>of</strong><br />
ttie hydrazine in the discharge be reduceLydegradative reactions should be<br />
and the yield increased correspondingly. Tha.t this is the case can be seen from<br />
Figures 7 and 8i The arrangement <strong>of</strong> the cross-flow/coaxial reactor employed in these<br />
runs is shown in Figure 9. The reactor tube was made from 1.60 m.thick quartz<br />
tubing which constituted the dielectric barrier and was 28.6 mm.0.D. The inner<br />
electrode consisted <strong>of</strong> a perforated stainless steel drum 18 mm O.D. which was<br />
mounted on a coaxial shaft so that it could be rotated at high speed. A liquid<br />
absorbent, in this case ethylene glycol, was fed to the drum so that, as the latter.<br />
rotated, the glycol was forced.through the drum perforations into the discharge<br />
annulus.in the form <strong>of</strong> a fine spray. The data shown in Fi,gure 7 were obtained at<br />
an average power density <strong>of</strong> 0.044 Kw/cc and illustrate the fact that not only can<br />
the energy yield <strong>of</strong> h7drazine be increased'by reducing the gas residence time<br />
but that a further substantial increase in yield can be obtained if the hydrazine<br />
is rapidly removed from the discharge by selective absorption (13). at<br />
The data shown in Figure 8 in which the energy yield is plotted versus the<br />
discharge duration for a constant gas flowrate were obtained in a cross-flow<br />
parallel plate reactpr using a pair <strong>of</strong> electrodes each measuring 12.5 x 6.3 mm.<br />
and located llmm. apart. 1n.thes.e runs the average power density was 0.01 Kw/cc<br />
using a pulsed D.C.discharge, the pulse duration ranging from 12 to 130 micro-<br />
seconds. Under these conditions, the effective,residence time <strong>of</strong> the reactant and<br />
products was no longer dictated by the gas flowrate but by the pulse duration.<br />
This technique is therefore a device for securing very low residence times without<br />
the inconvenience <strong>of</strong> high gas velocities. Again the data are in agreement with<br />
previous results in that the lower residence times favour higher energy yields<br />
because <strong>of</strong> the smaller loss <strong>of</strong> hydrazine through reverse and consecutive reactions.<br />
It is interesting to note that an alternative technique whereby atomic hydrogen is<br />
converted to less reactive molecular hydrogen through the agency <strong>of</strong> a platinum<br />
catalyst has'also shown increased yields <strong>of</strong> hydrazine ( k).<br />
.It has been assumed 60 far that the reactant flows as a coherent plug<br />
throu& the discharge. ?his concept <strong>of</strong> plug-flow is only an approximation at<br />
I<br />
1<br />
, I<br />
I<br />
\ '<br />
\c<br />
<<br />
4<br />
i<br />
i<br />
t<br />
c
05 1.0 15<br />
5<br />
Residence Time 7x10 hr.<br />
Fig.5. The effect <strong>of</strong> residence time on propane-ethane yield<br />
from methane.<br />
Hipher Mobcular Welght FrCCtia?<br />
1 2 3 4<br />
Rnldrncr flmr rlO' hr<br />
-<br />
Pressurr lOmm -<br />
rig.6. The cffr~t<br />
<strong>of</strong> rr,sidcnr.rA time on the yield <strong>of</strong> higher<br />
molrvular wc.ight fraction from methane.
- ;i<br />
Y .<br />
15. Pressure lOmm<br />
5<br />
> 10- 1<br />
--<br />
4 U _,<br />
a<br />
2-<br />
e<br />
c 5.<br />
N<br />
0<br />
k<br />
I ,.<br />
-.
J<br />
I<br />
i'<br />
i P<br />
L iqud<br />
i<br />
Gas
302<br />
the best and in many practical situations considerable divergences are observed I<br />
between the residen
Acknowledgements .<br />
303<br />
The author is indebted to a number <strong>of</strong> his colleagues and to Dr. C.A.Walley<br />
<strong>of</strong> the Department <strong>of</strong> Electrical Engineerlng for manv helpful discussions.<br />
Refcrcnrcs.<br />
(1) Andcrsen W . H . , Zwollnski B.J., Parlin R.B., 1nd.Eng.Cheni. ,1959.%.(4).527.<br />
Breirer A.L. , W~sthai cr J.W. , J .Phvs .Chem. , 1929.2.883 (Cf. also J .Phys .Chem.<br />
1930. '4. 153)<br />
Cotton W. J. , Trans .Elect rocheni.Soc. , 1947. 2, 407, 419.<br />
Devins .J .C. , Burton M. , J .Anier.Chem.Soc. , 1954., 3. 2618.<br />
Ilowatson A.M., "Introduction to Gas Discharges" (Pergamon. PreSS,1965).<br />
Imperial Chemical .Industries, U.K.Patents 948,772; 958,776; 958,777; 958,778;<br />
9GG,40G (all 1964).<br />
Kraaij\.cld Von H.J., Waterman J.I., Brennst<strong>of</strong>f-Chemic?, 1961.42.(12),369.<br />
Llewellyn-Jones F., "Ionisation an? Breakdpwn in Gases", (Methuen & Co.<br />
Lrd. 1966).<br />
AlcCarthy R.L., J.Chem.Phys., 1954, 22, 13FO.<br />
hliya-zaki, Takahashi, Nippon Ragaku Zasshi, 1958. 3. 553.<br />
Sergio R. Unpublished Work, University <strong>of</strong> 'Newcastle upon Tyne. England.<br />
Thornton J.D., Chem.Proccssing, 1966. (2), SG.<br />
Thornton J.D., Charlton W.D., Spedding P.L.., "Hydrazine,Synthesis in a<br />
Silent Electrical Discharge" (to be published).
GENERATION AND MEASUREMENT OF AUDIO FREQUENCY POWER<br />
FOR CHEMICAL-ELECTRICAL DISCHARGE PROCESSES \<br />
James C. Fraser '.<br />
Research and Development Center<br />
General Electric Company<br />
Schenectady.. New York<br />
In developing equipment for use in high voltage, high frequency<br />
<strong>chemical</strong>-electrical processing, we have built a number <strong>of</strong> different<br />
types <strong>of</strong> "corona" power supplies. At the start, from the standpoint<br />
<strong>of</strong> the electrical equipment, let us use "corona" in its broadest sense.<br />
We will define a "corona" discharge as an electric discharge produced<br />
by capacitively exciting a gaseous medium lying between two spaced<br />
electrodes, at least one <strong>of</strong> which is insulated from the gaseous medium<br />
by a dielectric barrier. A corona discharge may be maintained over<br />
wide ranges <strong>of</strong> pressure and frequency, although atmospheric pressure<br />
and frequencies in the audio range substantially above power trans-<br />
mission values are typically employed.<br />
Since the power which can be dissipated in a corona reactor or<br />
cell is a function <strong>of</strong> the supply frequency, we have settledarbitrarily<br />
on the audio range (3,000 to 10,000 cycles per second) as a desirable<br />
compromise. It is within the l i m i t s <strong>of</strong> rotating machinery and solid<br />
state components, presents no undue corona reactor heat dissipation<br />
problems, and provides a happy compromise between operating voltages,<br />
reactor size, and economy <strong>of</strong> operation.<br />
The ratings for the basic electrical components are dependent<br />
upon the characteristics <strong>of</strong> the corona reactor and the impedance load<br />
which it represents to the power supply. Corona reactor values which<br />
affect these ratings are:<br />
a. The corona power required<br />
b. The electrode corona power density and, thereby, the<br />
electrode cooling or heat dissipation capability<br />
C. The gaseous atmosphere present<br />
d. The gaseous gap spacing<br />
e. The barrier material and dielectric constant<br />
From these values we can determine:<br />
a.<br />
b.<br />
c.<br />
Peak voltage required to initiate corona<br />
Barrier thickness required to withstand the total voltage<br />
across the barrier in the event <strong>of</strong> an arc across the gap<br />
Barrier, gaseous gap, and total reactor capacitance<br />
d.<br />
e.<br />
f.<br />
Maximum operating voltage<br />
Capacitive charging, or displacement, current drawn by<br />
the reactor<br />
Corona resistive current .<br />
g.<br />
h.<br />
Resultant load current<br />
Power factor <strong>of</strong> the corona reactor load<br />
I .I<br />
1<br />
?<br />
\<br />
1<br />
\<br />
'\
1<br />
f<br />
/<br />
I 7<br />
\<br />
i/<br />
'1:s<br />
The objectives which,we attempted to meet in the design <strong>of</strong> our<br />
equipment were :<br />
a. Suitability for use with a wide variety <strong>of</strong> corona reactors<br />
b. Operation over a wide range <strong>of</strong> breakdown voltages<br />
C. Flexibility over a wide range <strong>of</strong> operating conditions<br />
d.<br />
e.<br />
Maximum electrical efficiency<br />
Trouble-free operation and minimum maintenance :over long<br />
periods <strong>of</strong> ,operation<br />
Among the types <strong>of</strong> equipment built were the inductor-alternator<br />
with step-up transformer and parallel-resonant tuned circuit, the<br />
SCR inverter with step-up transformer, and the mobile vacuum tube<br />
high power-amplifier.<br />
Motor-Alternator Set<br />
A typical power supply based on rotating equipment consists <strong>of</strong><br />
the following major components: an inductor-alternator, by means <strong>of</strong><br />
which 60 cycle power is converted to 10,000 cycles, at 500 volts.<br />
This output is fed to the primary <strong>of</strong> a high voltage, high frequency<br />
transformer rated for 25 or 50 KV at 10,000 cycles. The output <strong>of</strong><br />
this transformer is fed, in turn, to a parallel resonant tuned circuit,<br />
the discharge reactor, and the high volthge instrumentation.<br />
The KVA rating <strong>of</strong> the hl-G Set (inductor-alternator) is determined<br />
by the product <strong>of</strong> the corona operating voltage and the resultant load<br />
current (or the corona power divided by the power factor). Since the<br />
corona power generated in a gaseous gap in series with a dielectric<br />
is directly proportional to frequency, and the upper l i m i t for frequency<br />
for commercially available rotating equipment is about 10,000 cycles<br />
per second, this has generally been considered the optimum frequency.<br />
Also, the use <strong>of</strong> lower frequencies would require higher voltages or<br />
increased corona reactor capacitance for any given power level. The<br />
output voltage <strong>of</strong> the alternator can be any value consistent with usual<br />
generator practice and with the transformer primary voltage.<br />
The KVA rating <strong>of</strong> the transformer will usually be the same as<br />
that <strong>of</strong> the inductor-alternator. Its secondary voltage will be de-<br />
termined by the corona reactor requirements for voltage breakdown <strong>of</strong><br />
the gaseous gap. Secondary or load current will be the vector sum<br />
<strong>of</strong> charging, or displacement, current and resistive, or corona, current.<br />
Primary voltage must be consistent with alternator output voltage.<br />
If experimental flexibility is required, the transformer primary should<br />
be arranged for series-parallel connection.<br />
f The corona reactor represents a capacitive load on the major items<br />
1 <strong>of</strong> the power supply equipment, up to the time <strong>of</strong> corona initiation.<br />
5 Since this generally results in a poor power factor, there are two<br />
, possible means <strong>of</strong> solution:<br />
Y<br />
a. The motor-alternator set must be built with sufficient KVA<br />
\ capacity to meet the power factor and corona power requirements.
. The transformer secondary and the corona reactor can be tuned<br />
with a parallel resonant circuit consisting <strong>of</strong> a choke and<br />
high voltage, vacuum capacitors. This provides power-factor<br />
correction so that the high voltage transformer secondary sees<br />
mainly the resistive load represented by the corona power.<br />
The parallel-resonant circ.uit must be tuned to give maximum trans- 1<br />
former output voltage for some arbitrarily selected value <strong>of</strong> alternator<br />
field current. With a 0.16 henry choke, for example, the capacitance <<br />
required for tuning at 10,000 cycles can be found from the expression 1<br />
1 1<br />
f = , or C = = 1580 pic<strong>of</strong>arads<br />
2 f G 462 €%<br />
However, this value is reduced by the capacitance <strong>of</strong> the discharge react<br />
itself, by that <strong>of</strong> any instrumentation voltage dividers, and by stray “t;<br />
capacitances, so that the actual tuning capacitance used is somewhat<br />
lower and must be determined experimentally. 7<br />
The decision as to whether the motor-alternator should be built<br />
with increased KVA capacity or whether the secondary circuit should be<br />
tuned depends on the experimental I versatility required, and on economic<br />
considerations. In general, the dollars per KVA for high frequency<br />
motor-alternator sets are considerably higher than for tuning circuit<br />
components.<br />
An alternator field-control chassis supplies and regulates the current<br />
for the alternator field windings, and has a feedback system designea<br />
to meet varying conditions <strong>of</strong> high voltage output. Alternator output<br />
voltage and, thus, transformer high voltage are raised and lowered by<br />
changing alternator field current.<br />
A magnetic amplifier stabilizer<br />
circuit maintains constant high voltage output. In the event <strong>of</strong> a de-<br />
crease in high voltage, it calls for an increase in alternator field<br />
current, with a corresponding output voltage increase.<br />
I<br />
\<br />
1<br />
‘<br />
i<br />
Control devices and instrumentation for a ’<br />
corona power supply based<br />
on rotating equipment includes at least the following: 1<br />
a.<br />
b.<br />
C.<br />
d.<br />
e.<br />
Provision for varyi .ng the transformer output high voltage from<br />
zero to maximum by varying alternator field current, and a<br />
means <strong>of</strong> measuring the field current.<br />
Provision for measuring transformer primary voltage, current,<br />
and power.<br />
Provision for measuring the peak voltage applied to the corona<br />
reactor.<br />
Provision for measuring the power dissipated in the corona<br />
reactor.<br />
Some form <strong>of</strong> protective current overload relay, for removing<br />
high voltage from the corona reactor in the event <strong>of</strong> a short<br />
circuit in the reactor, or a low voltage,high current arc<br />
5
i I.<br />
r<br />
>-<br />
.i<br />
between the electrodes. This will prevent exceeding the current<br />
1' a t i n g o f t he t r a n s f o rme r s e c o n da r y .<br />
Since the use <strong>of</strong> this equipment involves working with high voltage.<br />
the control circuit includes provision for test area safety interlocks<br />
and flasher-warning lights.<br />
I nver t e r<br />
j. The solid state corona generator consists <strong>of</strong> a 3.000 cycle inverter<br />
I - utilizing silicon controlled rectifiers feeding a step-up transformer.<br />
A rectifier is a. device or circuit for changing alternating current to<br />
' direct-current. The inverter may be thought <strong>of</strong> as a rectifier operating<br />
ill reverse, changing d-c to a-c. A combination <strong>of</strong> the two achieves a<br />
q' change from 60 cycle a-c to approximately 3,000 cycle a-c.<br />
.-'<br />
Rectifier circuits occur in several configurations such as halfwave,<br />
full wave, bridge. etc. Inverter circuits may be gr0u.p ed in a<br />
/similar manner. The key feature <strong>of</strong> an SCR (silicon controlled rectifier)<br />
as used in an inverter circuit is that a small current from its "gate"<br />
4 element to the cathade can fire or trigger the SCR so that it changes<br />
8 from being an open circuit into being a rectifier.<br />
The inverter operates from a single phase, 220 volt, 60 cycle<br />
I source, and by means <strong>of</strong> a full-wave bridge rectifier, rectifies it to<br />
' 187 volts d-c which in turn is changed to 150 volts, 3,000 cycles,<br />
1 approximate sine wave through SCR's. Variac control <strong>of</strong> the 220 volt<br />
input provides a variable supply output.<br />
The inverter output is fed to a step-up transformer delivering up<br />
4 to 12,000 volts rms. This transformer is a dry type, potted with RTV,<br />
with integral high voltage terminals. The particular transformer used<br />
was built as a compromise between low cost, size, and electrical<br />
capability. Rated at 3 KVA, the'transformer was only 6" x 6" x 7"<br />
1 high overall.<br />
I What is noteworthy with this equipment is that the inverter,<br />
transformer, and corona reactor are not independently operating com-<br />
' ponents. The concentric cylinder- corona reactor was so designed that<br />
j its capacitance, refle'cted to the primary <strong>of</strong> the transformec formed<br />
a part <strong>of</strong> the inverter LC circuit, as did the inductance <strong>of</strong> the high<br />
' voltage transformer. The reactor gaseous gap conductance forms a<br />
/part <strong>of</strong> the inverter load circuit.<br />
1<br />
Since this is an untuned high voltage circuit, the inverter and<br />
) the transformer were designed to carry the power factor loss current.<br />
r'<br />
Y<br />
\<br />
Advantages <strong>of</strong> .the inverter supply over the motor-alternator is the<br />
absence <strong>of</strong> moving parts, ease <strong>of</strong> mounting and installation, and the<br />
"module" or building block concept whereby units can be stacked to in-<br />
crease voltage and current capability.
Vaccum-Tube Amplifier<br />
The mobile corona generating equipment was designed for operation<br />
ove mr a wide range <strong>of</strong> frequency and voltage. Corona power is generated<br />
by a vacuum tube, high-power amplifier driven by a stable low-power,<br />
audio-frequency sine-wave oscillator. Power is taken from the secondary 1<br />
winding <strong>of</strong> a high-voltage step-up transformer, with one end at ground<br />
potential.<br />
I $<br />
5<br />
The corona output voltage may be varied from zero to 50,00O:volts, 1<br />
depending on the impedance <strong>of</strong> the applied load, and on the settings \<br />
<strong>of</strong> the frequency and amplitude controls located on the master sine-wave<br />
oscillator panel. The frequency <strong>of</strong> the output voltage is determined<br />
by the master oscillator and is independent <strong>of</strong> load impedance or output !<br />
voltage level. The frequency may be varied instantaneously between ,<br />
the rated limits <strong>of</strong> 3 to 10 kilocycles per second.<br />
The a-c power from the 60 cps line is converted into the high<br />
voltage variable frequency output as follows:<br />
A master oscillator feeds a driver amplifier. This is turn drives<br />
a tetrode power amplifier stage and the output step-up transformer.<br />
A d-c plate supply source, a screen grid voltage supply, a control grid<br />
bias supply, and an a-c filament supply feed power at appropriate<br />
voltage levels to the tetrode amplifier stage. The power line control )<br />
circuitry incorporates on-<strong>of</strong>f switching, circuit breakers, and starting<br />
contactors for the power amplifier supplies.<br />
Power amplification is produced by four tetrode vacuum tubes con-<br />
nected in pushpull-parallel as a Class AB2 amplifier. Control <strong>of</strong> the *<br />
master audio oscillator signal amplitude is provided by a potentiometer<br />
on the master control panel. The output voltage <strong>of</strong> the equipment may<br />
be reduced to zero even when full voltage is applied to the plates <strong>of</strong> \<br />
the power tetrodes.<br />
Power delivered by the tetrodes to the output transformer, and<br />
thence to the corona reactor load, is controlled by the oscillator and )<br />
the variable autotransformer in the tetrode plate power supply. The<br />
autotransformer control keeps the total power consumed by the equipment<br />
during an experiment as low as possible, to minimize heat developed<br />
in the cabinet and to extend tube life. -.<br />
The corona cell impedance, which is mainly capacitive before corona '<br />
breakdown, acquires a resistive component after breakdown which depends (<br />
on the rate <strong>of</strong> production <strong>of</strong> ionized and electrically conducting I<br />
<strong>chemical</strong> entities. The a-c voltage at the plates <strong>of</strong> the power tetrodes 'i<br />
will lag the plate current by a large angle before power is consumed, 1<br />
and the plate current will be collected at a high rather than a low<br />
value <strong>of</strong> plate voltage. Thus, the power dissipated by the tetrodes<br />
will be high under no-load conditions.<br />
The high voltage transformer has an effective primary to secondary '<br />
step-up ratio <strong>of</strong> 11 to 1. The maximum a-c current that can be drawn<br />
from the secondary winding <strong>of</strong> the transformer is 60 milliamperes rms.<br />
$ 1<br />
\<br />
7<br />
\<br />
I<br />
P<br />
1<br />
8<br />
'u;<br />
\<br />
i<br />
\<br />
i
\ /<br />
’\<br />
i Reactor<br />
I<br />
300<br />
The corona reactor or cell represents a capacitive load to the power<br />
supply equipment up to the time <strong>of</strong> initiation <strong>of</strong> the electrical discharge.<br />
The total reactor capacitance consists <strong>of</strong> the series combination <strong>of</strong> di-<br />
electric barrier and gaseous gap capacitance. The voltages appearing<br />
across the barrier and the gap divide inversely as their, capacitance.<br />
’ Before corona starts, the reactor current consists <strong>of</strong> the charging,<br />
or displacement, current which is proportional to the applied voltage,<br />
/ frequency, and cell capacitance. After the start <strong>of</strong> corona, the cell<br />
I acts as an impedance load consisting <strong>of</strong> a conductance across the gaseous<br />
gap, in addition to the gap capacitance. The total current drawn by<br />
the reactor then is the vector sum <strong>of</strong> charging plus resistive currents.<br />
If i<br />
the corona reactor power supply has a tuned circuit in the transformer<br />
high voltage secondary, for power factor correction, the corona<br />
reactor capacitance functions as part <strong>of</strong> the tuning capacitance, and<br />
,’ the reactor charging current is a part <strong>of</strong> the circulating current in<br />
the parallel resonant circuit. The total load current through the transformer<br />
secondary winding then consists chiefly <strong>of</strong> corona current plus.<br />
whatever capacitive current might be present as a result <strong>of</strong> tuning unbalance.<br />
If the power supply is untuned, the total power available for<br />
<strong>chemical</strong> processing is the product <strong>of</strong> supply volt-amperes times the<br />
power factor <strong>of</strong> the load, and in this case transformer secondary wind-<br />
ing must carry the full reactor impedance current.<br />
The dielectric barrier in the corona reactor serves as a built-in<br />
current-limiting device, or ballast in the event <strong>of</strong> a short circuit or<br />
arc between the cell electrodes, so that the stored energy is dissipated<br />
in the barrier at breakdown.<br />
The maximum power density at which a corona reactor can be operated<br />
without puncturing the barrier is a function <strong>of</strong> the dielectric strength<br />
<strong>of</strong> the barrier material, the total thickness <strong>of</strong> the barrier, the ambient<br />
temperature in which it is operated, and the gap spacing between<br />
barriers. The dielectric constant <strong>of</strong> the barrier affects both the<br />
voltage gradient across the barrier, and the corona power which can be<br />
dissipated in the reactor. The dissipation factor is a measure <strong>of</strong> the<br />
electric losses which produce heating <strong>of</strong> the barrier.<br />
The voltage at which breakdown will occur between a set <strong>of</strong> electrodes<br />
in a corona reactor depends upon the gaseous atmosphere present, the<br />
density (pressure, temperature) <strong>of</strong> the gas between the electrodes, and<br />
the gap length.<br />
The corona power dissipated in a gaseous gap in series with one<br />
or more dielectric barriers can be calculated from the expression<br />
P = Corona power in watts
310<br />
f = Power supply frequency in cycles/second<br />
cb = Dielectric barrier capacitance<br />
Cgas = Capacitance <strong>of</strong> corona cell gas gap<br />
Ct = Total corona cell capacitance<br />
I<br />
Vgas = Peak voltage across the gaseous gap at the time <strong>of</strong> corona<br />
initiation<br />
Vmax = Total operating voltage applied to the corona cell<br />
This can be re-written as:<br />
Lgas<br />
Vt = - = voltage across the corona cell at the instant<br />
.Ct 'gas <strong>of</strong> corona initiation<br />
An- item <strong>of</strong> concern to all, in evaluating an experiment, or in<br />
scaling up for pilot plant operation, is the electrical efficiency <strong>of</strong><br />
a process. Care must be taken not to confuse equipment power factor<br />
with what might be called "electrical efficiency <strong>of</strong> conversion",<br />
both may contribute to the same end result. Power factor relates though\ to<br />
ability to convert equipment input volt-amperes (not necessarily in<br />
phase) to available <strong>chemical</strong>-result-producing "watts". This is a<br />
function <strong>of</strong> electrical equipment and electrical circuit design.<br />
i<br />
"Electrical efficiency <strong>of</strong> conversion" may be considered as the<br />
effectiveness in utilizing all or most <strong>of</strong> the available wattage in the<br />
corona reactor. This is a function <strong>of</strong> reactor design, with particular<br />
emphasis on relative barrier and gap capacitance and gap spacing. As (<br />
a matter <strong>of</strong> practical design, the cell geometry and gap spacing present ~<br />
limits to the power which can be dissipated in any given cell.<br />
Power Measurement<br />
The power consumed in the discharge cell is determined by means (<br />
I<br />
<strong>of</strong> the parallelogram-oscilloscope technique which shows the relation-<br />
ship between the voltage on the cell electrodes at any instant, and<br />
the charge flow in the circuit up to that instant. Before corona<br />
initiation, the corona null value is seen on the oscilloscope as a<br />
closed figure, or slant line. With the initiation <strong>of</strong> the electrical<br />
discharge, the figure opens up into a parallelogram, and the discharge<br />
power is represented by the area <strong>of</strong> the parallelogram.<br />
The bridge circuit for power measurement consists <strong>of</strong> two branches 1<br />
between the high voltage terminal <strong>of</strong> the reactor and ground, called<br />
"Voltage" and "Charge". The "Voltage" branch consists <strong>of</strong> a capacitance<br />
i<br />
\ !<br />
I<br />
t<br />
I<br />
h<br />
\<br />
\
I<br />
1<br />
311<br />
voltage divider giving an approximate lo4 to 1 voltage division. The<br />
"Charge" branch consists <strong>of</strong> the corona reactor in series with some<br />
value <strong>of</strong> Capacitance such that a voltage division <strong>of</strong> lo4 or lo3 to 1<br />
will be obtained across the divider. The outputs from the low voltage<br />
ends <strong>of</strong> these dividers are fed to the X and Y axes <strong>of</strong> the oscilloscope.<br />
A variable potentiometer across the lower end <strong>of</strong> the "Charge" divider<br />
provides phase shift control to bring the "Voltage" and "Charge" branch<br />
outputs into phase for a null reading on the scope.<br />
It should be noted that two signals, <strong>of</strong> equal amplitude, in phase,<br />
fed to the X and Y axes <strong>of</strong> a scope, give a 45-degree line on the scope.<br />
Any variation <strong>of</strong> relative amplitude shifts the angle <strong>of</strong> the line.<br />
The area <strong>of</strong> the parallelogram represents the total power dissipated<br />
in the cell when the parallelogram bridge circuit and the oscilloscope<br />
are calibrated in the following manner:<br />
The X-axis deflection is calibrated with a peak reading voltmeter<br />
/so that reactor voltage (peak-to-peak) is presented as volts per cm<br />
<strong>of</strong> scope deflection.<br />
/<br />
The Y-axis deflection is calibrated with the known value <strong>of</strong><br />
capacitance in the grounded end <strong>of</strong> the "Charge" branch so that the<br />
charge flowing through the reactor is presented as coulombs per cm<br />
/' <strong>of</strong> scope deflect ion.<br />
/<br />
The X - Y product, then, is:<br />
-<br />
) (vo:~s,)(cou:;bs 1<br />
volts - coulombs, watt-seconds<br />
cm2 or<br />
cm2<br />
/ . Since this is the energy per cm2 in one cycle, the power per cm2<br />
<strong>of</strong> parallelogram area is obtained by multiplying the energy per cycle<br />
2 by the frequency in reciprocal seconds. The parallelogram area can be<br />
, determined by planimeter measurement <strong>of</strong> a Polaroid picture, or by<br />
'J direct measurement.<br />
\<br />
With a dual beam oscilloscope it is also possible to view the<br />
a"/ voltage and charge waveforms simultaneously.<br />
\
FUFARCI! INSTITUTE OF TFYDLC UNIVERSTTY<br />
b150 KNRY P.VE.<br />
V'TIJDFLPPJA, PA.<br />
INTRODIJCTICN<br />
uses,<br />
Dlasma penerators, in general, have been found suitable for a variety <strong>of</strong><br />
Thev penerally Drovide an electric arc which is condensed or constricted<br />
into a smaller circular cross-section than would ordinarily exist in an onen arc-tyDe<br />
device. This constriction penerates a very hinh temperature (8,000-20,000°K) so<br />
that a sunerheated-Dlasna workinp fluid can he ejected throuph the nozzle and the<br />
comoosition <strong>of</strong> the Dlasma determine the use to which the plasma nenerator is put.<br />
Plasma Fenerators have been used for cuttinn, welding, metal spravine and <strong>chemical</strong><br />
orocessinr. For <strong>chemical</strong> processinp, plasma penerators have provided the possibility<br />
<strong>of</strong> the production <strong>of</strong> new aaloys and compounds and the processing <strong>of</strong> less commonly<br />
used materials, as well as the preoaration <strong>of</strong> certain common <strong>chemical</strong>s.<br />
nLAS#A JET EQUTPMrNT<br />
Two types <strong>of</strong> plasma generators are possible: the nontransferred and the<br />
transferred arc. A nontransferred arc consists <strong>of</strong> a cathode and a hollow anode<br />
where the arc is struck between the electrodes and the flame emerges through the<br />
orifice in the anode, In the transferred arc, the cathode is placed some distance<br />
away from the anode and an arc is passed between the electrodes. The nontransferred<br />
arc is the nost popular in the'<strong>chemical</strong> studies made to date.<br />
A plasma jet used in <strong>chemical</strong> synthesis can have varied desips to meet 1<br />
special requirements, such as<br />
oath at a particular point.<br />
the introduction <strong>of</strong> a reactant material into the flame<br />
Consumable cathodes have been used in experiments in which carbon was one <strong>of</strong> \<br />
the reactants, Carbon, vaporized from a graphite cathode, was used in the synthesis<br />
<strong>of</strong> cyanogen and hydrogen cyanide. powdered carbon introduced in a gas stream or as a<br />
constituent <strong>of</strong> a Kas has also been used as the source <strong>of</strong> carbon in a plasma flame.<br />
Electrodes <strong>of</strong> 2%-thoriated tungsten are the most frequently used water-cooled<br />
nonconsumable electrodes, Hater-cooled copper anodes have been widely used in experhens<br />
work. Figure 1 shows a typical plasma jet assembly. A reactor chamber may be <strong>of</strong> my<br />
confipuration desired to accomodate different feedinp: and quenching devices.<br />
PLASMA tTET REACTIONS<br />
GAS-CAS PEACTIONS TO PRODUCE A CAS AND CAS<br />
DFCO?'POS7TION PEACTIONS TO PFODUCF A GAS<br />
A considerable amount <strong>of</strong> research has been done by a number <strong>of</strong> people in<br />
the area <strong>of</strong> Dlasma jet eas-pas reactions.<br />
pases that have been investipated.<br />
The followinp is the gas reaction producing<br />
I<br />
\<br />
\<br />
i<br />
x<br />
\'<br />
x<br />
\<br />
I<br />
t<br />
\'
31 4<br />
,Tet.<br />
- ~2H2<br />
CK4 (or other hvdrocarbons) Ar ,?e?<br />
CTq + !I7--+ F3 + C=31!r2 (6) 'I<br />
Srs + h'2+!'F3<br />
t:P3 or CU,, crackinp rate studies (8)<br />
In the oast several vears considerable effort has been directed to the \<br />
investipation <strong>of</strong> the nroduction <strong>of</strong> acetylene from hvdrocarbons (reaction 1).<br />
"he Drnduction <strong>of</strong> acrtvlene hv reactinp Fethane in the flame cf an arpon<br />
!<br />
olasms iet vielded an R0% conversfan to acetvlene (1).<br />
Almost all <strong>of</strong> the methane was<br />
converted to acetvlene and hvdrogen with little formation <strong>of</strong> soot. Pisure 7 shows<br />
the Dower consumption VS. the feed ratio <strong>of</strong> arpon to methane. This ratio is the<br />
most imnortant naraveter in the acetylene yield. "he minimun power consurnntion 60<br />
Kwhr/100 CU. ft. acetvlene nroduced, corresDonded to a ratio <strong>of</strong> arpon to acetylene <strong>of</strong><br />
0.3. Danon and White (2) selected the manufacture <strong>of</strong> acetylene as am aoplicaticn<br />
for plasma processinp which mipht be <strong>of</strong> interest to the petroleum industry. Pethane<br />
was one <strong>of</strong> the pases nroposed with the use <strong>of</strong> recvcle procedure. Anderson and Case<br />
studied the methane decomDosition reaction and compared it to available themodvnamic<br />
data. In these exaeriments a hvdropen plasma torch was used coupled to a reaction<br />
chamber and water ouench svstem. ?he hot hydrogen stream emitted from the Dlasma jet, \<br />
entered the reaction chamber and mixed with a methane feed. The<br />
after affluinp from the reaction chamher and water auench svstem.<br />
conditions for methane produced a 30? yield <strong>of</strong> acetylene.<br />
In a report <strong>of</strong> the <strong>National</strong> Academv <strong>of</strong> Sciences the investigation by the<br />
Linde Company <strong>of</strong> the production <strong>of</strong> acetylene usinp a plasma jet and natural pas was<br />
reported. This process is said to have a more efficient transfer energy to the feed<br />
stream than does the open arc process used in Germany.<br />
Considerable research has been done on the methane-nitrogen and methaneammonia<br />
reactions (Tactions 7 t 3) to produce hvdroeen cyanide and as a bv product<br />
acetvlene. Leutner 6 6)reported up to 50% conversions were obtained based on<br />
carbon input (as methane) bv usinR either nitropen, arEon or nitroEen-argon mixtures<br />
as the plasma eas. fipre 3 shows a schematic <strong>of</strong> the apparatus used<br />
These experiments shoved that up to 75% <strong>of</strong> the carbon inDut as methane<br />
WN and acetylene for reaction 3 and 90% for reaction P. Ho other hydrocarbons beside!<br />
acetylene were found and cvanopen was present in only trace amounts.<br />
f<br />
Damon and Vhite (7) DroDosed the production <strong>of</strong> reducer gas by reaction 4<br />
Using natural eas or DroDane as a hvdrocarbon source. The proDosed process=for stem+<br />
methane refonninR would ooerate at a t emperature <strong>of</strong> 3000 to 60OOOF and Drovide a hi@<br />
temperature reducing gas for metals and other hip.h temperature processes.<br />
The fixation <strong>of</strong> nitropen (reaction 5) has been one <strong>of</strong> the major aDnlications/<br />
for arc induced reactions in the<br />
oxypen-nitro~en mixtures has beep<br />
input has been converted to NP (6<br />
\ !<br />
f<br />
q<br />
"i<br />
\<br />
1<br />
I<br />
\<br />
6<br />
\
J<br />
#<br />
I<br />
/<br />
4)<br />
I<br />
. .<br />
'8<br />
r4<br />
0<br />
r(<br />
P
W<br />
v)<br />
a<br />
m<br />
0<br />
0<br />
0<br />
I<br />
e<br />
z<br />
v,<br />
a<br />
?<br />
an<br />
a<br />
0<br />
I- O<br />
W<br />
J<br />
J<br />
0
317<br />
i Cuencb svsten w.nnar;itus use8 has been full11 characteri::ec! and shown to<br />
i be R verv COO(! Fit f ~ a r d;ifrlsi.onal -n*el.<br />
/<br />
DeceatlV iny~e~tj,patidns at the Pesiarch Institute <strong>of</strong> TemDle IJniversi.ty<br />
5ave shown that hvdro sulfide ccn be bvnthesiz.e(! fron its element, hvdropen and<br />
sulfur nowder fed in a heliur plasm ;et (12). Cnnversions as hish as 37: hased on<br />
/ thk sulfur innut have heen o?t?ined. riuure 4 sh&is hojh the ? conversion and the . '<br />
F/Y.whr <strong>of</strong> hyrdropcr siilf-ide fo6ed vs. Kwhr/p @f,kUlfur,iGput. As can be seen from the<br />
fipure the raximw conversion ?, dces not necessarily; hi,ve the maxivum nroduction<br />
eff ic ienclr.<br />
P consic2erable nurhei <strong>of</strong>, dvntheqi.ses ha& he& .carried out usinp solid carbon<br />
j powder or rranhite eleyent,s .as .a ,carhcn' soprc6 for reactj:ons with various materials<br />
includj.np h-ydroren , nitrocen , .hydroben-n'itrc%n and ammonia. reactions 1 thro~wh 5<br />
2 show the various producti.ons ,obtaine$ .hqi ihese reacti&s., Tn the case <strong>of</strong> acetylene<br />
. ' synthesjs (reaction 2) tt1.e h~.~~&t,ylq;d obtained bv pirect svnthesis from the<br />
elerents was 339 (11.<br />
for recction 11 have been obtained (5). . t complete studv <strong>of</strong> the synthesis <strong>of</strong> cyanopen<br />
\ fro* i.ts elenents was mrle tv !,eutne$-(K 6 13) .and,this-reaction Pave 15% conversion<br />
, ' hased on the carbon innut at the ojtilrun reaction conditions.<br />
v'<br />
.. . .. I<br />
r<br />
.. 1<br />
I+dropen cyanide yields up to 514; for reaction 3 and UD to 399<br />
Decently Craves , Yawa and 1iiteshu.e (I4) reborted investipations wine hituninous<br />
feE. into an aroon plasma. iet .,. .,Acqtvlene, , the princinal Drodiict , was obtained<br />
in vields <strong>of</strong> 15 wt. ?. -his work .s'tudie< the ,effects <strong>of</strong> coal feed rate, particle<br />
size and nlasna terperature on vieid- and nroducts' forned.<br />
.. . . , . - . , . . - .. .<br />
PAS-GAS ~~ACST.CIIS .TO .PAODIJC.C. A m r n<br />
Ilvdrccarbons 4 C (2)<br />
t'arnisch, Yemer and Cciailu: .(15) rknorted the preparation <strong>of</strong> titanjum<br />
' nitride (reaction 1) fror titanium tetrachloride pas with either nlasna
318<br />
FIGURE 4<br />
I
epcrte? the noss~kil.itv <strong>of</strong> nroduct-r carton bla ck fror hvdrocarbons usinp a nlnsna<br />
'et. !!ethane or other hvtrccarbons which would h e introduced intc the plasma flane,<br />
uoul? be cracke2 usir.;. tvCro.ren as the ooeratinr vas an? Droducirp carbon black.<br />
LicciC as i.re11 3s' Fzseous h-:?rncar!mns car te cse? 5s a source for carbon and the<br />
'.'itrc khoratories (4) have exnerinente? ;iith carton Slack production frnv linuic!<br />
h:idrocarhons.<br />
'P"?O5 + !Z1 j Ta (8)<br />
\!C3 t 4 :.: ' (9)<br />
~ 1 3 + 0 Y~/CE,,+ ~<br />
PI (10)<br />
re0 + U7+ ?e (11)<br />
The nroduction <strong>of</strong> metal nitrides (F! f* 17) from the elements has been<br />
invest ipated for three elements, titanium, -arrnesiun and tunpsten (reactions 1 to 3).<br />
The Droduction <strong>of</strong> titaniun nitride in the 1000. vield was accovplished bv usinp 200<br />
mesh titaniur Dowder fed into a nitrooen Dlasna ;et. The titanium nitride particle<br />
size was found to be 0.75 to 7.5 microns and the nroduct was also formed in larpe,<br />
corpact, Folden .rellov crvstals. In a like manner tunpsten nitride was formed in<br />
75p yield. 40% conversion to rnapnesiun nitride was obtained when nacnesium was<br />
fed into a nitropen olasma jet.<br />
The preDaration <strong>of</strong>'cetal carbiles has also been reDorted (12 6 17). Tipure<br />
5 shovs the 9 conversion VS. Kwhr/a tunasten input for reaction 4. As can be seen,<br />
the XC conversion is directlv Dronortional to the power input level. The hiRhest<br />
conversions obtained were 43? for \,!2r and 11% for %'C. reaction 5 aSove is shown in<br />
riplire 6 uhere .9. conversion is plotter] VS. Kwhr/v !XI3 innut. The three products <strong>of</strong><br />
the reaction, tunrsten (43 to Rl? cnnversicn), tunester. carhide (4 to 11% oonversion)<br />
and ditunesten carbide (9 to 35% conversion) are fomad in a total conversion <strong>of</strong> 81<br />
to 9UQ.. ?e naior Droduct is tiinosten which is'favored at higher Kwhr/p WOg inputs.<br />
The reaction <strong>of</strong> tantalum and methane in a helium nlasma jet is shown in Fipure 7 where<br />
a water-cooled cuenchinp nroba was ~ l ~ c at e d 1/3" and 5" below the plasma jet. The<br />
effect <strong>of</strong> the ruenchinr distance is dranatic. The annunt <strong>of</strong> Ta2C formed is notapDrecfahlv<br />
different in either case, however, the TaC vield chanped considerably bv<br />
the n1acerrer.t <strong>of</strong> the ouenchinp device. Conversions UD to 72% have been produced<br />
by this reaction. Tipure e shows a nlot <strong>of</strong> the tantalur-pentoxide plus nethane<br />
reaction carried o ut in a helitic Dlasma jet. The % conversion is plotted vs. Kwhr/y<br />
tantalun nentoxide input for two different ouenchinp nrobe distances frm the plasma<br />
iet. Jn the case where the auencher was 1/7" 5elow the jet the Droduction <strong>of</strong> TaC
0 0 0 0'0<br />
\ D M . S ( C ) h l l l ;<br />
c<br />
3<br />
H c<br />
L<br />
I<br />
\<br />
\<br />
t<br />
?
1'<br />
b<br />
t<br />
+
4<br />
10<br />
I,
323<br />
....... -<br />
. . . . . . . . . . . . . . . ...........<br />
i<br />
. .<br />
. . .<br />
. . . . . . . . . . . . . ................ -., ......................
324<br />
5<br />
Toes u? linearallv with the Kihr/p input. Where the Quenching distance was 5" a<br />
Deak was obtained which shows that adequate quenchinrr does not take place bevond a<br />
value <strong>of</strong> 0.4 for Kwhr/p. Note that in the second case the formation <strong>of</strong> Ta2C also<br />
peaks and then Falls <strong>of</strong>f rapidly, in contrast to the 1/2" distance. Tantalum metal<br />
is the favored product in the 5" case.<br />
and 18% %a for both cases.<br />
Maximum conversions are 24% TaC, 17% Ta2C<br />
The reduction <strong>of</strong> tantalum pentoxide with hvdroEen (12) in 7 helium plasma jet<br />
to produce tantalum metal (reaction A) is shown in Pipure 9. P.@n the ? conversion<br />
is plotted vs. Kwhr/p Ta2O5 input for two different quenching distances. As can be<br />
seen the more rapid quenching (1/2". case) gives the maximum conversion (42%).<br />
which peaks at 0.35 Kwhr/g Tap05.<br />
In a similar manner the reduction <strong>of</strong> tunpsten trioxide was carried out in a<br />
)<br />
'1<br />
\<br />
'<br />
helium plasma jet (17) with the quenching device 5" below the plasma jet. Conversions<br />
as hiKh as 959, were obtained carryinp the tunesten trioxide in hydroaen into the<br />
(<br />
flame <strong>of</strong> the 'et. The reduction <strong>of</strong> other metal oxides has also bean experimentally<br />
investigated 117). Ferric oxide was reduced to iron metal in a 100% conversion '\<br />
using a helium plasma jet and carrvinp the ferric oxide in hvdroEen (reaction 11).<br />
Titanium dioxide and zirconium dioxide reductions were also attempted hv the same<br />
method, however, no reduction was obtained in either case. The reduction <strong>of</strong> aluminum i ,<br />
oxide with hydropen in a helium plasma jet produced only a 2 to 5% conversion to<br />
aluminum metal usinp several different quenching methods.<br />
Pydrocarbons C2H2<br />
LIOUIDIGAS FEACTIONS PRODUCING A GAS<br />
The aroduction <strong>of</strong> ozone b means <strong>of</strong> a plasma jet was accomplished by feeding,<br />
liquid oxyEen into a helium jet (1* v , Figure 10 shows the schematic <strong>of</strong> the apparatus<br />
used and Fipure 11 shows the effect <strong>of</strong> liquid oxypen flow on ozone production under<br />
constant arc conditions. The liquid oxygen acts as both a reactant and quenching mediu<br />
Another example <strong>of</strong> this type reactant is the decomposition <strong>of</strong> hydrocarbons into<br />
acetylene by use <strong>of</strong> a plasma jet device. Thermodynamics Corporation (19) has proved \<br />
the feasibility <strong>of</strong> producinp acetylene from kerosene using a plasma torch. Preliminary<br />
runs gave yields <strong>of</strong> 18% acetylene.<br />
II II<br />
LIOUID-GAS REACTIONS TO PRODUCE A LIQUID<br />
\<br />
1
1<br />
\<br />
i<br />
I i<br />
,<br />
/<br />
/<br />
/<br />
8<br />
3<br />
1<br />
325<br />
To205 +5H2 -2Ta + 5H20<br />
50<br />
40<br />
30<br />
2<br />
0 20<br />
W<br />
><br />
z<br />
0<br />
V<br />
,\" 10<br />
0<br />
IN HELIUM PLASMA JET<br />
5 INCHES<br />
0.1 0.1 0.2 0.3 0.4<br />
KWHR. IC. To2 O5 INPUT
pl
- .-<br />
-<br />
- . - - . . . - .<br />
327<br />
n<br />
+<br />
c<br />
.r<br />
II . J<br />
&<br />
.r<br />
7<br />
.l<br />
3<br />
C<br />
c<br />
LL<br />
z<br />
c<br />
-1<br />
.-.
Under propran carried on at the.Research Institute <strong>of</strong> Temple Uni ersity for<br />
the Clidder. Company the reactions Of terpenes in a plasma jet were studied r ?O). The<br />
reactions shown above were investipated by use <strong>of</strong> the apparatus showed schematically<br />
in ripure 13. A 1 1 experimental data were obtained usinr a helium plasma jet where the<br />
terpene was added in a liquid state into the helium plasma flame. The products were<br />
analyzed by rems <strong>of</strong> chranatipraphic absorption. The most productive synthesis was the<br />
conversion <strong>of</strong> bpinene to mvrcene in 26% yield (reaction 1). Although this is a normal'<br />
Dyrolsis product <strong>of</strong> A-pinene it is the first time that a complicated'molecuje has been \<br />
croducsd. hy yeans <strong>of</strong> a nlasma iet. Other reactions included the preparation <strong>of</strong> limonene,<br />
frorn ,>-ainene in 1% conversion a nd para-cymene<br />
yield fmr? ;y-terninene (reactions 2 to Q).<br />
in 2 1/29. yield and *-pinene in 2 1/2%<br />
SUMMARY -<br />
The cheTica1 reactions discussed above summarize the synthesises that have<br />
been accorolished thus far using a plasma jet device and are by no means all the<br />
synthesises studied using a plasma iet. The plasma jet has shown itself to be a useful'<br />
tool in the area <strong>of</strong> synthesis <strong>of</strong> corpounds and has moved from the preparation <strong>of</strong> simple<br />
materials to more canplex ones recently, With the ever increasing number <strong>of</strong> investiga- \<br />
tions heing carried out by private industry, there is no doubt that the plasna let<br />
will becone a cmnercial <strong>chemical</strong> nmcess device. Its potential has been just touched<br />
and with each new use a whole field <strong>of</strong> investipation is onencd. \.<br />
,
'<br />
POWER<br />
PLASMA GAS<br />
l e , '<br />
I ! I<br />
I<br />
329<br />
- PLASMA JET GENERATOR<br />
j-~ &TERPENE IN<br />
! !<br />
I 1 /- OUENCHING CHAMBER<br />
/ I<br />
.<br />
4<br />
i<br />
1<br />
I<br />
'<br />
:.<br />
-<br />
:<br />
j<br />
:<br />
1- H20 IN<br />
I '<br />
I<br />
I r- ' PLASTIC<br />
I<br />
I<br />
I<br />
1 BAG<br />
+LIQUID<br />
COLLECTOR<br />
I
330<br />
PPTRFKCCS<br />
1. Leutner, 1'. l!. & Stokes, C. c. , Ind. Pnp. Chem. 2 341 (1961)<br />
2.<br />
3.<br />
4.<br />
Damon, F.A. F, thite, D.E., "roposec! "lasma Chemical Processes <strong>of</strong> Interest to the<br />
Petroleurr Industrv, Fenort KO. PLR-115, Plasmadyne Corp. , Santa Pna, Calif.<br />
(Feb. 8, 1962)<br />
I \<br />
Anderson, .l.E. E Case, L.K., I & F.C. Procoss Desipn and Develop. i(1962)<br />
Development and Possible Applications <strong>of</strong> Plasma and Related High Temperature<br />
. Ceneratinp: Uevices, Deport !JPP-l67-!! , Pivision <strong>of</strong> Jhpi.neerinp and Tndustrial \<br />
Research , Hational Pcadeav <strong>of</strong><br />
(PUP. 30, 1960)<br />
5. I,eutner, F.W.<br />
6.<br />
I.<br />
R.<br />
9.<br />
Sciences , Fational Research Council , l?'ashineton. D.C.<br />
I .E F.P., Process Design and Develop. 2 315 (1963)<br />
Crosse , /I..V., Leutner, H.W. &. Stokes, C.S. , Plasma iTet Chemistry, First Pnnual Feport<br />
The Research Institute <strong>of</strong> Tenple University,. Office <strong>of</strong> :!aval Reseerch Contract<br />
K!':!?-3085(02), (Dec. 31, 1961)<br />
DaRon, R.A. & G!hite, D.F., Typical InorFanic and Inorganic Miph Temperature plasma '\<br />
,?et Reactions, Peport ?!o, PLR-119, Plasmadvne Corp. , Santa Ana, Calif. (Sept. 20, 196:<br />
Stokes, C.S. F, Knipe, W.H., Ind. rng. Chem. 52, 287 (1960)<br />
1<br />
Office <strong>of</strong> Naval Research<br />
Grosse, P.V., Stokes, C .S., Cahill, J.A. & Correa, ,T..T., Plasma Jet Chemistry,<br />
Final Feport, The Sesearch Institute <strong>of</strong> Temple Universitv,<br />
Contract NONR-3085(02) ,(dune 30, 1963)<br />
10. Bronfin, D.?. & Hazlett, P.H., I & C.C., Fundamentals 2 472 (1966)<br />
\1<br />
. 11. Freeman, H.P. & Skrivan, ,?.I". , A .1 .Ch.E. Journal 5 450 (1962) c<br />
12. Stokes, C.S. & Cahill, ,T.A., Plasma Jet Chemistry, Final Report, The Research<br />
Institute <strong>of</strong> Temple University, A i r Form Office <strong>of</strong> Scientific Research Grant<br />
775-65, (December, 1965)<br />
13. Leutner, R.W., Ind. Ene. Chem. Process Design &-Develop. 1166 (1962)<br />
I&. Graves, R.D. , Kawa, W. & Hiteshue, R.W. , I t E.C. Process Desi,qn E, Develop.<br />
15.<br />
16.<br />
17.<br />
18.<br />
19.<br />
\<br />
\<br />
\<br />
\<br />
\I<br />
'.<br />
f<br />
s'<br />
59 (1 6<br />
Parmisch, F., Kevmer, C. & Schallus, E., Anaew, Chem. Intemaut.' Edit. 2 238 (1963) \<br />
Carbon-Elack Formation Usinp the Plasma Flame in Yicro-Chemical Reactions, Plasma-Fax,<br />
Bulletin PF-3, Thermal Dynamics Corp. Lebanon, K.H. (Oct. 1960)<br />
k<br />
Stokes, C.S., Cahill, J.A., Correa, J.J. & Grosse, P.V., Dlasma Jet Chemistry, Final<br />
Report, ?he Research Institute <strong>of</strong> Temple University, Air Force Office <strong>of</strong> Scientific<br />
Research, Grant 62-196 (Dec. 1964)<br />
Stokes, C.S. & Strenp, T,.A., I & L.C. Product Research & Develop. 5 36 (1965)<br />
Plasma Flame Used in Formation <strong>of</strong> Acetylene from Kerosene, Plasma-Fax Bulletin<br />
Pr-2, Thermal Dynanic CorD., Lebanon, N.H. (kt. 1960) c<br />
50. Stokes, C.S. & Correa, ,?.J., Terpene Reaction in a Plasma Jet. Final Report. The<br />
Pesearch Institute <strong>of</strong> Temple Univ. The clidden Company (spnsor)(Dec, 19610 \<br />
-<br />
?
331<br />
f'ENI?AL PF'FCLCCFC<br />
Dernis, P.R., qmith, r.D., Cates, n.1'. F. Sond, T.O., Dlasna tTet Technolow<br />
'.ASP CD-5033, "ct. 1965<br />
Stokes, C.C., Chenical Peactinns with the plasma Jet, Chev. Cnp. PDril 17, 1965,<br />
D. 191<br />
tlulburt, I..)!. & Freeman, K.O., Chepjcal "eactions in the Plasma tTet, Trans. V.Y.<br />
Pcad. <strong>of</strong> Sciences E 770 (1963)<br />
5taff Feature, Plasma - Fourth State <strong>of</strong> Vatter, Ind. I'np.. Chem. 55 16 (1963')<br />
$lohn, R.R. t nade, W.L. rlecent Advances in Flectric Arc Dlasna Generator Technology<br />
APS .Taurnal<br />
Creenlee, R.L., '"he Dlasma ,'et in Chemical Processinp. Clyde hrilliams & Co.,<br />
Columbus, Chi0 ("arch 1, 19631, Creenlee, R.E. & Fickley, W.H., Potential<br />
Pcplications for plasma %'et Drocessinp in the petro<strong>chemical</strong> Industry, Clyde<br />
Villiams & CO., Coiumbus, Ohio (April 30, 1963)<br />
Peed, 7 .Pa , Dlasma "orches , International ccience and Technolop, June, 1962, p. 42 '<br />
Parynawski, C.W., Phillips, V.C., Phillips, tT.R. F, I'iester, N.K., Thermodynamic <strong>of</strong><br />
Selected Chenical Systems Potentially Applicable to Plasma .let Synthesis, I & E.C.<br />
Fundmentals 157 (1967)<br />
I
INTRODUCI'ION<br />
332<br />
PRODUCl'ION OF BYDROGEN CYANIDE FROM METHANE I N A NITROGEN PLASMA JET<br />
I. Reactive Species Titration; -her Q,uantitative Studies<br />
Hark P. Freeman<br />
Central Research Division, American Cyanad Co., Stamford, Cmnecticut<br />
The reaction <strong>of</strong> nthane vith a nitrogen plasma to make hydrogen cyanide and acet-<br />
ylene is <strong>of</strong> considerable interest. Conversions are high enough t o be <strong>of</strong> commercial<br />
interest on the one hand,(l,2) vhile the formation <strong>of</strong> H a in particular proceeds in<br />
(1)<br />
(2)<br />
E. M. Hulburt and M. P. Freeman, Trans. N.Y. Acad. Sci., 2, No. 25, 770 (1963).<br />
E. W. Leutner, Ind. Ehg. Chem. Process Design Develop., 2: 315 (1963).<br />
such an interesting and reproducible way that clarification <strong>of</strong> the details <strong>of</strong> the<br />
reaction should considerably advance the use <strong>of</strong> the plesma jet in 8ynthOtiC chew<br />
istry, and further might be expected to contribute rlgniflcantly to baaic chendcal<br />
knowledge.<br />
The forrmlly identical synthesis <strong>of</strong> BCB from "active Ditrogon" aad methane has<br />
been extensively studied(~5) for 'mre than a half century. %at the mysteam are<br />
(3) K. R. Jenninge and J. Y. Llnnett, Quart. Rev. (London) l2, 116 (1958).<br />
(4) G. G. Manella, Chem. Rev. 63, 1 (1963).<br />
(5) 1. E. V. Ewms, C. R. Freeman, and C. A. Winkler, Can. J. Chem. 2, 1271<br />
(1956).<br />
different is apparent, for the high-voltage discharge Is Z high excitation &rice,<br />
whereas the plasma jet is thought to be nearly in local therml equillbrlum .nd<br />
hence a l w excitation device (spectroscopically speaking interredlate betveen 'arc<br />
and spark.(6)) Furthermore, the plasaa jet experiments an perforad at one-half at-<br />
(6) F. A. Kovoler and Yu. K. Kvaratskheli, Opt. Spectr., (IkSR) (Eugllgh Trml.),<br />
- io, 200 (1961).<br />
I
i<br />
J<br />
333<br />
; mosphere (as opposed to about 1 torr) and at an average temperature tventy times<br />
as high on the absolute scale as room temperature, vhere the bulk <strong>of</strong> active nitrogen<br />
experiments have been performed. Finally, in the plasma jet the carbonaceous<br />
species reacting is evidentially not methane. That is, the products other<br />
than HCN are acetylene and higher acetylenes vith various degrees <strong>of</strong> saturation.<br />
These are the same products that would form if the jet vere, say, argon. It has<br />
b been shown elsevhere(7) that the precursors for these products form rapidly compared<br />
I<br />
/ (7) M. P. Freeman and J. F. Skrivau, A.1.Ch.E. (Am. Inst. Chem. Engrs.) J., g,<br />
450 (1962).<br />
I<br />
><br />
,' to the time for mixing <strong>of</strong> the methane vith the jet.<br />
I<br />
,'<br />
There is no question that the plasma Jet provides a difficult environment in which<br />
to do "good vork" in the usual sense. The enonnous temperature gradients and consequent<br />
inhomogeneities are generally thought <strong>of</strong> as being a sort <strong>of</strong> physical <strong>chemical</strong><br />
bar sinister. On the other hand, perhaps because <strong>of</strong> their extreme magnitude, the<br />
i effects due to the temperature gradients are found to be sufficiently reproducible<br />
to allow systematic investigation <strong>of</strong> the jet as a whole, vhich in its hotter parts re-<br />
' presents in a steady state flow situation a <strong>chemical</strong>ly unique environment found only<br />
/ in high intensity arc devices. As ccsnpensation for tackling this difficult environ-<br />
ment, the investigator need not vork with trace quantities, but instead vith partial<br />
pressures and relative conversions at least tvo orders <strong>of</strong> magnitude higher than those<br />
I encountered in active nitrogen research.<br />
) The vork reported here represents a systematic continuation <strong>of</strong> a study performed<br />
some t ime ago.(8) The procedure followed then 8s nov is to add methane through an an-<br />
I<br />
* (8) M. P. Freeman, "The Nature and Quantitative Determination <strong>of</strong> the Reactive<br />
$'<br />
5 /<br />
'<br />
Species In A Nitrogen Plasma Jet," presented at the 147th Natl. Meeting, Amer.<br />
Chem. SOC., April 5-10, 1964, Philadelphia, Penn.<br />
4 calibrated flov rate and a measured heat flow). It has been shovn that under these<br />
\ condition xing is very rapid, as is the drop in temperature deflncd from average<br />
nular slot to a confined nitrogen jet <strong>of</strong> precisely deflned average enthalpy (i.e., a<br />
enthalpy. 17Y Aiter about one millisecond the resulting flow <strong>of</strong> high temperature species<br />
t/ is further chilled by the entrainment <strong>of</strong> cold product gas (the fastest <strong>of</strong> several<br />
\ quench methods investigated on the basis <strong>of</strong> its efficacy in quenching the ammonia<br />
decomposition reaction)(T) and the flov <strong>of</strong> HCN in the product gas is <strong>chemical</strong>ly de-<br />
f termined. Except as noted, the data are taken at 350 2 20 torr chamber pressure as<br />
1 this pressure is found to reduce the formation <strong>of</strong> solid product to an insigniflcant<br />
) level. As the methane is added at various flw rates the corresponding rate <strong>of</strong> production<br />
<strong>of</strong> HCN is noted.<br />
{<br />
'<br />
Just 88 in e.g., Winkler's work vith active nitrogen, a<br />
plateau is observed vhich seem to indicate that some active species is indeed being<br />
titrated (Figure 1). Of utmost interest in'the older study vas the provocatively<br />
simple dependence <strong>of</strong> the plateau level on jet power level (Figure 2). Because this<br />
may in turn be shown to correlate with the rate <strong>of</strong> production <strong>of</strong> ions in the arc<br />
!
335<br />
I I I I I I I I 1<br />
1<br />
0.12 - -<br />
- -<br />
y 0.08 -<br />
1.<br />
Y<br />
.t<br />
- -<br />
0 0<br />
0<br />
-O- -<br />
I I I I I I<br />
0 0.2 0.4<br />
%CH4/G<br />
0.6 0.8 1 .O<br />
?
; it raised the question, still &?zesolved, as to whether the ions might<br />
’ not in some way be responsible for the chemistry. As a direct consequence there is<br />
a long-term quantitative spectroscopic study <strong>of</strong> plasma jets(9) currently in progress<br />
/(9) M. P. Freeman, “A Quantitative Examination <strong>of</strong> the LTE Condition in the Ef-<br />
1 fluent <strong>of</strong> an Atmospheric Pressure Argon Plasma Jet,“ CE-JILA-ONR Symposium<br />
on the “Interdisciplinary Aspects <strong>of</strong> Radiative Energy Transfer,”. Philadelphia,<br />
Penn., Feb. 24-26, 1966. In Press.<br />
/<br />
/<br />
J’<br />
that is expected to ultimately yield information on the nature and quantity <strong>of</strong> ions,<br />
atoms and high temperature molecular species flowing in jets <strong>of</strong> common plasma ma-<br />
terials.<br />
Although thermodynamic calculations had previously been performed for the nitro-<br />
gen-carbon-hydrogen system(l0) they were not in a form easy to compare with plasma<br />
/<br />
‘ (10) C. W. Marynmski, R. C. Phillips, J. C. Phillips, and N. K. Hiester, Ind.<br />
Eng. Chem. Fundamentals, 1, 52 (1962).<br />
/<br />
1 jet results obtained under normal operating constraints. The calculations were there-<br />
; ,fore reproduced(11) with the pertinent parameters varied to conform to the exigencies <strong>of</strong><br />
;I<br />
i<br />
1 (11) B. R. Bronfin, V. N. DiStefano, M. P: Freeman, and R. N. Hazlett, “Thermo-<br />
\<br />
1<br />
<strong>chemical</strong> Equilibrium in the Carbon-Hydrogen-Nitrogen System at Very High Tem-<br />
peratures,” Presented at the 15th CIC Chem. Engr. Conf., Q,uebec City, Quebec,<br />
Oct . 25-27, 1965.<br />
7<br />
j<br />
,/ plasma jet operation. Figures 3 and 4 show the results obtained when solid carbon<br />
is suppressed in the calculations to be consistent with its absence in the observed<br />
I/ products. An important fact immediately emerges. Neither at any experimentally<br />
/ attained average enthalpy, nor at any experimentally used ratio <strong>of</strong> methane to nitrogen,<br />
has the thermodynamically expected yield <strong>of</strong> HCN for a well-mixed system been<br />
(Note that the apparent fall-<strong>of</strong>f in calculated yield at high power levels<br />
/ ~ ~ ~ % methane w flow rates is due to the competitive formation <strong>of</strong> cyano, CN, which<br />
, may be presumed to be an HCN precursor.)<br />
7 The relationship between the observed and calculated equilibrium yield for the<br />
CHb-N2 System is strikingly similar to that recently reported for the F-C-N system by<br />
1 aronfin(l2) who thereupon advanced the plausible hypothesis that in either case the<br />
I<br />
r‘<br />
(12) B. R. Bronfin and R. N. H,azlett, Ind. Eng. Chem. F’undamentals, 5, 472 (1966).<br />
#<br />
i<br />
observed yield is in some way a consequence <strong>of</strong> equilibrium considerations. Now,<br />
from earlier work on the acetylene system(7) it would seem that the composition and
3.0<br />
2.5<br />
2.0<br />
H2'N2<br />
15<br />
3. -<br />
I .o<br />
Q5<br />
'0 02 0.4 0.6 OB u) L2 1.4 16 W u)<br />
Calculated yieldr <strong>of</strong> HCH, .c&yl.no urd 4drg.n rrlatim to<br />
initial nitrogen renilting f'ra tho indicatd nitraeon-uthw rirhP0r.<br />
The methane ir initially aold md the nltr0e.n initi&lly .t tho indicated<br />
onWpy. Solid cubon hu boat mgpro8r.d u a rp.cior in thlB adslatian.<br />
'\<br />
'\<br />
'\<br />
i<br />
?,<br />
r<br />
<<br />
t<br />
r<br />
,<br />
i
p .I<br />
V<br />
I<br />
.=<br />
-<br />
- HCN/C(CALC.<br />
WITH C (SI )<br />
337<br />
100<br />
ENTRANT HEAT FLOW KCALIMOLE Nz<br />
4. Old HCN plateau yield data plotted y~ initial nitrogen enthalpy.<br />
Also shown are the appropriate theoreticilly calculated curves for HClf<br />
(W aquillbrlum), HCH (cubon suppressed) and A at-. The dMhed<br />
line indicates where the stmewhat more scattered experl,mentrl data<br />
would have fallen if plotted y~ 2" reactor cL enthalpy.<br />
HEAD<br />
INTERMEDIATE<br />
SECTION
8<br />
enthalpy dependence are not what one would%xpect for mixing and subsequent freezing<br />
<strong>of</strong> a reaction in a confining tube and further for the shorter reactors the enthalpy<br />
at the exit is still very high. We shall therefore disregard this possibility here.<br />
(Note however, that Bronfln is currently testing this nodel by means <strong>of</strong> computer<br />
(13) B. R. Bronfln, personal cammunication.<br />
I<br />
i<br />
$<br />
I \<br />
simulation.) The expected way in which equilibrium could control the reaction is for<br />
the product distribution to be a function <strong>of</strong> the enthalpy and canposition pr<strong>of</strong>iles<br />
at the <strong>of</strong> the reactor where there is an onset <strong>of</strong> rapid quenching. The calcula- \<br />
tions indicate that composition is only <strong>of</strong> secondary importance in the "plateau"<br />
region so that presumably the enthalpy pr<strong>of</strong>lle would be controlling. This differs<br />
\<br />
from true titration in the important respect that in titration one in effect irre- (<br />
versably consumes a certain potential <strong>of</strong> the nitrogen Jet to form HCH that is clearly<br />
a function only <strong>of</strong> the temperature and/or composition and velocity pr<strong>of</strong>ile in the<br />
\<br />
nitrogen at the point <strong>of</strong> mixing but before mixing occurs. If we a~sumc the velocity \<br />
pr<strong>of</strong>ile <strong>of</strong> a plasma jet is uniquely determined by the enthalpy pr<strong>of</strong>lle,(g) then the<br />
reacting potential as a consequence would in turn be uniquely related to the heat<br />
flow in the gas.<br />
1<br />
Despite this difference, there is no way to distinguish unambiguously between these<br />
two possibilities using but one reactor. This is because fractional heat loss from a<br />
plasma in a particular reactor type has been shown to be primarily a function <strong>of</strong><br />
reactor length and arc unit design,(14) so that the ratio <strong>of</strong> exit heat flow to inlet heat \<br />
(14) J. P. Skrivan and W. VonJaskowaky, Ind. Eng. Chem. Process Design Develop. 4,<br />
371 (1965).<br />
flow is nearly constant. The primary objective <strong>of</strong> the work reported here vu there-<br />
fore to carefully distinguish between these possibilities by performing identical<br />
titration experiments in tvo or =re reactore that differ signiflcantly in length in<br />
order to determine unambiguously whether inlet or exit heat flaw controls the<br />
reaction.<br />
It had been inferred fran the earlier work by rather incomplete evidence that smhow<br />
the capacity to make HCN is primarily dependent on arc conditions no matter hav<br />
far removed the injection point is fran the arc unit itself. A further objective <strong>of</strong><br />
this experiment wat# therefore to systematically check this tentative conclusion by<br />
careful control and variation <strong>of</strong> the injection point on a losg nactor. Ran the<br />
standpoint <strong>of</strong> the titration hypothesis, if the arc unit conditions are indeed controlling,<br />
then a very long-lived reactive species is Implied; such a species is hardly to<br />
be expected under these experimental conditions.<br />
Finally, at the same time the older vork VY being done, Leutner(l5) using a very<br />
short tubular reactor <strong>of</strong> othervise the sam design found he vu able to work at<br />
(15) E. H. kutner, Ind. Eng. men. Procesm Design Develop., 2, 3 5 (1963).<br />
,
1<br />
1<br />
339<br />
atmospheric ressure and achieve a nitrogen fixation <strong>of</strong> 12.5% (erroneously reported<br />
as 21.9WP in a nitrogen (62%)-argon jet (erroneously reported as pure nitrogen)<br />
(16) C. S. Stokes, personal co&unication.<br />
.J with a total flow <strong>of</strong> 5.0 l(STP)/min. and a power flowing in the gas <strong>of</strong> 11.5 kw X<br />
55%(16) = 6.32 kw. It was deemed desireable to reproduce his reactor as nearly as possible<br />
to attempt to see if the two sets <strong>of</strong> results were consistent. Such compari-<br />
'<br />
son will be made in Part I1 <strong>of</strong> this paper, where the effect <strong>of</strong> argon dilution<br />
<strong>of</strong> the plasma will be discussed in some detail. The contribution such a reactor<br />
makes to the present work is <strong>of</strong> course to extend the range <strong>of</strong> reactor lengths studied.<br />
J<br />
EXPERIMENTAL<br />
1<br />
/ Apparatus<br />
i<br />
(<br />
1<br />
3<br />
Plasma-jet reactors consist <strong>of</strong> three parts, head or arc unit, intermediate section,<br />
and quenching section. The plasma-jet heat unit used for this study is a Thermal Dynamics<br />
640 Plasma-jet with "turbulent nitrogen" electrodes, powered by two 12 kw welding<br />
power supplies, open circuit voltage 160 volts, connected in parallel but with<br />
opposite phase rotations on their 3 $ input so as to minimize line frequency ripple in<br />
the output. The intermediate sections (Figure 5) are made <strong>of</strong> copper and are fully<br />
water-cooled, as is the head. The three intermediate sections are themselves modular.<br />
Of length 2". 2", and b", they are mutually compatible and can be joined in any order<br />
to make a reactor <strong>of</strong> length 2, 4, 6, or 8 inches with a feed port at any multiple <strong>of</strong><br />
2 inches. Note that the feed rings are not exactly reproducible in that there are<br />
residual gaps left when the surrounding gaskets are tightly compressed by the joining<br />
threaded parts. The "Leutner reactor" standard Thermal Dynamics spray nozzle with the<br />
solids injection port, which is about l/h" from the nozzle exit, opened up to a 180'<br />
slot.<br />
The spray nozzle and the turbulent nitrogen electrode used with the intermediate<br />
reactors has a 7/32" diameter as do the intermediate reactors. Ins<strong>of</strong>ar as the various<br />
reactor configurations vary only in length and feed point, it w ill suffice to distin-<br />
guish between them with a bracket specifying first the distance from the point <strong>of</strong> heat<br />
balance to the point <strong>of</strong> methane feed, and second the distance from the point <strong>of</strong> heat<br />
balance to the exit <strong>of</strong> the reactor. Thus the 8" reactor fed at the 2" point would be<br />
designated (2"; B"), vhile the Leutner reactor is (-1/4"; 0").<br />
The quenching section where the hot stream <strong>of</strong> plasma and reaction products are quenched<br />
by entrainment <strong>of</strong> cold product gas is simply a stainless steel pot 11 inches long and<br />
11 inches in diameter sparsely wound with soldered-on copper tubing. All parts subject<br />
to heat damage are vell-cooled, but between the windings the pot may get hot enough to<br />
cause flesh bums. At the outlet <strong>of</strong> the quenching section is six feet <strong>of</strong> 1 inch thick<br />
rubberized acid hose. This in turn is fastened to the bottom <strong>of</strong> a vertical mixing section<br />
consisting <strong>of</strong> a three foot long 2" diameter pipe loosely packed with glass wool.<br />
The carbon dioxide is mixed with the product stream at the inlet to the mixing section.<br />
The top <strong>of</strong> the mixing section is connected to a high capacity steam vacuum jet with an<br />
automatic control valve for maintaining desired pressures.<br />
A Toeppler pump is arranged to withdraw 522 ml <strong>of</strong> gas from the top <strong>of</strong> the mixing section<br />
at room temperature and at the reactor pressure. This aliquot is then collected<br />
for analysis in a suitable gas collection system.
340 .<br />
Gas flaws except for methane are metered by orifice gages calibrated by water dis-<br />
1<br />
placement to within 1% for CQp and N2, respectively. Methane flow, much less critical,<br />
is determined by a rotameter calibrated by calculation. All cooling water flows are<br />
i<br />
determined by experimentally calibrated rotameters. Cooling water temperature rise<br />
is determined by suitably graduated, interconsistent mercury thermometers.<br />
Procedure<br />
Heat flaw in the nitrogen plasma at the point <strong>of</strong> methane introduction is determined<br />
by subtracting from the voltage-current product in the arc the heat ldst to all cooling<br />
water supplies up to that point. For the most part, just the heat fldng at the<br />
exit <strong>of</strong> the head is required. For data taken at a particular heat flow an attempt is<br />
made to keep the heat flaw constant. In this endeavor the relatively great intrinsic<br />
stability <strong>of</strong> plasma jets made by this manufacturer help, but especially in runs lasting<br />
for several hours it is necessary to continually introduce small corrections. TNs<br />
control is greatly facilitated thmugh use <strong>of</strong> an analog camputer that continuourly<br />
monitors voltage, current, and cooling water temperature rise and either directly controis<br />
the rectifiers or displayo the net heat flow in killowatts continuously on a recorder<br />
chart so that manual corrections mag be introdvced as needed. Heat levels<br />
given are generally correct to within 25%.<br />
Except where noted, the pressure in the quenching chamber is kept at 350 2 20 torr.<br />
. The actual pressure <strong>of</strong> each sample is known to fl torr but that is not a significant<br />
datum in the analysis and is used only as a consistency check. Quench section pressure<br />
measures intermediate section pressures fairly vell, but probably not the arc<br />
pressures because <strong>of</strong> the pressure drop through the front orifice <strong>of</strong> the plasma jet.<br />
These arc units are not instrumented to measure the pressure inside the head. J<br />
Whenever methane flow rate, paver level and/or pressure conditions are changed, the<br />
system is operated for eight minutes before taking a sample. This is found to be<br />
sufficient time to establish a constant composition.<br />
The collected gas aliquot is slowly bubbled through 200 ml <strong>of</strong> ice cold-caustic con- ,<br />
The half liter space over the caustic is initially<br />
1<br />
taining 12.5 millimoles <strong>of</strong> base.<br />
evacuated so that the entire sample, together vith the air used to flvsh the lines,<br />
might be collected in the caustic and the space over it. This is followed by one minute<br />
<strong>of</strong> Vigorous shaking. This procedure has been found satisfactory for the quantitative<br />
recovery <strong>of</strong> Cog and HCR. Total acid insthe gam is then determinod by back-titration<br />
with 0.500 HC1 until all the carbonate haa been converted to bicarbonate (pH = 8.3). 1<br />
Ammoniacal M is then added aa an indicator and cyanide determined by precipitametflc ,<br />
titration with 0.0100 J silver ion. This permits the initial ratio <strong>of</strong> ECIi to CO2 to<br />
be determined. Because the absolute flaw rate <strong>of</strong> Co2 is known, the absolute production<br />
rate <strong>of</strong> HCN follavs directly. Hote that for convenience in preeentation the flow rate \<br />
<strong>of</strong> HCR is nlwws presented as some fraction <strong>of</strong> 0.383 l(STP)/sec.(0.0171 gram moles<br />
sec-1) so that it mey conveniently be compared to the most <strong>of</strong>ten used flw rate <strong>of</strong> nitrd<br />
RESULTS 341<br />
Composition Dependence<br />
Figure 6 shows typical "titration" curves for the different situations <strong>of</strong> interest.<br />
In every case a plot <strong>of</strong> HCN produced vs. methane added (both normalized to the "standard<br />
flow rate" <strong>of</strong> 0.0171 l(STP)/sec) divides cleanly into two renimes separated by a<br />
break to which we refer as the "equivalence point." To the left <strong>of</strong> the equivalence<br />
1 point, the yield is simply dependent on methane feed rate but not on the power level.<br />
1 Lines Of slope 1/3 and 2/3 are included on the graph to facilitate intercomparison in<br />
'<br />
)<br />
\<br />
this region.<br />
Although the data are too scattered to draw firm conclusions, it is clear<br />
that the break quite generally occurs along the line correspondine to a slope <strong>of</strong> 113.<br />
In this region it is as though the nitrogen were somehow present in excess. To the<br />
right Of the break the HCN throughput is seen to depend only veakly on methane but, as<br />
is demonstrated below, is a strong and simple function <strong>of</strong> heat flow in the nitrogen<br />
before mixing. Excluding for the moment the (-1/4"; 0") data (Figure 6-d), the methane<br />
dependence on the right shows a real linear increase with methane flow rate in<br />
every case, but this slope is not found to be particularly reproducible since it is apt<br />
to change slightly when the apparatus is demounted and reassembled. Hence the scale<br />
has been chosen to emphasize the plateau-like character <strong>of</strong> the curve. Note that the<br />
data excepted may be explained by the fact that in this case the exit rather than the<br />
lentrant heat flow is regulated. Because <strong>of</strong> the small heat loss in this short section<br />
only a second order effect <strong>of</strong> this magnitude would be expected.<br />
Power Level Dependence<br />
To take account <strong>of</strong> the residual slope <strong>of</strong> the "titration" curves, intercomparison <strong>of</strong><br />
power-level-dependence studies is done at the same methane flow rate corresponding to<br />
one half the "standard" flow whether it is an interpolated point taken from a full<br />
titration curve as shown in Figure 6 (filled points) or an isolated measurement (open<br />
points). The best line through the oid data (Figure 2) appears in Figures 7 through 9<br />
as a reference to aid in intercomparison.<br />
Note that Figure 6-a should be exactly con-<br />
sistent with the older data <strong>of</strong> Figure 1 while the data <strong>of</strong> Figure 7-b should exactly<br />
reproduce Figure 2. The extent that they fail to do this is a fair measure <strong>of</strong> the<br />
nonreproducibility <strong>of</strong> this experiment when performed in different laboratories, with<br />
different equipment, vith the <strong>chemical</strong> analyses performed by different persons.<br />
In Figure 7-b data from two different 2" reactors are intermingled as shown. This<br />
is to be compared with the (0"; 8") data <strong>of</strong> Fieure 7-a. It is quite clear that within<br />
the experimental scatter it would indeed be difficult to improve the agreement.<br />
Now the heat leaving the 8" reactor for a given input is only about one half that<br />
leaving a 2" reactor for the same input, so that there can be no question but that the<br />
HCN production depends only on the heat flow at the point <strong>of</strong> mixing (or at the exit Of<br />
the head) and not at all on that at the exit <strong>of</strong> the reactor.<br />
Mixing Point Dependence<br />
Figure 8 demonstrates clearly that the ability <strong>of</strong> the nitrogen to make HCN does not<br />
persist d m the tube at its high initial level, but rather decays as the heat flowing<br />
in the gas decays. Shown are a particular set <strong>of</strong> (6"; 8") data plotted on the left<br />
- VB heat flow at the exit <strong>of</strong> the head, and on the right as a function <strong>of</strong> the heat flow-<br />
ing at the point <strong>of</strong> mixing, the 6" point. Hence that part <strong>of</strong> the earlier work indicating<br />
the existence <strong>of</strong> a "long-lived" reactive species is wrong.<br />
Leutner Reactor<br />
Data for the simulated Leutner reactor are shown in Fi&ure 9. Despite the fact that<br />
these data were taken at atmospheric pressure it is clear that the results are completely<br />
consistent with those <strong>of</strong> the other reactors. This atzrees with an observation<br />
made in the earlier work that there is but slight pressuke dependence for this reaction.<br />
.._ . _,.-. ..<br />
., .... ..~. - ...<br />
'
*<br />
L 1 1 1 1 I I 1 1 I I I<br />
U<br />
-<br />
0.10 1 / 6099 kw. (0";8") -<br />
0.Q) - I /<br />
0.09<br />
0.m<br />
0.07<br />
u<br />
6 0.06<br />
X<br />
.e - 0.05<br />
9<br />
2 0.04<br />
p 0.03<br />
0.02<br />
O.O1 0 L<br />
342<br />
-<br />
QW<br />
1 1 1 1 1<br />
.I<br />
-<br />
METHANE FLOW (hCHJC)<br />
- o '0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 a9 1.0<br />
' ATMOSPHERIC<br />
l/l I I I I 1 1<br />
4770k~(-1/4";0")<br />
PRESSURE 1<br />
- rn A .<br />
-<br />
- " -<br />
1 1 1 1 1 ' 1 1<br />
6. Typical titratiaa curre. for the cue notad. nomd.i;ml productim<br />
rate or HCR V. flow rat. or m e . DMII.~ liar. or ~iape i/3 .nd 2/3<br />
ue inciw~~-?or referonce.<br />
1<br />
0 I 2 3 4 5 6 7 8 9 IO<br />
HEAT FLOll (kw)<br />
-<br />
Y<br />
0.09 I<br />
0.08<br />
0.07<br />
3 0.06<br />
I<br />
'5<br />
0.05<br />
2<br />
0.04<br />
5) 0.03<br />
0.02<br />
0.01 1<br />
-<br />
!<br />
I<br />
~
0.09<br />
0.08<br />
0.07<br />
9 0.06<br />
2<br />
U<br />
-<br />
.$ 0.05<br />
0<br />
0.04<br />
><br />
2<br />
U<br />
I 0.03<br />
0.02<br />
0.01<br />
0<br />
0.13<br />
0.12<br />
0.1 I<br />
- 0.10<br />
W<br />
\<br />
z 0.09<br />
U<br />
0.08<br />
v<br />
2 0.07<br />
ul<br />
;<br />
0.06<br />
'5 0.05<br />
I<br />
0.04<br />
. 0.03<br />
-<br />
-<br />
-<br />
-<br />
-<br />
-<br />
343<br />
1 1 1 1 1 1 1 1 1 1<br />
( - I /4 'I ; 0 'I)<br />
0.09<br />
0.08 -<br />
0.07 -<br />
0.06 -<br />
0.05 -<br />
1 I<br />
(b)<br />
I I 1 I<br />
-<br />
0.04 - -<br />
0.03 - -<br />
0.02 - -<br />
0.01 -<br />
-<br />
0 1 2 3 4 5 6 7 0 1 2 3 4 5 6<br />
ENTRANT HEAT FLOW (kw) , FEED POINT HEAT FLOW (kw)<br />
8. Aorrallzed production rate <strong>of</strong> HCN fo- the eight inch reactor fed<br />
methane at the sFx inch point, (6"; 8") data, v8 heat nm at (a)<br />
the arit <strong>of</strong> the hend end (b) at the 6" feed poGt. Fill.& points<br />
were intarpolated trca full titration curves. The "nomd" line <strong>of</strong><br />
Figure 2 is included in either cam for reference.<br />
n<br />
i<br />
:::: 1 , ', I , I ,' , I , ,<br />
0<br />
0 I 2 3 4 5 6 7 8 9 1011<br />
HEAT FLOW (kw)<br />
9. Romsllxed production rate <strong>of</strong> HClO in the Lautner reactor vs heat<br />
flow at the <strong>of</strong> the reactor. The "noxmd" curve <strong>of</strong> rigur2 is<br />
included for reference.
344<br />
Because <strong>of</strong> the use <strong>of</strong> diluent argon in his vork, comparison with Leutner's results must<br />
await Part I1 <strong>of</strong> this paper which will explicitly treat this complication. OS significance<br />
here is the fact that reactors from 1/4" to 8" in length give the same result,<br />
dependent only on entrant heat flow. (Actually for the 1/4" reactor the exit heat flow<br />
is measured, but presumably in this case there is a nearly negligible difference.)<br />
Product Distribution<br />
Plateau Region - Mass spectrometer checks made on product formed i<br />
the "plateau"<br />
region show 12, H2, HCN, C2H2 and CHI, together with some small quantities 4 <strong>of</strong> higher a<br />
\\<br />
acetylenes to be the only species present to any appreciable extent. The R2 is present<br />
<strong>of</strong> course as a solvent while the H2 is simply the balance <strong>of</strong> the hydrogen. The HCN<br />
is apparently fo2med by some sort <strong>of</strong> irreversible process to be discuased further belov<br />
while the relative amounts <strong>of</strong> methane and acetylene seem to conform to considerations<br />
investigated previously(7) for the cracking <strong>of</strong> methane in an argon jet.<br />
\<br />
\<br />
\<br />
1<br />
Initial Region - To the left <strong>of</strong> the break region where the potential <strong>of</strong> the nitrogen<br />
Jet to react is in excess, one might expect all <strong>of</strong> the methane to be converted to<br />
HCN, i .e. , an initial straight line <strong>of</strong> unit slope, but -such is not the caae. Earlier \<br />
work seemed to favor an initial slope <strong>of</strong> 1/3 for all titration curves. This vould \.<br />
imply that one mole <strong>of</strong> acetylene is formed for each mole <strong>of</strong> HCN produced. At the tima,<br />
however, admittedly crude mass spectrometer checks shoved no more than two-thirds to<br />
three-quarters mole <strong>of</strong> acetylene to each mole <strong>of</strong> HCN. The work reported here indicates<br />
an initial slope corresponding to one-quarter to one half mole <strong>of</strong> acetylene for each<br />
mole <strong>of</strong> HCH. At the breakpoint, however, equimolar quantities <strong>of</strong> Ha and acetylene<br />
would still seem to be the rule. The analytical md sampling apparatuses were neither '<br />
designed for accuracy nor high precision in this low yield region and it is possible<br />
that the differences in the low HCN yield region might have reflected some small change<br />
in analytical procedure. It seema more probable, though, #at this region is indeed -<br />
not reproducible and that product distribution here reflect8 some intangible <strong>of</strong> the<br />
process such as "mixing efficiency", etc. (Note that the reactors are constructed 80<br />
that the widths <strong>of</strong> the slots through vhich the methane ?love art not precisely reproducible.<br />
)<br />
DISCUSSIOB<br />
The Titration Currc<br />
It is clear that the reactor length md hence the heat flw at the onset <strong>of</strong> sudden<br />
quenching is irrelevant; the heat Zlw at the mixing point evidentially gov~ln~ the<br />
extent <strong>of</strong> reaction. It is -her seen that the ability <strong>of</strong> the nitrogen jet to react<br />
vith the methane decreases as the nitrogen flow6 dawn the reactor in such a vay that<br />
its potential to react with methane, at least with this reactor geometry, is a function<br />
only <strong>of</strong> heat flow in the nitrogen before mixing.<br />
Initid Region - Roln a different perspective, with no methane ?laving in a long<br />
tube the reactive potential <strong>of</strong> the nitrogen jet is seen to persist to 8- appreciable<br />
extent almost indefinitely, but &cays an the heat flow decays, pres-4 throw<br />
heat conduction processes. As the methane flow is introduced, 8- <strong>of</strong> this "potentid<br />
to read" (PR for brevity) is now rued up by the methane, vhile the balance decqys by<br />
the heat conduction process. This gives rise to the "initial regIo2l.l'<br />
Equlrslence Point - As the =thane flow is increased, more <strong>of</strong> the PR <strong>of</strong> the nitro-<br />
gen jet is wed by the =thane uatilthe equivrlence point is reached. At this point<br />
nane <strong>of</strong> the PR sunires the mixing <strong>of</strong> the Jet with the methane md the break in the<br />
cUP7C occurs.<br />
~<br />
Plateau Region - Because mixing is not itmtantaneoum at the point <strong>of</strong> mixing, sane<br />
<strong>of</strong> the PR is still used up by heat conduction processes at the equivalence point. k<br />
the =thme flw is further Increased, the equivalent amount rixes closer and claret<br />
I<br />
(<br />
[<br />
\<br />
~
II<br />
\ to the Slot So that less <strong>of</strong> the residual d 2s lost to heat conduction. This latter<br />
process probably gives rise to the observed residual slope in the "plateau reuion" <strong>of</strong><br />
\ the titration CUES.<br />
/<br />
'<br />
5<br />
I<br />
I<br />
/<br />
The Potehtial To React<br />
It remains to propose an explanation for the "PR." Although something is being<br />
titrated, it is by no means clear just what it is. Nor, as <strong>of</strong> the present time, has<br />
anyone experimentally characterized a nitrogen Jet sufficiently well to clearly distinguish<br />
between likely alternatives. Nonetheless, it is instructi& to examine some<br />
<strong>of</strong> these possibilities for their heuristic value.<br />
Heat Balance - From Figure 2 a fairly constant heat requirement <strong>of</strong> 1280 kcalfmole<br />
may be obtained. This corresponds very nicely (and. probably fortuitously) to the<br />
endothermic heat <strong>of</strong> the most probable reaction at 6000°(17):<br />
(17) "Janaf Thermo<strong>chemical</strong> Tables .I1 Dow Chemical Company, Midland, Michigan, Dee-<br />
ember 31, 1960.<br />
b<br />
I<br />
, or that <strong>of</strong> the equally probable reaction at 4500':<br />
f<br />
1<br />
AH = 1281 kcal<br />
AH = 1225 kcal<br />
I From this point <strong>of</strong> view it would appear that the heat <strong>of</strong> the jet is in some way being<br />
> titrated by the methane.<br />
Active Species - One might well ask however, if it is Just the heat being titrated<br />
why docs the reaction apparently stop when the core <strong>of</strong> the Jet is still 4000-6000'K?<br />
And why is the amount <strong>of</strong> cyano (CN) formed so sharply limited and constrained so far<br />
below the equilibrium value? Note that below 4000' strong exothermic reactions occur<br />
that delay further cooling, so that fast quenching cannot be the answer.<br />
The most<br />
ready answer to these questions is that the HCN reaction is far too slow to equilibrate<br />
in Jet residence times. Of the manifold complex <strong>of</strong> forward reaction paths lead-<br />
'ing to equilibrium, only a few <strong>of</strong> them will be fast enough to produce HCN in the time<br />
available. But these fast reaction paths might well involve nitrogenous species which<br />
at thermal equilibrium corresponding to the average enthalpy <strong>of</strong> the jet, would flow in<br />
trace amounts but which, aa a consequence <strong>of</strong> the hot core, are present in the plasma<br />
jet at many orders <strong>of</strong> magnitude higher throughout. Thus we are led naturally to consider<br />
this as the titration <strong>of</strong> some sort <strong>of</strong> especially reactive species in the jet.<br />
Consider for example a Jet producing 0.00171 moles sec-1 <strong>of</strong> HCN. From Figure 2, we<br />
1 I can see that this requires about 9 kw. OS heat flowing in the gas. If we assume there<br />
f ia a small core to the jet at 12,000' (enthalpy at one-half atmosphere = 500 kcal/<br />
L mole),(l8) then the heat flaw can be accolmted for by assuming the hot core occupies<br />
i<br />
1 -<br />
4<br />
t<br />
I (18) F. Martinek, "Thermodynamic and Transport Properties <strong>of</strong> Gases, Liquid and Solids"<br />
I'<br />
J<br />
j<br />
3<br />
J<br />
I<br />
McCraw-Hill Book Co., Inc., New York, N.Y., 1959, p. 130.
about half the dlamcter <strong>of</strong> the jet (one fourth 345 the area) and that the nitrogen flowing /<br />
outside this core carries negligible enthalp . In this calculation is the unproven<br />
assumption carried over from the argon jet(9 J that the mass flux <strong>of</strong> nitrogen is constant (<br />
over the entire cross-section. Under the assumed core conditions the Set is 100%<br />
disssociated and the atoms 17% ianized. Tfie fluxes are then:<br />
atom flow: 1/4 X 2 X (1 - 0.17) X 0.0171 = 0.0071 moles sec-1<br />
ion flov: 1/b X 2 X 0.17 X 0.0171 = 0.00145 moles sec-1<br />
I<br />
The ion flw is thus seen to be in excellent agresment vith the 0.00171 moles sec-1<br />
production rate <strong>of</strong> HCN. To some extent this may be due to a fortunate choice <strong>of</strong> hot<br />
core temperature but is nevertheless a promising possibility.<br />
On the other hand there seems to be no good way to account for the reaction on the<br />
basis <strong>of</strong> nitrogen atoms, in this case present in large excess, suggesting that the<br />
nitrogen atoms are somehow deactivated before the carbonaceous species enters the hot<br />
core. Ins<strong>of</strong>ar M the hydrogen from the dissociation <strong>of</strong> methane must completely flll<br />
the reactor in a very short vhile, it seems probable that tha nitrogen at- u e<br />
deactivated fram this initial inmion, and that the carbonaceous species reacWwith<br />
the nitrogen ions (vhich might well be molecular ions by this time) or aome other<br />
species sometime later.<br />
CONCLUSION<br />
By systematic variation <strong>of</strong> 'reactor geometry it has been conclusively demonstrated<br />
that methane added through a peripheral slot to a nitrogen jet titrates, apparently<br />
in the true meaning <strong>of</strong> the word, some potential <strong>of</strong> the nitrogen Jet to react with the<br />
thermal decomposition products <strong>of</strong> methane to form HCN. It is further demonstrated<br />
that the potential to react is simply related to heat flow even far down the reactor<br />
and is therefore probably the consequence <strong>of</strong> some steady-state temperature and/or<br />
composition pr<strong>of</strong>lle in a flw with local thermal equilibrium.<br />
For their heuristic value, tvo superficially different explanations are proposed to<br />
explain the "potential to react." On the one hand, the experimental endotherm <strong>of</strong> the<br />
reaction at the equivalence point is ehom to be quite consistent vith the heat flov-<br />
ing in the hot core <strong>of</strong> the jet, for a jet model consistent with vhat ve light expect.<br />
On the other hand, for the same heat flov and jet model, the yield is shown to be con-<br />
sistent vfth the flw rate <strong>of</strong> e.g., ions at the point <strong>of</strong> mixing and it may equilly<br />
well be postulated that the ions or some other identifiable species are in fact an<br />
active ingredient being titrated.<br />
It is clearly pointless to attempt to conjecture further on t he mechanism invol-d<br />
in the titration vithout more direct evidence on the species and relocity pr<strong>of</strong>llee in<br />
the jet. A spectroscopic program currently undenqy in there laboratories dl1 pre-<br />
sumably help to flll this gap.<br />
Acknowledgement<br />
Usnp people have contributed in nome vay to the success <strong>of</strong> this work, but in pu-<br />
ticular the author wishes to express his appreciation to Mr. Charles Mentzer<br />
(Chemical Engineering Department, Princeton University) who helped perform some <strong>of</strong> the<br />
experiments , and to Pr<strong>of</strong>essor Hugh Hulburt (Chemical Engineering Department, Iorthvesfern '<br />
UnivCmity) and Dr. B~rrg R. Bronfln (United Nrcraf't <strong>Laboratory</strong>) for their invaluable<br />
di8cunsions and continuing Interest.<br />
1<br />
I<br />
I
i<br />
4<br />
2<br />
HYDROCARBON-NITROGEN REACTIONS IN A TRERMAL INDUCTION PLASMA<br />
Bary R. Bronfin<br />
United Aircraft Research Laboratories<br />
I East Hartford, ConneLticht 06108<br />
I<br />
4, Gas temperatures anove l5,OOO K have been observed in plasma generateddby<br />
radio-frequency induction coupling'. This high thermal erlergy regirre becon?es <strong>of</strong><br />
iqcerest to the chemist for the investigation <strong>of</strong> highly endothermic reactions.<br />
I?. particular, this paper will report reactions between methane and nitrogen fed<br />
to a thermal induction plasma ar,d subsequently quenched to yield hydrogen cyanide,<br />
acetylene and hydrogen.<br />
/<br />
PREVIOUS STUDIES OF THE E-C-N SYSTEM<br />
Lar Pressure Discharges. Winkler and his co-workers2-5 have systematically<br />
studied the reactions <strong>of</strong> nitrogen, activated by passage through a high-voltage<br />
discharge, with various hydrocarbons. These studies were carried out at pressures<br />
near 1 torr with very low reagent flow rates. Gas temperatures were also low,<br />
typically 300 C. In this nonequilibrium system the reaction betw-een methane and<br />
"active" nitrogen yielded hydrogen cyanide, acetylene and hydrogen2.<br />
High Pressure Arcs. More recently high-power thermal arcs were used for the<br />
study <strong>of</strong> the H-C-N system near atmospheric pressure. Leutrer' operated 2 nitrogen<br />
plasma jet into which methane was mixed. In his briefly reported results, up to<br />
LO percent conversion <strong>of</strong> the nitrogen to HCN was found'. -At this symposium<br />
Freeman? has reported an extensive study <strong>of</strong> the synthesis <strong>of</strong> HCN, from EL nitrogen<br />
plasma jet intermixed with methane. Typically, a 7 percent conversion <strong>of</strong> N2 to<br />
HCN was observed. In both studies &, C2IJ2, and unreacted CHI, were also identified<br />
as major constituents in the cooled product stream.<br />
If the maximum mean temperature attainable in these previous studies is cal-<br />
cdated, one finds that enthalpy input was insufficient to generate mean tempern-<br />
f Caes greater than - 5000 K.<br />
\<br />
J<br />
,'<br />
B<br />
d<br />
P<br />
Dissociating Plasmas. Upon considering the various species possible in.the<br />
H-C-N system, the strongest bond found is N E N (226 kcal/g-mole). Thermal-dissociation<br />
<strong>of</strong> N2 is essentiaU.y complete at temperatures in excess <strong>of</strong> 8000 K". Orv.<br />
car. envision a high-pressure, stable plasma fed with any <strong>of</strong> e. variety <strong>of</strong> carbon,<br />
hydrogen, or nitrogen compounds and supplied with sufficient erthalpy to reach<br />
this high temperature range. The constituent species <strong>of</strong> srlch a plasma would be<br />
prinarily atomic nitrogen, atornic hydrogen, and atomic carbon near lo1-( cm-3 concentration.<br />
The result <strong>of</strong> rapidly quenching such! highly reactive atom mixture<br />
nas been generally Lnexplored. &ann and TimminsJ,lO, however, did study the<br />
rapid quenching <strong>of</strong> the simpler atonic-nitrogen/atormc-oxygen mixtures at 10,Oc)O K
348<br />
and 1 atm, which were generated by a constricted d.c. arc. The study which is<br />
reported in this paper was undertaken to explore the <strong>chemical</strong> composition <strong>of</strong><br />
mixtues formed. by the rapid . .quench . <strong>of</strong> atomic-nitragen/atomic-carbcn/atomichydrogen<br />
rnixtures.<br />
PLASMA REACTOR<br />
Radio-frequency Induction Plasma. One convenient approach to achieve the<br />
reqdred temperatures in excess <strong>of</strong> 10,000 K at atmospheric pressure is generation<br />
<strong>of</strong> a thermal plas,m by radio-frequency induction heating. The techniques for<br />
generating and containing a stable high-temperature induction plasma have been<br />
described in detail by Reedljll, Mironeru, Marynowski and Monroe'3, Freeman and<br />
ChaselL, and Thorpel?. Figure 1 presents a diagram <strong>of</strong> the plasma generator used,<br />
A few turns <strong>of</strong> copper tubing were wound around the outside <strong>of</strong> a water-cooled<br />
quartz reactor tube. This coil inductively coupled the supplied r.f. power into<br />
the gaseous reactants sent through the reactor. A water-cooled sampling tube with<br />
very small internal diameter which served to quench rapidly the reactive plasma<br />
species is shown. Efficient cooling allowed the entrance tip <strong>of</strong> the quench tube<br />
tc be placed directly into the plasma core.<br />
A cross-sectional diagram <strong>of</strong> the reactor is presented in Fig. 2. The iso-<br />
therms indicated are those determined spectroscopically by Reed' and' are repre-<br />
sentative <strong>of</strong> conditions in this study. No direct temperatwe measurements were<br />
attempted in the present study.<br />
The Induction Plasma as a Chemical Reactor. Several characteristics <strong>of</strong> this<br />
system can be exploited for <strong>chemical</strong> synthesis:<br />
1. Average temperatures in excess <strong>of</strong> 10,000 K allar essentially complete molecular<br />
dissociation. (Lower power input can reduce the specific enthalpy to<br />
preserve desired free radicals.)<br />
2.<br />
Plasma stability is maintained while operating at very low gas velocities<br />
(
I 1<br />
349<br />
c<br />
M<br />
m<br />
;<br />
a<br />
e,<br />
k<br />
M<br />
x
350<br />
PLASMA COMFOSITION 1<br />
Thermo<strong>chemical</strong> Equilibrium. One <strong>of</strong> the unique features <strong>of</strong> the thermal induction<br />
plasma is stable operation with low gas flow. Since flow velocities in the<br />
plasma zone are typically a few centimeters per second, residence times on the<br />
order <strong>of</strong> a second are associated with plasma species. In a plasma near atmospheric<br />
pressure the particle mean free path is very short and the collislpn rate very high.<br />
At plasma temperatures (c. 10,000 K) gas phase reactions can be expected to proceed<br />
quite rapidly. These facts lead to the initial assumption that local plasma<br />
composition approaches thermodynamic equilibrium.<br />
1<br />
,<br />
\<br />
The H-C-N System. The theoretical compositions <strong>of</strong> hydrogen-carbon-nitro en<br />
mixtures at thermod namic equilibrium have been computed by Kroepelin, et al. f6 ,<br />
Mrynowski, et al.'f, and Bronfin, et a1.l8, for a variety <strong>of</strong> conditions. Computations<br />
were based on free energy minimization using tabulated thermo<strong>chemical</strong> datalg.<br />
Figure 3 presents calculated equilibrium cornposition data for a typical stoichiometry:<br />
H:C:N = 4:1:2, (CH4/N2 = l), at 1/2 atm (380 torr). Only species whose concentration<br />
is greater than 1.0 mole-percent are shown on this plot; however, twentyone<br />
different <strong>chemical</strong> species were considered. In Fig. 3a, full equilibrium was<br />
considered for a two-phase system which included graphite. Figure 3b shows that<br />
temperature segment which is altered by the exclusion <strong>of</strong> the solid-phase, C(s).<br />
Molecular nitrogen, N2, is a major constituent over a broad range up to 8000 K;<br />
thereafter thermal dissociation results in the predominance <strong>of</strong> atomic nitrogen, N.<br />
As mentioned above, at temperatures over 8000 K, not shown <strong>of</strong> the graph, the system<br />
becomes completely dissociated into a simple three-component atomic state: H, C, N.<br />
At temperatures greater than 7000 K significant thermal ionization occurs, gener-<br />
ating significant concentrations <strong>of</strong> singly-ionized atoms, e.g., C , H , N'. As<br />
noted in both plots, CHq dissociation is well underway at 1000 K, resulting in the<br />
formation <strong>of</strong> I+ and C(s) in the two-phase system (a); but the formation <strong>of</strong> HCN and<br />
C2& in the single-phase system (b). At temperatures above 3000 K, Q% begins to<br />
fragment to QH and H, and with increasing temperature, to the atomic species. At<br />
temperatures above 3800 K, HCN begins to fragment to CN and H, and with increasing<br />
temperature, to the atomic species. Nitrogen-hydrogen species, e.g., NIQ, .occur<br />
at concentrations below 0.01 mole-percent over the entire range plotted.<br />
'\<br />
i<br />
'I<br />
'<br />
'\-<br />
+ + I<br />
In comparing the common temperature segments <strong>of</strong> the single-phase and two-phase<br />
composition diagrams, important differences are noted in that: (1) higher concentrations<br />
<strong>of</strong> species like HCN and C2& ar~ preserved at lower temperatures in the<br />
single-phase case, and (2) the maximum concentration <strong>of</strong> these species is somewhat<br />
higher in the single-phase case. Hence in the lower temperature range, the pre-<br />
dlcted yleld <strong>of</strong> HCN and QIQ is enhanced by retarding carbon nucleation. This .; ,<br />
'<br />
kinetic limitation acts to freeze the mixture composition at around 2700 K so that<br />
negligible composition change is predicted over a fairly broad temperature interval,<br />
down to 1500 K.<br />
Composition <strong>of</strong> the Plasma. No direct determination <strong>of</strong> the plasma composition<br />
was attempted in the present study. Only a few efforts in this direction have 1<br />
,<br />
1<br />
\<br />
'<br />
\<br />
\<br />
\<br />
)<br />
'<br />
1<br />
P<br />
5<br />
i<br />
1<br />
i
1.0<br />
I<br />
- a.<br />
I<br />
n<br />
TEMPERATURE - OK<br />
Figure 3 Equilibrium conposition <strong>of</strong> equimolar CH&-N* mixture. (a) Including solid carbon; (b) Excluding solid<br />
carbor.. Region above T, common.<br />
I<br />
0<br />
70<br />
60<br />
x)<br />
40<br />
30<br />
20<br />
IO<br />
NITROGEN GONVCRSK)N<br />
0 10-1 2 5 100 2 5 IO' 2 5<br />
P41'IW -<br />
-<br />
Fig'xe 4 yieid cf HCI:, expressed as percent Pi2 converted, as a function <strong>of</strong> input stoichiometry. Solid lines<br />
shov .win-- yield predicted by thermodynaniic equilibriwn. Lata points shov experimental results<br />
frx. the plas-a reactcr: ~r flow rate 42 std cc/sec,. total reagent flow rate 2 std cc/sec, net<br />
pmer - 3.5 kv. Reactor pressure identified with key. Points labeled x from plasma Jet studies6J7.
352<br />
appeared in the literature. O'Halloran, et al?', have directly sampled an argon<br />
plasma jet using a specially designed entrance cone opening to a time-<strong>of</strong>-flight<br />
mass spectrometer. Raisen, et a1.21J have made spectroscopic identifications <strong>of</strong><br />
species in an air plasma. Unlike the mass spectrometer measurements, however,<br />
emission spectroscopy 1s difficult to quantify due to the wide variance in the<br />
oscillator-strengths <strong>of</strong> the likely emitters.<br />
I<br />
Faced with the difficulty <strong>of</strong> determining plasma composition directly, one is<br />
prone to apply equilibrium predictions as 8 guide to local plasma composition.<br />
Referring both to Figs. 2 and 3, a highly dissociated composition can be expected<br />
in most <strong>of</strong> the plasm region. Temperatures in excess <strong>of</strong> 10,000 K, expected over<br />
most <strong>of</strong> the central region <strong>of</strong> the plasma, would dictate that the atomic species<br />
H, C, and N, and their ions would predominate.<br />
QUENCHING<br />
At this juncture it is important to ask what Changes in composition would be<br />
encountered on cooling the €I-C-N atom plasma. A slow, gradual cooling <strong>of</strong> the<br />
labile intermediates resident in the plasma zone may well allow the system to<br />
revert to the original reactants along an equilibrium path.<br />
Kroepelin and Kippid2<br />
found tNs effect in their study <strong>of</strong> a 10 amp, 40 volt d.c. arc burning in 8 hydro-<br />
carbon-nitrogen atmosphere. The composition <strong>of</strong> the slowly cooled arc-heated gases<br />
was found to be mainly w, &, and C1 and C2 hydrocarbons. No HCN was observed.<br />
Under extremely rapid cooling, however, kinetic limitations can interpose along<br />
the reaction path to yield different, uwre interesting or valuable products.<br />
The Cold-Wall Tube. A small-diameter, water-cooled tube was selected from<br />
the variaty <strong>of</strong> available high-cooling-rate devices, to provide rapid and convenient<br />
quenching <strong>of</strong> the plasm species. Figure lb shows a diagram <strong>of</strong> the simple design<br />
<strong>of</strong> the three-concentric tube arrangement <strong>of</strong> the quenching probe23. The overall<br />
outside diameter <strong>of</strong> the probe was 318 in. The internal diameter <strong>of</strong> the quenching<br />
tube which receives the hot plasma was 0.032 in. The surfaces <strong>of</strong> the inner tube<br />
were stainless steel; other parts were fabricated from copper. The effect <strong>of</strong> a<br />
variation in the composition <strong>of</strong> the cold surface waa not studied.<br />
Cooling Rate. Due to the Ngh rate <strong>of</strong> heat transfer from plasma to adjacent<br />
cold wall, rapid cooling occurs in the quenching tube. Freeman and ~ltrivan~~,~5<br />
have measured an initial temperature decay rate <strong>of</strong> 5 x 107 Vsec in water-cooled<br />
tubes. In their model <strong>of</strong> the heat-transfer process occurring in very smaU tubes,<br />
Anmann and Timminsm have calculated cooling rates greater than lo9 Vsec. *obably<br />
quenching rates in this range were associated with the quenching tube used<br />
in. this study.<br />
Reaction Path During Quench. Unfortunately, there is a sparsity <strong>of</strong> hightemperature<br />
kinetic data for the H-C-N system. Hence one is unable to predict with<br />
certainty the reaction path followed as the atomic species H, C, and N, are cooled<br />
from 15,000 K to 500 K in 10 11tI~iBeCOnd6, for example. After an examination <strong>of</strong>
353<br />
the experimental results in the succeeding sections, it may be possible to infer<br />
th important steps in the reaction sequence.<br />
EXPERIMENTAL CONDITIONS<br />
Radio-frequency SuppQ, A commercially 12 kw (n&mind) induction<br />
heater, oscillating at 4 mHz, was the pmer source for the experiment. The load<br />
coil, shown in Fig. 1, consisted <strong>of</strong> five turns <strong>of</strong> 1/4-in. o.d., water-cooled, copper<br />
tubing wound tightly around the quartz reaction tube.<br />
coil was 1 1/2 in., with a central diameter <strong>of</strong> 3 in.<br />
The overall height <strong>of</strong> the<br />
Reactor. Containment <strong>of</strong> the plasma was accomplished within a 35 cm long,<br />
47 mm 1.d. quartz tube, mounted vertically. The high-power loadings necessitated<br />
cooling which was afforded by flowing l/3 gpm <strong>of</strong> water into a cooling jacket surrounding<br />
the central reaction tube. The plasma-forming gases were premixed and<br />
sent into the tube through a brass fitting sealed onto the tube base. 0a6 flow<br />
rates were monitored with calibrated rotameters, Plasma-heated gases left the<br />
reaction tube through a second brass fitting sealed onto the top <strong>of</strong> the tube. This<br />
cap, which WBB <strong>of</strong> approximately 1 liter volume, was water-cooled. In this configusation<br />
the top fitting functioned as a cooling chamber to reduce the average<br />
gas temperature to within a few degrees <strong>of</strong> ambient. The system pressure was controlled<br />
by a high-capacity regulated vacuum line attached to the upper cap.<br />
A sliding O-ring seal was provided at the center <strong>of</strong> the upper cap for positioning<br />
<strong>of</strong> the 3/8-in. 0.d. by O.032-ln. i.db quench probe along the central axis<br />
<strong>of</strong> the reaction tube. Cooling water was supplied to the quench probe at a metered<br />
rate <strong>of</strong> l/3 gpm. The entrance tip <strong>of</strong> the probe WBB typically located at the center<br />
<strong>of</strong> the uppermost winding <strong>of</strong> the load coil.<br />
The flow rate through each cooling water line was metered with calibrated<br />
rotamet.era wNch were placed downstream <strong>of</strong> liquid pressure regulators. Interconsistent<br />
mercury thermometers were mounted at each cooling water line inlet and<br />
outlet.<br />
total enthalpy delivery rates could be determined.<br />
& measurement <strong>of</strong> the temperature rise and flow rate in each cooling line,<br />
Chemical Analysi.8. The gae stream withdrawn through the quench probe wae sent<br />
to an on-line gas chromatograph for quantitative analysie. A triacetin column,<br />
recommen ed by IsbeU27, followed by a molecular sieve column, recommended by<br />
Purnel128, was ueed for reeolution <strong>of</strong> chromatogram peaks. This configuration<br />
allowed the detection <strong>of</strong> the following compounds with a thermal conductivity cell:<br />
HCN, I@, Ar, N2, CQJ and various higher hydrocarbons. A parallel flame-ionization<br />
detector was useful for detecting low concentrations <strong>of</strong> wdrocarbons, e,g., Cq,<br />
CzQ.<br />
Experimental Variables. Total pressure in the plasma reactor was typically<br />
380 torr; some data were also acquired in a range from 160 to 760 torr. For the<br />
data reported here the argon feed rate was 42 std cm3/sec (6.5 g-mole/hr).<br />
This
354<br />
flow rate insured good plasma stability for the available r.f. power and remained<br />
with the capacity <strong>of</strong> the subatmospheric pressure regulating equipment. Added<br />
reagent gases were fed at 1150th to l/lOth that rate, with the molal ratio varied<br />
over a broad range: 0.1 5 CH4/N2 5 25. The power coupled into the gas mixtures<br />
was -3.5 kw maximum, as determined from the summation <strong>of</strong> cooling water heating<br />
rates.<br />
I<br />
In the later stages <strong>of</strong> the experiment various nitrogen-substituted hydrocarbon<br />
liquids were fed to the argon plasma, namely: CH3CN, CH3C&CN, and CQCHCN. Special<br />
modifications to the gas-flow system were made to insure injection <strong>of</strong> these<br />
nltriles into the plasma. A part <strong>of</strong> the argon feed stream was split <strong>of</strong>f to a<br />
sealed gas-liquid bubbler containing warmed reagent. Since these compounds are all<br />
relatively volatile , a substantial mount <strong>of</strong> nitrile entrainment occurred. Rather<br />
than introduce the nitrile-laden argon stream into the relatively cool gas region<br />
at the base <strong>of</strong> the reaction tube, a special injection probe was provided to introduce<br />
the stream into the hot plasma region. This second water-cooled probe was<br />
positioned through an O-ring seal in the base cap. The probe design was identical<br />
to that previously described for quenching (cf. fig. lb). The tip <strong>of</strong> the injector<br />
probe was placed at the center <strong>of</strong> the lareramst winding <strong>of</strong> the load coil.<br />
rial exiting the injector thus was assured <strong>of</strong> entering the plasma zone.<br />
kte-<br />
EXPERIMENTAI, RESULTS<br />
Plasma Stabilitx. Over the entire range <strong>of</strong> experimental. variables the plasma<br />
was stable and brightly luminous. The luminous region <strong>of</strong> the plasma appeared to<br />
fill about 90 percent <strong>of</strong> the cross-sectional area <strong>of</strong> the cooled quartz tube and<br />
extended from the bottom to about 3 in. above the r.f. load coil. With high hydrocarbon<br />
fluu rates a gradual build-up <strong>of</strong> soot occurred on the quartz tube walls.<br />
Methane-Nitrogen Reactions<br />
Products <strong>of</strong> Reaction. A quantitative analysis <strong>of</strong> the gas stream withdrawn<br />
through the quenching probe was made for each run. Over the entire range <strong>of</strong><br />
stolchlometric studies, the methane fed to the plasma wea completely reacted; no<br />
methane was detected in the 'quenched .gas stream. The following major reaction<br />
products were identified: HCN, C2&, 9. Also present were Ar and unreacted 5.<br />
Nitrogen Conversion. Since essentially complete conversion <strong>of</strong> methane was<br />
observed for each run, the varying conversion <strong>of</strong> nitrogen was selected 88 an important<br />
datum. If the mole fraction <strong>of</strong> a species i found in the quenched product<br />
atream is defined ea xi , then the conversion <strong>of</strong> nitrogen, defined BB a is given<br />
bY :
,)<br />
355<br />
The ceaswed conversion cf ni;rsgc.n xa-ied from 3 psrcent at the lowest<br />
stoichionetry cocsidered, CFU.+/Id2 = j.1, 'io I niaximum <strong>of</strong> 70 percent at a methanerick,<br />
stc' -b.~lo:.-.ctr;, -;.. CH4/32 = 2j. Phis higii vaiuc <strong>of</strong> iiitrogen fixation is considerablj<br />
1:: excess <strong>of</strong> prs-,.ioGsly reported nitrogen conversion levels observed in<br />
therclai piasna reactions. Prcsswe variaiions had no significant effect.<br />
Qpi?al Product Conpcsition. Tabie I, below, presents the composition <strong>of</strong> the<br />
prcd..ct stream produced under typicel conditions.<br />
Teble I<br />
Typical Product Composition<br />
Heactor Pressure - 3S0 torr Net Power Input - 3.5 kw<br />
Feed Rate Feed Composition<br />
InpLt (std cc/sec) (mole-percent)<br />
97.6<br />
1.2<br />
1.2<br />
-<br />
Total 42.8 100.0<br />
Composition<br />
(mole-percent 1 (mole-percent, &-free)<br />
90.5<br />
2.0a<br />
1.0<br />
0.3<br />
0.2<br />
trace<br />
-<br />
-<br />
57<br />
29<br />
9<br />
5<br />
-<br />
-<br />
190 .0 100<br />
:;cte e: Ccnpositioiis dcterzi:ied fri:n gas chrcmatogra:r,i; - + 10 percent.<br />
As wzs seen in Fig. h, Table I shcws that about l2 percent <strong>of</strong> :!,e nitrogel1 fed at<br />
tbls stoici:icnetry vas corlverted to hydrogen cyanide. Ilinety-nine percent <strong>of</strong> the<br />
,?!ethane fed was convertei :o the gaseoiis prccl.icts hydrogen and acetylene and also
..<br />
to an unmeasured small qusntity <strong>of</strong> solid product which deposited on the wall.<br />
The minor disperity between the nitrogen and carbon material balances indicates<br />
the small fraction <strong>of</strong> material lost from the reaction zone to deposition on the<br />
reactor walls.<br />
Nitrile Reactions<br />
Products <strong>of</strong> Reaction. Sdl quantities <strong>of</strong> nitriles could be continuously fed<br />
to the argon plasma by using the modified injection arrangement (described in the<br />
previous section).<br />
cooled probe was analyzed with no change in the gas chromatograph. Essentially<br />
Mixed plasma which was quenched by withdrawal through the water- ,<br />
complete conversion <strong>of</strong> the lower molecular weight compounds , acetonitrile , CH3CN,<br />
and acrylonitrile, C&CHCN, was observed. A minor amount (-10 percent) <strong>of</strong> propionitrile,<br />
CH3C+CN, was observed in the quenched gas stream, perhaps due to a<br />
bypassing <strong>of</strong> the plasma core. Each <strong>of</strong> the nitriles produced the same reaction<br />
products, a repeat <strong>of</strong> those produced in the methape-nitrogen study. High concentrations<br />
<strong>of</strong> HCN, Q+, I@, and N;! were observed dong with the argon diluent. A<br />
't<br />
'<br />
complete quantitative <strong>chemical</strong> analysis <strong>of</strong> these nitrile runs has not, as yet, been<br />
performed.<br />
1<br />
DISCUSSION OF RESULTS<br />
Variance in the Conversion Percentage. Considerable variance in the f'raction<br />
<strong>of</strong> nitrogen converted was observed among repetitions <strong>of</strong> runs with constant stoichiometry,<br />
net power input, and pressure. The data points plotted in Fig. 4 include<br />
the maximum observed conversion levels for each stoichiometry. Sources <strong>of</strong> error<br />
in the experimental procedure which could have scattered the data darn from maximum<br />
conversion were :<br />
1.<br />
2.<br />
3.<br />
Improper adjustment <strong>of</strong> r.f. supply controls so 88 to mismatch the supply and t<br />
plasma impedance, perhaps generating plasma irregularities<br />
Minor air leaks which would add additional nitrogen to the plaema to altec<br />
the stoichiometry from preset values<br />
Deposition <strong>of</strong> carbon or hydrocarbon material on the reactor walls or, the<br />
reverse, significant vaporization <strong>of</strong> carbon from the walls, to alter the pre-$ ,<br />
set stoichiometry.<br />
I<br />
i<br />
Thermo<strong>chemical</strong> Equilibrium Predictions. As described in the "Plasma Composi-<br />
tion" section, above, a full range <strong>of</strong> composition calculations were made assuming<br />
thermo<strong>chemical</strong> equilibrium. These calculations predicted that the plasma composi- '<br />
tion within the r.f. coil- would be dominated by atomic species for the specific<br />
power input used in this study. Perhaps the following additional consideration Of<br />
these theoretical predictions can elucidate the processes occurring during rapid ~<br />
quench. 'I<br />
i<br />
',<br />
I<br />
\<br />
I<br />
\<br />
\<br />
l,<br />
<<br />
,<br />
\<br />
\, i<br />
I<br />
i<br />
\<br />
i c
1<br />
6<br />
357<br />
Freezing. 3jpotnesise thzf eqxi1ii.ri.m is followed du.ri!ig the initial stages<br />
I'\<br />
cf coccling withir, the quench t.&e. For he~xistic pill'poses,, assaw that equilib-<br />
I ,.i.... - k.. IS cbeyed dwing te:nperat:ce decay tc\ epprosicately 3300 K, or more specifi-<br />
I<br />
-,. -G~L;, tc that temperat-re vh5r-e ?qKilibri,x:i predicts the maximum number <strong>of</strong> moles<br />
cf FCII tc be famed.<br />
P<br />
Then assux tiiat eli reactions involving nitrogen species<br />
, are froze:: at that point, sc chat even as ccoling continues negligible decomposi-<br />
1 tio!? I;f the occurs.<br />
i Chemical Reaction Kicetics. In :w,kiiig this series <strong>of</strong> seemingly unwarranted<br />
ass.mptions, sone specific reacCiori retes have been implied to be very fast, while<br />
, .otiisrs, very slow. Focuskg C.!I nitrzgen-containing species, the fcllowing types<br />
cf reactio:. nave been ass;;risd tc be rapid iinaer the conditions developed in the '<br />
1 ..<br />
Q',ik:.x:iLng tube:<br />
I<br />
\<br />
Y<br />
i<br />
1<br />
i<br />
14' + e- M<br />
Further, the following have been assumed to be very slow:<br />
HCD; - many steps - €I! + N2 + C (5)<br />
To date a full description <strong>of</strong> the reaction kinetics <strong>of</strong> the H-C-N system, even<br />
at moderate temperatures, is unavailable. In the recent shock tube study <strong>of</strong><br />
hhrshall, Jeffers, and Bauer29, preliminary results indicate that the equilibrium<br />
in Reaction 4 may be achieved rapidly at elevated temperatures. However, the<br />
evidence points to a rather complex reactior. mechanism for the thermal dissociation<br />
<strong>of</strong> HCN, wherein many more steps are involved than mentioned here. The cornplexity<br />
<strong>of</strong> this system was encodntererl in the earlier study <strong>of</strong> Robertson and<br />
Pease3', and iil similar systems explored by Goy, Shaw and Pritchard3l.<br />
Rapid dissociation/recombination rates have been determined for nitrogen,<br />
ReactiOn 3, by Wray3*. Reactisn rate data is not available for Reacclon 2, nor<br />
for otker reactions likely to be quite important in the H-C-8 system.<br />
Atomic-ion/electror. recoxbination reactions for each <strong>of</strong> the three atoms in<br />
the plasma are likely to be rapid33 relative to the quenching time.<br />
In surnniary, existing reaction rate data is fa- from sicfficient to check the<br />
assu-,pticns made abcut the reaction niectanisms apprcpriate to this H-C-N system.<br />
kdiiti onal kinetic studies which generate presently unl.own reaction rates are<br />
needed before an accurate reaction path cari be predicted theoretically for the<br />
reacting H-C-N system.<br />
I
358<br />
Ccxposition at the Assumed Freezing Point. The validity <strong>of</strong> this freezing<br />
approacn can be found comparing the predicted composition at the freezing temperature<br />
witc the sbserved composition <strong>of</strong> tne quznched gas stream. The follcwing<br />
variaoles are defined for Table 11, whlcn facilitates the comparison.<br />
For the equilibrium calculatians let the input stoichiometry be charac-<br />
terized by the molar ratio CHq] /[N2 , set equal to + ; let nN21be the<br />
number <strong>of</strong> moles <strong>of</strong> N;1 intro6uced; ani let n: be the number <strong>of</strong> moles OP<br />
species i present at equilibrium 2t a pressure, P, and a temperature, T .<br />
Then Tf is establisned for a given and P, as the temperature at which<br />
equilibrium predicts the quantity nlCN/nN2 is a maximum.<br />
Table I1 shows the mole fraction <strong>of</strong> the predominant species for a Tf value<br />
found in an equimolar input stoichiometry. As mentioned in the "Plasma Composition"<br />
section, above, solid-carbon formation may be retarded in this system. Therefore,<br />
a second set <strong>of</strong> mole fraction data have been presented in Table I1 which<br />
excludes the species C(s) from the calculation.<br />
Table I1<br />
Calculated Equilibrium Composition at the Temperature<br />
where HCN Yield is Maximized<br />
Pressure - 380 torr - 4 P [CHJ+]/ [Np] = 1.0<br />
x2<br />
*e<br />
HC N<br />
0.421<br />
0.252<br />
0.097<br />
others 0.030<br />
Including C( s)<br />
Mole Fractions<br />
0.543<br />
0.283<br />
0.109<br />
0.038. 0.065<br />
0.144 -<br />
0.020<br />
0.497<br />
0.266<br />
0.120<br />
0.073<br />
0.025<br />
0.005<br />
Note a: Adjusted to allow for the reactions: C2H + H - C2%, 2H - h.<br />
0.522<br />
0.274<br />
0.124<br />
0.080<br />
-
359<br />
\ Frozen Compositior: Calcillation v5rsus Expcri;:icntally. 0bscrvt.d Ccwposition.<br />
IC, indeed, the oriSinal assuption that important reactiotis occurrirg in the gas<br />
'<br />
withdrawn from the plasma into the cold quenching tube are .frozen.in ths vicinity<br />
1 1 ,<strong>of</strong> 3000 K is, valid, then agree'ment should be found betveeri the cc::!pssitions listed<br />
in Tables I and 11. Ir. comparing the argon-fret coinposition listed in Table I with<br />
PO~UIM 2 in Table 11, rciatively $cod agrement is found.<br />
This comparison has been extended to thc' ikll ranee <strong>of</strong> stsichiomctries studied.<br />
i)n Fig. 4, the maximum nitrogen'conversion, amar-l , and am0,-2, has been plotted<br />
' against input CH)+/N2-molc ratio, 4,. The s.dlscript 1 refers to calculations includ-<br />
'<br />
ing C(s); subscript 2, to excludine, C(s). Agrceinunt between the freezing mcdel and<br />
crbserved results is very good over more than a two-decade range in stoichiometry<br />
variation. These results strongly support .the freezing model in the overall reac-<br />
;<br />
tion path. With the existing scatter <strong>of</strong> the data, a delineation between the<br />
single-phase and two-phase equilibrium predictions <strong>of</strong> composition at Tf is not<br />
possible. Additional sxperimental runs may lessen this ambiguity. The retarda-<br />
/ tion <strong>of</strong> carbon solid fcrmation, however, accoiints fur Less than a 10 percent change<br />
in predicted maximum nitrogen conversion, an the average.<br />
Freezing Temperature. The data do not allow an t.xact determination <strong>of</strong> a frcczing<br />
temperature appropriate to the quenching process. keferring back to Fig. 3a, a<br />
change <strong>of</strong> a few hundred degrees in the assumed Tf could account for the data points<br />
which indicate 10 to 20 percent less than the two-phase equilibrium predictions <strong>of</strong><br />
maximum nitrogen conversion. Fig. 3b shows the HCN concentration plateaus to be<br />
quite broad. Hence the nitrogen conversion prediction would be relatively insensitive<br />
to variation in freezing temperature over the range from 1500 to 3000 K.<br />
Future experimental refinenents may allow a mvre exact determination <strong>of</strong> Tf .<br />
Observed Compositions in Nitrile Experiments. An analysis <strong>of</strong> the maximum<br />
nitrugen convcrsion predicted by tiiernochenical equilibrium has not been nlade fi.r<br />
the different nitrile inputs desckibed in tne "Expcrirnental Conditions'' zectiuri.<br />
The similarity cjf .product ciistri!iiition and uf the rxtio <strong>of</strong>. HCN to !Q in the prcduct<br />
poitito to the same reaction mechanisms as iii tile CiU+/l\I2 studics.<br />
Correlatiuri with Studies by Other Investigators<br />
If the proposed reaction mechanism is correct, then the yields found in other<br />
studies <strong>of</strong> the synthesis <strong>of</strong> HCN by thermal reactions <strong>of</strong> hydrocarbons with riitrogeri<br />
should not exceed the predicted values <strong>of</strong> a . Some results <strong>of</strong> the production <strong>of</strong><br />
HCN in plasma jets have been reported, as was mentioned in the "fieviolis Studies"<br />
section. While the differences in design between the plasma jet and induction<br />
plasma reactors are not detailed here, sample results from these other studies are<br />
plotted on Fig. 4. The maximum nitrogen conversion vdues observed 'in the nitrogen<br />
plasma jet experiments <strong>of</strong> Leutnerll (designated XL) and FreemanE (designated xF )<br />
appear. These experimental data points are seen to lie below the maximum nitrogen<br />
conversion prediction.<br />
I
360<br />
The phenomena ir. t:te pias::.& ::et experiments which convert less nitrogen than<br />
the ,;axinurn pred3ct.Yd 6). the ?'re:czilig rnuuel my well be l.ir,ked to a [nixing or diff'L;sian<br />
rate (CHq a:. i 32 were !~dt prcfliixta) or' tc temperatures 4nsur'ficient to reach<br />
Thi;.qsesticn is ~ i ~ sub,).:et l : 12 a separ-te theoretical analysis now in progress<br />
Tf .<br />
by the author .<br />
THE REACTION SEQUENCE<br />
The experimental data support the fallowing description <strong>of</strong> the overall reac-<br />
tion seqdence:<br />
i. Plasma Reactic;;w. ' Feed reagerits. are dissociated to their atomic constitu-<br />
ents in a therim, plasma. Some Tthermai ioiiizatiQn accurs at sufficiently<br />
high ternperatu.-s.<br />
CF& i 92 - c + H + N + C+ + H+ + N+ + e-<br />
2. Initial Coohnt Plasma taken inco a small-diameter cold tube rapidly<br />
cools witn <strong>chemical</strong> reactio:is following equlibriwn.<br />
C + H + N + ions - H + CIV + C2H + N2 - HCN + @ + N2 + C2w<br />
3. Frozen Reaction Emetics. Rapid cooling continues, but at a rate much<br />
greater than the progress <strong>of</strong> the apparently complex reaction sequence<br />
necessary to des%roy trle species HCN, C2H2, and %. Hence the composi-<br />
tion <strong>of</strong> the cooiliig gas is frozen at the end <strong>of</strong> Step.2.<br />
Evidence fyr the Freezing llbdel in Other Reacting Systems. The freezing model ,<br />
likely is applicable to a wide variety <strong>of</strong> reacting thermal plasma systems which '<br />
emplcy a rapid quench. hmnann and. Timiinsl5 found a mechanism involving the freezin&<br />
<strong>of</strong> equilibratiag reactiuns 'ta be applicable to their study <strong>of</strong> the quenching <strong>of</strong> \<br />
nitrogen-oxygetl plasmc. Ahed by a wealth <strong>of</strong> published rate data on <strong>chemical</strong> reactiotis<br />
in air, they were ~ble to model thc time-temperature-composition history <strong>of</strong><br />
M-0 plasma cooling within mail-diameter tubes. A quite similar freezing tempera- 1<br />
ture, Tf , was found at 35Oi;'K 1'c.r that system. The equilibrium composition <strong>of</strong> nitric<br />
uxi?e, NO, at Tf was preserved during continued rapid coolfng.<br />
\<br />
CONCLUSIONS ' . , . . \<br />
Mixtures <strong>of</strong> mechane and nitrogen can be fed continuously to a thermal argon<br />
indluctiori plasma maintaified abcve 13,300 K. .A rapid quer.ch <strong>of</strong> the heated plasma<br />
kxture produces HCN, C2@, anc &.<br />
the nitrogen converted, range between 3 and 70 percent, a function <strong>of</strong> the input<br />
,<br />
Final yields <strong>of</strong> HCN, expressed'as a fraction Of I<br />
stoicbiometry . i<br />
1<br />
I<br />
'';<br />
<<br />
ti<br />
c
I<br />
1<br />
\<br />
361<br />
-.<br />
ne reaction proceecis 1.; t:ie :cap;ccc intion on <strong>of</strong> the feed reactants in<br />
-rj ..._ pias:% to yield a mixt,ae cf 3, C, ani 3. As this nixt.xe is quenched, the<br />
rsectia:: initially procee5s 2;cr.g ar. ey;llibri,.it? pth, for1:;ir.g HCN, CZiI2, H2: and<br />
*.4,-,. EC~GW the tenperat33y whey- cli? equ:-ibriL;:. :field OS 'TCN is maximized, the<br />
;?owcr r-;lrra:;gclrient reacsions etlci carto:. ::x ieatiori are f'rozen by the rapid cool-<br />
;:.&, s- tha: negligible citeraticn in the des cc!:ipositic!-. occurs upon further<br />
c 15.<br />
I<br />
i<br />
11.<br />
12.<br />
/ 13.<br />
P. R. hm&nn and R. S. Timnlns, A.1.Ch.E. J., 17, 956 (1966).<br />
T. B. Reed, J. Appl. Phys., &, 3146 (1963).<br />
A. Mironer, A.I.A.A. J., L, 2638 (i963).<br />
C. W. IWynuwski and A. G. Mc mJe, "R-f Generation <strong>of</strong> Thermal Plasmas," in<br />
Sigh Teaperatue Technolcgy ( .
14.<br />
15.<br />
Lc .<br />
17.<br />
18.<br />
19 *<br />
20.<br />
21.<br />
22.<br />
23<br />
24.<br />
25.<br />
26.<br />
27.<br />
362<br />
y. P. Freeman and J. D. Chase, "Energ- Transfer Plechanism <strong>of</strong> and Typical<br />
Gperarink Cnarazteristics for thi. T!it.riial Radio-Frequency Piasma Generator, "<br />
Paper No. 216, 133th Meeting, TiIc EJectrxhaniical Society, Oct. 12, 1966,<br />
Philadelphia, Pa.<br />
M. L. Thorpe, Research/Devel~~:;ient, 2 (L), 28 (Jan. 1966). ,<br />
I<br />
H. Kroepelin, D. E. Kipping, and H. Pietruck, "Equilibrium Partial Pressures<br />
in the Systems C + H2 + N2 and C + Ii;! .+ iJH3 ar Temperatures from io00 to<br />
I2000 K,," in J. F. Masi and D. H. Tsai, eds., Progress in International<br />
Research on .Thermodynamic anii 1'rni:sport Properties, Proceedings <strong>of</strong> 2nd Symposiu?<br />
on Thermcphysical Properties , Jan. 24-26, 1962, Princeton, N. J.<br />
(Amer. SOC. Mech. Eng./Acade:nic PI'css, N. Y., 1962), pp. 626-48.<br />
C. W. Marynowski, et al., Ind. El)&. Chen. Fund., 1, 52 (1962).<br />
B. R. Bronfin, et al., "Therrric<strong>chemical</strong> Equilibrium in the Carbon-IIydrogen-<br />
Nitr.ogen System at Very High Temperatures, " 15th Chemical Institute <strong>of</strong><br />
Canada Chemical Engineering Conference, Oct. 25-27, 1965, Universitf Laval,<br />
Quebec.<br />
JANAF Tables <strong>of</strong> Thermo<strong>chemical</strong> Data, Dow Chemical Co., Midland, Mich., Dec. 31,<br />
1964.<br />
G. J. O'Halloron, et al., "Determination <strong>of</strong> Chemical Species prevalent in a<br />
Plasna Jet, " Report No. ASD-Tm-62-644, Bendix Corp. Research Leboratories,<br />
Southfield, fich., 1964.<br />
E. Reisen, et al., Appl. Spectr., 2, 41 (1965).<br />
H. Kroepelin and D. E. Kipping, "Teniperature Distribution in an Electric Arc<br />
Operated ir! B Iiydrocar.bon-Nit1.0221) Atmosphere," in J. F. Masi and D. H. Tsai,<br />
e&., Prorress in International Research on Thermodynamic and Transport Prop-<br />
-, erties Proceedings <strong>of</strong> 2nd Symposium on Thermophysical Properties, Jan. 24-26,<br />
1962, Princeton, N.<br />
pp. 649-55.<br />
J. (Amer. SOC. Mech. Eng./Academic Press, N. Y., 1962),<br />
M. N. Piooster and T. B. Reed, J. Chem. Pliys., s, 66 (1959).<br />
M. P. Freenaq and J. F. Skivan, A.1.Ch.E. J., 8, 450 (1962).<br />
J. F. Skrivan and W. Von Jaskowsky-, Ind. Eng. Chem. Proc. Design Devel., I,<br />
371 (1965)-<br />
Lepel High Frequency Labs, Inc., New York, N. Y., Model T-5-3.<br />
R. E. Isbeil, Anal. Chem., 2, 255 (1963).
\ 36 3<br />
'<br />
h,<br />
I<br />
26. H. Purnell, Gas Chronatogaphy (John Wiley, N. Y., 1962) pp. 371-3.<br />
29. D. ;&shall, P. Jeffers, S. H. Ba,Jer, Chenistry Dept., Cornell Univ., Ithaca,<br />
M. Y., personal comnvAnicaticn, NOV., 1966.<br />
33. 1:. c. Rcbertson and R. N. Pease,.J. Amer. Chem. Soc., &, 1880 (1942).<br />
31. C. A. Goy, D. H. Shaw, arid H. 0. Pritchard, J. Phys. Chein., 9, 1504 (1965).<br />
, 32. K. L. Wray, Res. Rept. 104, Avco Everett Res. 'Lab., Everett, Mass. (June,<br />
1961).<br />
J<br />
,<br />
33. D. R. Bates, A. E. Kingstcn, and'R. W. P. McWhirter, hoc. Roy. Soc. A., 3,<br />
, 297 (1962).
THE CONVERSION OF i.iEEIHAiW IN AN ARC REACTOR<br />
P. R. Anunann*<br />
R. S. Timmons<br />
V. Krukonis<br />
The conversion <strong>of</strong> hydrocarbons into other products, namely acetylene,<br />
has been <strong>of</strong> considerable commercial and academic interest during the past<br />
fzw decades. Several types <strong>of</strong> arc devices have been reported for carrying<br />
out ?;he pyrolysis <strong>of</strong> methane.<br />
Thermodynanic calculations show that if the acetylene is to be re-<br />
covered r'rori an equilibrium <strong>chemical</strong> mixture, high temperatures in the<br />
range <strong>of</strong> 2000 to 35000K are required, depending on the carbon-hydrogen<br />
ratio and the pressure <strong>of</strong> the system. Furthermore, kinetic considerations<br />
indicate that the pyrolysis <strong>of</strong> methane is fast relative to the decomposi-<br />
tion <strong>of</strong> acetylene, and that there is an optimum reaction time to maximum<br />
both the conversion <strong>of</strong> the methane and the yield <strong>of</strong> acetylene.<br />
The most desireable accomplishment <strong>of</strong> high temperature methane pyroly-<br />
sis would be to effect the endothermic reaction.<br />
2CH4 = C2H2 + 3 H2<br />
High temperature <strong>chemical</strong> equilibria for the carbon-hydrogen system have been<br />
computed by Blanchetl and Bauer and Duff2. Assuming that <strong>chemical</strong> equilibrium<br />
is achieved in an electric arc reactor, the data indicate that the temperature<br />
at which the acetylene concentration is a maximum. The kinetics <strong>of</strong> methane<br />
pyrolysis and acetylene decomposition during heating and cooling ultimately<br />
determine the yield <strong>of</strong> acetylene.<br />
Anderson and Case3 developed a kinetic model to describe the reactions<br />
between arc heated hydrogen and methane. ' Calculations provided information<br />
on conditions, such as He/CHI+ ratio, and reaction time, to achieve optimum<br />
conversion <strong>of</strong> methane to acetylene. The analyses indicated that conversion<br />
is not sensitive to contact time, within 0.1 and 1.3 milliseconds, and that<br />
speed <strong>of</strong> mixing is the most important parameter.<br />
Reactions <strong>of</strong> methane and hydrogen with carbon vapor produced by an electric<br />
arc were reported by Baddour and Blanchetl. In their experiments, high<br />
acetylene concentrations <strong>of</strong> 20 to 40 percent in the product gas were found.<br />
In an arc process, the reactions which determine the success <strong>of</strong> the process<br />
occur during the heating step and also during the cooling step. Heating<br />
<strong>of</strong> the reactant may proceed either as it passes through the discharge, or<br />
when it mixed with arc-heated gases. Cooling <strong>of</strong> the high temperature gases<br />
is usually carried out quickly, either by contact with cold surfacestop mix-<br />
ing with cold fluids.<br />
This paper describes an experimental study on the con-<br />
version <strong>of</strong> methane which either (1) passed through a confined arc discharge,<br />
or (2) was mixed with an arc-heated gas. In this investigation, an attempt<br />
was made to understand the factors which affect the extent <strong>of</strong> methane pyro-<br />
lysis ana the yield <strong>of</strong> acetylene.<br />
-c Avco Space Systems Division, 201 Lowell Street, Wilmington, Mass. 01887
In experiments on the direct conversion <strong>of</strong> methane, the reactant was<br />
psssed through a confined arc discharge and quenched by mixing hydrogen or<br />
helium at several different quench to methane flow ratios. The net enthalpy<br />
Of the gas from the arc discharge varied between 78 and 150 kca$ per gram<br />
mole which correspond to equilibrium temperatures <strong>of</strong> 2500 to 33000K. The<br />
conversion <strong>of</strong> methane, that was observed, varied from 65 percent at the low<br />
enthalpy to 85-100 percent at the high enthalpy. With a helium quench, the<br />
methane conversion varied between 90 and 98 percent, but at low He/CH4 ratios,<br />
a large fraction <strong>of</strong> the methane went to carbon black, due principally to lower<br />
quenching rates and greater acetylene decomposition. Methane conversion<br />
was slightly lower with a hydrogen quench and the latter probably affected<br />
the kinetics <strong>of</strong> methane decomposition.<br />
The conversion <strong>of</strong> methane by mixing with arc-heated hydrogen was studied<br />
in a plug-flow type reactor. Hydrogen was passed through a confined arc discharge.<br />
Islethane was mixed with the hydrogen leaving the arc zone, and heated<br />
and reacted as the gases flowed co-currently through a cooled pipe., The conversion<br />
<strong>of</strong> the methane was correlated with the enthalpy <strong>of</strong> the hydrogen arc<br />
gas. Conversions were low (15-20 percent) at low enthlpies, and were nearly<br />
complete at high enthalpies (go-100 percent). These energies corresponded to<br />
50-60 kcal per mole <strong>of</strong> methane at the low enthalpies to 220 kcal at high values,<br />
and were very Large for the degree <strong>of</strong> methane reaction. Two effects<br />
were important. The arc gas lost energy to the cool reactor walls and the<br />
rate <strong>of</strong> energy transfer decreased with time due to a decrease in the energy<br />
driving force. By allowing for energy losses, the conversion was correlated<br />
and fair agreement was obtained between the data from (1) cracking methane in<br />
the arc discharge, (2) cracking methane in an arc-heated hydrogen stream, and<br />
(3) the experbents <strong>of</strong> Anderson and Casel.<br />
The yield <strong>of</strong> acetylene was also<br />
observed to be a function <strong>of</strong> the available energy, and under sane conditions,<br />
acetylene yields <strong>of</strong> go to 100 percent& theoretical were achieved.<br />
References<br />
1. Blanchet, J. L.,"Reactions <strong>of</strong> Carbon Vapor with Hydrogen and with Methane<br />
in a High Intensity Arc, " Sc. D. Thesis, M.I.T., June, 1963.<br />
2.<br />
Bauer, S. H., and Duff, R. E., The Equilibrium Composition <strong>of</strong> the C/H<br />
System At Elevated Texperatures, LA-2556, Los Alamos Scientific Labora-<br />
tory, Los Alamos, N. M., September 18, 1961.<br />
3. Anderson, J. E., and L. K. Case, An Analytical Approach to Plasma Torch<br />
Chemistry, 1, 3, 161-165 (1962).
_r<br />
333<br />
THERMAL CRACKING OF LOW-TEMPERATURE<br />
LIGNITE PITCH<br />
John S. Berber, Richard L. Rice, and Delmar R. Fortney '<br />
U. S. Department <strong>of</strong> the Interior, Bureau <strong>of</strong> Mines,<br />
Morgantown Coal Research Center, Morgantown, West Virginia<br />
INTRODUCTION<br />
The tar used in this study was produced by the Texas Power & Light Company<br />
from a Texas lignite carbonized at about 500" C in a fluidized bed. The<br />
pitch, as used in this research program, is the tar distillation residue boiling<br />
above 350" C. This pitch is about 40 to 50 percent <strong>of</strong> the crude tar. Pitch is a<br />
complex resinous mass <strong>of</strong> polymerized and polycondcnsed compounds (1). It is<br />
an amorphous solid material and quite brittle at room temperature. Tce pitch<br />
analyses average 84 percent carbon, 8 percent hydrogen, 5 percent oxygen, and<br />
1 percent each oi nitrogen and sulfur. It is <strong>chemical</strong>ly similar to the tar from<br />
which it is prepared, .being mainly mixtures <strong>of</strong> the higher homologs <strong>of</strong> the compounds<br />
contained in the distillable fractions <strong>of</strong> the tar.<br />
The pitch is potentially useful as a binder in the manufacture <strong>of</strong> products<br />
such as ro<strong>of</strong>ing cement, metallurgical electrodes, asphalt paving, and pitch fibre<br />
pipe, if its characteristics can be modified by physical and <strong>chemical</strong> techniques to<br />
approximate those ol. asphalt and bituminous binders (2).<br />
Researchers <strong>of</strong> the Bureau <strong>of</strong> Mines, U. S. Department <strong>of</strong> the Inter.ior, have<br />
investigated three methods for changing the lignite pitch characteristics.<br />
Air-blowing, which is used successfully to treat bituminous pitch by lower-<br />
ing the hydrogen content and increasing the s<strong>of</strong>tening point and the penetrability,<br />
was not too effective with lignite pitch (1, 2).<br />
--<br />
Catalytic dehydrogenation (a) was found to be effective in reducing the<br />
hydrogen content <strong>of</strong> the pitch, but catalyst cost per pound <strong>of</strong> treated pitch is high,<br />
and the catalyst recovery cost would be prohibitive.<br />
Thermal cracking has proven to be the most effective in changing the pitch<br />
characteristics. This' report covers the preliminary work done on thermal cracking<br />
<strong>of</strong> pitch and shows the variety <strong>of</strong> products that may be obtained.<br />
Thermal cracking is widely used in the petroleum industry, particularly for<br />
the production <strong>of</strong> olefins. Thermal cracking has also been used in the treatment<br />
<strong>of</strong> coal tar.(A, ?), but seldom has been used in the treatment <strong>of</strong> pitch. The successful<br />
treatment <strong>of</strong> pitch by this process could upgrade the pitch into metallurgical<br />
coke, binders, oxidation feedstock for phthalic and maleic anhydrides, and gases<br />
such as hydrogen, methane, and ethylene.<br />
The binding ability <strong>of</strong> the cracked pitch and oil distillation.pitch can be<br />
determined by the quality <strong>of</strong> metallurgical electrodes produced using the pitches<br />
as binders. Many factors affect the binding properties' <strong>of</strong> the materials being
367<br />
used as blnders, with different producers and consumers using entirely different<br />
standards, so the electrode quality tests were used to help evaluate the effective-<br />
ness <strong>of</strong> thermal cracking <strong>of</strong> the lignite pitch.<br />
EQUIPMENT<br />
The thermal cracking system is shown in figures 1 and 2. The feed tank<br />
(figure 2, A) is made from a 13-in. length <strong>of</strong> 10-in. diameter schedule 40 pipe,<br />
to which a cone has been added for the bottom. The tank is heated by commercial<br />
electric heaters. The feed pump (B) is a small gear pump with a variable speed<br />
drive. The thermocracker (D) is a 4-1/2 ft-length <strong>of</strong> 2-1/2-in. diameter schedule<br />
40, type 304, stainless-steel pipe, heated by external electric heaters. The<br />
heaters are controlled by outside surface thermocouples welded to the reactor.<br />
The receiver (E) is a 20-in. length <strong>of</strong> 4-in. diameter schedule 40 pipe. The<br />
receiver is flanged so that it can be bolted to the bottom <strong>of</strong> the reactor. The condenser<br />
(F') is a 30-in. length <strong>of</strong> 6-in. diameter pipe, swaged at the bottom end to<br />
I-in. diameter and flanged at the top. The top flange supports a water-cooled<br />
tube which inserts into the condenser body in the manner <strong>of</strong> a cold finger. The<br />
knockout (G) is a 34-in. length <strong>of</strong> a 2-in. diameter pipe having a tangential gas<br />
inlet about 8-in. from the bottom. The scrubber (H) is a 30-in. length <strong>of</strong> a 4-in.<br />
diameter pipe having a water-spray nozzle near the top. The water from the<br />
scrubber drains into a standpipe water-seal tank (I). The water-seal tank is a<br />
piece <strong>of</strong> 6-in. dlameter pipe. The gas meter (J) is a laboratory-type wet-test<br />
meter capable <strong>of</strong> metering 1,000 cu ft <strong>of</strong> gas at standard conditions.<br />
PR OC E DURE<br />
Lignite pitch at 200" C was pumped from the feed tank through electrically<br />
heated lines into the top <strong>of</strong> the thermocracker. The temperature <strong>of</strong> the thermocracker<br />
was maintained at approximately 790" C as indicated by the outside<br />
surface thermocouples. At the end <strong>of</strong> a run, the flow <strong>of</strong> pitch was stopped and the<br />
pump was' flushed with a low-boiling tar fraction to keep the impeller <strong>of</strong> the pump<br />
from freezing. Reactions within the cracker produced coke, cracked pitch, oil,<br />
and gas. Both the top and bottom <strong>of</strong> the cracker were removed, and coke was<br />
removed from the sides and weighed. Cracked pitch caught in the receiver was<br />
weighed and analyzed. The oil removed by the condenser and knockout chamber<br />
was weighed and then distilled to 400" C, leaving a pitch residue. This pitch<br />
residue was analyzed and, if found to have the desired carbon-to-hydrogen atomic<br />
ratio (1. 20 to 1.80), was used as a binder for electrodes. Distillate from the oil<br />
may be oxidized to phthalic and maleic anhydrides or separated into acids, bases,<br />
and neutral oils. Gas from the cracker was cooled and scrubbed, then metered,<br />
sampled, and analyzed by gas chromatography.<br />
RESULTS AND DISCUSSION<br />
Results <strong>of</strong> the thermal cracking tests are given in tables 1 to 4. Of the four<br />
major products obtained from the pitch--coke, cracked pitch, oil, and gas--the<br />
coke averaged about 15 to 25 percent <strong>of</strong> the feed to the cracker, the cracked pitch<br />
amounted to about 20 to 40 percent, the oil was about 20 to 30 percent, while the<br />
balance <strong>of</strong> the feed to the cracker consisted <strong>of</strong> gas.
Distribution and yield rates <strong>of</strong> products (figures 3 to 6) are as expected considering<br />
the reaction within the cracker at different crude,pitch.feed rates. As<br />
the pitch is heated it begins to vaporize, and the turbulenc'e <strong>of</strong> the vapors splashes<br />
some <strong>of</strong> the iluid onto the hot wall where it sticks and is coked. If enough heat is<br />
available, the rest <strong>of</strong> the pitch is vaporized and cracked. ' As the cracked<br />
materiais leave the hot zone, heavy high-b
369<br />
'<br />
i<br />
1.<br />
REFERENCES<br />
Berber, John S., and Richard L. Rice. Oxidation <strong>of</strong> a Low-Temperature<br />
Lignite Tar Pitch. Preprints, Div. <strong>of</strong> Fuel Chemistry, Am. Chem. Soc.,<br />
v. 8, No. 3, Aug. 31-Sept. 3, 1964, pp. 145-151.<br />
'<br />
,<br />
2. Chelton, H. M., R. N. Traxler, and J. W. Romberg. Oxidized Asphalts in a<br />
Vertical Pilot Plant. Ind. and Eng. Chem., v. 51, No. 11, dovember 1959,<br />
pp. 1353-1354.<br />
I<br />
' 3. Greenhow, E. J., and Calina Sugowdz. The Chemical Composition <strong>of</strong> Coal-<br />
I Tar Pitch. Coal Research in CSIRO, No. 15, November 1961, pp. 10-19.<br />
' 4. Montgomery, R. S., D. L. Decker, and J. C. Mackey. Ethylene and<br />
Aromatics by Carbonization <strong>of</strong> Lignite. hd. and Eng. Chem., v. 51, No. 10,<br />
October 1959, pp. 1293-1296.<br />
I'<br />
1<br />
I<br />
i<br />
J<br />
I 'I<br />
\<br />
J<br />
5. Naugle, B. W., C. Ortuglio, L. Mafrica, and D. E. Wolfson. Steam-<br />
Fluidized Low- Temperature Carbonization <strong>of</strong> High Splint Bed Coal and Thermal<br />
Cracking <strong>of</strong> the Tar Vapors in a Fluidized Bed. BuMines Rept. <strong>of</strong> Inv. 6625,<br />
1965, 22 pp.<br />
6. Rice, Richard L., and John S. Berber. Catalytic Dehydrogenation <strong>of</strong> Low-<br />
Temperature Lignite Pitch. Presentation, Div. <strong>of</strong> Fuel Chemistry, Am. Chem.<br />
Soc., March 22-31, 1966.
367<br />
used as blnders, with different producers and consumers using entirely different<br />
standards, so the electrode quality tests were used to help evaluate the effective-<br />
ness <strong>of</strong> thermal cracking <strong>of</strong> the lignite pitch.<br />
EQUIPMENT<br />
The thermal cracking system is shown in figures 1 and 2. The feed tank<br />
(figure 2, A) is made from a 13-in. length <strong>of</strong> 10-in. diameter schedule 40 pipe,<br />
to which a cone has been added for the bottom. The tank is heated by commercial<br />
electric heaters. The feed pump (B) is a small gear pump with a variable speed<br />
drive. The thermocracker (D) is a 4-1/2 ft-length <strong>of</strong> 2-1/2-in. diameter schedule<br />
40, type 304, stainless-steel pipe, heated by external electric heaters. The<br />
heaters are controlled by outside surface thermocouples welded to the reactor.<br />
The receiver (E) is a 20-in. length <strong>of</strong> 4-in. diameter schedule 40 pipe. The<br />
receiver is flanged so that it can be bolted to the bottom <strong>of</strong> the reactor. The condenser<br />
(F') is a 30-in. length <strong>of</strong> 6-in. diameter pipe, swaged at the bottom end to<br />
I-in. diameter and flanged at the top. The top flange supports a water-cooled<br />
tube which inserts into the condenser body in the manner <strong>of</strong> a cold finger. The<br />
knockout (G) is a 34-in. length <strong>of</strong> a 2-in. diameter pipe having a tangential gas<br />
inlet about 8-in. from the bottom. The scrubber (H) is a 30-in. length <strong>of</strong> a 4-in.<br />
diameter pipe having a water-spray nozzle near the top. The water from the<br />
scrubber drains into a standpipe water-seal tank (I). The water-seal tank is a<br />
piece <strong>of</strong> 6-in. dlameter pipe. The gas meter (J) is a laboratory-type wet-test<br />
meter capable <strong>of</strong> metering 1,000 cu ft <strong>of</strong> gas at standard conditions.<br />
PR OC E DURE<br />
Lignite pitch at 200" C was pumped from the feed tank through electrically<br />
heated lines into the top <strong>of</strong> the thermocracker. The temperature <strong>of</strong> the thermocracker<br />
was maintained at approximately 790" C as indicated by the outside<br />
surface thermocouples. At the end <strong>of</strong> a run, the flow <strong>of</strong> pitch was stopped and the<br />
pump was' flushed with a low-boiling tar fraction to keep the impeller <strong>of</strong> the pump<br />
from freezing. Reactions within the cracker produced coke, cracked pitch, oil,<br />
and gas. Both the top and bottom <strong>of</strong> the cracker were removed, and coke was<br />
removed from the sides and weighed. Cracked pitch caught in the receiver was<br />
weighed and analyzed. The oil removed by the condenser and knockout chamber<br />
was weighed and then distilled to 400" C, leaving a pitch residue. This pitch<br />
residue was analyzed and, if found to have the desired carbon-to-hydrogen atomic<br />
ratio (1. 20 to 1.80), was used as a binder for electrodes. Distillate from the oil<br />
may be oxidized to phthalic and maleic anhydrides or separated into acids, bases,<br />
and neutral oils. Gas from the cracker was cooled and scrubbed, then metered,<br />
sampled, and analyzed by gas chromatography.<br />
RESULTS AND DISCUSSION<br />
Results <strong>of</strong> the thermal cracking tests are given in tables 1 to 4. Of the four<br />
major products obtained from the pitch--coke, cracked pitch, oil, and gas--the<br />
coke averaged about 15 to 25 percent <strong>of</strong> the feed to the cracker, the cracked pitch<br />
amounted to about 20 to 40 percent, the oil was about 20 to 30 percent, while the<br />
balance <strong>of</strong> the feed to the cracker consisted <strong>of</strong> gas.
Distribution and yield rates <strong>of</strong> products (figures 3 to 6) are as expected considering<br />
the reaction within the cracker at different crude,pitch.feed rates. As<br />
the pitch is heated it begins to vaporize, and the turbulenc'e <strong>of</strong> the vapors splashes<br />
some <strong>of</strong> the iluid onto the hot wall where it sticks and is coked. If enough heat is<br />
available, the rest <strong>of</strong> the pitch is vaporized and cracked. ' As the cracked<br />
materiais leave the hot zone, heavy high-b
369<br />
'<br />
i<br />
1.<br />
REFERENCES<br />
Berber, John S., and Richard L. Rice. Oxidation <strong>of</strong> a Low-Temperature<br />
Lignite Tar Pitch. Preprints, Div. <strong>of</strong> Fuel Chemistry, Am. Chem. Soc.,<br />
v. 8, No. 3, Aug. 31-Sept. 3, 1964, pp. 145-151.<br />
'<br />
,<br />
2. Chelton, H. M., R. N. Traxler, and J. W. Romberg. Oxidized Asphalts in a<br />
Vertical Pilot Plant. Ind. and Eng. Chem., v. 51, No. 11, dovember 1959,<br />
pp. 1353-1354.<br />
I<br />
' 3. Greenhow, E. J., and Calina Sugowdz. The Chemical Composition <strong>of</strong> Coal-<br />
I Tar Pitch. Coal Research in CSIRO, No. 15, November 1961, pp. 10-19.<br />
' 4. Montgomery, R. S., D. L. Decker, and J. C. Mackey. Ethylene and<br />
Aromatics by Carbonization <strong>of</strong> Lignite. hd. and Eng. Chem., v. 51, No. 10,<br />
October 1959, pp. 1293-1296.<br />
I'<br />
1<br />
I<br />
i<br />
J<br />
I 'I<br />
\<br />
J<br />
5. Naugle, B. W., C. Ortuglio, L. Mafrica, and D. E. Wolfson. Steam-<br />
Fluidized Low- Temperature Carbonization <strong>of</strong> High Splint Bed Coal and Thermal<br />
Cracking <strong>of</strong> the Tar Vapors in a Fluidized Bed. BuMines Rept. <strong>of</strong> Inv. 6625,<br />
1965, 22 pp.<br />
6. Rice, Richard L., and John S. Berber. Catalytic Dehydrogenation <strong>of</strong> Low-<br />
Temperature Lignite Pitch. Presentation, Div. <strong>of</strong> Fuel Chemistry, Am. Chem.<br />
Soc., March 22-31, 1966.
. .<br />
I<br />
. I<br />
370<br />
. . . .<br />
., . . ..<br />
. .<br />
'',..,' , :.,<br />
. . ..<br />
. .<br />
i<br />
1<br />
't
I<br />
7<br />
/*<br />
i<br />
,I<br />
{<br />
\<br />
J<br />
7<br />
1/<br />
r-'<br />
I<br />
Ia Jd - I<br />
PI -------<br />
I<br />
I
4<br />
372<br />
090-<br />
> E w - m c<br />
N O - 0<br />
z : 0000<br />
. . . .<br />
L o<br />
3
I'<br />
p'<br />
/<br />
!<br />
2'<br />
9'<br />
c<br />
u<br />
.r( Y<br />
a<br />
e,<br />
.r( Y<br />
c<br />
e0<br />
d .r(<br />
e,<br />
I4<br />
* 1<br />
m<br />
0<br />
8<br />
Y<br />
I<br />
: d<br />
u<br />
0<br />
M<br />
c<br />
.r(<br />
5<br />
m<br />
u<br />
d<br />
g<br />
e,<br />
5<br />
k<br />
0<br />
w<br />
e,<br />
2<br />
d<br />
4<br />
n<br />
4<br />
m<br />
.r(<br />
I4<br />
0)<br />
Y<br />
2<br />
I<br />
I<br />
373<br />
. . .<br />
. . . . . . . .<br />
0 - N N N N m m<br />
I 1<br />
I<br />
. ,
A-Feed tank<br />
6-Thermocracker<br />
GReceiver<br />
D-Condenser<br />
374<br />
FIGURE 1. - Pitch Thermal Cracking Apparatus.<br />
. . A.F.ed lank<br />
B.Pumo and drih<br />
C.Purnp (lush tank<br />
D.Thermocracker<br />
J<br />
my water<br />
FIGURE 2. - Thermal Cracking System.<br />
I 1<br />
\<br />
\<br />
\<br />
\<br />
\<br />
t<br />
i<br />
\<br />
Y<br />
1<br />
Y<br />
'r<br />
c
FIGURE<br />
375<br />
I I I I I I I I I I I<br />
0 4 8 12 16 20 24<br />
FEED RATE, Ib/hr<br />
3. - Coke Rate Based on Crude Pitch Feed<br />
Rate - Thermal Cracking.<br />
14<br />
12<br />
10<br />
2<br />
0 4 a 12 16 20 24<br />
FEED RATE, Ib/hr<br />
FIGURE 4. - Product Pitch Rate Based on Crude Pitch<br />
Feed Rate - Thermal Cracking.<br />
I
80<br />
\<br />
0<br />
70<br />
60<br />
e 40<br />
a U<br />
VI<br />
9 30<br />
20<br />
10<br />
0 4 8 12 16 20 24<br />
FEED RATE, Ib/hr<br />
FIGURE 5. - Cas Production Rate Based on Crude Pitch<br />
Feed Rate - The'rmal Cracking.<br />
8<br />
- e4<br />
0a i k6: 2 -<br />
I I 1 - 1 1 I I I I I '<br />
_,J: -<br />
-0 I I I I I I I I I I<br />
i<br />
-
I<br />
,<br />
1<br />
1<br />
I<br />
4 9<br />
I<br />
1'<br />
i<br />
J<br />
20<br />
0<br />
377<br />
Llllilllrlrll<br />
4 8 12 16 20 24<br />
FEED RATE, Ib/hr<br />
FIGURE 7. - Carbon-to-Hydrogen Atomic Ratio <strong>of</strong> Oil<br />
Distillation Residue Based on Crude Pitch<br />
Feed Rate.
COAL DEASH& AND HIGH-PURITY COKE<br />
Ward J. Rloamer , The Lummus Company, Newark, Ne J, and<br />
F, L, Shea, Great Lakes Rasearch Corp., Elizabethton, Tennereea<br />
I--" tLC A O ~ Ut C 132<br />
In 1957, The Lummus Company and Great Lakee Carbon<br />
Corporation began a joint investigation into the qse <strong>of</strong> coal<br />
as a source <strong>of</strong> high-purity coke. The process involved the pro-<br />
duction <strong>of</strong> a low-ash, low-sulfur deaohed coal ao1,uaion from<br />
high volatiie bituminous coal, and the conversion <strong>of</strong> this coal<br />
solution into coke using a modification <strong>of</strong> the Lummus delayed<br />
coking operation,<br />
Experimental work on a bench scale was initiated in<br />
1953 and completed in 1961. The experimental stope was expanded<br />
to include the production <strong>of</strong> deashed coal as well ae the high-<br />
purity coke in either 'combined or alternate operetianso<br />
Bench programs were carried oat on several scales <strong>of</strong><br />
ogeration. A contifiuous (block operation) pilot plant was<br />
built and operated to conf%rm the bench work. This unit was cap-<br />
able <strong>of</strong> processing coal continuously at approximately 200 pounds<br />
per hour through the filtration step. At this point, the filtrate<br />
was stored for further continuous proceseing to either drashed<br />
coal product (performed in 150-200 pound quantities) or high-<br />
purity cake (500 pound quantities) .,<br />
-<br />
S unna rv<br />
A process has been developed to upgrade the quality <strong>of</strong><br />
coal to a low-ash, low-sulfur, low-chloride product, As a fuel,<br />
this material would result in reduced capital and operating coati<br />
for power stations, Investment costs would be reduced, due to<br />
the higher heating value and cleaner fuel. Operating costs would<br />
be reduced by charges associated with the transportatfon <strong>of</strong> ash,<br />
ash hacdling and disposal costs, lower maintenance coetr, and fly<br />
ash removal costsc The substantial reduction in eulfur, ash, and<br />
volatile matter would greatly minimize air pollution.<br />
Other potential applications are for use in gas turbine#,<br />
as a reductant, as a raw material for synthesis gas, aa an ingredien<br />
ia preparing coal blends for metallurgical coke, or for high-purity<br />
coke.<br />
)'<br />
The economic potential <strong>of</strong> the process in its present stage '<br />
<strong>of</strong> development is covered in Table 5. The process has certain i<br />
aspects requiring sustained demonstration efforts to obtain firm<br />
econonic bases.<br />
1.
Process Descrivtion<br />
379<br />
Figure 1 shows a schematic,flowsheet for the production <strong>of</strong><br />
either deashed coal or high-purity coke as investigated in this pro-<br />
gram,<br />
I<br />
Deashed Coal - Crushed, ground and dried raw coal is fed to<br />
agitated premix tanks where it is mixed with warm solvent. The<br />
slurry is pumped through an extraction heater and thence through<br />
either in-line reaction tubes or to agitated digestion tanks. Diger-<br />
tion was investigated over the temperature range <strong>of</strong> 650 to 850'F and<br />
over a pressure range <strong>of</strong> 75 to 135 psig. Residence times varied from<br />
three minutes to two hours. Solvent-to-coal ratios were varied from<br />
l r l to 4:l<br />
From the digestion tanks, the coal dispersion 1s fed to a<br />
rotary pressure precoat filter designed for operating at.100 paig<br />
and 700°F. Washing provisions are included.<br />
The filter zake (L)fr discharged into a cake receiver,<br />
Filtrate and wash are collected and fed to the product and rolvent<br />
recovery operations, Vapors from the filtrate receiver are cornprersed,<br />
heated and recycled to maintain pressure on the feed side <strong>of</strong> the fil-<br />
ter drum,<br />
Filtrate flows through the filtrate heaters into a vacuum<br />
product recovery unit, Vapors from this unit pass to a flash tower<br />
where the bottom stream is all solvent for recycle, A side stream<br />
is taken <strong>of</strong>f and in part is used for solvent make-up with the re-<br />
mainder processed for by-product recovery.<br />
Overhead vapors from the flash tower are condensed, and a<br />
low boiling fraction recovered as a by-product. Vapors flashing<br />
from the filtrate receiver are condensed and introduced as reflux into<br />
the bottom section <strong>of</strong> the flash tower,<br />
Filter cake is calcined and the epent cake used as fuel for<br />
the process, Gas and distillate vapors from the kiln combined with<br />
gaseo from the filtrate receiver and flash tower overheads are passad<br />
into a light oil scrubber. The absorption oil -- the intermediate8<br />
cut from the flash tower -- is stripped in the flash tower, Light<br />
oil fractions leave the rystem from a decanter on the flash tower<br />
overhead stream. Gas from the light oil scrubber is ured for procesr<br />
heat<br />
High-Purity Coke - For coke product the same flow pattern<br />
prevails through the filtrate receiver. The filtrate is pumped to"<br />
a coking heater and thence to coke drums. Overhead vaporr from the<br />
coke drums pars to a combination tower where recycle rolvent and by-<br />
product fractions are recovered, Overhead vapors from the tower parr<br />
I
3Pn<br />
_. -. - L -J:,J.-;~~~L, ):as separator, and thence to a light oil scrubber.!<br />
at~ave operations were investigated on both 8 bench ,<br />
..:.- ;i;: ?L;,nt scale with the exception <strong>of</strong> cake crlcining which was<br />
?cr:azn~2 oniy on bench scale, Variables investigated are discusred ,<br />
I<br />
... i.16 :oiAowing sections.<br />
'racLDJ ,nd Oneratint! Variables for Deashed Coal<br />
- I-<br />
,I reviev <strong>of</strong> our work on solvent deashing indicated the<br />
07crntiny. conditions necessary to optimize the process. These<br />
Z L ~ be briefly summarized:<br />
.~<br />
i: .The development <strong>of</strong> a stable, high-extractive-efficiency<br />
i~iviii: cripabla <strong>of</strong> essentially complete recovery in the vacuum recov-<br />
2:y uniz 'ca yreld ueashed coal was accomplished by special treatment<br />
sz L i~i:~::-b~iii~g coal tar distillate. The required solvent having<br />
ascelia:-.: ccmperacure ~ t a blity i and high solvent power was attained<br />
by rcfra-:otrLing the hifih-boiling coal tar distillate, employing<br />
" -LL>c>e ,I. t:iarmal decomposition and fractionation to eliminate the leas<br />
- -Li-4L-o--y , .- . -. and low-bailing constituents. Gratuitously, these are the<br />
;CL= cffectivi ercraccion components,<br />
2 Xicsve:y <strong>of</strong> from 88 to 95 weisht percent <strong>of</strong> the avail-<br />
3bie ~arbsnac-s~s matter at extractor preesures in the range <strong>of</strong> 5 to 8<br />
.i;-isJ?heiLs and ri;h solvent-to-coal ratios in the range <strong>of</strong> 2 or 311<br />
was Ychi?ve:d using ;he iefractorized solvent with A boiling range <strong>of</strong><br />
600 ~a 900-F Typical extraction conditione were 750°F, 75 psig, at<br />
a asLven;-io-co~i :de10 ot 311.<br />
2, Extraction at residence times <strong>of</strong> 5 to 20 minutes in a<br />
-.c-;:r3u s~i~tizer vessel orB preferably, in continurvs in-line<br />
ssi~rroa n~~c:ng' coils which would permit a direct circuit from the<br />
ao:vrct-csz. mixing tank through a conventional heater with a discharge<br />
to dr~her a rotary pressure precoat filter or a suitable<br />
tyCro:lond,% i3i Bench extractions were successfully conducted at<br />
no1G:ng t.irnSs ;f as low as 3 minutes. There were duplicated in a<br />
piic: f ~ ~ i i - hiater ~ e coil discharging the hot coal rolution continuously<br />
~:hr~,gh cr, expanded section <strong>of</strong> the transfer line to the filter<br />
charga tank,<br />
4. Ash removal from the coal solution WAO accomplished at<br />
i:-ir.i~ror, r.i~cs <strong>of</strong> 200 to 300 lbsfhrleq. ft, <strong>of</strong> filter surface,<br />
?=el:cindry rates <strong>of</strong> 200 KO 250 were secured in the pilot rotary<br />
.,;J;OL~ prcss~re filter at low pressure differentiala, Small-scale<br />
:-zb an cocit: washing and calcining were performed to obtain closediycie<br />
data an soibent and distillate distribution. It was anticipa~od<br />
that the filter cake would be washed with a light oil, calcined<br />
zni the spexit cake used as nteam fuel for the procars in A full-scale<br />
Tlznt,<br />
\
381<br />
The bench and pilot plant operations in general Closely<br />
approached the goal5 set at the start <strong>of</strong> the work for the deashed<br />
coal produstiJn phase <strong>of</strong> the program. In Table I, there are pro-<br />
vided examples <strong>of</strong> the data relative to the composition <strong>of</strong> the raw<br />
coal, coal-to-solvent ratios, temperatures and pressures and analyses<br />
<strong>of</strong> the coal extract solutions, Yields are shown on he basia <strong>of</strong><br />
both raw and ash-free coals. Typical coal deashing results are<br />
shown in Table 2,<br />
Avai lable Coal Charge Stocks<br />
Extraction <strong>of</strong> coal with phenanthrene by earlier workers<br />
related the degree <strong>of</strong> extraction to the rank <strong>of</strong> the Thus,<br />
about 95 percent <strong>of</strong> the organic material in bituminous coal was<br />
dispersed as compared to 27 percent for subbituminous coal and 23<br />
percent for lignite Bench studies using a preprocessed coal tar<br />
distillate solvent marched these extraction efficiencies for the<br />
bituminous coal and lignite studied,<br />
The coals extracted ranged in ash content from 5 to 20<br />
weight percent and in sulfur values from 1 to over 4 percent. In<br />
each case, it was possible to secureexcelknt removal <strong>of</strong> the ash<br />
and a subsranrial portion <strong>of</strong> the sulfsr, chlorides and other con-<br />
t aminan t s - Sulfate and pyritic sulfur were fully eliminated with the<br />
ash and the organic sulfur forms were noticeably reduced., It was<br />
also possible to reduce the volatile matter contents from original<br />
values <strong>of</strong> near 40 percent to around 15 to 25 percent by distillation<br />
or by solvent precipitation (5) <strong>of</strong> the deashed coal solution,<br />
Improvement <strong>of</strong> Solvent for Coal Deashing<br />
Ihe wide-range heavy anthracene oil fraction originally<br />
used in the coal solution studies boiled between 600'F and 1050°F.<br />
Bench delayed coking <strong>of</strong> the deashed coal solutions gave solvent<br />
losses ranging as high as 30 to 60 volume percent through polymer-<br />
ization <strong>of</strong> the heavy ends and decomposition <strong>of</strong> the less refractory<br />
components <strong>of</strong> the solvent.<br />
Accordingly, studies were initiated to produce a more<br />
stable solvent, As a result, the efficiency <strong>of</strong> the coal solvent<br />
was improved to a point where 85 to better than 90% solution is<br />
achieved <strong>of</strong> the total extractable carbonaceous material <strong>of</strong> the coal<br />
(l.e., excluding the nonsoluble fusain)<br />
This preferred polvent, freed <strong>of</strong> easily polymerizable<br />
materia1,is capable <strong>of</strong> essentially complete recovery from either<br />
the cos! solution by vacuum stripping or from the delayed coking<br />
operation at reduced pressure. .<br />
F
S ~ l ~ o n~pg:ading r is the result <strong>of</strong> two important factors.<br />
r l ~ s t , the 201 irrit holling range has been narrowod and, secondly,<br />
the 5ol~ent has S E ~ ~ ,cabilised I<br />
by removal <strong>of</strong> its less refractory<br />
(readllv crached, ,,mponents.<br />
qolvent improve 1cb stahilitv.<br />
Continued use and recycle-<strong>of</strong> this<br />
A hciff dlscuaslon <strong>of</strong> the variables in coal solution and<br />
coking <strong>of</strong> its deasned ~olutlon wlll outline the reasons for the<br />
improvemenrs -e:.ireri by the channes In solvent boiling range and<br />
composition noted
383<br />
ash content <strong>of</strong> the initial high volatile bituminous coal. Thus,<br />
it has been repeatedly demonstrated in the bench and pilot solvent<br />
deashing studies on coal samples with ash contents varying from 5<br />
to 20 weight percent that final ash values <strong>of</strong> 0.1 to 0.3 could be<br />
obtained in the treated coal extracts.<br />
Lisewise , inorganic chlorides have been iargely eliminated<br />
with the ash. Coals containing 0.25 and 0.15 weight percent chlorine<br />
were reduced to values <strong>of</strong> 0.004 to 0.003, respectively. It is<br />
assumed that coals <strong>of</strong> considerably higher inorganic chloride contents<br />
could be reduced to similar low values. The harmful corrosive<br />
effects <strong>of</strong> ash and chlorides on furnace operation are well known and<br />
their effective reduction is obviously very desirable.<br />
Sulfur Reduction<br />
The degree <strong>of</strong> bituminous coal desulfurization by solvent<br />
deashing has proven to be a function <strong>of</strong> the original ratio <strong>of</strong><br />
inorganic sulfate and pyritic sulfur to the organic forms. Original<br />
total sulfur contents <strong>of</strong> 1 t o 4 percent are reduced to values <strong>of</strong><br />
0.4 to 1 weight percent in the extracts.<br />
It is known that this ratio increases with coals <strong>of</strong><br />
increasing total sulfur content, i.e., in the lower grade coals <strong>of</strong><br />
correspondingly higher ash. The final sulfur <strong>of</strong> the deashed coal<br />
therefore relates to the original organic sulfur which is only<br />
partially removed in the process, whereas the inorganic sulfur is<br />
removed substantially quantitatively.<br />
Thus, in Table 1, complete elimination <strong>of</strong> the pyritic<br />
and sulfate sulfurs has been achieved with an accompanying 26<br />
percent reduction in organic sulfur, for an overall sulfur reduct-<br />
ion <strong>of</strong> about 50 percent.<br />
Delayed Coking<br />
The original 1957 concept <strong>of</strong> the coal deashing process<br />
was to prepare a coal solution which, after filtration for removal<br />
<strong>of</strong> its ash, sulfur and other contaminants, would be charged to the<br />
conventional Lummui delayed petroleum coking process. The coke<br />
drum vapor would pass to the combination fractionation tower for<br />
separation <strong>of</strong> gas and light and medium distillate from a tower<br />
bottom cycle oil which would be re-used for fresh coal solution.<br />
The bench and pilot coking <strong>of</strong> the deashed coal Solution<br />
proved to be almost routine. This charge stock resembled in many<br />
<strong>of</strong> its characteristics a low temperature carbonization pitch.<br />
Lummus had previously in bench scale and pilot plant delayed coking<br />
apparatus successfully coked both low and high temperature carbon-<br />
ization pitches as well as Gilaonite and Athabasca tar sand pitches.<br />
n
The range ~f temperatures and general operating characteristics<br />
proved similar to those employed for the stocks noted above.<br />
(<br />
Reduced press~re is necessary for high solvent (600-900'F) recovery.<br />
Thus, it is anticipated that full scale plant operation would<br />
produce cokes with volatile matter contents approaching those<br />
normally encountered wich pecroleum stocks. Such cokes would be <strong>of</strong><br />
\<br />
low ash and satisiactorv sulfur contents and would be secured at<br />
high yields. \<br />
The product distribution on coal solution and on coal are<br />
summarized in Table 3, based on a 2:1 solvent to coal ratio. It \<br />
was found on examination <strong>of</strong> the coker total liquid products that<br />
the extraction solvent had been recovered without significant chnage. '<br />
The properties <strong>of</strong> the coker feed and product coke are listed in 1<br />
Table 4.<br />
F<br />
An alternate mechod <strong>of</strong> producing high purity coke utilizes ,<br />
the pure coal excract as the raw material. This may be pulverized<br />
in a ball and ring or xing roller type m i l l , such as is used for<br />
powdered coal burners. The milled pure coal is then used to form a<br />
slurry with a refractory recycle stock, having a boiling range <strong>of</strong><br />
about 500-700'F, The boiling range <strong>of</strong> this stock will vary somewhat,<br />
depending upon conditions in the heoter, the coke drum, and the ratio<br />
\<br />
<strong>of</strong> solids to liquid, but is selected so that sufficient liquid phase 1<br />
remains in the heater coils so as to convey the milled pure coal s<br />
without coke build-up in the heater.<br />
A third method <strong>of</strong> pure coke production considered,<br />
particularly where production <strong>of</strong> pure coke from coal would be<br />
conducted in canjunction wich the operation <strong>of</strong> a refinery, involves<br />
utilization <strong>of</strong> petroleum coker feedstock as the liquid in which the (<br />
slurry <strong>of</strong> pure coal is formed. Where a low coke yield feedstock is<br />
used, the coke produced would be predominantly from pure coal. \<br />
However, che proportion might be varied, depending upon the type <strong>of</strong><br />
coker feedstock employed and the ratio <strong>of</strong> solids to liquid in the<br />
slurry charged LO the delayed coker.<br />
Alternate Processes for Coal Extract Recovery \<br />
As an alternate to the recovery <strong>of</strong> coal extracts <strong>of</strong><br />
original or decreased volatile matter contents by distillation, as<br />
noted in Tables 1 and 3, it was determined that these products could .<br />
be recovered readily from solution by precipitation with hydrocarbon ,<br />
solvents. Thus, a paraffinic solvent yielded the complete deashed<br />
coal extract, whereas it was possible to recover a deashed coal with<br />
a volatile matter content 60 weight percent below that <strong>of</strong> the original<br />
coal with control <strong>of</strong> intermediate product values by manipulation <strong>of</strong><br />
the aromaticity <strong>of</strong> the solvent blends.<br />
c<br />
4<br />
\<br />
\<br />
,<br />
,
385<br />
Referencee<br />
1. Bloomer, W. J., and Martin. S. W.. U. S. Patent 3,109,803<br />
(November 5, 1963).<br />
2. Dicalite 40, General Refractories Company, used for bench and<br />
pilot filtrations.<br />
3. Patent pending,<br />
4. Orchin, M., Golumbic, C., Anderson, J. E.,<br />
U. S. Bur. Mines Bull. 505, 1951.<br />
and Storch, 8. H.,<br />
5. Patent pending.<br />
I<br />
. . .
. ..<br />
386<br />
9<br />
* -<br />
r.<br />
Y<br />
2<br />
.'<br />
d<br />
a<br />
f'<br />
r.<br />
*<br />
0<br />
..<br />
mo<br />
00<br />
..<br />
m<br />
m N<br />
90<br />
..<br />
m<br />
0<br />
00<br />
A0<br />
In<br />
O H .. *o<br />
..<br />
MN<br />
dln<br />
NN<br />
I ,<br />
N<br />
mm<br />
rc<br />
IC-<br />
-I M<br />
a m<br />
'i<br />
c'<br />
J<br />
h<br />
.<br />
u? b<br />
-<br />
r,<br />
C<br />
i:<br />
0)<br />
C<br />
..<br />
ON<br />
om<br />
0-6<br />
..<br />
m.m<br />
tnm<br />
rn'<br />
..<br />
Inm<br />
IC-.<br />
.or-<br />
..<br />
,.<br />
, .c<br />
'.O<br />
1<br />
\
I<br />
.-<br />
4 -C .-<br />
.-<br />
J<br />
u<br />
0<br />
-- 0<br />
N -<br />
In<br />
VI<br />
m l -<br />
NVI VI<br />
0<br />
d<br />
. \*<br />
.. .<br />
. .<br />
- L (<br />
Nlin<br />
- 0<br />
I l l<br />
I l l
.)<br />
.L<br />
m<br />
388<br />
I
.<br />
UI<br />
m<br />
rs<br />
m<br />
389<br />
. . . .<br />
t u 0 - r -<br />
tu CI 0 9.<br />
N 9<br />
4<br />
$<br />
0,<br />
Y<br />
s<br />
c<br />
C<br />
0)<br />
d<br />
0<br />
ln<br />
cr(<br />
N<br />
00.<br />
0<br />
N O<br />
y;d<br />
N 2<br />
0<br />
CI<br />
I
TABLE 4<br />
390<br />
DELAYED COKING OF DEASHED COAL SOLUTION<br />
RESULTS OF COKER TEST RUN DC-13<br />
Properties <strong>of</strong> Feed.(’)<br />
Specific Gravity, 100/60*F<br />
S<strong>of</strong>tening Point. *F (R&B)<br />
Benzene Insoluble. Wt.70<br />
Properties <strong>of</strong> Coke<br />
Wt. Percent <strong>of</strong> Feed<br />
Density, lbs/ft3<br />
By Weight <strong>of</strong> Volume 50.1<br />
Occupied in Coke Drum.<br />
Coke Drum Cokc Drv.7<br />
so. 1 Kc?. 2<br />
1.1926 l.!6!l<br />
158<br />
22.2<br />
25.7<br />
By Weight <strong>of</strong> Water 66.7<br />
’ Displaced by Sample<br />
Volatile Matter, Wt. % 9.17<br />
Fixed Carbon, Wt. % 90.22<br />
Ash, Wte% 0.61<br />
Sulfur. Wt. % 0.37<br />
Heating Value, BTU/lb. 14,520<br />
(1) Weight percent coal extract<br />
Benzene insoluble, weight p<br />
. . .. . . . .<br />
. .<br />
. . .<br />
-. .. . .<br />
r<br />
-26.2<br />
ent- 2 I., 0<br />
ion. 5<br />
I?. 6<br />
22.3<br />
38.5<br />
51.3<br />
10.06<br />
89.’28<br />
0.66<br />
0.38<br />
I<br />
;<br />
1<br />
1<br />
\<br />
\
9'<br />
-7<br />
.I<br />
\<br />
f-<br />
P<br />
,<br />
/<br />
,/-<br />
391<br />
TABLE5<br />
rr-ovx'rr STUDY - DTASHED COAL PRODUCT<br />
D:ar.t Cost 513,400,000 d 24 000s OOO<br />
Cost/Ton //MM BTU CostrTon<br />
\<br />
C'MM BTU<br />
Product Product<br />
Raw b! ate ria; ( Cc L 1)<br />
Lzkor, SILT) e r ~ s : ion,<br />
56.50<br />
:. 14<br />
21.6<br />
3.8<br />
$6.50<br />
'<br />
. .9R<br />
, 21.6 . . ,<br />
. . .<br />
. ' 3;2<br />
.<br />
R Over.':cai<br />
Ctiiitics 1.30 4.3 ,' . 1;30 ,<br />
4.3 . .<br />
De ?- .. ,. : .c..-..on . ,:<br />
\4 a. ..., -- .-. - - ..-r.ce -<br />
1.19 '<br />
.54<br />
3.9<br />
1.8<br />
1.06<br />
.40' '<br />
3.5, '<br />
-1.6.<br />
Tnsc... . A-.,cc - 4 :>:erest . .A9 2.9 , .80 .: . . 2,7<br />
-<br />
s;;i.- , J4!S - .I5 - .5 - .IS<br />
.5 .<br />
CY,,- LT?cra? ng Costs S11.71 38.R $11.27 ' 37.4<br />
-<br />
3y-7 :occct c red its 2.60) (8.6) (2.60) - (8.6)<br />
yet C:c:ati'r.z Costs<br />
q:c-z; . 'r.vcst-ncnt Sc',irn<br />
$9. ! 1<br />
2.24<br />
30.2 $8.67<br />
- 7.4 2.00<br />
2A. 8<br />
- 6.6<br />
s2*c, ->:.ce, 911.35 37.6 $10.66 35.3<br />
-.<br />
d- :L ?rice,<br />
= 54.00 'tor.:<br />
39.72 31.9 $9.03 29.6<br />
' 7 ~ 1 $j.?O,ton) ,'<br />
Sz.-s: :,<br />
:.<br />
Plant is a 'grass-rcots' facility capable <strong>of</strong> hahdling run-<strong>of</strong>*minc cool<br />
from s?ockpile.<br />
LsSor - SS.OO/rnan hour including supervieton and overhead.<br />
2. Utilities - Electricity 3.006/KMrH<br />
Steam Q$ .5O/M Ibe.<br />
4.<br />
Cooling Water B$.O2/M Gallons<br />
Deoreciation - Straight Line over 15 year period.<br />
5. Vaintrnance - 3OL <strong>of</strong> investment.<br />
6. Insurance & Interest - 5% <strong>of</strong> Plant Colt.<br />
7. By-product Credits - 8.01 I.pound average for net execs. dirtillate.<br />
8. Pay-Out - R Years. - - - -<br />
/<br />
I
,<br />
!<br />
I- on<br />
'---
THE HEAT OF REACTION?% HYDROGEN AND COAL<br />
A. L. Lee, E. L. Feldkirchner, F. C. Schora,and J. J. Henry*<br />
Institute <strong>of</strong> Gas Technology<br />
Chicago, Illinois<br />
This investigation is a part <strong>of</strong> the study conducted at the Institute<br />
<strong>of</strong> Gas Technology (1,2,4,5,6,7,9,10,11) to obtain the necessary infor-<br />
mation for designing an efficient coal hydrogasification plant. A thor-<br />
mgh literature search reveals that there is no reported data on the<br />
heat 3f reaction <strong>of</strong> hydrogen and coal. Therefore, two calorimeters were<br />
desisned, constructed, and operated, one to measure the heat <strong>of</strong> reaction<br />
md the other to meesure heat capacity by the drop method. The heat-<strong>of</strong>-<br />
reaction calorimeter can be operated at temperatures up to 1500'F and<br />
pressures up to 1500 psia; the drop calorimeter operates at atmospheric<br />
pressure and temperatures up to 1500°F. This paper reports the results<br />
<strong>of</strong> the following investigations :<br />
APPARATUS<br />
1. The heat <strong>of</strong> reaction <strong>of</strong> hydrogen with coals<br />
and coal chars after various degrees <strong>of</strong> gasi-<br />
fication.<br />
2. The heat <strong>of</strong> reaction <strong>of</strong> coa1,pretreatment.<br />
3. The heat capacity <strong>of</strong> coals and coal chars.<br />
The heat-<strong>of</strong>-reaction calorimeter built by Dynatech Corporation is<br />
shown in Figure 1. The sample is placed in the upper portion <strong>of</strong> the<br />
neck which is cold. The calorimeter body is filled with hydrogen and<br />
heated. The temperature <strong>of</strong> the calorimeter body is measured by foi2<br />
thermocouples and two platinum resistance thermometers. The sample is<br />
lowered into the body after the temperature has remained steady for 2<br />
hours. The change in temperature due to a reaction is then measured as<br />
a function <strong>of</strong> time.<br />
The drop calorimeter, which was also built by Dynatech, is shown in<br />
Figure 2 . The sample is placed in the top f'urnace until it reaches the<br />
desired temperature. It is then dropped into the copper receiver,and<br />
the heat capacity <strong>of</strong> the sample is determined from the temperature rise<br />
and heat capacity <strong>of</strong> the copper receiver.<br />
EXPERIIQCNTAL FESULTS<br />
'J<br />
I' The major problem encountered in determining the heat <strong>of</strong> coal reactisns<br />
at high temperature and high pressure is prereaction, since coals<br />
1 Ceconpose when heated. Meaningful results can be obtained only if the<br />
A'<br />
czal and hydro&en react at conditions at which the desired temperature<br />
I and pressure are stabilized. Therefore, the method <strong>of</strong> introducing the<br />
sayple t2 the reaction conditions is critical. The coal must not react<br />
I befsre the conditions are set, and the pressure must not be disturbed<br />
when the coal is introduced.<br />
!:<br />
1 ' * Dynatech Corp. , Cambridge, Massachusetts.<br />
(<br />
><br />
\<br />
c<br />
f
394<br />
The present method is to keep the sample at room temperature inside<br />
the calwimeter drop tube so that the reaction will not take place prematurely.<br />
Convectim shields are installed to prevent a large heat loss<br />
:rm convectim and to ensure that the.sample is in a cold zone while<br />
L l<br />
L..c cel?rizeter is being stabilized at the reaction cmciitions.<br />
Establishing ki heat balance around the calorimeter was necessary in<br />
order to calculate the heat <strong>of</strong> reaction from experimental 'data. The<br />
imzt b?Jance is best expressed by:<br />
-<br />
where: AHR = heat <strong>of</strong> reaction<br />
' !I, =<br />
in<br />
heat input from the neck heater<br />
m = mass<br />
C = heat capacity<br />
P<br />
AT = temperature change<br />
chain<br />
3?c!i term in Equation 1 must be either established by calibration or<br />
acccrately measured. The heat capacity <strong>of</strong> the calorimeter metal was de-<br />
tr'rixined in the drop calorimeter. These data are shown in Figure 3,<br />
The effective nCp <strong>of</strong> the reaction calorimeter was calibrated by a ,-*'<br />
constant-heet-input technique in a hydrogen atmosphere at 1000 psia and<br />
4 ,~~.?cratures<br />
q-, from 840° to 1460°F. The results are presented in Figure 4. jy<br />
After the Calorimeter constants were determined, the effective (mCpdT)'s r<br />
<strong>of</strong> the convection shield, the chain, and the empty sample basket were<br />
czlibrated in the calorimeter to determine the change <strong>of</strong> heat input wjth<br />
ti-", as shown in Equation 2:<br />
where : K(A) = a constant dependent only on time<br />
4 = time<br />
Combining Equations 1 and 2, we have:<br />
Eq-dation 3 was used to calculate the heat-<strong>of</strong>-reaction data reported in<br />
this paper.<br />
(1)<br />
\<br />
\s<br />
\
1<br />
I<br />
i Design <strong>of</strong> a hydrogasification $??ant requires data on the heats <strong>of</strong><br />
I reaction <strong>of</strong> raw coal in the coal pretreatment process, the pretreated<br />
Coal in the low-temperature gasifier, the residue from the low-tempera-<br />
'I twe gasifier in the high-temperature gasifier, and the residue from<br />
/ iA? high-temperature gasifier (4,6). The heat-<strong>of</strong>-reaction and heat capa-<br />
city measurements are given in Tables 1 and 2. Figure 5 shows the tem-<br />
j perature and pressure pr<strong>of</strong>iles <strong>of</strong> a typical experimental m.<br />
DISCUSSION<br />
><br />
Until now, the heat <strong>of</strong> the coal hydrogasification reaction has only<br />
been determined by calculation. These calculations have become more<br />
c Precise as more data became available, but no measurements were made<br />
to check the validity <strong>of</strong> the calculated data.<br />
J<br />
/ Initially (in the absence <strong>of</strong> accurate pilot plant yield data), the<br />
, :;eat <strong>of</strong> reaction was estimated by assuming that coal and carbon were<br />
, ewivalent and that the hydrogasification reaction could be approxi-<br />
:-ited. by (j, 8) :<br />
i' C + 2H2 - CHq<br />
' This approach, <strong>of</strong> course, is very crude and could not be expected to<br />
give a reliable answer, but it could be usef'ul for comparing the therf<br />
rial efficiencies <strong>of</strong> various gasification processes.<br />
The next approach, using pilot plant data (7), was to calculate the<br />
'heat <strong>of</strong> reaction from the heats <strong>of</strong> combustion <strong>of</strong> the reactants and pro-<br />
,'ducts. The heats <strong>of</strong> combustim <strong>of</strong> various coals could be obtained by<br />
a Parr-bomb calorimeter or calculated by the modified Dulong formula.<br />
'But if one attempts to calculate the heat <strong>of</strong> reaction <strong>of</strong> hydrogen and<br />
7 coal from heats <strong>of</strong> combustion, there axe two drawbacks.<br />
First, be-<br />
)cause the calculation involves taking the differences <strong>of</strong> large numbers<br />
<strong>of</strong> the same order <strong>of</strong> magnitude, <strong>chemical</strong> analyses <strong>of</strong> all reactants and<br />
1 products must be very accurate and pilot material balances must be quite<br />
) clme to 100 percent or else the balance must be forced. If this is<br />
i/not the case, large errors can be made in this calculation. This makes<br />
the calculated heat <strong>of</strong> reaction dependent on the quality <strong>of</strong> the analy-<br />
' tical data and on the method used to force the balance. The second<br />
'drawback is that the calculated heat <strong>of</strong> reaction is determined for 25'C,<br />
9 so no information is obtained on the reaction heat at actual reaction<br />
' temperatiires.<br />
v<br />
, <strong>of</strong> cc',irse, if the heat <strong>of</strong> reaction could be determined at one temperature,<br />
the heat <strong>of</strong> reaction at various temperatures could be cal-<br />
Iculated from heat capacity data. Some heat capacity data are available<br />
in the literature for coals and cokes, but none are available for<br />
' the particular coals used here. Moreover, the measurement methods<br />
',u'sed so far are not very accurate. In most cases the gaseous decom-<br />
, position products were allowed to escape from the calorimeters during<br />
I coal hcatup, and, consequently, the heat capacity data are rather<br />
, douhtP,l.<br />
\<br />
2 from the heat <strong>of</strong> formation data for C + 2H2 - CH4 and the pilot plant<br />
ddata,with that obtained from the calorimetry studies is shown in Fig-<br />
A conprison <strong>of</strong> the heats <strong>of</strong> reaction <strong>of</strong> hydrogen and coal obtained<br />
J WE<br />
i<br />
c. Iiote that the pilot plant data were based on a 77'F reference<br />
,<br />
J
396<br />
‘c erperature, while the present experimental data were obtained at opereti:i;<br />
conditions. Noreover, the experimental data were obtained from<br />
coals at four different stages <strong>of</strong> reaction: raw coal, pretreated coal,<br />
lw- t emperature gasification residue, and high- t emperature gasification<br />
residue. Ash balances were used to put these results on a common basis.<br />
The ash balance calculations gave the percent <strong>of</strong> carbon gasified in<br />
each cDal or char. Raw coal was asswed t3 have 0s carbon gasified.<br />
7-L~s Figure 6 shows the general trend <strong>of</strong> the heat <strong>of</strong> reaction.<br />
Accurate heat-sf-reaction data are given in Table 1. Although the<br />
pilot plant data are considerably scattered, the average value is not<br />
tm different from that obtained by the other methods. The calorime-<br />
try data also show some scattering, which is due to the hetemgeneous<br />
nature <strong>of</strong> the mal, and the characteristics <strong>of</strong> the calorimeter and the<br />
sensiw instruments. Examinations <strong>of</strong> the temperature measurement, the<br />
pressure measurement, the temperature distribution in the calorimeter,<br />
the total mass balance, and the calibration results obtained from the<br />
cmstant-heat-input method and experimental runs on hydrogen and 2-<br />
aecane reactions indicate that the data reported in Table 2 should not<br />
::zve T: deviation greater than 10%.<br />
This work is jointly sponsored by the American Gas Association and<br />
tile U.S. Department <strong>of</strong> the Interior, Office <strong>of</strong> Coal Research. Their<br />
suppopt is gratefilly acknowledged. Valuable advice and discussions<br />
riere provided by Drs. B. S. Lee, S. A. Weil, and C. W. Solbrig <strong>of</strong> the<br />
Institute <strong>of</strong> Gas Technology. J. R. WSando assisted in the experimen-<br />
tal :.:3rl.;.
i<br />
Ij<br />
EPERENCES CITED<br />
307<br />
HJ7drOgen Mixtures at Eigh Temperatures and Pressures, " Ind. E~P.<br />
Chex Process Desim Develop. 4, 134-42 (.1965) April.<br />
Huebler, J. and Schora, F. C., "Coal Hydrogasification, Chem.<br />
Erg. Prom. 62 87-91. (1966) February..<br />
,'<br />
' JANAF Thermo<strong>chemical</strong> Tables. Distributed by: Clearinghouse for<br />
Federal Scientific and Technical Information, Springfield, Va.<br />
KaVlACkr V. J. and Lee, B. S., "Coal Pretreatment in Fluidized<br />
Bed, he??. Chem. SOC. Div. Fuel Chem. Preprints 10, No. 4J<br />
131-39 (1966 September.<br />
Lindentl H. R., "Pipeline Gas From Coal: Status and Future Prospects,<br />
Coal Ape 5, 64-71 (1966) January.<br />
Pyrcioch, E. J., Lee, B. S. and Schora, F. C., "Hydrogasification<br />
<strong>of</strong> Pretreated Coal for Pipeline Gas 'Production, It her. Chem. SOC.<br />
Div. Fuel Chem. Preprints 2, No. 4, 206-23 (1966) September.<br />
Pyrcioch, E. J. and Linden, H. R., ";ipeline Gas by High-Pressure<br />
Fluid-Bed Hydrogasification <strong>of</strong> Char, Ind. Erg . Chem. E,, 590-94<br />
( 196 0 ) July.<br />
Rossini, F. D. et al., Selected Values <strong>of</strong> Physical and Thermo-<br />
dynamic Properties <strong>of</strong> Hydrocarbons and Related Compounds.<br />
Pittsburgh: Carnegie Press, 1953.<br />
Tajbl, D. G., Feldkirchner, H. L. and Lee,IIA. L., "Cleanup<br />
Methanation for Hydrogasification Process her. Chem. SOC.<br />
Div. Fuel Chem. Preprints l.0, No. 4, 235-45 (1966) September.<br />
Tsaros, C. L. and Feldkirchner, H. L., "Production <strong>of</strong> High Btu<br />
Gas, 'I Chem. -*62, 49-54 (1966) August.<br />
von Fredersdorff, lIC. G. and Vandaveer, F. E., "Substitute Natural<br />
Gas From Coal, in Segeler, C. G., Ed., Gas Wineers Handbook,<br />
Section 3, Chap. 9, 3/100-3/123. New York: The Industrial<br />
Fress, 1965.<br />
I
2,<br />
'!<br />
!<br />
Proximate Analysis, wt $<br />
Moisture<br />
Vo lat i les<br />
' Fixed Carbon ,<br />
Ash<br />
Total<br />
ntimte Analysis, wt %<br />
Cerbon<br />
Hydrogen<br />
Nitrogen<br />
Oxygen<br />
Sulrur<br />
Ash .<br />
Total<br />
Reactyon Temperature, OF<br />
Reaction Pressure, psia<br />
Coal Reacted (Av J, %<br />
Carbon Gasified YAvg), %<br />
Avg AHR, Btu/lb coal<br />
rkacted<br />
3P8<br />
Table 1. ANAE$SIS OF COATl AND COAL CHARS<br />
Raw Pretreated Low-Temp FTigh-Temp<br />
Coal Coal .' Residue Residue<br />
-<br />
1.3<br />
34:6 '<br />
52.0<br />
-&&<br />
0.5<br />
23.3<br />
63.5<br />
d%<br />
0.6<br />
4.6<br />
77.6<br />
1oo.o 17*2<br />
0.6<br />
3.3<br />
71.6<br />
24.5<br />
m<br />
71: 2<br />
5.14<br />
1.23<br />
6.03<br />
70.1 ~<br />
3.70 '<br />
1.37<br />
* '76.9'<br />
2.05<br />
1.01<br />
0.55 ,<br />
4.19<br />
12!21.<br />
100 I 00<br />
1300<br />
1000<br />
$49.7<br />
50.6<br />
181% .<br />
*' 8.30<br />
-. 3.80<br />
-<br />
12 * 73<br />
100.00<br />
1300<br />
1000<br />
47.2<br />
41.0<br />
I<br />
1919 ',<br />
2 .&I<br />
17 30<br />
m37<br />
1300<br />
1000<br />
27.3<br />
29.5<br />
2432<br />
Table 2. HEAT OF REACTfm OF COAL. . \<br />
I<br />
72.6<br />
1.08<br />
0.54<br />
0.00<br />
1.24.<br />
24.62<br />
100..00<br />
1300 \<br />
1000<br />
28.8 ,<br />
26.0,<br />
3078<br />
Temperature, Pres sure, Coal 'Carbob mR,, Btu/lb'<br />
Material OF pia I Reacted,% Gasified,$ coal' reacts<br />
d<br />
Raw coal<br />
1000 41.5 . -- 1300 /<br />
Raw coal<br />
1000 .52.4 50.7 8 1951 \<br />
Raw coal . 1300 1000 52.2 '52.0 1723<br />
RZIT coal ,1300 1000 48.1 55.6 1788 c<br />
Raw coal 1300 1000 46.1 44.2 18 07 1<br />
Raw coal 1400 1000 51.7 52.3 1775<br />
?retreated coal 1300<br />
Pretreated coal 1300<br />
Pretreated coal 1300<br />
Pretreated coal 1300<br />
Pretreated coal 1300<br />
bw-temp residue 1300<br />
bw-temp residue 1300<br />
HiZh-temp residue 1300<br />
r.<br />
1000<br />
1000<br />
1000<br />
1000<br />
4000<br />
1000<br />
1000<br />
1000<br />
7 -<br />
*<br />
46.1 37.6 1935 $\<br />
47.3 41.2 1717<br />
47.3 41.5 1844<br />
47.1 40.0<br />
48.3 45.0 212:; *<br />
\<br />
\<br />
?<br />
\<br />
- \
I<br />
i /<br />
- .<br />
399<br />
. . .<br />
I .<br />
2’<br />
0<br />
H<br />
fi<br />
rl<br />
a)<br />
h<br />
J
0 IS<br />
0 14<br />
LL<br />
9 013<br />
e<br />
\<br />
3<br />
b-<br />
m<br />
*- 012<br />
t<br />
V<br />
a<br />
4<br />
V<br />
b- 0 I1<br />
a<br />
W<br />
I<br />
0 IO<br />
0 09<br />
200 400 600 eo0 1000 1200 1400 1600<br />
TEMPERATURE . OF A-Ile14OS<br />
Figure 3. MEAN HEAT CAPACITY OF CALORIMETER<br />
(From 70°F to Temperatures Indicated)<br />
800 900 1000 1100 1200 1300 1400 600<br />
, TEMPERATUREPF L-IIII404<br />
Figure 4. CALIBRATION OF HEAT-OF-REACTION CALORIMETER
I20 0 zo -0 SO 00 100 110 110 160 In0 200 I10 140 1.0 zoo<br />
TIME, min<br />
Figure 5. TIME-rTEMPERATLTRE DATA FOR HEAT OF REACTION OF<br />
HYDROGEN AND COAL<br />
3900<br />
3700<br />
3wx)<br />
3300<br />
w c<br />
y 3100<br />
Y<br />
22.00<br />
d<br />
0 2700<br />
m<br />
2<br />
2 2500<br />
+<br />
m<br />
,i 2=<br />
0 NOT PLANT DATA<br />
? Zloo b RAW COAL .<br />
RlETREAIED COIL<br />
1000 V LOW-TEMP RESIOVE<br />
0 MIGM-TEMP RESIDUE<br />
1700 MAT-Of-FORMATION<br />
1300<br />
0 IO 20 30 - 40 50 60 m m<br />
CARBON GASIFIED. Y .<br />
,101.s*<br />
Figure 6. COMPARISON OF DATA FOR HEAT OF REACTION<br />
OF HYDROGEN AND COAL<br />
J
402<br />
HYDROGEN CYANIDE PRODUCED FROM COAL AND AMMONIA<br />
, .-.<br />
G. E. Johnson, 'W. A. Decker, A. .J. Forney, and J. H. Field ' ,':'<br />
Pittsburgh Coal Research Center, U. S. Bureau <strong>of</strong> Mines,<br />
4800 Forbes Avenue, Pittsburgh, Pennsylvania 15213<br />
' INTRODUCTION, .<br />
Hydrogen cyanide (HCN) has been one <strong>of</strong> the country's strongest growth<br />
petro<strong>chemical</strong>s in recent years. U. S. production has increased from 174 million<br />
pounds in 1960 to 350 million pounds in 1964, a 100-percent increase over the<br />
4-year period. The growth <strong>of</strong> production <strong>of</strong> hydrogen cyanide has been directly<br />
'related to the expansion in production <strong>of</strong> synthetic textiles from acrylonitrile.<br />
Relative growth and production <strong>of</strong> hydrogen cyanide and acrylonitrile is as<br />
follows :.<br />
Production <strong>of</strong> Hydrogen Cyanide and Acrylonitrile<br />
Hydrogen cyanide,?' Acrylonitrile,<br />
Year million lb million lb<br />
1960 174 229- lo/<br />
1961 211 25%;<br />
1962<br />
1963<br />
1964<br />
1965<br />
266<br />
293<br />
350<br />
---<br />
360-<br />
10/<br />
455-<br />
593-<br />
10/<br />
371 (6 months)- 4/<br />
About 50 percent <strong>of</strong> the total output <strong>of</strong> hydrogen cyanide goes into the<br />
production <strong>of</strong> acrylonitrile; most <strong>of</strong> the remainder is use in production <strong>of</strong><br />
adiponitrile and the manufacture <strong>of</strong> methyl methacrylate.1' However, in recent<br />
years acrylonitrile and adiponitrile are being produced by processes which<br />
generate hydrogen cyanide as a byproduct.?/ The bulk <strong>of</strong> acrylonitrile is a,<br />
used in production <strong>of</strong> acrylic fiber (Orlon, Acrilan, Dynel, Zefran, etc.),-<br />
a smaller amount in production <strong>of</strong> nitrile rubber, the adiponitrile in manu-<br />
facture <strong>of</strong> Nylon.<br />
The manufacture <strong>of</strong> sodium cyanide utilizes about 7 percent <strong>of</strong> hydrogen<br />
cyanide production. The remaining hydrogen cyanide goes to a large number <strong>of</strong><br />
relatively small uses including ferrocyanides, acrylates, ethyl lactate, lactic<br />
acid, chelating agents, optical laundry bleaches, and pharmaceuticals.<br />
The Andrdssod process is the major commercial process used for producing<br />
hyjrogvli :/aiide. It invo1v:s the reaction <strong>of</strong> methane, ammonia, and air over<br />
a platinum catalyst at l,OOOo to 1,200' C.21 The platinum catalyst is usually<br />
alloyed with rhodium (10 to 20 percent).<br />
Conversion by the Andrussow process in a single pass is limited to about<br />
69 percent <strong>of</strong> the ammonia (about 75 percent with gas recycle) and 53 percent<br />
<strong>of</strong> the methane. A typical analysis <strong>of</strong> the reaction gases leaving a catalytic<br />
reactor is as follows in volume-percent: Nitrogen 56.3, water vapor 23.0,<br />
hydrogen 7.5, hydrogen cyanide 6.0, carbon monoxide 4.4, ammonia 2.0, methane<br />
0.5, carbon dioxide 0.2, and oxygen 0.1.<br />
a/ Reference to trade names is made for identification only and does not imply<br />
endorsement by the Bureau <strong>of</strong> Mines.<br />
I
3<br />
1<br />
403<br />
In the Catalytic Degussa process which is not in general use but is similar<br />
to the Bureau <strong>of</strong> Mines method in that heat is provided externa.lly, the <strong>of</strong>fgas<br />
from the ammonia-methane reaction contains more than 20 percent hydrogen,cyanide.<br />
Utilization <strong>of</strong> methane and ammonia are reported as 91 and 85 percent, respect-<br />
ive ly .<br />
In its search for new uses for coal the Bureau <strong>of</strong> Mines has been .investigating<br />
the production <strong>of</strong> hydrogen cyanide from cpa.1. Although hydrogen cyanide<br />
is present in coke oven gases, and at one time was recovered as a byproduct,'<br />
this source <strong>of</strong> the gas has not been commonly used in the United States since the<br />
development <strong>of</strong> the newer methane-ammonia.processes. Although the production <strong>of</strong><br />
hydrogen cyanide from coal is technically feasible, production in yields that<br />
could be competitive is a problem <strong>of</strong> major concern.<br />
..<br />
. .<br />
EQUIPMENT AND PROCEDURE<br />
.. .<br />
Figure 1 is a flowsheet <strong>of</strong> the experimental unit. Coal ground to minus 300<br />
mesh is dropped in free-fall through a heated reaction zone in the presence <strong>of</strong><br />
ammonia at rates up to 1.10 lb/hr. A revolving-disk feeder especially designed.<br />
by the Bureau <strong>of</strong> Mines to feed coal at low rates was constructed; it delivered "<br />
to within 5 percent <strong>of</strong> the desired feed rate.<br />
A special coal feed system is used to prevent agglomeration and possible<br />
plugging <strong>of</strong> the reactor by heating the coal rapidly through its plastic range<br />
(about 400' C).<br />
The reactor itself is a &-foot length <strong>of</strong> vitreous refractory mullite,<br />
1-1/8 inches ID and 1-3/8 inches OD, jacketed with two electrical resistance<br />
heaters. The top heater (maximum temperature 850" C) is 12 inches long and is<br />
wound with Nichrome wire. It serves as preheater for the coal and gas. A<br />
Kanthal heater (Al-Cr-Co-Fe alloy, maximum temperature 1,250' C) encloses the<br />
center 20 inches <strong>of</strong> the tube, or the reaction zone. The bottom section <strong>of</strong><br />
the reactor is exposed to the atmosphere for rapid cooling <strong>of</strong> the product gases.<br />
The bottom <strong>of</strong> the reactor tube fits into a 4-liter side-arm flask or char<br />
receiver in which the heavier solids are collected. The fine solids and carbon<br />
black produced are collected in an electrostatic precipitator. After the<br />
product gases leave the precipitator they pass through a cooler, then they are<br />
either metered or sent through absorbers to remove the hydrogen cyanide for<br />
analysis.<br />
All <strong>of</strong> the piping and vessels are stainless steel or glass in order to<br />
counteract the corrosive nature <strong>of</strong> the gases. Since the gases are toxic, the<br />
unit is completely enclosed, and the enclosure is well ventilated to prevent<br />
any accumulation <strong>of</strong> escaped gases. The whole structure (6 ft x 6 ft x 15 ft<br />
high) is covered with steel sheeting. It has an exhaust blower (400 cfm) on the<br />
ro<strong>of</strong> and access doors at both ground and 8-foot levels. Figure 2 shows the<br />
exterior <strong>of</strong> the unit and figure 3 shows its interior.<br />
Product gas can be recycled to the top <strong>of</strong> the reactor adjacent to the<br />
cooled feed tube along with part <strong>of</strong> the feed gas. This flushes away any tar<br />
vapors which might adhere to the walls and cause plugging. The remainder <strong>of</strong><br />
the feed gas (0 to 10 scfh) enters the reactor with the coal. Ammonia, helium,<br />
methane, nitrogen, or air, or mixtures <strong>of</strong> these gases fed from cylinders have<br />
been used as feed gas.
404<br />
Before startup, the system is purged with inert gas. After the reactor<br />
has been heated to 1,250' C the desired flows <strong>of</strong> coal and gas. are started.<br />
Ammonia and nitrogen OK helium are the gases usually used. The gas flow is<br />
generally split, part entering the top <strong>of</strong> the reactor adjacent to the cooled<br />
feed tube, and the remainder entering with the coal.<br />
The powdered coal is fed through a steam-jacketed tube (5/16Linch OD) which<br />
extends into the preheat zone <strong>of</strong> the reactor. .The coal leaves the end <strong>of</strong> the<br />
feed tube which is at the temperature <strong>of</strong> the steam to enter the preheat zone <strong>of</strong><br />
850' C. The temperature <strong>of</strong> the coal rises very suddenly to 850" C because <strong>of</strong><br />
the high heat-transfer rate to the small particles. The carrier gas (usually<br />
helium, an inert gas) fed with the coal keeps the particles in motion and helps<br />
prevent agglomeration as the coal rapidly passes through its plastic range.<br />
Proximate and. ultimate analyses-/are made <strong>of</strong> the char and heavier<br />
solids collected in the char receiver and <strong>of</strong> the lighter solids collected by<br />
the electrostatic precipitator. Mass spectrometric and chromatographic analyses<br />
are made on spot samples <strong>of</strong> the product gas. For cyanide determinations,<br />
metered amounts <strong>of</strong> product gas are bubbled through two scrubbers in series<br />
containing solutions <strong>of</strong> sodium hydroxide. Titration with silver nitrate solu-<br />
tion determines the total cyanide pre sent .i/<br />
EXPERIMENTAL RESULTS AND DISCUSSION<br />
The initial tests were made with a metallic reactor tube, but because <strong>of</strong><br />
low yields <strong>of</strong> hydrogen cyanide and failure <strong>of</strong> the metal at the temperatures<br />
employed, the metal tube was replaced by a ceramic reactor tube.<br />
In all the tests <strong>of</strong> this report with hvab coal, Pittshrgh seam coal from<br />
Bruceton, Pa., was used. Its ultimate analysis is as follows in'percent:<br />
Carbon 7.5.6, ash 8.4, oxygen 8.0, hydrogen 5.1, nitrogen 1.6, and sulfur 1.3.<br />
The effect <strong>of</strong> varying the coal-ammonia feed rates is illustrated in table 1.<br />
Hydrogen cyanide yields were computed from the wet-<strong>chemical</strong> method <strong>of</strong> analysis<br />
which is considered the more reliable method since it was determined from pro-<br />
portionated gas samples taken continuously throughout the test (sample volume<br />
<strong>of</strong> 0.2 to 2 cu ft). Only spot gas samples (sample volume 0.01 cu ft) were used<br />
for the chromatograph and mass spectrograph analyses.<br />
In test C-241 the'coal-feed rate was 0.37 lb/hr and the ammonia-feed rate<br />
was 1 cu ft/hr. (All process gas volumes reported are corrected to standard<br />
.conditions <strong>of</strong> 0" C and 760 mm mercury pressure.) A hydrogen cyanide yield <strong>of</strong><br />
0.6 cu ft per cu ft <strong>of</strong> ammonia reacted was obtained, corresponding to about<br />
12 percent hydrogen cyanide in the product gas.<br />
In test C-243 the hvab coal-feed rate was,reduced to 0.19 lb/hr, while the<br />
ammonia-feed rate was increased to 2.3 cu ft/hr. A yield <strong>of</strong> 0.4 cu ft hydro-<br />
gen cyanide per cubic foot <strong>of</strong> ammonia consumed was obtained, corresponding to<br />
about 13 percent hydrogen cyanide in the product gas.<br />
The yield <strong>of</strong> hydrogen cyanide per pound <strong>of</strong> coal was approximately doubled<br />
when the coal-feed rate was halved and the ammonia-feed rate doubled (tests<br />
C-241 to 243), while the hydrogen cyanide yield per cubic foot <strong>of</strong> ammonia con-<br />
sumed decreased by one -third.
’f ’f<br />
\<br />
?<br />
i<br />
i<br />
k<br />
><br />
/<br />
f<br />
j<br />
I<br />
405<br />
Table 1.- Data from Tests with hvab Coal and Ammonia with Helium at 1,250° C<br />
Product gas, percent<br />
Test HCNL’ H,S N& H2 O-, N, CO ’ C02 C& CZ% C3H8 C8H10<br />
C-241 11.8 0.0 0.0 77.7 0.1 6.7 9.6 0.3 5.1 0.4 I 0.1 0.0<br />
C-243 13.1 .O 17.5 68.6 .9 6.8 4.0 .1 1.0 .o .o 1.1<br />
C-239 13.2 .1 .5 75.9 .1 4.4 10.6 .5 6.9 .6 .I .3<br />
C-198 5.4 .3 .O 78.3 .3 3.5 11.1 .3 5.7 .5 .o .o<br />
Feed Yield, cu ft<br />
Total<br />
Coal <strong>of</strong>f gas Length HCNIC~<br />
HCNICU<br />
He 3 NY3r feed, He-free, <strong>of</strong> run, HCN/lb ft N& ft N b<br />
cu ft/hr cu ft/hr lb/hr cu ft/hr min coal feed consumed<br />
C-241 1-00 1.00 0.37 5.06 30 1.59 0.60 0.60<br />
C-243 0.49 2.35 .19 4.90 30 3.31 .27 .40<br />
C-239 .98 1.00 .73 5.59 15 1.01 .74 .76<br />
C-198 2.12 0.20 .37-.40 3.61 15 0.51 .98 .98<br />
- 11 Wet-analytical method <strong>of</strong> analysis for HCN only; other components are the<br />
average <strong>of</strong> 2 chromatograph and 2 mass spectrometer analyses, HCN-free basis.<br />
In test C-239 (table 1) the coal-feed rate was increased to 0.73 lb/hr,<br />
while the ammonia flow was maintained at 1 cu ft/hr. A hydrogen cyanide yield<br />
<strong>of</strong> about 0.8 cu ft per cubic foot ammonia reacted was obtained, equivalent to<br />
about 13 percent hydrogen cyanide in the product gas.<br />
In the next listed test, C-198, the coal-feed rate was 0.37 to 0.40 lb/hr,<br />
while the ammonia flow was only 0.2 cu ft/hr. The hydrogen cyanide content in<br />
the product gas was only 5 percent, table 1, but conversions <strong>of</strong> about 100<br />
percent <strong>of</strong> the ammonia were obtained with 1 cu ft <strong>of</strong> hydrogen cyanide formed<br />
per cubic foot <strong>of</strong> ammonia used. This yield <strong>of</strong> hydrogen cyanide approximates<br />
the stoichiometric yield according to the following reactions:<br />
C + N& - HCN + H2<br />
C& + HCN + 3H2.<br />
In typi cal commercial units using catalysts for the methane-ammonia reaction,<br />
the ammonia conversion attained is about 75 percent. Yield values<br />
include .the slight amount <strong>of</strong> hydrogen cyanide that may be formed when coal is<br />
heated to hi gh temperatures without adding ammonia.<br />
In general, the tests <strong>of</strong> table 1 indicate that an excess coal feed is<br />
desirable in order to attain maximum utilization <strong>of</strong> the ammonia since the<br />
ammonia is by far the more expensive raw material.
4 OL.<br />
'<br />
,<br />
I.<br />
.<br />
TesCs. With.Coals .<strong>of</strong> Different Rank<br />
. . .<br />
In addition to the h,yab -coal, lignite, subbituminous, low-volatile bituminous,<br />
and anthracitc coals. coal char, and activated carbon were' tested as<br />
raw materials for producing hydrogen cyanide. It is thought that the volatile<br />
matter in.coa1 reacts with the ammonia to form hydrogen cyanide, therefore<br />
coals with higher volatile content should produce more hydrogen cyanide.<br />
The<br />
volatile-matter contents on a moisture-free basis <strong>of</strong> the various materials<br />
tested are as follows:<br />
Volatile<br />
matter,<br />
Tes t Identification <strong>of</strong> coal Source <strong>of</strong> coal percent<br />
C-123, 124,. Hvab, Pittsburgh seam Bruceton, Pa. 34.0<br />
125, 198,<br />
239, 241,<br />
243<br />
C -207 Activated carbon, Union Carbide an,d 2.0<br />
Grade SXWC Carbon Corp.<br />
c -2 10 Anthracite Anthracite Research 7.6<br />
Center, Schuylkill<br />
Haven, Pa.<br />
C -246 Subb, Laramie seam Erie, Colo. 38.5<br />
C -249 Lvb, Pocahontas #3 seam Stepheson, W. Va. 17.5<br />
C-252 Lignite, unnamed seam Beulah, N. Dak. 41.1<br />
C-254<br />
1/<br />
Pretreated hvab,-<br />
Pittsburgh seam<br />
Bruceton, Pa. 32.6<br />
- 1/<br />
Treated with air at ZOOo C.<br />
Table 2 shows the results <strong>of</strong> these tests. Lignite with 41 percent volatile<br />
matter produced the most hydrogen cyanide, 0.4 cu ft per cubic foot <strong>of</strong> ammonia<br />
consumed; activated carbon, containing the least volatile matter (2.0 percent),<br />
produced the least hydrogen cyanide, 0.007 cu ft per cubic foot <strong>of</strong> ammonia<br />
consumed.<br />
The <strong>chemical</strong> nature <strong>of</strong> the volatile matter and the oxygen content <strong>of</strong> the<br />
coal may also affect the hydrogen cyanide yield. The ratio <strong>of</strong> H, to CO in the<br />
<strong>of</strong>f gases varied from 2.3 to 1 for subb coal and 2.4 to 1 for lignite to 7 to 10<br />
to 1 for hvab. The higher carbon monoxide values obtained uith subb coal and<br />
lignite are due to the higher oxygen contents <strong>of</strong> these coals, being 17.1 and 20.3<br />
percent, respectively, compared with 8.0 percent for the hvab coal.
I<br />
b<br />
1<br />
I<br />
Table 2.- Data from Tests with Various Coals and Ammonia with Helium at 1.250" C<br />
I<br />
I Product gas, percent<br />
I Test HCNL' H2S & H2 02 N2 CO C02 Ct4 C2H4 C3H8<br />
I<br />
' C-246 Subb 5.4 0.0 0.0 64.2 0.0 4.9 28.0 0.5 2.4 0.0 0.0<br />
, C-249 Lvb 4.2 .O .O 78.9 .1 14.2 3.6 .O 3.0 .2 tr.<br />
C-252 Lignite 6.4 tr. .O 64.6 .1 6.1 26.3 .5 2.4 .O .O<br />
C-254 Pretreated, hvab 3.8 .1 .O 72.3 .8 10.6 14.3 .O 1.9 .O .O<br />
j C-210 Anthracite 0.2 .o .o 73.4 .3 21.4 2.9 .4 1.5 .o .o<br />
C-207 Activated carbon .25 .O .O 70.0 .O 23.0 6.6 .1 0.3 .O .O<br />
9 Feed<br />
i<br />
I<br />
He 9<br />
cu ft/hr<br />
NH3 9<br />
cu ft/hr<br />
Sol ids<br />
feed,<br />
lb/hr<br />
Total<br />
<strong>of</strong>f gas<br />
He-free,<br />
cu ft/hr<br />
'Length<br />
<strong>of</strong> run,<br />
min<br />
1 C-246 Subb 0.91 1.00 0.42 6.88 30<br />
C-249 Lvb .97 1.00 .47 3.92 30<br />
C-252 Lignite 1.06 1.00 .37-.40 6.38 15<br />
C-254 Pretreated, hvab 1.04 1.00 .35 5.80 30<br />
C -210 Anthracite 1.02 1.15 -37-.40 4.15 15<br />
f C-207 Activated carbon 1.06 1.15 .62 3.23 15<br />
/ Yield, cu ft<br />
1 HCN/cu ft HCN/cu ft<br />
J HCN/lb coal feed consumed<br />
,I C-246 Subb 0.88 0.37 0.37<br />
C-249 Lvb<br />
/ C-252 Lignite<br />
! C-254 Pretreated, hvab<br />
C-210 Anthracite > C-207 Activated carbon<br />
.34<br />
1.05<br />
.63<br />
.02l2/<br />
.013-<br />
.16<br />
.41<br />
.22<br />
.007<br />
.007<br />
.16<br />
.41<br />
.22<br />
.007<br />
.007<br />
I<br />
)<br />
L/<br />
- 21<br />
Wet-analytical method <strong>of</strong> analysis for HCN only; other components are the<br />
average <strong>of</strong> 2 chromatograph and 2 mass spectrometer analyses, HCN-free basis.<br />
Per pound <strong>of</strong> carbon.<br />
Tests were made to determine the effect that oxygen in the treating gas<br />
would have on the yield <strong>of</strong> hydrogen cyanide. It was thought that the heat for<br />
raising the temperature <strong>of</strong> the reactants to reaction temperature could be supplied<br />
by direct contact with a hot flue gas containing oxygen instead <strong>of</strong> by electric<br />
heating (table 3). In test C-125, a maximum yield <strong>of</strong> hydrogen cyanide was pro-<br />
duced for tests with air in the treating gas--0.12 cu ft <strong>of</strong> hydrogen cyanide per<br />
cubic foot <strong>of</strong> ammonia consumed, or 9.2 percent hydrogen cyanide in the product<br />
gas. When more than 0.5 percent. air was fed into the reactor, moisture condensed<br />
on the walls <strong>of</strong> the solids-collection flask; the yield <strong>of</strong> hydrogen cyanide de-<br />
creased. The moisture could have been absorbing the hydrogen cyanide since<br />
hydrogen cyanide is highly soluble in water. This approach was abandoned.
Table 3.- Product Gas Analyses and Yields <strong>of</strong> Hydrogen Cyanide from<br />
Tests with hvab Coal. Ammonia, and Air at 1,250" C<br />
Product gas, percent Length Feed<br />
<strong>of</strong> run, N&, Air, Coal,<br />
Test HC&' C& NH3 H2 N2 CO min cu ft/hr du ft/hr lb/hr<br />
C-123 10.3 1.1 10.2 62.8 16.2 9.7 20 5.90 0.52 0.17-0.20<br />
C-124 10.9 0.6 6.9 62.1 17.2 13.2 20 2.96 .53 .17- .20<br />
C-125 9.2 .4 9.6 48.4 31.5 10.1 20 3.08 1.08 .17- .20<br />
Total Yield, cu ft<br />
. <strong>of</strong>f gas, HCN/cu ft HCN/cu ft N&<br />
cu ft/hr HCN/lb coal NKq feed c on s ume d<br />
C-123 4.18 2.30 0.073 0.078<br />
C-124 3.05 1.77 .112 .120<br />
C-125 3.70 1.82 .110 .123<br />
- 1/ Wet-analytical method <strong>of</strong> analysis for HCN only; other components are the<br />
average <strong>of</strong> 2 chromatograph and 2 mass spectrometer analyses, HCN-free basis.<br />
Tests With Methane and Ammonia<br />
Tests were made without coal feed %ut with amnonia and methane to determine<br />
the resulting hydrogen cyanide yields for comparison. In one series <strong>of</strong> tests<br />
2.0 cu ft/hr <strong>of</strong> methane was reacted with ammonia in flows varying from 0.3 to<br />
2.5 cu ft/hr at 1,250' C. As illustrated in figure 4, yields <strong>of</strong> 0.18 to 0.61<br />
cu ft <strong>of</strong> hydrogen cyanide per cubic foot <strong>of</strong> ammonia consumed were obtained,<br />
equivalent to 1.2 to 13.8 percent hydrogen cyanide in the product gas. The<br />
yield <strong>of</strong> hydrogen cyanide reached a maximum at a feed ratio <strong>of</strong> methane to amonia<br />
<strong>of</strong> about 1 to 1.<br />
' A series <strong>of</strong> tests was made in which the methane and ammonia flow rates<br />
were maintained at 2 and 1 cu ft/hr, respectively, while the reactor temperature<br />
was increased from l,OOOo to 1,275' C. Hydrogen cyanide yields are plotted<br />
with temperature in figure 5, indicating increased hydrogen cyanide yields with<br />
increased temperature. Maximum yields <strong>of</strong> about 0.6 cu ft <strong>of</strong> hydrogen cyanide<br />
per cubic foot <strong>of</strong> ammonia consumed were obtained at the higher temperatures,<br />
while at 1,000" C only 0.07 cu ft <strong>of</strong> hydrogen cyanide per cubic foot <strong>of</strong> amnonia<br />
consumed was formed.<br />
To explore the use <strong>of</strong> coal-derived gases in the formation <strong>of</strong> hydrogen<br />
cyanide from amnonia, synthetic mixtures <strong>of</strong> a low-temperature carbonization<br />
gas, a coke oven gas, and a producer gas were prepared and reacted with amnonia<br />
at 1.250" C. Gas flows were adjusted to give a minimum methane-to-ammonia<br />
ratio <strong>of</strong> 1 to 1. In figure 6 the hydrogen cyanide yields obtained are plotted<br />
with the methane' contents <strong>of</strong> the coal gases. Increasing hydrogen cyanide yields<br />
were produced with increasing methane contents <strong>of</strong> the feed gas.<br />
\<br />
\
409<br />
ECONOMIC EVALUATION<br />
The Bureau <strong>of</strong> Mines Process Evaluation Group: Morgantown, W. Va., made a<br />
preliminary cost study <strong>of</strong> an integrated plant to produce hydrogen cyanide<br />
by reaction <strong>of</strong> ammonia with coal. The cost study was based on experimental<br />
results including a yield <strong>of</strong> 0.6 cu ft <strong>of</strong> hydrogen cyanide per cubic foot <strong>of</strong><br />
ammonia. Electrical heating was assumed as in the becch-scale tests; a plant<br />
capacity <strong>of</strong> 40 million pounds per year was chosen. The total estimated<br />
capital investment was $12,930,000 including costs for power generation.<br />
Based on a coal cost <strong>of</strong> $4.00 per ton and an ammonia cost <strong>of</strong> $100.00 per<br />
ton, the operating costs before pr<strong>of</strong>it and taxes would be 5.82 cents per<br />
pound <strong>of</strong> hydrogen cyanide product allowing byproduct credit. Addition <strong>of</strong><br />
12-percent gross return on investment would give production costs <strong>of</strong> 9.7 cents<br />
per pound <strong>of</strong> product when $4.00 per ton coal is used. The current market<br />
price is 11.5 cents per pound.81<br />
Credit has been allowed in the cost figures for a 7.6-percent yield <strong>of</strong><br />
carbon black and the excess char produced in the process. Some <strong>of</strong> the char<br />
and the scrubbed product gas (containing about 75 percent hydrogen) are<br />
consumed in the steam plant for power generation. Electrical heating, which<br />
was used in the test unit and also in the cost figures, is one <strong>of</strong> the most<br />
expensive types <strong>of</strong> heating, accounting for greater than 40 percent <strong>of</strong> the<br />
capital costs in the estimate. If cheaper conventional heating could be used,<br />
production costs would be lowered considerably.<br />
CONCLUSIONS<br />
Hydrogen cyanide has been produced from coal and amnonia at 1,250" C in<br />
bench-scale studies. The use <strong>of</strong> a metal reactor was unsuccessful because the<br />
metal failed at the temperatures required, and the yield <strong>of</strong> hydrogen cyanide<br />
was low. The yield was improved greatly when a refractory ceramic reactor was<br />
used.<br />
Hydrogen cyanide yields approximating stoichiometric <strong>of</strong> 1 cu ft <strong>of</strong> hydrogen<br />
cyanide per cubic foot <strong>of</strong> ammonia reacted were obtained at low flows <strong>of</strong> ammonia.<br />
At higher amnonia flows, ammonia conversion <strong>of</strong> about 75 percent was obtained,<br />
which is the usual conversion attained in commercial units using natural gas<br />
and a platinum catalyst.<br />
The low-volatile coals gave low yields <strong>of</strong> hydrogen cyanide; the high-<br />
volatile coals gave the best yields. The results indicated that the hydrogen<br />
cyahi.de is produced by reaction <strong>of</strong> amnonia with the hydrocarbons in the coal.<br />
Yields <strong>of</strong> hydrogen cyanide from reaction <strong>of</strong> ammonia with gas mixtures con-<br />
taining methane are directly related to the methane content <strong>of</strong> the gas.<br />
Cost studies indicate that hydrogen cyanide can be produced from coal<br />
and aunnonia at a price approximating the posted sales price.
410<br />
BIBLIOGRAPHY<br />
1. American Public Health Association, Inc. Standard Methods, for the<br />
Examination <strong>of</strong> Water and Wastewater. 11th ed., 1960, pp. 355-,356.<br />
2. A.S.T.M. Standards with Related Material. Gaseous Fuels, Coal and Coke.<br />
Part 19, 1966, 470 pp.<br />
3. Chemical Week, Market Newsletter, v. 99, No. 23, Dec. 3, 1966, p. 65.<br />
4. Chemicals, Quarterly Industry Report, U.S.. Business and Defense Services<br />
Admin., v. 12, No. 4, Dec. 1965, p. 40.<br />
5. Current Industrial Reports, Inorganic Chemicals and Gd'ses 1964. U. S.<br />
Commerce Department, Series M-28A, Released Jan. 28, 1966, p. 6.<br />
6. Fieldner, A.C., and W. A. Selvig.. Met!iods <strong>of</strong> Analyzing Coal and Coke.<br />
BuMines Bull. 492, 1951, 51 pp.<br />
7. Fugate, W.O. Hydrogen Cyanide, pp. 574-585 in v. 6 Encyclopedia <strong>of</strong><br />
Chemical Technology. Interscience Publishers, New York, N.Y., 1962,<br />
2d ed., ed. by R. E. Kirk. and D.O. Othmer.<br />
8. Oil, Paint and Drug Reporter. V. 189, No. 23, June 6, 1966, p. 20.<br />
9. Sherwood, Peter W. Synthesis <strong>of</strong> Hydrogen Cyanide by Autothermic Reaction.<br />
Refining Engineer, v. 31, No. 2, February 1959, pp. C-22-26.<br />
10. U. S. Tariff Commission, Synthetic Organic Chemicals, U.S. Production<br />
and Sales:<br />
1960 - Tariff Comission Publication No. 34, p. 53, published 1961.<br />
1961 - ditto 72, p. 53, published 1962.<br />
1962 - do. 114, p. 56, published 1963.<br />
1963 - do. 143, p. 55, published 1964.<br />
1964 - do. 167, p. 55, published, 1965.<br />
All'published in Washington, D. C.<br />
\<br />
,<br />
1<br />
i
W<br />
0
Figure 2. Enclosure surrounding hydrogen cyanide unit.<br />
1<br />
‘1
Figure 3. Hydrogen cyanide reactor.
0 0<br />
f v)<br />
C<br />
0<br />
u<br />
m<br />
I<br />
z<br />
2<br />
c<br />
2<br />
r<br />
d<br />
J<br />
w<br />
*<br />
z<br />
V<br />
I<br />
0.f<br />
5 ..<br />
.4<br />
7 .-<br />
.2<br />
.I<br />
CG-6<br />
- _ ..<br />
I I<br />
44<br />
C%-3<br />
/<br />
0 0.5 I .o 1.5 2 .o<br />
NH, FLOW,scfh<br />
12 -7- 66 L-9726<br />
I
C<br />
:G-l I<br />
I<br />
00 1,100 1,200<br />
TEMPERATURE, OC<br />
1<br />
0<br />
CG-3 /<br />
12-8- 6 6 L-9727
0. E<br />
c<br />
.4<br />
7 ..<br />
.c<br />
. I<br />
(<br />
-<br />
Low temperature<br />
carbonization gas<br />
/<br />
I I<br />
20 40 60 80 IO0<br />
CH, IN FEED GAS, percent.<br />
I2 -7- 66 L-3728<br />
\<br />
a<br />
\<br />
i
417<br />
h6SSBAUER SPECTROSCOPY OF IRON IN COAL<br />
John F. Lefelhocz*, Robert A. Friedel' , and Truman P. Kohmd<br />
*Depafiment <strong>of</strong> Chemistry, Carnegie Institute <strong>of</strong> Technolow<br />
'Pittsburgh Coal Research Center, Bureau <strong>of</strong> Mines, U.S. Department <strong>of</strong> the Interior<br />
Pittsburgh, Pennsylvania 15213<br />
INTRODUCTION<br />
Metallic elements (Na, Mg, Al, Si, K, Ca, Ti, Fe) which occur abundantly in<br />
the ash obtained fron the combustion <strong>of</strong> coals are present in the original coals<br />
partly as inorganic constituents or minerals. The identification <strong>of</strong> minerals<br />
present usually does not account quantitatively for several <strong>of</strong> these elements, and<br />
it is supposed that some sort <strong>of</strong> organic bonding with the coal is involved. The<br />
nature <strong>of</strong> this bonding in most coals has not been determined. Changes in the infrared<br />
spectra <strong>of</strong> lignites and brown coals following acid and alkali treatment indicate<br />
the presence <strong>of</strong> metallic salts <strong>of</strong> carboxylic acids (1,2,3). For higher rank coals,<br />
iittle or nothing is known about the siructures <strong>of</strong> the metallic elements<br />
organicalljr bound". In the case <strong>of</strong> iron, for example, it has not been determined<br />
whether the so-called "organict1 iron is in the ferrous or ferric state, to what<br />
Element fr elements the iron is bonded, or whether there is more than one form <strong>of</strong><br />
,<br />
organic iron.<br />
The term "organic" must be interpreted with care; the nature <strong>of</strong><br />
the bonding is uncertain, and presumbly could be ionic, coordinate, or organo-<br />
metallic.<br />
Mineral components oontaining iron can <strong>of</strong>ten be identified petrographically.<br />
Pyrite (FeS2) is common, both as distinct nodules and as veins, <strong>of</strong>ten intertwined<br />
with carbonaceous macerals. An electron microprobe study <strong>of</strong> several coals (4)<br />
revealed, however, many examples which lacked a correlation between Fe and S<br />
distributions. In some cases Fe was distinctly associated with Si and AI.,<br />
suggesting incorporation in an aluminosilicate gel or kaolinite. A strong<br />
parallelism <strong>of</strong> Fe and Ca (but not Si, Al, K, or S) in one specimen suggested the<br />
presence <strong>of</strong> Fe as carbonate or possibly oxide. In some cases the Fe shared<br />
fairly uniform distribution with no ftpparent correlation with other ash-forming<br />
elements. This presumably could be organic" iron.<br />
M'dssbauer spectroscopy has been employed to study the <strong>chemical</strong> properties <strong>of</strong><br />
iron in a great variety <strong>of</strong> natural materials: oxides and oxyhydroxides (5,6),<br />
sulfides (7,8) , numerous silicate minerals (5,6,9,10,11,~)~. ilmenite and related<br />
titanium minerals (5,13), siderite (6), jarosites (5,14), lollingite (15); ordinary<br />
chondrite meteorites (16) , carbonaceous chondrites (13, an achondrite (16), and<br />
several tektites (11).<br />
Recent general articles on Mzssbauer spectroscopy (18,19,20,21,22) discuss<br />
the interpretation <strong>of</strong> spectra in terms <strong>of</strong> the <strong>chemical</strong> state <strong>of</strong> iron, including<br />
its oxidation state, bonding, and environmental symmetry. The isomer shift (d),<br />
quadrupole splitting (A), line width (r), line intensities, and a comparison <strong>of</strong><br />
the spectra obtained at room temperature and at liquid-Ng temperature are useful.<br />
We have undertaken Mgssbauer studies to characterize non-mineral iron in
418<br />
cual. A mjor aa\;antage or' the bf6ssbauer technique is that the iron is observed<br />
without alteri:ig its <strong>chemical</strong> state or eniiiroluuent. The relatively low atonic<br />
nilnber Oi' the mjor deinents present :Bakes it possible for small: amounts <strong>of</strong> iron<br />
to >e obser.;ed in rather large amoun':,s 01' cml. Sa:nples <strong>of</strong> whole coal and vitmin<br />
as free as 2ossible from ninerals xere selected in wder to iyiinimize mineral-iron<br />
interzerence, and a I'ew saiqles vhose <strong>chemical</strong> analysis indicated "organic" iron<br />
were included.<br />
I<br />
ES'ERIMENTAL PROCEDURES<br />
Nine specixens, yritrain or vhole-coal, <strong>of</strong> seven coals with rank from lignite<br />
(72:. C) to anthracite (93;: C) were ased. Their designations and geographic origins<br />
are given in Table 1, in order <strong>of</strong> increasing carbon content.<br />
Table 2 is a compilation <strong>of</strong> the analytical data for C, H, N, S, Fe, ash, and<br />
0 (by difference) on samples <strong>of</strong> these materials, as determined by the Coal Analysis<br />
group at the Bureau <strong>of</strong> Mines. The total iron in the coal was determined by ash<br />
analysis. HC1-soluble iron was determined by treating a separate powdered sample<br />
with 23;; HCl to extract any iron present as carbonates, oxides, sulfates, etc.<br />
Pyrite iron was then removed by treating the HC1-leached coal with 25;; €IN& to<br />
dissolve the iron conbined with sulfur; the extract was evaporated to dryness to<br />
expel oxides <strong>of</strong> nitrogen, and the residue was dissolved in HC1. In each case the<br />
iron was reduced with SnC12, the slight excess <strong>of</strong> which was eliminated with HgC12.<br />
"he reduced iron was titrated with potassium dichromate. "Organic" iron was then<br />
calculated by subtracting the two acid-soluble iron contents from the total iron.<br />
The Zesulting iron contents are listed in Table 2 as: total, HC1-soluble, pyrite,<br />
and organic .<br />
MEssbauer spectra were obtained on coal samples that were ground in a mortar<br />
and pestle. A cylindrical cell, open on one end with a circular aperture <strong>of</strong><br />
7.14 em2, was made by gluing a sheet <strong>of</strong> lucite 1/52" thick with Duco cement to<br />
the bottom <strong>of</strong> a circular wall mde from a paper card. A 3.50-g sample was then<br />
placed into this cell. The top window, also 1/32" lucite, was then cemented in<br />
place. The sample mass per unit area was thus held oonstant at 0.4% g cmm2.<br />
Inorganic iron compounds and mineral samples were ground and mounted either<br />
between two lucite sheets held together with Duco cement, or by mixing the solid<br />
with acetone and Duco cement and alluwing this mixture to harden on a lucite sheet.<br />
This latter method was used only when the sample would not interact with acetone<br />
or the cement. The area for these samples was also 7.14 cm2. All <strong>of</strong> the materials<br />
used in this investigation contained natural iron with presumably 2.19 atom $ FeS7.<br />
The M'dssbauer spectrometer incorporates a Nuclear Science and Engineering<br />
Corporation Model-B lathe-type drive modified in this laboratory for automatic<br />
operation. The operating mode employs constant velocit advanced in increments<br />
<strong>of</strong> C.05 mm s-'. The detector <strong>of</strong> the 14.4-kev Co57 -. Fegf gamm radiation is a<br />
Reuter-Stokes proportional tube containing lo$ methane in krypton, feeding through<br />
a single-channel analyzer set at 11 - 17 keV. Absorbers were mounted on the<br />
moveable table pe endicuLar to the radiation beam.<br />
The stationary source consists<br />
<strong>of</strong> - 5 di <strong>of</strong> co5Fdirfused into chromium metal. A Bird-Atcrmic scanning count<br />
integrator and Varian chart recorder plot the number <strong>of</strong> counts in a constant time<br />
interval at each velocity in succession.<br />
Powdered samples <strong>of</strong> sodium nitroprusside gave an isomer shift, relative to<br />
the CoS7-Cr source, <strong>of</strong> -0.11 mm s-l, and a uadrupole splitting <strong>of</strong> 1.68 m &I-'.<br />
With this source a line width <strong>of</strong> 0.25 mm s-' was obseNed with an absorber containing<br />
5 ny cmS <strong>of</strong> Fe as K4Fe(CN)e.3&0. The estimated uncertainty in the velocity<br />
setting is about 2: <strong>of</strong> the velocity and the estimated uncertainty in derived<br />
spectral parameters is - 0.05 mm sL.<br />
The counting time at each velocity was - 5
I n<br />
\D<br />
r-<br />
m<br />
d<br />
c<br />
d<br />
rl<br />
rl<br />
n .<br />
aJ<br />
2<br />
C<br />
W<br />
c,<br />
d<br />
X . a<br />
d<br />
t-<br />
0<br />
2<br />
Po<br />
.><br />
E.<br />
r: .<br />
2<br />
c,<br />
d<br />
><br />
r:<br />
aJ<br />
c,<br />
E<br />
d<br />
PI<br />
? ?<br />
0 0<br />
no\<br />
(d'<br />
?<br />
5<br />
P<br />
d<br />
F O x n<br />
(v<br />
k<br />
4<br />
2<br />
d<br />
u<br />
0<br />
c,<br />
c<br />
I1<br />
?I<br />
t
420 Ir<br />
?Y ? ? 9 9 9<br />
pc rod n o c o o 0<br />
Y<br />
0 00 M O<br />
. o<br />
0<br />
-f.<br />
H ' O 9<br />
0 I O I O<br />
0 0 0<br />
0;'<br />
N<br />
c9<br />
0<br />
9<br />
rl<br />
;?<br />
N<br />
9<br />
rl<br />
m<br />
0.<br />
rl<br />
f co'O<br />
? E??<br />
0 0 0<br />
f n o<br />
9 ?Y<br />
0 0 0<br />
O F lnd O d<br />
'?? ?cu. ? ?<br />
0 0 0 0 0 0<br />
I
L.<br />
421<br />
minutes, yielding c 600,000 counts and a relative standard deviation < 0.135.<br />
The velocity region scanned for each sauple was from -2.OC mm s-l to about<br />
+ 4.00 mm s-'.<br />
Most measurements were made a rooi; temperature. A cryostat mde from<br />
I' S t y r O f a $as mounted on the moveable table 0: the instrument ;or low-terqerature<br />
I measurements. The sample was placed in this and submerged in liquid N2. The<br />
, spectrum <strong>of</strong> each coal sample was run twice at room temperature, befcke and after<br />
, the spectm was taken at liquid42 temperature.<br />
Spectral parameters were generally read from plotted spectra. The data from<br />
sample F(a) were fitted to a double Lorentz curve by a least-squares program using<br />
J an IB1 7C40 computer. Isomer shifts are reported (in m s-') with respect to the<br />
, ismer shift <strong>of</strong> sodium nitroprusside. Quadrupole splittings are reported (in mm s-')<br />
as the velocity differences between the minima <strong>of</strong> two associated absorption lines.<br />
I DATA<br />
/<br />
Room-temperature spectra for the nine coal samples are illustrated in Figure 1.<br />
J Each spectrum shows neither, either, or both <strong>of</strong> just two components: (1) a close<br />
doublet similar to those <strong>of</strong> pyrite and mrcasite, and (2) a wide doublet similar<br />
to those <strong>of</strong> many ferrous compounds. Table 3 lists the Mossbauer paramters,<br />
including the fractional peak absorptions, for what we will call respectively the<br />
pyrite and non-pyrite resonances. Isomer shifts and quadrupole splittings obtained<br />
/ with our instrument on sane powdered iron campounds and minerals which were<br />
regarded as possibilities for the non-pyrite spectrum appear in Table 4.<br />
J<br />
/ The parameters observed for the pyrite iron are: isomer shift (d) = +0.54<br />
mm s-l; quadrupole splitting (A) = 0.58 m s-I. All <strong>of</strong> the coals having a non-<br />
' pyrite absorption as shown in Figure 1 have: d = +l.38 mm s-'; A = 2.62 mm s-'.<br />
p' The computer analysis <strong>of</strong> sample F(a) indicated both lines in the spectrum had<br />
c widths (r) <strong>of</strong> 0.39 mm s-l; their intensity ratio is within 57; <strong>of</strong> unity.<br />
I<br />
i Liquid-N2 spectra showed the same absorption peaks as at room temperature,<br />
f with d = +0.65 m s-l and A = 0.58 mm s-l for pyrite and d = +1.47 mm s-' and<br />
> A = 2.78 mm s-l for the non-pyrite iron.<br />
INTERPRETATION<br />
i<br />
Y The isomer shift is a function <strong>of</strong> nuclear properties and the electron density<br />
at the absorbing nucleus relative to that <strong>of</strong> the source or a standard absorber,<br />
such as sodium nitroprusside (23,24). As the electron density at the iron<br />
nucleus, due essentially only to s electrons, increases, the isomer shift decreases<br />
'[algebraically. Thus, ferrous compounds have a more positive isomer shift than<br />
ferric compounds, as the additional 3d electron <strong>of</strong> the former increases the<br />
d shielding effect on the 3s electrons and thereby decreases their density at the<br />
f nucleus (18). For ferric iron, isomer shifts (relative to sodium nitroprusside)<br />
\ have been observed in the range 4.1 to +1.1 mm S'l, and for ferrous iron from<br />
-0.1 to +1.6 mm S-'.<br />
i;<br />
I<br />
Quadrupole splitting into a two-line spectrum occurs when the iron nucleus<br />
he field consists <strong>of</strong> two parts, (1) that<br />
f is in an asymmetric electric field.<br />
i produced by the electrons <strong>of</strong> the iron atom, including those shared with adjacent<br />
1 atoms, and (2) that resulting from charges <strong>of</strong> the surrounding atoms; each part<br />
J can contribute to asymmetry at the nucleus. Ferric compounds shm quadrupole<br />
splittings from zero to about 2.3 nun s-l; here the asymmetry is caused chiefly by<br />
charges <strong>of</strong> the surrounding atoms. Ferrous compounds show larger values, frm zero<br />
Ferrous iron can exist in either a high-spin or a low-spin<br />
J<br />
1<br />
i<br />
I<br />
r<br />
I<br />
to - 3.3 mm s-I.
422<br />
Table 3. Room-texperature M'dssbaaer spectral parameters for coal samples"<br />
-<br />
?,TiL c iron I'Jon-Pyrite iron<br />
Saqle 1so:ner Quadru2ole Fractional Is omer Quadrupole Fractional<br />
No. shift I s2licting absorption shift t<br />
abs orpt ion<br />
w&iyy<br />
(Ilm s-1 j ('L"1 s-1 ) 0 (m 3-1) (:?I<br />
A (a) - - N.O.* - - N.O.*<br />
B (a) C.->, ./ 1.5 - - N.O.<br />
c (3) - -<br />
N.O. 1 *39 2.65 2 -3<br />
(b) - N.O. 1.3LJ 2.63 2.1<br />
- - N.O.<br />
D (a) G.;l 0.>5 /.J<br />
(b) 0.23 0.4. 3.6 - - N.O.<br />
E (a) 0.3G 0.32 22.7 1.41 2.70 2.6<br />
(b) 0.A G.3 2 -3<br />
- - N.O.<br />
(c)<br />
F (a)<br />
(b)<br />
o.>c<br />
-<br />
-<br />
0.,3<br />
-<br />
-<br />
1.7<br />
N.O.<br />
N.O.<br />
-<br />
1.38<br />
1-39<br />
-<br />
2.62<br />
2.65<br />
N.O.<br />
11.0<br />
5.5<br />
\<br />
\<br />
\<br />
G (a) - -<br />
N.O. 1.38 2.65 1.5<br />
(b) - - N.O. 1.38 2.65 1.0<br />
H (a) 0.54 0.60 1.3 1-39 2.65 1.5<br />
(b) 0.59 0.66 1.7 1.36 2.60 1.7<br />
- - -<br />
I (a) - N.O. N.O.<br />
(b) 0.56 0.65 1.0 - N.O.<br />
* Weighing 3.50 and distributed evenly over -(.14 em2.<br />
May include marcasite.<br />
t With respect to center <strong>of</strong> sodium nitroprusside spectrum.<br />
i- Zero-point uncertain by 0.10 m s-'.<br />
t N.O. = not observed; in general, < O.y{.<br />
..<br />
i<br />
\<br />
\<br />
\<br />
\<br />
!<br />
\<br />
\
I 1 1 \<br />
......... ...U ...... .."."..-*....'-... .U!.:; ........ .............. a;. ...... (a) *<br />
. .<br />
. .............. ................<br />
...........................................<br />
;(a)<br />
.. ....<br />
.... ................................... .............<br />
'(a) ............. ... .<br />
..<br />
....<br />
.....<br />
........ . .'<br />
)(b) .......................... ............. ........ '.'.: .............. I.;.<br />
.......<br />
..................<br />
.......<br />
.. ... ..<br />
...<br />
' . .."*. '.<br />
*.<br />
:(a)<br />
.....<br />
.'<br />
' *<br />
..<br />
..<br />
,- ........... -4. .........<br />
c(b)**...,.**, .*.... ...... ............... . -..- ..... ..:.. .........<br />
... ......<br />
3( c) . ..-..-- ... r. ................ .............. ....,\. " ...................... . ............ ':."'...... ...<br />
qa) . :. ... .**'..'...... ................ .e.'.* .<br />
.. e.<br />
o(a) .- .a, . .............<br />
. .<br />
.* .* .... .'<br />
. '<br />
9 .<br />
. .<br />
* .<br />
9 .<br />
............................... .... *...........<br />
.......... . ";* ...,... ....<br />
....... .*L ......" ..... .. ...... ...... ................. . ..................<br />
.'.a ., ,.e<br />
ab) '*"'"' ' -<br />
9. .*. .,.+ 99.- .<br />
.....<br />
I (*) ................. ............., ........ ......... .:*.e :-* . .....<br />
-2<br />
1 I<br />
I I<br />
vgIx)cITY (mm e-')<br />
. ...*.-....*<br />
.. . ...-.* :*.:9.*,<br />
Figure 1. M'ksbauer spectra <strong>of</strong> coal samples. In maet -see, sach point<br />
represents - 600,000 counts. The m c e was Cos' diffused into chromlm<br />
metal, To convert the velocity scale to the sodium nitroprusside scale,<br />
add 0.11 mm 8' to the indicated values.<br />
I<br />
I<br />
b<br />
1
424<br />
state, depending on the strength <strong>of</strong> the ligand field around the central iron<br />
atom. In the high-spin configuration the large asymmetry <strong>of</strong> the valence shell<br />
results in considerable asymmetry at the nucleus; low-spin ferrous compounds have<br />
smaller field asynrmetries, which are due chiefly to the external environments only.<br />
Pyrite and mrcasite, respectively stable and metastable Tom <strong>of</strong> FeS2, are<br />
low-spin ferrous compounds. Isomer shifts and quadrupole splittings observed in<br />
this laboratory (Table 4) are in agreement with those <strong>of</strong> Temperley add Lefevre (8).<br />
The parameters <strong>of</strong> the two minerals are so similar that the spectra cannot be<br />
resolved when both are present. A comparison <strong>of</strong> the data in Tables 3 and 4<br />
confirms the conclusions fran petrographic (25) and x-ray diffraction (26) studies<br />
that the iron sulfide in coal consists minly <strong>of</strong> pyrite.<br />
From the Mksbauer data on the non-pyrite iron Observed in several coal<br />
samples (Figure 1 and Table J), it can safely be concluded that this iron is in a<br />
high-spin ferrous state. Furthermore, the combination <strong>of</strong> such high values <strong>of</strong><br />
both isomer shift and quadrupole splitting has been reported only in octahedrallycoordinated<br />
high-spin ferrous compounds, so it is highly probable that the nonpyrite<br />
iron in coals is in octahedral coordination. Neither the analytical nor<br />
the Mksbauer data yields a distinction between inorganic or orgsnic minerals or<br />
compounds.<br />
The natural line width (r,) <strong>of</strong> the FeS7 gamma radiation corresponds to a<br />
velocity dlfference <strong>of</strong> 0.0973 mm s-' (27), and the minimum observable line \width<br />
(I') in a MGssbauer spectrum is twlce this, 0.195 mm s-'. Inhomogeneities in the<br />
source and absorber, instrumental "noise", unresolved quadrupole splitting, and<br />
atomic spin-spin relaxation effects (22,28,29) increase the apparent line widths,<br />
and further widening occurs with thick absorbers (30). The minimum width ye have<br />
observed with our instrument is 0.25 mm s-l for a very thin absorber.<br />
The value<br />
<strong>of</strong> r = 0.39 mm sa observed for the strongest non-pyrite iron spectrum (sample F(a))<br />
indicates that this doublet is caused by a fairly well-defined iron compound or<br />
mineral, though some inhomogeneity may be present.<br />
A large number <strong>of</strong> iron compounds have strong M&sbauer absorptions (large<br />
recoil-free fractions) at room temperature. Some ferrous compounds show little<br />
or no absorption at roan temperature, but at liquid42 temperature the intensity<br />
is strongly enhanced (31J2). We have observed tNs effect with ferrous stearate,<br />
which must be cooled to liquid-N, temperature before resonant absorption is<br />
detected. Eerzenberg and Toms (5) have observed that samples <strong>of</strong> y-Fe;?% and<br />
d-FeOOH give nonresonant absorption at roan temperature. These would probably<br />
exhibit an effect at liquld-N2 temperature. Lack <strong>of</strong> such an effect in the coals<br />
is interpreted to man that there are no compounds present in significant amounts<br />
that do not have appreciable resonant absorption at room temperature.<br />
Isomer shifts and quadrupole splittings depend on the temperature. A<br />
second-order Doppler effect (22) decreases the isomer shift 8s the temperature is<br />
increased. Quadrupole splittings for high-spin ferrous canpounds are affected<br />
by temperature much more strongly than those <strong>of</strong> other iron ccmpounb because the<br />
population <strong>of</strong> the dc levels <strong>of</strong> iron in an octahedral field is determined by a<br />
BoltzlarM distribution (18,22). For example, FeS04"&0 shows a change <strong>of</strong> d<br />
from +1.53 to t1.56 mm s-l and <strong>of</strong> A frm 3.19 to 3.47 mm s" in going from roam<br />
to liquid-nitrogen temperature (18).<br />
The observed change in the non-pyrite iron<br />
spectrum in car1 <strong>of</strong> d from +l.38 to +1.47 mm 6-l ana <strong>of</strong> A fran 2.62 to 2.78 m sa<br />
is in agreement with our assignnent to this class <strong>of</strong> compounds.<br />
In some iron compounds where the M&mbauer absorption ordinarily shows a<br />
6-line hyperfine structure as a result <strong>of</strong> a magnetic field at the nucleus, very
;<br />
i 1<br />
1<br />
1<br />
finely conminuted samples show instead a two-line pattern at room temperature as 8<br />
resilt <strong>of</strong> them1 disruption <strong>of</strong> the cacroscopic magnetic domains. KKndig et al.<br />
(33) observed this effect in a-Fe203 in particles <strong>of</strong> - 50-8 diameter, but at<br />
1iqdid-N~ teqerature the b-line pattern was observed. The absence <strong>of</strong> any magnetic<br />
splitting in the coal spectra at roo-or 1iq~id-N~ temperatures probably rules out<br />
the possibility that the non-pyrite doublet is caused by a magnetically ordered<br />
msterial (such as Fe203, Fea04, FeC3, FeS, or metal) present as very1 small particles.<br />
, Inequality in the intensity <strong>of</strong> the components <strong>of</strong> a doublet my result frm<br />
the ardsatropy <strong>of</strong> the absorption cross section relative to the crystal axes when<br />
(1) a single-crystal absorber is oriented prererentially with respect to the<br />
d optical axis, or (2) there is snisotropy in the recoilless fraction <strong>of</strong> the split<br />
312 state; the latter condition results in unequal absorption even with powdered<br />
' samples (Coldanskii effect) (20,21). Most <strong>of</strong> the samples used in this work were<br />
'<br />
powdered, so only the second effect could be operative, except for biotite, which<br />
was mounted as a sheet. The essential equality <strong>of</strong> intensity <strong>of</strong> the two nonpyrite<br />
coal spectrum lines would eliminate any compounds showing unequal absorption<br />
I in powdered samples as possible causes <strong>of</strong> this absorption.<br />
( At the present state <strong>of</strong> Mgssbauer spectroscopy, the identification <strong>of</strong> an<br />
unknown compound from its spectrum can only be made by finding a ham compound<br />
havlng the same spectrum and temperature dependence. We have been unable to find<br />
1 any published combination <strong>of</strong> spectral parameters, for either natural or synthetic<br />
iron compounds, which match that <strong>of</strong> the non-pyrite iron in coal. Ankerite<br />
,I<br />
(Table 4), which has not been reported previously, likewise does not match.<br />
I '<br />
In Figures 2 and 3 we have plotted A versus d for all single- or two-line iron<br />
spectra for which we have been able to obtain data (5,6,~,9,10,11,14,18,31,34,35,<br />
+,36,37,38,39,40). Attention is called to the variations in d and A within isomorphous<br />
silicate series (olivines, pyroxenes) in which the Fe/Mg ratio varies. Many<br />
silicate minerals show two or more coupled Mossbauer doublets, ellnlnating from<br />
consideration nany points near the coal point on the plots. Biotite gives an<br />
apparent 2-line spectrum with d and A similar to the coal spectrum, but with<br />
,I decidedly unequal intensities (our measuement), which persist in powdered samples<br />
'<br />
(6), and which can be resolved into a coupled doublet (g), whereas the coal<br />
/ spectrum is symmetrical. Even considering the compositional variations and the<br />
uncertainties <strong>of</strong> measurement, it is apparent that the non-pyrite iron in coal is<br />
r' distinct from any compound whose Mossbauer spectrum is known. Minerals excluded<br />
>, include oxides and oxyhydroxides, sulfides and related compounds, the carbonates<br />
I<br />
siderite and ankerite, titanian minerals, and the many silicates examined.<br />
4'<br />
Some ferrous complexes <strong>of</strong> pyridines (35) and 1,lO-phenanthrolines (37) have<br />
similar thmgh non-matching d and A values. These cmparisons suggest that the<br />
non-pyrite iron could be bound to heterocyclic nitrogen aromatic groups in the<br />
coal macemls, or possibly in a clay-like silicate mineral or gel.<br />
f<br />
,<br />
. An attempt was made to correlate the Mzssbauer absorption intensities in the<br />
coals wlth the analytical data. M'dssbsuer spectra were determined on mixtures <strong>of</strong><br />
pyrite in carbon, and the fractional peak absorption was plotted awnst the mas8<br />
fraction <strong>of</strong> pyrite iron. In a few coal samples the M'dssbauer absorption and the<br />
1; analytically determined amount <strong>of</strong> iron as "pyrite" did agree with the above plot,<br />
but in others the agreement was poor. This may be due to the inadequscy <strong>of</strong> the<br />
I <strong>chemical</strong> method for determining pyrite iron, and to differing mrtrix effects in<br />
the coah and the standards. Likevise, a poor correlapn WBB found between the<br />
non-pyrite iron absorption in coals and the amount <strong>of</strong> organic'' iron deduced fran<br />
the <strong>chemical</strong> analyses, although the sample with the highest 'organic" iron,<br />
Jewell Valley coal F(a), showed the strongest non-pyrite Morrsbauer spectrum.<br />
i
Table 4. Room-?;ergerature isomer shifts and quadrupole splittings for<br />
po1ycr;stalline iron compounds and n;inerals<br />
This Work Gt her Work<br />
Compound or hlineral d*> hS d+; AS Ref. (<br />
Pyrite, FeS2<br />
Marcasite, FeS,<br />
Siderite, (Fe,Mg)Ch<br />
Ankerite , ( Ca, Mg , Fe )COS<br />
Olivine, (Mg,Fe)zSiOr<br />
Biotite, K(Mg,Feh(AlSbOlo)(OH)2<br />
Tomline, Na(Mg,Fe )3Als(BO3 13 (Sieole) (OH14<br />
Ferrous sulfate, FeSO4'7HzO<br />
Ferrous acetate, Fe( C2H302 )Z<br />
Ferrous stearate, Fe(C1&502)2<br />
Non-pyrite iron in coal<br />
0.54<br />
0.51<br />
1.47<br />
1.46<br />
1.39<br />
1.36<br />
1.30<br />
1.56<br />
1.45<br />
o.66t<br />
1.38<br />
0.60 0'- 55<br />
0.50 0.52<br />
1.78 1.47<br />
1.50 -<br />
2 -95 1.39<br />
2.50<br />
1.27<br />
1.46<br />
Fe+2 1.19<br />
2.38 Fe" 1.44<br />
Fe*3 0.99<br />
3 -05 1-33<br />
2 -23 -<br />
0.70t -<br />
2.62 -<br />
0.61<br />
0.51<br />
1.80<br />
-<br />
3.04<br />
2.41<br />
2.81<br />
2.10<br />
2.61<br />
0.91<br />
* Respect to center <strong>of</strong> sodium nitroprusside spectrum. ,<br />
t A t liquid nitrogen temperature; no resonant absorption observed at room temperature. ]<br />
9 In m s-'. \.<br />
3<br />
3 e 1 9<br />
-<br />
-<br />
4<br />
i
,'<br />
I<br />
t<br />
f<br />
I<br />
i<br />
I<br />
1<br />
$-<br />
i<br />
h<br />
4<br />
m<br />
3<br />
1<br />
d = ISOMER SHIFT (mm s-')<br />
aigure 2. Representati.re plot <strong>of</strong> quadrupole splitting versus isomer shift<br />
(relati-re to sodium nitroprusside) for: - ferric compounds (A)<br />
and ninerals (A); ----- ferrous compounds (0) and minerals (0);<br />
ferrate compounds (x), and the non-pyrite iron found in coal (*).<br />
+1<br />
i2
I' 1.3<br />
. /o<br />
9<br />
IO<br />
9<br />
6<br />
a<br />
425<br />
F<br />
F<br />
33 I<br />
3<br />
*9 1 - 1 a . a ,<br />
1'. 0 1'. 1 1'. 2 1'- 3- 1'. 4 1'.5 1:6<br />
d = ISOMER SHIFT (mm S-l)<br />
Figure 3. Enlarged section <strong>of</strong> Figure 2, showing quadrupole splitting versus isomer shift<br />
relative to sodium nitroprusside for two-line ferrous spectra. Lines connect pairs <strong>of</strong><br />
points which are observed together in the same mineral or compound. Dsshed lines indicate'<br />
that a ferric spectrum is associated with the plotted ferrous point. Synthetic compounds:<br />
1-ferrianite (synthetic mica), 2-synthetic pyroxenes, 3-synthetic olivines, 4-welding glass<br />
>-germanium spinel, 6-oxy-salts, 7-carboxylates, 8-halides, 9-pyridine complexes, 10- \<br />
phenanthroline complexes, ll-ferrous hemoglobin. Natural mterials: A-sctinollte, B-<br />
biotite, C-clay minerals, F-fayalite and olivine, M-cllnopyroxenes, P-orthopyroxenes, \<br />
R-ankerite, S-siderite; T-tourmaline, X-tektites, *-non-pyrite iron in coal.<br />
'I<br />
8<br />
6<br />
6<br />
&<br />
8
CONCLUSION<br />
429<br />
In sumnary, several coal sanples contain a form <strong>of</strong> iron which exhibits a<br />
hitherto unobserved M'dssbauer spectrum, in addition to the well-known pyrite<br />
spectm. Tne non-pyrite iron is ferrous, in a high-spin configuration, in<br />
cctahedral s;metry, and apparently in a rather well-defined state Comparisons<br />
with known compounds suggest that this iron my be bound in the cdl macerals to<br />
heterocyclic nitrocen aromatic groups, though a clay-like silicate mineral or gel<br />
is a possibility. iicssbauer spectrometry can at the very least indicate the<br />
most suitable coal samples for further studies <strong>of</strong> this iron by <strong>chemical</strong> and other<br />
metbds.<br />
coal with synthetic ccapounds or analogs. We intend to pursue this line Of<br />
inrestiGtion.<br />
ACKNOWLEKmNTS<br />
It should ultimately permit unamoimous identification <strong>of</strong> the iron in<br />
'vle wish to ttank Drs. Maurice Deul and Bryan C. Parks <strong>of</strong> the Bureau <strong>of</strong> Mines<br />
for their assistance in obtaining some <strong>of</strong> the coal samples. We are especifrllly<br />
indebted to Mr. Michael Toia for the construction and msintenance <strong>of</strong> the Mossbauer<br />
spectrometer.<br />
The work at Carnegie,Institute <strong>of</strong> Technology was performed under<br />
the auspices <strong>of</strong> tke U.S. Atomic Energy Commission under contract AT(30-1)-844;<br />
Report No. NYO-&44-63.
Brooks J.D. and Sternhell S. (1957) Brown coals. I. Oxygen containing<br />
functional Groups in Victorian brarn coals. Australian J. A S . g. 5<br />
206-221.<br />
Brooks J.D., Durie R.A. and Sternhell S. (19%) Chemistry <strong>of</strong> brown coals.<br />
11. Infrared spectroscopic studies. Australia? J_. A B . E. 1% 63-80.<br />
El<strong>of</strong>son R.M. (1957) Infrared spectra <strong>of</strong> humic acids and related materials.<br />
Can. J. G. 3 3 926-931.<br />
Dutcher R.R., White E.W. and SpaclmBn W. (1964) Elemental ash distribution<br />
in coal components--Use <strong>of</strong> the electron probe. Proc. 22nd Ironmakin<br />
Conf., Iron and Steel Div., Metallurgical E. % n I . M X E d .<br />
-5) Sprenkel-Se@ E.L. and Hanna S.S. (l'961t) M'lssbauer analysis <strong>of</strong> iron in<br />
stone xeteorites. Geochin. Cosnochim. Acta 2 ~ 1313-1931.<br />
,<br />
-7) Gerard A. and Dehelle M. (19s~) Effet M'dssbauer et caractere ionique<br />
des atones de Fer dans les metkorites Orgueil et Cold Bokkweld. Compt.<br />
Rend. Paris a, 17;6-17:.3.<br />
--<br />
.E) Fluck E., Kerler W. and Neuwirtil Vf. (1963) The M'dssbauer effect and its<br />
sipificance in chemistry. Anhew. Chen. Internat. EL. 5 277-22{.<br />
-3) Wertheim G.K. (1964) M'dssbauer Effect, Principles and Applications.<br />
Academic Press, Inc., New York.<br />
IC) Goldanskii V.I. (1964) M&sbauer Effect and its Applications<br />
Chemistri. Consiltants Bureau, New York.<br />
'1) Herber R.H. (195;) Introduction to M'dssbauer spectroscopy. J. Chem. Educ.<br />
fil, 130-1C7.<br />
J2) DeBenedetti S., Barros F. des. and Hoy G.R. (1966) Chemical and structural<br />
effects on nuclear radiations. A=. E. E. g. & 31-82.<br />
'3) Herber R.H. and Hayter R.G. (19&) Mgssbauer effect in, cis-trans isomers.<br />
J.. &. Chem. SOC. g, 301302.<br />
'4) Spijkemn J.J., &egg F.C. and DeVoe J.R. (1965) Standardization <strong>of</strong> the<br />
differential <strong>chemical</strong> shift for FeS7, M'dssbauer Effect Methodology,<br />
pp. 113-l2@. Plenum Press, New York.<br />
j5) Thiessen C. (1945) Chemistry <strong>of</strong> Cos1 Utilization (Ed. H.H. Lowery),<br />
Vol. 1. John Wiley, New York.<br />
36) Mitra G.B. (195h) Identification <strong>of</strong> inormic impurities in coal by the<br />
X-ray diffraction method. Fuel 3& 316-330.<br />
'7) Ecknause M., Harris R.J., Shuler W.B. and Welsh R.E. (1966) Measurement<br />
<strong>of</strong> the lifetime <strong>of</strong> the 14.4-keV level <strong>of</strong> 57Fe. Proc. Phys. e. London<br />
a 187-1i36.<br />
~ 8 Obenshain ) F.E., Roberts L.D., Colemn C.F., Forester D.W. and Thomson<br />
J.O. (1965) Hyperfine structure <strong>of</strong> the 14.4-keV y-ray <strong>of</strong> s7Fe in hydrated<br />
ferric smmonium sulfate as a function <strong>of</strong> the magnetization <strong>of</strong> the salt.<br />
~2. E. Lett. & 36j-369.<br />
'9) Wignall J.W.G. (1966) M'dssbauer line broadening in trivalent iron compounds.<br />
5. =. P*. 4k, 2462-2467.<br />
iC) Margulies S. and Ehrrcan J.R. (1961) "ransmlssion and line brcadening <strong>of</strong><br />
resonance radiation incident on a resonance absorber. Nucl. Instr. Meth.<br />
131-137-<br />
j1) Epstein L.M. (1962) MGssbauer spectra <strong>of</strong> some iron complexes. 5. E.<br />
P2. 2731-2737-<br />
1
(52) Gib3 T.C. and Greenwood N.N. (19%) The Fidssbauer spectm <strong>of</strong> the tetra-<br />
chlor<strong>of</strong>errate(I1) ion. >.<br />
(j;) Lang G. and Marshall W. (1966) M'dssbauer effect in some haerno~lo3in<br />
corzpounds. Proc. Phys. E. London 87, 3-34.<br />
(j3) t4atr.u- H.B., Sinha A.P.B. and Yagnik C.M. (L465) The M'dssbauer spectra <strong>of</strong><br />
spinel oxides containing ferrous ion. S a<br />
State C=. & 401-403.<br />
(4G) Frank E. and Abeledo C.R. (1566) Mzssbauer effect in iron (111)<br />
dithiocar3amates. Inorg. z. & 1433-145>.
1133<br />
REACTIONS OF COAL AND RElATED MATERIALS IN MICRWAVE DISCHARGES IN H2, H20 AND Ar<br />
Yuan C. Fu and Bernard D. Blaustein,<br />
U. S. Bureau <strong>of</strong> Mines, Pittsburgh Coal Research Center, .<br />
4800 Forbes Avenue, Pittsburgh, Pennsylvania 15213<br />
INTRODUCTION I<br />
Recent investigations on the reaction <strong>of</strong> carbon in a high frequency discharge have<br />
shown that it produces methane, acetylene and minor amounts <strong>of</strong> other hydrocarbons<br />
in the hydrogen dischar e,=/ but produces primarily an H2-CO mixture downstream<br />
from a water discharge.-/ 9 Similar vork with respect to coal, however, is almost<br />
non-existent except that carried out by Letort et a1.4/ and by Pinchin.'/ Though<br />
some work on coal in a plasma jer6-91 has been reported, the characteristic <strong>of</strong><br />
those reactions is generally the thermal decomposition <strong>of</strong> coal by rapid heating<br />
to high temperatures in an inert atmosphere.<br />
In a microwave-generated discharge, hydrogen or water vapor can be excited or<br />
dissociated into atoms, ions and electrons at temperatures much lower than those<br />
attained in a plasm jet. The present study is concerned with the reactions <strong>of</strong><br />
various coals, a polynuclear hydrocarbon, and graphite, in microwave <strong>discharges</strong><br />
in H2, water vapor and Ar. The results are compared in terms <strong>of</strong> the product yield<br />
and distribution in each type <strong>of</strong> discharge. Differences observed between the<br />
reactions <strong>of</strong> the various coals and the other materials suggest that the amount and<br />
type <strong>of</strong> volatile material, carbon content, and the type <strong>of</strong> the carbonaceous material,<br />
as well as the type <strong>of</strong> the gas discharge, are all factors affecting the product yield<br />
and distribution. The difference between coal and graphite - . in their behavior toward<br />
water vapor in the microwave discharge is <strong>of</strong> particular interest.<br />
discharge, the graphite yields almost no hydrocarbons but only an H2 + CO mixture<br />
while the coal yields appreciable amounts <strong>of</strong> C2H2 and CH4 in addition to H2 + CO.<br />
Experiments using DzO and Ar.<strong>discharges</strong> ,indicate that the D20 and the water actually<br />
do participate in the reactions with coal to form hydrocarbons. It is also demon-<br />
strated that the C2H2 yield from coal in the hydrogen discharge can be drastically<br />
increased by condensing at a low temperature part <strong>of</strong> the primary products formed<br />
during the discharge reaction.<br />
ACKNOWLEDGEMENTS<br />
1 ,<br />
In the water<br />
The authors wish to thank Dr. Irving Wender for his valuable discussions, Gus<br />
Pantages for his technical assistance, A. C. Sharkey, Jr. and Janet Shultz for<br />
their mass spectrometric analyses, and John Queiser for his infrared analysis.<br />
EXPERIMENTAL<br />
The experiments were carried out in a static system 4th the discharge produced by<br />
a Raytheon microwave generator in the air-cooled Ophthos coaxial cavity at 2450Mcl<br />
sec. Chemical analyses and origins <strong>of</strong> the vitrains <strong>of</strong> the different coals used are<br />
given in table 1. All the vitrains were -200 mesh. Ultra purity spectroscopic<br />
graphite powder (325 mesh, United Carbon) and chrysene (c18H12) were used for<br />
comparison. As reactant gases, a 9.7:1 H2-Ar mixture, H20-Ar mixtures and Ar were<br />
used. The H2 and At were obtained from cylinders and passed through a liquid N2<br />
trap prior to storage. The uater vapor was obtained from distilled water degassed<br />
in high vacuum.<br />
In the experimental procedure, a knom weight <strong>of</strong> graphite or coal powder was placed<br />
in a cylindrical Vycor tube (ID - 11 m, vol - 32 ml) and degassed in a high vacuum<br />
at an elevated temperature-(150°C for graphite and 100°C for coal) for a half to<br />
one hour to remwe the moisture and the air adsorbed on the carbon. ' A known<br />
pressure <strong>of</strong> the reactant gas or gas mixture measured by a Pace Engineering pressure
434<br />
transducer vas then introduced into the reactor tube. For the H20-Ar mixture, a<br />
known pressure <strong>of</strong> the water vapor was first introduced to the reactor containing<br />
the degassed carbon, and then a known pressure <strong>of</strong> the argon was introduced while<br />
the water was condensed in the end <strong>of</strong> the tube by dry ice and acetone. The portion<br />
<strong>of</strong> the tube cooled by the dry ice was so small compared to the total volume that no<br />
corr=ctim CI? preee~re reeding spp~~r! to he necensary. The dinrhnrgc wan then<br />
produced with the carbon in the discharge zone. The gaseous products were analyzed<br />
by -8s spectrometer.<br />
1<br />
'Lhe solid product obtained from the high volatile bituminous coal in the H2 discharge<br />
was analyzed by infrared spectrometer. The sample was obtained either by scraping<br />
the reactor wall or by solvent (benzene or acetone) extraction. In some instances, a<br />
reactor consisting <strong>of</strong> a tube divided by a fritted Vycor disc was used. The coal vas<br />
placed on the disc and KBr powder was introduced on the other side <strong>of</strong> the disc.<br />
During the discharge, the lovcr end <strong>of</strong> the tube was immersed in liquid N2, thus<br />
permitting the condensable low volatile products to pass through the disc end be<br />
adsorbed on the surface <strong>of</strong> the KBr powder. This was later removed, pressed into<br />
a pellet, and examined by infrared analysis.<br />
TABU 1. - Analysis <strong>of</strong> vitrainr (moisture free basis, percent)<br />
0 Vola ti le I\<br />
C H N S (by diff.) mtter<br />
Anthracitdl<br />
Low volatile<br />
91.06 2.49 0.96 0.83 2.89 1.77 6.1<br />
pninou&l 89.57 4.67<br />
4 . ,,&k~$~IA<br />
81.77 5.56<br />
1.25<br />
1.71<br />
.81<br />
.97<br />
2.17<br />
5.87<br />
1.53<br />
2.06<br />
20.2<br />
39.2<br />
9<br />
. -5/ Lignite21 66.45 5.40 .31 1.40 22.84 3.06 44.0<br />
2/<br />
3/<br />
4'<br />
Dotrance nine, &high Valley Coal Co., Luzerne County, Pennsylvania.<br />
Pocahontar lo. 3 Bed, Buckeye No. 3 Mine, Page Coal and Coke Co.,<br />
Stephenson, Wyoming County, West Virginia..<br />
Bruceton, Pittsburgh Bed, Allegheny County, Pennsylvania.<br />
Beulah-Zap Bed, North Unit, Bculah Mine, Knife liver Coal Ubi- Co.,<br />
Beulah, Mercer County, North Dakota.<br />
RESULTS AND DISCUSSIOH<br />
All result. were obtained from experiments using 5 mg <strong>of</strong> the carbonaceour arterial<br />
in each discharge at 35 watts <strong>of</strong> power input. For consistency, an initial total<br />
pressure <strong>of</strong> about 25 UD y1s used for most runs, but in the runs with H20 vapor it<br />
tar varied in the range <strong>of</strong> 12 to 25 m.<br />
The time <strong>of</strong> the discharge reaction -8<br />
intended to be 60 seconds in mort NIIl, but the dirchrge would 80t rustaio itrelf<br />
in s oy runs. It ir rurpected that the rolid product (probably polymuclur hydro-<br />
carbons) formed u y teed to draw away the electroar in the dircharge to form negative<br />
ions and create a rhortage <strong>of</strong> energetic species in the gas phase. &never, under the<br />
experimental condition. crployed, a large part <strong>of</strong> the reaction occurred within 30-60<br />
seconds, and prolaged treatment resulted .ply in IQY chnge in the product dirtri-<br />
bution and a little lacreare in the extent <strong>of</strong> the garification. A few rum were<br />
ude for ar long .E 3 rinutes.<br />
Hydrogen-Argon and Argon Dirchrges<br />
In prelirinary runr with graphite in an 82 dirchrge, we attempted to interpret<br />
the data on the baris <strong>of</strong> the hydrogen balance before and after reaction. It<br />
appear., horcver, that appreciable w n t r <strong>of</strong> hydrogen were consumed in the<br />
fotration <strong>of</strong> rolid product or were taken up by the carbon. Therefore, to allov
435<br />
interpretation <strong>of</strong> the data, Ar was introduced into the reactant gas as an internal<br />
standard. The presence <strong>of</strong> Ar in the system gives no noticeable effect on product<br />
type or distribution.<br />
Table 2 summarizes the results obtained from the reactions <strong>of</strong> each material in<br />
microwave <strong>discharges</strong> in an H2-Ar (9.7:l) mixture and in Ar. In the Ar discharge,<br />
coal is partially gasified to give Hq, CO, and C2H2 as the major gaseous products,<br />
CQ2, CHq, and minor amounts <strong>of</strong> other hydrocarbons (C2 and C3 hydrocarbons.<br />
biacetylene and benzene were detected), in addition to a solid prodkt and residual<br />
char. In the H2-Ar discharge, the percent <strong>of</strong> carbon forming gaseous hydrocarbons is<br />
increased as compared with the Ar discharge; the total amount <strong>of</strong> carbon gasified is<br />
also increased. In this discharge, C2H2, CO, and CH4 are the main constituents <strong>of</strong><br />
the gaseous product, and minor amounts <strong>of</strong> other hydrocarbons are also formed as in<br />
the Ar discharge. Comparison <strong>of</strong> the several 60-second runs indicated that the net<br />
amount <strong>of</strong> hydrogen remaining after the reaction was decreased, except for high<br />
volatile bituminous coal and lignite. Besides that converted to hydrocarbons, part<br />
<strong>of</strong> the hydrogen was probably consumed in the formation <strong>of</strong> solid product or was<br />
taken up by the coal residue. With high volatile bituminous coal and lignite,<br />
gasification was extensive and resulted in a net increase <strong>of</strong> hydrogen.<br />
The percent <strong>of</strong> carbon converted to gaseous hydrocarbons increases with the volatile<br />
matter <strong>of</strong> coal as shorn in figure 1, suggesting that a rapid release <strong>of</strong> the volatile<br />
matter and its subsequent gas phase reaction in a discharge may be the determining<br />
factors. Figure 1 includes the data for graphite. Lignite, with the highest<br />
volatile matter, however, gave only small amounts <strong>of</strong> hydrocarbons in both <strong>discharges</strong>.<br />
The high oxygen content <strong>of</strong> the lignite results in higher yields <strong>of</strong> carbon oxides and<br />
water but apparently inhibits hydrocarbon formation. Water vapor, in a discharge,<br />
can reverse the reactions <strong>of</strong> hydrocarbon fomtion by reacting with the hydrocarbon<br />
species to form carbon oxides and hydrogen.-<br />
Chrysene did not behave particularly different from coal in both <strong>discharges</strong>; the<br />
extent <strong>of</strong> gasification was appreciably small, approximately the same as that for<br />
anthracite. Interestingly, the carbon contents <strong>of</strong> the chrysene and the anthracite<br />
are also close to each other and are much higher than the other coals.<br />
For coal in general, the higher the carbon content the lower the volatile matter.<br />
The extent <strong>of</strong> the reaction <strong>of</strong> graphite with H2 was very much smaller under<br />
comparable conditions, probably due to the absence <strong>of</strong> volatile matter. The main<br />
products from graphite were C% and C2H2; however, the hydrogen balance Indicated,<br />
for example, that with an initial pressure <strong>of</strong> the H2-Ar mixture <strong>of</strong> 25 m Hg, the<br />
percentage <strong>of</strong> the initial hydrogen present in each component <strong>of</strong> the product is H2,<br />
77.6; CH4, 9.2; C2H2, 3.3; C2 + C3 hydrocarbons, 1.6; and the remainder (8.3%) was<br />
apparently consumed in the formation <strong>of</strong> polymer or was taken up by the graphite.21<br />
It is interesting to note that C2H2 accounted for 75-92% <strong>of</strong> the gaseous hydrocarbons<br />
produced from coal in both <strong>discharges</strong>. Hydrogasification or carbonization<br />
<strong>of</strong> coal usually gives CHq as the major hydrocarbons. It has been suggested that, in<br />
a microwave hydrogen discharge system, transport <strong>of</strong> carbon from the solid to the<br />
vapor phase takes place by bombardment <strong>of</strong> the carbon by energetic ions and electrons,<br />
and that the ’pr”seous carbon species could react with H atoms or CH species to form<br />
hydrocarbons . 2 1 A sfmilar mechanism appears co apply to the system containing<br />
coal except that H and CH species would also be evolved from the coal in addition<br />
to gaseous carbon species, even in the absence <strong>of</strong> an initial hydrogen. The<br />
predominance <strong>of</strong> C2R2, however, is also observed in rapid heating <strong>of</strong> coal by various<br />
high temperature methods such as plasma jet,=/ laser be&/ and flash heating.Ef<br />
etc. A cornpariron <strong>of</strong> our experimental results with those obtained vlth an Ar plasma<br />
jet by Bond et al.l/ indicates that the percentage <strong>of</strong> carbon converted to C2H2 In<br />
our system is somewhat lower, but the effect <strong>of</strong> volatile matter on the hydrocarbon<br />
4
436<br />
yield is quite similar. The conversion <strong>of</strong> carbon to C2H2 for the high volatile<br />
bituminous coal (VM 5 39.2%) in table 2 is 9.2% in the H2-Ar discharge as compared<br />
to 12.5'1. in the Ar plasma for a coal <strong>of</strong> similar volatile matter content.<br />
TABLE 2. - Reactions <strong>of</strong> coal and related materials<br />
in microwave <strong>discharges</strong> <strong>of</strong> H? and Ar 1<br />
Pressure,<br />
Percent <strong>of</strong> c present as ,/<br />
Uaterial<br />
m<br />
H,, Ar<br />
Time, Yield x lo4, mole/g <strong>of</strong> solid<br />
sec H? clll C2H2 CO CO?<br />
Gasebus Gaseous<br />
products hydrocarbons<br />
hvab 22.7 2.3 60 63.9 4.7 31.1 21.1<br />
22.7 2.3 60 53.6 10.2 28.6 20.2<br />
23.4 2.4 60 12.5 4.5 27.0 23.6<br />
22.8 2.4 90 11 3,9 29.8 22.2<br />
21.8 2.3 105 L/ 4.8 21.0 ' 16.9<br />
21.9 2.3 110 11 3.6 20.0 16.0<br />
- 25.7 17 3.3 .1 1.2 7.1<br />
- 24.0 60 56.4 .9 13.4 23.0<br />
- 23.6 180 44.0 1.1 16.5 22.6<br />
lvb 23.8 2.5 34 1/ 4.1 15.5 7.3<br />
21.6 2.2 60 1/ 3.8 17.5 8.1<br />
- 23.5 60 15.3 .3 3.6 7.7<br />
Lignite 21.6 2.2 60 25.4 2.1 9.3 66.2<br />
22.6 2.3 180 22.9 2.1 7.7 80.3<br />
20.7 2.1 180 26.0 2.1 7.3 69.2<br />
- 24.7 32 50.4 .7 6.9 61.7<br />
- 23.0 60 18.4 .6 5.0 68.4<br />
Anthracite 21.6 2.2 60 11 4.2 8.3 4.0<br />
22.6 2.4 60 11 4.6 9.6 3.0<br />
- 24.0 60 .3 trace trace 2.0<br />
Chrysene 22.7 2.4 10 11 6.5 12.8 2.0<br />
20.0 2.0 30 11 10.4 5.8 trace<br />
- 22.0 30 lz.6 .8 2.6 trace<br />
Graphite 22.4 2.3 60 11 3.5 2.5 trace<br />
22.6 2.4 60 11 3.4 1.8 trace<br />
11 Net decrease <strong>of</strong> hydrogen was indicated.<br />
0.2<br />
.2<br />
.4<br />
.2<br />
.2<br />
.2<br />
.1<br />
.2<br />
.2<br />
.1<br />
.1<br />
trace<br />
3.5<br />
1.8<br />
1.7<br />
3.2<br />
3.2<br />
trace<br />
trace<br />
.1<br />
trace<br />
trace<br />
trace<br />
trace<br />
trace<br />
13.6<br />
13.1<br />
12.8<br />
13.1<br />
9.9<br />
9.4<br />
1.5<br />
7.7<br />
8.7<br />
6.1<br />
6.7<br />
2.1<br />
16.7<br />
18.3 '<br />
16.1<br />
14.1<br />
15.1<br />
3.6<br />
3.9<br />
.3<br />
4.6<br />
3.4<br />
.9<br />
1.2<br />
1.0<br />
I<br />
10.4<br />
10.3 \<br />
9.4<br />
9.9 4<br />
7.4<br />
7.0<br />
-4 \<br />
4.3<br />
5.3 '<br />
5.1 \<br />
5.6<br />
1.1<br />
4.1<br />
3.3<br />
::: /<br />
3.1<br />
3.4 ~<br />
4.4<br />
3.4 \<br />
.9 I<br />
1.2<br />
1.0 \<br />
Solid Product I<br />
The solid product obtained from coal is brownish and is similar to that usually<br />
observed from the thermal treatment <strong>of</strong> coal. Uo appreciable amount <strong>of</strong> solid product<br />
was formed from anthracite or from lignite. Though the extent <strong>of</strong> the gasification<br />
for the chrysene was not appreciable, the original white powder was instantaneously<br />
converted to a broun solid upon the initiation <strong>of</strong> both <strong>discharges</strong>. The infrared <<br />
spectra <strong>of</strong> the solid product and the residual char obtained from the high volatile<br />
bituminous coal in the H2-Ar discharge showed the usual aliphatic C-R bands and<br />
some weak aromatic bands which are typical <strong>of</strong> pitch and coal.<br />
3<br />
* 7<br />
1<br />
Effect <strong>of</strong> Cooling by Liquid Nitrogen<br />
Since the indications are that the wnter formed can retard the hydrocarbon formation<br />
and that the hydrocarbons produced may undergo further destructive reactions, it can<br />
be expected that a rapid quenching <strong>of</strong> the primary products should give a pronounced<br />
effect on the result. Experiments were carried out in a reactor consisting <strong>of</strong> the<br />
i t
437<br />
tube divided by a fritted Vycor disc. The coal was placed on the disc, and was<br />
subjected to reaction in the H2 discharge while the lower end <strong>of</strong> the tube was cooled<br />
in liquid N2. Though the process <strong>of</strong> condensing water and some hydrocarbons at this<br />
temperature is diffusion controlled, the effect is pronounced, a's shown in table 3.<br />
For both the bituminous coal and the lignite, the yield <strong>of</strong> the hydrocarbons, CqH2<br />
in particular, was greatly increased. The amount <strong>of</strong> Hg remaining and the amount <strong>of</strong><br />
CO produced after the reaction were also decreased. Therefore, the increase <strong>of</strong><br />
hydrocarbon yield can be attributed mainly to subsequent hydrocarbon formation by<br />
reaction <strong>of</strong> H2 and co; this hydrocarbon yield is greatly enhanced b$ rapid removal<br />
<strong>of</strong> H20 formed.<br />
Water-Argon Discharge<br />
There is a marked difference between the products obtained from graphite and coal in<br />
a water discharge. In the discharge in H20-Ar mixtures, as shown in table 4, graphite<br />
yields hydrogen and CO but practically no hydrocarbons; while the coals yield an<br />
appreciable amount <strong>of</strong> CZHZ and some CH4 in addition to H2 and CO. The amounts <strong>of</strong><br />
Hq and carbon oxides produced in the reaction with graphite rere stoichiometric.<br />
The extent <strong>of</strong> gasification was also rpuch greater for the coals than for the graphite.<br />
The active hydrogen species produced in the water discharge did not react further<br />
with the graphite or the CO formed from it to produce significant amounts <strong>of</strong> hydro-<br />
carbons. This is further evidence demonstrating that the presence <strong>of</strong> water vapor<br />
retards hydrocarbon formation.<br />
For the reaction <strong>of</strong> a given coal in aR2aAr discharge, an initial H20 pressure <strong>of</strong><br />
less than 12 mm Hg appears to give the optimum gasification end hydrocarbon<br />
production. Higher initial H20 pressures cause a decrease in both gasification<br />
and hydrocarbon production. So long as the H20 pressure is not at its highest<br />
values, more hydrocarbons are formed than in the Ar discharge. (Also, with the<br />
higher initial Hfl pressures, the discharge could not be initiated readily and<br />
would not sustain itself for as long as 60 seconds, perhaps due to too large an<br />
increase in the total gas pressure in the reactor.) The data seem to indicate<br />
that 60 seconds may have been too long a period for the maximum production <strong>of</strong><br />
hydrocarbons. The formation <strong>of</strong> the hydrocarbons should be a maximum at the time<br />
when a plateau <strong>of</strong> the extent <strong>of</strong> coal gasification is attained, and prolonged<br />
treatment probably allows the remaining H20 vapor to diffuse into the discharge<br />
zone, giving an adverse effect. It was also noticed that, at an initial H20<br />
pressure <strong>of</strong> less than 12 mm, the hydrogen content <strong>of</strong> the products exceeded that<br />
which could possibly be derived from the stoichiometric amount <strong>of</strong> the H20<br />
initially present.<br />
These results seem to indicate the following. The active hydrogen species formed<br />
in the Hq0 discharge participate in the reactions which lead to the formation <strong>of</strong><br />
hydrocarbons from (some <strong>of</strong>) the species derived from the coal. This occurs<br />
despite the retarding effect <strong>of</strong> H20 on hydrocarbon formetion. (If the H20<br />
pressure is too high, this latter effect decreases the hydrocarbon yield.)<br />
Presumebly, the coals when gasified supply enough CB species to allow hydro-<br />
carbons to be formed,even though the active oxygen species present react with<br />
some <strong>of</strong> the gaseous carbon and CH species. On the other hand, graphite produces<br />
negligible amounts <strong>of</strong> CH species in a H20-Ar discharge, and therefore cannot<br />
produce hydrocarbons since the reaction <strong>of</strong> the CO formed with the active hydrogen<br />
species is retarded by the H20 present.<br />
Lignite, with its high volatile matter content, was extensively gasified but gave<br />
a relatively low hydrocarbon yield, presumably due to the inhibition by its high<br />
oxygen content. For lignite, the production <strong>of</strong> COq was also higher in the R20-Ar<br />
discharge than in the Hq-Ar or Ar discharge. Again, the extent <strong>of</strong> gasification<br />
for chrysene was rather small.
?-!???<br />
0 mv,<br />
Uh\OhIe<br />
. . . .<br />
eirlrl<br />
00000<br />
wwwww<br />
r l r l<br />
I<br />
*<br />
438 ,<br />
I<br />
,
439<br />
Deuterium Oxide-Argon Discharge<br />
It is <strong>of</strong> interest to obtain further evidence as to whether water is actually involved<br />
in the reaction with coal to form hydrocarbons in addition to the production <strong>of</strong><br />
H2 + CO. The gaseous product obtained from the reaction <strong>of</strong> the high volatile<br />
bituminous coal in a D20-Ar mixture was analyzed by the high resolution mass<br />
spectrometer. At a resolution <strong>of</strong> 1 part in 20,000, precise masses for doublets and<br />
in some instances triplets that occurred at the same nominal masses could be used<br />
to identify completely deuterated, monodeuterated and nondeuteratedl species present<br />
in the product. For example, peaks due to C2H2 (mass = 26.0156), C2D (26.0141) and<br />
CN (26.0031) were observed at the nominal mass <strong>of</strong> 26 and peaks due to C2D2 (28.0282),<br />
N2 (28.0061) and CO (27.9931) were observed at the nominal mass <strong>of</strong> 28. The whole<br />
spectrum up to the nominal mass <strong>of</strong> 44 contained peaks attributed to all species<br />
present including minor amounts <strong>of</strong> oxygenated and N-containing compounds, but the<br />
major products were H2, HD, D2, C2H2, C2HD, C2D2 and partially deuterated methanes<br />
in addition to D20, DHO, H20 and carbon oxides. Thus it is apparent that D20 is<br />
initially dissociated into D, OD and possibly an active oxygen species which in<br />
turn recombine to give D20 and D2, or react with the active species derived from<br />
coal such as H, CH, C1, Cg and CO, etc., to give DHO, HD, CO, C02 and deuterated<br />
hydrocarbons.<br />
CONCLUSIONS<br />
In the microwave <strong>discharges</strong> <strong>of</strong> H2, H20 and Ar, coal is gasified to give gaseous<br />
hydrocarbons and carbon oxides plus a solid product and residual char, Hydrogen<br />
I is also produced either by dissociation <strong>of</strong> the water vapor or by devolatilization<br />
<strong>of</strong> the coal in the H20 and/or At <strong>discharges</strong>. But, in most cases in the hydrogen<br />
discharge, hydrogen is consumed rather than produced (except for hvab coal and<br />
lignite). The yield <strong>of</strong> the hydrocarbons is highest in the hydrogen discharge and<br />
that <strong>of</strong> carbon oxides is highest in the water discharge. Both the hydrogen and<br />
{ the water <strong>discharges</strong> give greater extents <strong>of</strong> gasification and yield more hydro-<br />
/ carbon products than the Ar discharge, indicating the occurrence <strong>of</strong> gas phase<br />
1 reactions <strong>of</strong> H, OH, and active oxygen species with the active species derived<br />
from the coal. The gasification <strong>of</strong> coal in the water discharge is <strong>of</strong> particular<br />
' interest because it produces H2 + CO ( - 1:l) plus C2H2 and CQ. The high oxygen<br />
J content <strong>of</strong> the lignite results in a higher yield <strong>of</strong> carbon oxides but apparently<br />
inhibits the hydrocarbon formation in the discharge. The hydrocarbon yield from<br />
the lignite or other coals in the hydrogen discharge, however, can be increased<br />
,' dramatically by rapidly condensing part <strong>of</strong> the product species produced, especially<br />
i H20, during the discharge reaction.<br />
L<br />
C2H2 accounts for as nuch as 9ZZ <strong>of</strong> the gaseous hydrocarbons produced, and, excepting<br />
for lignite, the amount <strong>of</strong> the hydrocarbons is related to the volatile matter <strong>of</strong> coal.<br />
The extent <strong>of</strong> nasification. however. is increased with the volatile matter content<br />
<strong>of</strong> coal including lignite.<br />
Thus, if the hydrocarbons are formed by the recombination<br />
<strong>of</strong> the species derived from the volatile material and other species present in the<br />
discharge, another intererting study would be to employ a flow system in khich coal<br />
is devolatilized by ordinary means and the evolved gases are subsequently reacted<br />
in a discharge.<br />
REFERENCES<br />
1. Blackwood, J. D. and McTaggart, F. K. Reactions <strong>of</strong> Carbon with Atomic Gases.<br />
Australian J. Chem., 12, 533 (1959).<br />
1. Shahin, n. n. Reaction <strong>of</strong> Elementary Carbon and Hydrogen in High-Frequency<br />
Discharge. Nature, 195, 992 (1962).<br />
3. Vastola, F. J., Walker, P. L. Jr., and Wightman, J. P. The Reaction Between<br />
Carbon and the Products <strong>of</strong> Hydrogen, Oxygen and Water Microwave Discharges.<br />
Carbon, 1, 11 (1963).
4.<br />
5.<br />
6.<br />
7.<br />
8.<br />
9.<br />
10.<br />
11.<br />
12.<br />
13.<br />
440<br />
Letort, M., Boyer, A. F., and Payen, P. Attempting Hydrogenation <strong>of</strong> Coal with<br />
Atomic Hydrogen. Bull. SOC. Chim. France, 1589 (1963).<br />
Pinchin, F. J. The Reaction <strong>of</strong> Coals with Atomic Hydrogen. Private<br />
Comnication.<br />
Bond, R. L., Galbraith, I. F., Ladner, W. R., and McConnell, G. I. T.<br />
Produccion <strong>of</strong> Acecyiene from Cuai, iising a Piasma iec. Nacure, =, 1313<br />
(1963).<br />
Bond, 8. L., Ladnei, W. R., and McConnell, G.<br />
a Plasma Jet. Fuel, 45, 381 (1956).<br />
I. T. Reactions <strong>of</strong> Coal in<br />
l<br />
Graves, R. D., Kawa, W., and Hiteshue, R. W. Reactions <strong>of</strong> Coal in a Plasma<br />
Jet. Ind. Eng. Chem., 5.59 (1966).<br />
Kawana, Y., Makino, M., and Kimura, T. Formation <strong>of</strong> Acetylene from Coal by<br />
Argon Plasma. Kogyo Kagaku Zasshi, 2, 1144 (1966).<br />
Blaustein, B. D. and Pu, Y. C.<br />
Formation <strong>of</strong> Hydrocarbons from H2 + CO in<br />
Microwave-Generated Electrodeless Discharges. Paper to be presented at 153rd<br />
<strong>National</strong> ACS Meeting, Miami Beach, Fla., April 1967.<br />
Montet, G. L. Retention <strong>of</strong> Protons by Graphite. Proc. Fourth Conf. on Carbon,<br />
Pergamon, New York, pp. 159-164 (1960).<br />
Sharkey, A. G., Shultz, J. L., and Friedel, R. A. Gases from Flash and Laser<br />
Irradiation <strong>of</strong> Coal. Nature, 202, 988 (1964); Coal Science, Advances in<br />
Chemistry Series 55, R. F. Gould, Editor, Am. Chem. SOC., Washington, pp- 643-<br />
649 (1966).<br />
Rau. E. and Seglin, L. Heating <strong>of</strong> Coal with Light Pulses. Fuel, 2, 147 (1964).<br />
1<br />
i
I2<br />
c<br />
C<br />
01<br />
? IO<br />
e 6<br />
LL<br />
0<br />
z<br />
13<br />
0<br />
0 hvab vitroin<br />
A Ivbvitroin<br />
X Anthrocite vitroin<br />
0 Graphite<br />
0 Lignite vitroin<br />
I<br />
Flgure I -Effect <strong>of</strong> volotile matter on yield <strong>of</strong> hydrocorbons in H2-Ar discharge<br />
(60 second run)<br />
I<br />
12-29-66 L-9741
442<br />
CHEMICAL REACTIONS IN A CORONA DISCHARGE I. BENZENE<br />
1<br />
. .. M. W. Ranney'.<br />
Wm. F . O'Connor<br />
Department <strong>of</strong> Chemistry<br />
Fordham University<br />
New York, New York 10458<br />
INTRODUCTION<br />
Corona <strong>discharges</strong> (silent) have been used to initiate <strong>chemical</strong> reactions for<br />
well over 150 years'with much <strong>of</strong> the early work being confined to inorganic gases.2 However,<br />
the only commercially significant development over' the years has been the process<br />
for synthesizing ozone by subjecting oxygen to a corona discharge3 (ozonizer). Most early<br />
investigators encountered serious difficulties in studying the interaction <strong>of</strong> high voltage<br />
electricity with organic molecules. Equipment was unreliable and dangerous, and the<br />
complexity <strong>of</strong> the reaction product mass precluded definitive performance evaluation. Slnce<br />
World War 11, technology has advanced to the point where high frequency power can be<br />
generated and controlled at reasonable cost and many new dielectric materials such as<br />
fused quartz, alumina and mica mat are a~ailable.~ Modern analytical techniques afford<br />
the opportunity to determine the composition <strong>of</strong> the product mix.<br />
Corona electrons are accelerated by the applied voltage to an energy.level<strong>of</strong> 1<br />
10-20 electron volts, which is sufficient to break the covalent bond. This represents a very<br />
efficient approach when compared with high energy radiolysis (m.e.v. range) where the<br />
actual <strong>chemical</strong> work is effected by secondary electrons with an energy <strong>of</strong> 10-25 e.v.,<br />
formed after a series <strong>of</strong> energy-dissipating steps. Potentially, corona energy can deliver<br />
electrons to the reaction site at the desired energy level to give products which are not<br />
readily obtainable by more conventional means. The energy available in the corona dis-<br />
charge is somewhat above that commonly encountered in photochemistry (up to 6 e.v. ).<br />
Bertholot reduced benzene to c@8 in 1876 in what appears to be the first<br />
exposure <strong>of</strong> an aromatic compound to the corona discharge. Losanitsch obtained the<br />
following products from benzene in an electrical discharge: ( CgH6)2, biphenyl, (C72Hgg)<br />
and (C6H6)90,6 Benzene vapor treated at 300° in an ozonizer tube gave resinous products<br />
with a 6/4 carbon-hydrogen ratio.7 Linder and Davis exposed benzene vapor to 37,000<br />
volts and found biphenyl, gaseous products and evidence <strong>of</strong> polymerization.8 The early<br />
work yith benzene reactions in various ty s <strong>of</strong> electrical <strong>discharges</strong> is reviewed in<br />
considerable detail by Clocker and Lind. 'Brown and Rippere investigated the hydrogenation<br />
<strong>of</strong> benzene flowing down the walls <strong>of</strong>,an ozonizer tube and found 1,3-and 1,4-<br />
cyclohexadiene, biphenyl and a resinous mass which gave an infrared spectrum con- i'<br />
sistent with polystyrene. !4 (<br />
In recent years, benzene has been subjected to direct electrode discharge<br />
10 1<br />
and glow <strong>discharges</strong> induced by microwave'' and radi<strong>of</strong>requency'l energy. In this paper, I<br />
we report some <strong>of</strong> our observations for the reaction <strong>of</strong> benzene in a corona discharge.<br />
I<br />
'7<br />
1<br />
/<br />
!<br />
'I<br />
1<br />
)<br />
><br />
i<br />
1,<br />
\d t<br />
!'<br />
\
443<br />
A PPA RA TUS<br />
The corona reactor system used in this study is shown in Figure 1 and the reactor<br />
/details are given in Figure 2. The system is designed to run at atmospheric or reduced pres-<br />
'*sure and, with slight modification, under recycle conditions. The use <strong>of</strong> the threaded rod<br />
;center electrode affords a more uniform and higher treating potential than is obtained with a<br />
'cylindrical electrode. l3 The electrode threads concentrate surface irregularities thereby<br />
I<br />
'eliminating severe discharge points. The copper electrode serves as a cooling coil and<br />
affords direct observation <strong>of</strong> the corona. In a typical run, the benzene reservoir temper-<br />
-ature is set at 53O, the helium is adjusted to 100 ml/min. and after a two minute purge, an<br />
electrical potential <strong>of</strong> 15,000 volts is applied. A brilliant blue corona is established, the<br />
intensity being a.function <strong>of</strong> helium flow, pressure, benzene content and the applied voltage.<br />
!Benzene traverses the corona reactor as.a vapor and is condensed by the cold water trap<br />
nd collected. Yellow solids gradually deposit in the flask at the bottom <strong>of</strong> the reactor and ,<br />
's eventually adhere to the dielectric surfaces in the reactor. Some benzene and the more<br />
volatile reaction products are condensed in the -70 and -195" traps. See experimental<br />
section for further details.<br />
,(<br />
RESULTS AND DISCUSSION<br />
t . J<br />
' .<br />
The excitation <strong>of</strong> benzene vapor in a corona discharge provides an 8.5% conversion<br />
to identifiable products. The major products are a benzene soluble polymer (6%):<br />
la benzene insoluble polymer (0.7%), biphenyl (0.3%), ,and acetylene (1%). The composition<br />
<strong>of</strong> the product mix obtained is given in Table 1. The overall yield obtained under<br />
'our experimental conditions is similar to that produced in a radi<strong>of</strong>requency glow discharge<br />
12<br />
I;( lo%, with longer residence time) and somewhat higher than Streitwieser obtained using a<br />
microwave'' induced glow discharge (5%). The corona discharge gives a considerably<br />
I higher proportion <strong>of</strong> polymer than is obtained from these other energy sources. Benzene<br />
ubstitution products such as toluene, phenylacetylene and ethylbenzene were observed in<br />
he microwave discharge but were not noted in this study or in the radi<strong>of</strong>requency invesigation.<br />
The high yield <strong>of</strong> fulvene in the radi<strong>of</strong>requency discharge is surprising in view <strong>of</strong><br />
ts tendency to polymerize or add oxygen under rather mild conditions. The sum <strong>of</strong> the<br />
arbon and hydrogen analyses for the polymeric fraction obtained by Stille in the glow disharge<br />
<strong>of</strong> benzene (86-94 per cent) suggests some fixation <strong>of</strong> oxygen and/or nitrogen during<br />
ischarge or product work-up. l2 The pressure for these electrodeless glow discharge<br />
tudies was <strong>of</strong> the order <strong>of</strong> 20 mm. or less, while the corona reactor was operated at<br />
tmospheric pressure. 'Schiiler, using an electrode discharge in benzene, obtained proucts<br />
similar to those produced in the microwave glow discharge. lo<br />
1<br />
The reaction products obtained in this study are roughly categorized as the<br />
cnzene fraction, biphenyl fraction and polymeric material for purposes <strong>of</strong> discussion.<br />
,fter 'initial studies indicated that the components <strong>of</strong> the -7O.and -195O traps were<br />
imilar to those in the benzene trap, these samples were combined. The acetylene<br />
ontent <strong>of</strong> the low temperature traps was determined by weight loss prior to mixing<br />
iith the benzene.<br />
cnzene Fraction<br />
The exposed benzene, as collected in the cold traps, is bright yellow. The<br />
allowing reaction products have been identified in this fractlon: I, 3-cyclohexadiene,<br />
,4-cyclohexadiene, fulvene and cyclohexene. The approximate per cent yield for each<br />
f these consntuents is given in Table 2. The data were determined using a 100 foot
helium<br />
444<br />
CORONA REACTOR SYSTEM<br />
Figure I<br />
condenser 400 mm<br />
threaded steel rod<br />
electrode 5/16"x 36"<br />
DETAIL OF CORONA REACTOR<br />
Figure 2<br />
7740 Boroslltcote gloss<br />
dielectric, llmm OD<br />
thermocouple well<br />
,1/4" copper tubing electrode<br />
and cooling coil<br />
Gap-4mm<br />
Internal volume -164 ml<br />
thermocouple well<br />
24/40 ground gloas joinf<br />
10 wet<br />
I<br />
\<br />
\<br />
'1<br />
%<br />
I<br />
1<br />
F
Product<br />
445<br />
TABLE 1<br />
Product Dstribution in the Corona Discharge <strong>of</strong> Benzene, 15KV<br />
Benzene insoluble polymer 0.7<br />
Benzene soluble polymer (a) 6.0<br />
Biphenyl 0.3<br />
0-. m-, and p-Terphenyl<br />
Phenyl, benzyl substituted<br />
0.1<br />
cyclopentenes (b) 0.3<br />
1,3-and 1,4-Cyclohexadiene 0.1<br />
Fulvene 0.01<br />
Acetylene 10<br />
TOTAL 8.5<br />
Yield-Per Cent Yield-Per Cent Radi<strong>of</strong>requency@)<br />
Benzene Exposed <strong>of</strong> Product Mix Yield, Per Cent<br />
-<br />
8.2<br />
70.5<br />
3.5<br />
1.2<br />
3.5<br />
1.2<br />
0.1<br />
11.8<br />
100.0<br />
2.0<br />
N.D.<br />
N.D.<br />
N.D.<br />
I,O<br />
2.0 (f)<br />
10.0<br />
(a) Polymer fraction includes materials from 250 molecular weight and up.<br />
(b) Includes several phenyl and benzyl substituted cyclopentenes similar to biphenyl and<br />
o-terphenyl in vapor pressure. See discussion.<br />
(c) See reference No. 12.<br />
(d) N. D. -- not determined.<br />
(e) Benzene and toluene soluble polymer.<br />
(f) Acetylene l%, allene 1%.<br />
support-coated open tubular column with a squalane liquid phase and appropriate calibration<br />
standards (Figure 3). No significant change is noted in the cyclohexane content when<br />
compared with starting benzene. The increase in the methyl cyclopentane peak is ascribed<br />
'to its higher vapor pressure thus affording some accumulation <strong>of</strong> this material in the<br />
benzene distillate and a concomitant depletion in the benzene feed stock. Cyclohexene is<br />
produced in very low yield, as expected, because <strong>of</strong> the degree <strong>of</strong> hydrogenation required<br />
during its short residence time in this single pass reactor.<br />
The reduction <strong>of</strong> benzene to the cyclohexadienes is interesting as<br />
1,3-cyclohexadiene is therm'odynamically unstable with respect to benzene, cyclohexene<br />
and cyclohexane. l4 The synthesis <strong>of</strong> 1,3-cyclohexadiene under these conditions is<br />
analogous to the conversion <strong>of</strong> oxygen to ozone which also represents an energetically<br />
unfavorable process. The momentary energy input and the rapid removal <strong>of</strong> the activated<br />
species from the reaction zone affords this material. in low yield., As expected, the<br />
more stable 1,4-cyclohexadiene is produced in higher yeld (3/1). Additionally,<br />
1,3-cyclohexadiene would be expected to polymerize at a rapid rate under these conditions.<br />
The thermodynamic instability <strong>of</strong> 1,3-cyclohexadiene is demonstrated by its<br />
gradual disappearance from the sample after a few days standing at room temperature.<br />
The yields <strong>of</strong> both the l,4-and 1,3-cyclohexadiene are somewhat higher than previously<br />
reported by Brown and Rippere. The yields reported by these workers after<br />
24 hours exposure to a 15 KV'corona discharge in a counter-current hydrogen stream<br />
were 0.02 and 0.01 per cent respectively. The major variable in technique which likely<br />
accounts for this difference.in yields is the use <strong>of</strong> benzene vapor in our work, versus<br />
the hydrogenation <strong>of</strong> a thin liquid benzene film running down the annular dielectric sur-<br />
.<br />
face by Brown and Rippere.
446<br />
The ultraviolet spectrum. for the benzene fraction was obtained by running<br />
differentially against pure benzene in isooctane. A broad absorption band with a maximum<br />
at 258 rnp is ascribed to the 1,3-cyclohexadiene. An additional absorption at 242 rryl and<br />
a tailing into the visible region with a broad maximum at 360-370 r y is also observed.<br />
These spectral features are consistent with those reported for fulvene, the non-aromatic<br />
isomer <strong>of</strong> benzene.15 Blair and Bryce-Smith found fulvene in the photo<strong>chemical</strong> irradiation<br />
<strong>of</strong> benzene in what appears to be the first direct conversion <strong>of</strong> an aromatic hydrocarbon to<br />
a non-aromatic hydrocarbon. l6 Fulvene co-distilled with benzene, but could be separated<br />
from benzene using the 100 ft. squalane column (Figure 3). Fulvene prepared by the<br />
method <strong>of</strong> Meuche gave the same retention time and spectral features.I7 Additional evidence<br />
for assignment <strong>of</strong> the fulvene peak included its disappearance from the chromatogram after<br />
the sample was refluxed with maleic anhydride (color disappears ) or exposure to a free<br />
radical catalyst. The Diels-Alder reaction product with maleic anhydride was isolated and<br />
hydrolyzed to give the adduct, 7-methylene-5-norbornene -2, 3-dicarboxylic acid. The<br />
melting point and infrared data for this adduct were identical with the sample prepared<br />
using fulvene synthesized from cyclopentadiene and formaldehyde. l2<br />
The formation <strong>of</strong> the 1,3,5-hexatrienyl diradical has been suggested as an<br />
intermediate in the photo<strong>chemical</strong> decomposition'' and the radi<strong>of</strong>requency discharge12 <strong>of</strong><br />
benzene. Material isolated in the photo<strong>chemical</strong> excitation gav.e a W spectrum similar<br />
to 1,3,5-hexatriene, but displaced by 7.5 mp and, more disturbing, the investigators<br />
were able to separate the material from benzene by fractional distillation. No experi-<br />
mental evidence was given in the radi<strong>of</strong>requency study. In our work, 1,3,5-hexatriene<br />
could not be detected (less than 100 ppm) in the benzene fraction using the squalane<br />
column. No spectral evidence was noted in the ultraviolet although the region <strong>of</strong> in-<br />
terest is complicated by the presence <strong>of</strong> 1,3-cyclohexadiene. l9<br />
The unidentified peak in the chromatogram (Figure 3) gives a negative<br />
test for an acetylenic hydrogen (ammoniacal cuprous chloride solution) and does not<br />
disappear after the sample is subjected to free radical catalysis for several hours. Thus,<br />
the open chain, acetylenic isomers <strong>of</strong> benzene, hexa-I, 3-diene-5-yne. 1,4-hexadiyne,<br />
1,s-hexadiyne and l.S-hexadien-3-yne do not appear to account for this peak. The<br />
possibility <strong>of</strong> valence isomers <strong>of</strong> benzene such as bicyclo (2.2.0) hexa-2, 5-diene,<br />
"Dewar benzene", were excluded based on the observation that the peak was stable to<br />
prolonged heating at looo. The valence isomers would be expected to convert to<br />
benzene under such conditions. 2o<br />
TABLE 2<br />
(a)<br />
Product Distribution in Dscharged Benzene Fraction<br />
Product Yield % Benzene Exposed<br />
1.3 -Cyclohexadiene 0.03<br />
1.4 -Cyclohexadiene 0.09<br />
Fulvene 0 01<br />
Cyclohexene 0.01<br />
Unidentified 0.02<br />
(a) Quantitative data obtained by GLC using squalane column and appropriate calibration<br />
standards. I<br />
I li<br />
\<br />
\
I<br />
f I<br />
I<br />
" I<br />
I<br />
-<br />
447<br />
~.<br />
Figure 3<br />
Analysis <strong>of</strong> Benzene Fraction Column<br />
I00 f t a 0020 In ID Support-coated open<br />
Tubular with Squalane Liquid Phase<br />
Temp 45': Isothermal<br />
Biphenyl Fraction - Silicone GumRubber Column<br />
ERM$L TO 300.C AT 6OVYIN , ISOTHERMAL X)O*<br />
' 185.C ' 300 4YIN<br />
45 SEC<br />
-.<br />
Figure 4<br />
I
Biphenyl Fraction<br />
448<br />
The low molecular weight compounds, soluble in isooctane and hot methanol,<br />
were primarily biphenyl and 0-, m-, and p-terphenyl. These products were identified by<br />
gas-liquid chromatography using a silicone gum rubber column (Figure 4 ) and a Carbowax<br />
20M column with appropriate standards. The peaks were trapped and analyzed by infrared<br />
and NMR spectroscopy for confirmation. Biphenyl was present in sufficient quantity to be<br />
readily detected in the initial infrared spectrum and was isolated by suhimation. \<br />
Quantitative data, obtained using the silicone column with appropriate calibration curves<br />
are presented in Table 3. With the possible exception <strong>of</strong> biphenyl, the yields are very<br />
low considering the overallconversion noted. The 0-, m- and p-terphenyl ratio (1/0.4/1)<br />
indicates a preference for the para position beyond that expected for random attack.<br />
-__ - -.- . -~ - - -_<br />
If hydrogenation <strong>of</strong> the polyphenyls by hydrogen radicals generated "in situ" I<br />
is to be considered a primary reaction mechanism, one would expect to see components<br />
corresponding tothe possible hydrogenated species <strong>of</strong> biphenyl and the terphenyls . This<br />
does not appear to be the case, however, as no significant peak can be ascribed to a<br />
hydrogenated o-terphenyl compound. The small peaks before biphenyl and after o-terphenylt<br />
in the chromatogram (Figure 4 ) actually include four or more, components present in \I<br />
small proportions which have not been completely identified. The first peak, (eluted<br />
before biphenyl) amounting to less than 0.1 per cent <strong>of</strong> the total yield, was initially<br />
ascribed to a mixture <strong>of</strong> the possible hydrogenation products <strong>of</strong> biphenyl. On the Carbowax'<br />
'><br />
column at lower temperatures this peak is eluted between phenylcyclohexane and I-phenylcyclohexene.<br />
Initial infrared examination <strong>of</strong> this peak after trapping indicated an intriguing<br />
similarity with the peak (s) noted after o-terphenyl and with the polymer fractions. The \<br />
possibility that this component was a low molecular weight precursor to polymer formation<br />
prompted further study. Infrared indicates a phenyl substituted aliphatic compound containing<br />
some olefinic unsaturation, and the spectrum is not consistent with the anticipated<br />
phenyl cyclohexenes . Careful examination <strong>of</strong> the spectrum indicates that the following<br />
phenyl substituted compounds are not present on the basis <strong>of</strong> the indicated missing<br />
absorptions - phenyl cyclohexane ( 1010, 1000, 888, 865 cm-' ), 1-ppylcyclohexene<br />
(922, 805 cm-l), and 3-phenylcyclohexene (855, 788, 675 cm-l). The spectrum<br />
is consistent with a non-conjuga'ted benzylcyclopentene system with absor tions at<br />
1070, 1030, and 960 cm-' and the expected aromatic substitution bands.' Two<br />
additional absorptions are noted, one at 850 cm-l which may be attributed to vinyl<br />
protons and a phenyl substitution band at 730 cm-l. This likely results from biphenyl<br />
contamination, but it is interesting to note that benzylidenecyclopentane has an absorption<br />
at 732 cm-' with the remainder <strong>of</strong> the spectrum being similar to the benzylcyclopentenes.<br />
NMR data provides strong evidence for a rigid ring system (non-olefinic<br />
protons poorly resolved 1-3 ppm ), non-conjugated olefinic protons at 5.7 ppm and phenyl<br />
protons at 7.1 ppm. The olefinic protons are complex, giving a closely-spaced doublet<br />
superimposed on a broad absorption, suggesting the presence <strong>of</strong> both ring and exocyclic<br />
unsaturation. Additional sharp proton resonances are noted at 1.2 ppm (methyl) and a<br />
doublet at 2.7 ppm which is attributed to benzylic protons. The lack <strong>of</strong> olefinic protons<br />
downfield from 5.7 ppm would rule out the benzylidene compounds and likely any sim-<br />
ilar structures wherein -the unsaturation is conjugated with the phenyl moiety. The<br />
olefinic protons in cyclopentadiene are found at 6.42 ppm and were not obvious in this<br />
fraction. However, absorptions due to a minor component could have escaped detection<br />
because <strong>of</strong> limited sample size. The difficulty encountered in isolating the components<br />
<strong>of</strong> this peak precludes definite structural assignments, but the reaction <strong>of</strong> phenyl<br />
radicals with fulvene followed by H radical termination and hydrogenation to give<br />
~<br />
4<br />
I<br />
'<br />
,%<br />
'i
j<br />
,<br />
i 449<br />
i<br />
1'<br />
phenyl or benzyl substituted cyclopentenes is clearly indicated. The following, reaction<br />
scheme is proposed to account for the experimental observations:<br />
The inherent complexity <strong>of</strong> the product mix is illustrated by IV above which<br />
could be 1-, 3-and 4:benzylcyclopentene. Preliminary mass spectral data for this sample,<br />
as isolated from the chromatographic separation, indicates m/e values <strong>of</strong> 156 and 158,<br />
I corresponding to the cyclopentadienes I, I1 and the cyclopentenes IV, V. Ring substituted<br />
compounds such as I11 would also have a mass <strong>of</strong> 156. Mass 91, corresponding to the<br />
benzyl radical is prominent as is mass 77, the phenyl radical, although this would be<br />
!,'expected to arise from.the biphenyl (154) contamination. Other features <strong>of</strong> the spectrum<br />
are being evaluated. The low yield is reasonable in view <strong>of</strong> the limited probability for<br />
radical termination and hydrogenation compared with the high reactivity <strong>of</strong> these olefins<br />
(or the diene precursors)'under the reaction conditions. The glow discharge work <strong>of</strong><br />
Schzler indicates the absence <strong>of</strong> any products with vapor pressure similar to biphenyl<br />
and the other related literature generally does not @ve details beyond biphenyl analysis<br />
and gross polymer properties .'<br />
The peak after o-terphenyl (Figure 4) actually consists <strong>of</strong>. 3-4 components<br />
when analyzed by varying the chromatographic conditions. This peak has the, same retention<br />
time as diphenyl fulvene and is in the same general range as noted for a commercially<br />
available hydrogenated terphenyl, Monsanto HB-40. This complex peak was trapped (with<br />
trace o-terphenyl contamination) as a yellow liquid which gave an infrared spectrum<br />
identical-to the polymer fractions and very similar to the peak.before biphenyl. Again,<br />
phenylknzylcyclopentenes are indicated and the spectrum bears little resemblance to<br />
the hydrogenated terphenyl spectrum. The NMR spectrum in carbon tetrachloride showed<br />
aromatic protons at 7.1 ppm,, a broadened olefinic proton resonance at 5.7 ppm and non-<br />
olefinic protons from 0.8-3.5 ppm with poor resolution. These extremely complex<br />
olefinic and non-olefinic resonances are typical <strong>of</strong> protons in a fixed ring system such<br />
as cyclopentene. The i,ntegration for these resonances allows approximation <strong>of</strong> an<br />
average structure containing a cyclopentene ring system substituted with one phenyl and<br />
one benzyl group. The reactions appear analogous to those responsible for the components<br />
<strong>of</strong> the peak before biphenyl with termination being with a phenyl radical rather than the<br />
hydrogen radical. The mass spectrum for this material as trapped from the chromatographic<br />
analysis shows prominent peaks at mass 232 and 234 corresponding to VI and, VU below.<br />
The benzyl and phenyl radical peaks are also evident at mass values <strong>of</strong> 91 and 77<br />
respectively. Evaluation <strong>of</strong> the find structure details is continuing. The following is<br />
believed to be representative <strong>of</strong> the many reactions which are possible under the<br />
experimental conditions, to give a variety <strong>of</strong> closely related compounds:
LUd<br />
The actual structural definition must await isolation <strong>of</strong> sufficient quantities <strong>of</strong> the com-<br />
ponents for NMR studies under various conditions. The possibility <strong>of</strong> a hydrogenated<br />
p-terphenyl was carefully considered, but the only structure even remotely consistent<br />
with the infrared and NMR data would require that the center ring <strong>of</strong> p-terphenyl be<br />
non-aromatic with m'ono-substituted phenyl groups attached. Such a material could be<br />
formed by preferential hydrogenation <strong>of</strong> the center ring <strong>of</strong> p-terphenyl (statistically<br />
unlikely ) or by the reaction <strong>of</strong> phenyl radicals and 1,3 -cyclohexadiene as follows:<br />
This mechanism requires the formation <strong>of</strong> hydrogenated ortho terphenyl derivatives such<br />
as VIII, which were not noted by gas-liquid chromatography or infrared.<br />
TABLE 3<br />
Biphenyl Fraction (a)<br />
X<br />
Yield %<br />
Exposed Benzene<br />
Biphenyl 0.30<br />
o - Terphenyl 0.05<br />
m - Terphenyl 0.02<br />
p-Terphenyl<br />
Benzyl and phenyl cyclopentenes (C12 1<br />
0.05<br />
0.05<br />
Phenylbenzylcyclopentenes ( Cle) 0.25<br />
TOTAL 0.72<br />
VI1<br />
(3)<br />
Yield<br />
% Reaction Products<br />
3.5<br />
0.6<br />
0.2<br />
0.6<br />
0.6<br />
-%<br />
(a) Quantitative data were obtained by GLC, using a silicone gum rubber column with<br />
appropriate standard calibration curves based on peak height.
Polymeric Products<br />
The material which adheres to the glass dielectric surface in the reactor is<br />
a high melting solid ( 3 320°), insoluble in benzene and all common solvents. The infrared<br />
spectrum and the carbon-hydrogen ratio are essentially the same as noted for the benzene<br />
soluble material. The benzene soluble polymer was fractionated into three molecular<br />
weight ranges based on solubility in isooctane. The polymers are all yellow with the in-<br />
tensity increasing as the molecular weight decreases. The ultraviolet spectrum for the<br />
low molecular weight polymer shows a gradual tailing into the visible region. The<br />
physical property data for the polymeric fractions are summarized in Table 4.<br />
Infrared data indicate that the polymeric fractions are structurally similar<br />
to the low molecular weight products identified as benzyl and phenyl substituted cyclopentenes.<br />
The infrared evidence already presented for the low molecular weight products is applicable<br />
to the polymeric products and need not be repeated. The data are consistent with an average<br />
repeating unit containing the cyclopentene ring structure substituted with phenyl or benzyl<br />
groups. NMR data for the polymers were obtained in carbon tetrachloride at the cell holder<br />
temperature (40"). The spectrum obtained for the 300 molecular weight polymer fraction<br />
is presented in Figure 5. The extreme broadening <strong>of</strong> the proton resonances is associated<br />
with the complex, long range roton coupling in a risd system and the motional averaging<br />
commonly noted in polymers .'2 Scanning the same sample at 90°-in tetrachloroethylene did<br />
not significantly improve the resolution. The NMR spectrum <strong>of</strong> the- polymer in .pyridine<br />
(Figure 5 ) gives some improvement in the high field proton resolution, indicating a doublet<br />
at 2.6 ppm (benzylic protons ) and a complex methyl "proton resonance. The spectra are<br />
similar to those obtained for the low molecular weight precursors containing unresolved ,<br />
ring protons. The NMR spectra generally eliminate polymer formation by:way <strong>of</strong> phenyl<br />
and hexatrienyl radicals as suggested for the radiolysis <strong>of</strong> benzene.23 The aliphatic<br />
proton portion <strong>of</strong> the spectrum is very similar -to that reported for cyclopentadiene polymers<br />
by Davies and Was~ermann.~~ The cyclopentadiene polymers had a molecular weight<br />
range <strong>of</strong> 1200-2300, ah'max.at 320-360 mp and non-olefinic proton to olefinic proton ratio<br />
<strong>of</strong> approximately 3/1.<br />
The data are consistent with polymer formanon by way <strong>of</strong> phenyl radical<br />
(or excited benzene) reaction with the fulvene produced to give phenyl or benzyl substituted<br />
cyclopentadienes which then polymerize to gve a polycyclopentene chain with pendant<br />
phenyl and/or benzyl groups. The average non-olefinic to olefinic proton ratio <strong>of</strong> 2.9<br />
indicates that the many possible structures similar to XI (5/2) predominate over the<br />
alternate type structures, XI1 (7/0), 24 assuming our analogy to cyclopentadiene type<br />
polymers is valid.<br />
Q<br />
&,<br />
XI<br />
Many similar structures must be considered, including those derived from phenyl attack<br />
on the ring with polymerization through the exocyclic vinyl group <strong>of</strong> fulvene.<br />
(4)
I<br />
I<br />
I<br />
I<br />
I<br />
In<br />
m<br />
9<br />
In<br />
2<br />
,<br />
I:<br />
r-<br />
In<br />
m<br />
\o<br />
al<br />
"0.<br />
In<br />
N<br />
In<br />
ln<br />
Q\<br />
\o<br />
In<br />
In<br />
PI<br />
hi<br />
9<br />
.-.I<br />
-.<br />
0<br />
r-<br />
In<br />
cv<br />
In<br />
In<br />
m<br />
9<br />
m<br />
9<br />
H<br />
0,<br />
9<br />
c\<br />
in<br />
N<br />
in<br />
In<br />
m<br />
9<br />
In<br />
z!<br />
m<br />
P-<br />
9<br />
m<br />
N<br />
r-<br />
m<br />
i "<br />
c<br />
.d<br />
\<br />
\ I
453
’.<br />
454<br />
Mechanism I<br />
The available evidence is thus not consistent with the generally proposed<br />
i-iiechaii;si;i 0: i;~:j.~~i f~iii~atisii based ~ ithe i iaiidoiii bui:d-i;p d 2 PO:? (pheiijjl~iiej chain<br />
1<br />
accompanied by hydrogenation, or the interaction with the hexatrienyl diradi~al.~’ In the<br />
radi<strong>of</strong>requency discharge <strong>of</strong> benzene, evidence was presented which clearly indicated that<br />
the polymer contained consecutive para linkages suggesting poly (p-phepylenes ) as the<br />
predominant structure.12 Schcler observed that the polymer (C H 1.03, M.W. 503) was<br />
a phenyl substituted aliphatic chain based on infrared evidence.16 Patrick and Burton have ‘<br />
demonstrated that hydrogen atoms are not involved to any significant extent in polymer<br />
formation when liquid benzene is irradiated with a 1.5 m.e.v. source. 24<br />
\<br />
In the corona discharge many types <strong>of</strong> energy transfer are occurring, varying<br />
from photolysis (visible corona) to relatively high energy electrons responsible for frag- ‘i<br />
mentation. The low yield <strong>of</strong> biphenyl in this single pass reactor suggests that phenyl radical<br />
’<br />
production through loss <strong>of</strong> a hydrogen radical is not the primary reaction route leading to<br />
polymer formation. Considerable energy should be available to excite benzene to relatively<br />
high vibrational levels which would be somewhat below that energy required to actually -\<br />
separate. the H radical. At any given time the number <strong>of</strong> these excited benzene molecules<br />
should greatly exceed the phenyl radical population. Fulvene is produced from benzene by<br />
ultra-violet energy (200 mp, about 112 kcals, or 4.9 electron volts).25 Thus, the formation<br />
<strong>of</strong> this benzene isomer with a resonance energy (11-12 kcals/mole) intermediate between<br />
benzene and 1,3 cyclohexadiene may be energetically favorable in the corona environment. \\ 1<br />
Fulvene has been shown to polymerize rapidly under similar conditions with no reversion<br />
to benzene.25 The uniformity <strong>of</strong> the phenyl (benzyl) -cyclopentene ratio throughout all<br />
polymer fractions makes it difficult to accept a mechanism based solely on random attack 1<br />
by phenyl radicals on the growing fulvene polymer. The initial synthesis <strong>of</strong> a monomeric<br />
unit comprised <strong>of</strong> the benzylcyclopentadiene system with subsequent diene type polymerization<br />
is consistent with all our observations. The phenyl radical produced in the discharge<br />
should collide with the nearest benzene molecule which will be excited to a \<br />
relatively high vibrational energy level. The following reaction sequences are suggested<br />
for the phenyl radical:<br />
U<br />
Re*=+<br />
Reaction (5) is the typical termination reaction by radical coupling leading to biphenyl.<br />
The intermediate radical in (6) would be expected to lose hydrogen mo e readily than<br />
it would add to another benzene molecule to give polymer formation.2d The possibility<br />
<strong>of</strong> ring contraction in the excited intermediate, accompanied by an intramolecular shift<br />
<strong>of</strong> a hydrogen atom is suggested in reaction (7). The benzylcyclopentadiene XV so<br />
obtained would be a very active monomer leading to the polymeric products observed.<br />
Although our data support polymer formation by the mechanism indicated, we cannot<br />
rule out participation by the cyclohexadienes and acetylene, both <strong>of</strong> these being noted<br />
a - e n e - CkEketewr-eentper-at-.<br />
XIV
1'<br />
b<br />
Scheme I<br />
fulvene<br />
455<br />
FIGURE 6 PROPOSED REACTION MECHANISMS<br />
I<br />
iHO<br />
1,4 1.3<br />
biphenyl<br />
A\ biphenyl '. 0.- terphenyls<br />
cyclohoxodienes cycloherene cyclohexane<br />
@z, /I P ) 7 3 C H 3<br />
. . .. . . .. . benzyl cyclopentenes<br />
/ \ 05CH3-<br />
phenyl, methyl<br />
substituted<br />
cyclapentenis<br />
phenyl, benzylrubstlhlted<br />
u cyclopentadiene type polymers<br />
/
456<br />
, , .<br />
The relatively low yield and the complexity <strong>of</strong> the reaction products does not<br />
allow an unequivocal designation <strong>of</strong> reaction mechanism or <strong>of</strong> the mode <strong>of</strong> energy transfer in<br />
the corona environment. However, where possible. we have proposed reaction mechanisms<br />
which are consistent with our results to date. These mechanisms are summarized in Figure<br />
6. Studies are underway to obtain deuterium exchange data in the corona reactor, and mass<br />
spectral data are being evaluated. Investlgation <strong>of</strong> the utility <strong>of</strong> corona chemistry will be<br />
' I<br />
extended to other volatile compounds.<br />
EXPERIMENTAL<br />
Materials: Fisher thiophene-free benzene contained trace quantities <strong>of</strong><br />
methyl cyclopentane, cyclohexane and toluene. Chromatogram (squalane ) <strong>of</strong> original<br />
material was used as reference for irradiated benzene evaluation under the same GLC<br />
conditions.<br />
Reference materials for chromatographic identification were generally used<br />
as obtained from commercial sources, Most <strong>of</strong> these materials were <strong>of</strong> sufficient purity to<br />
identify the major peak. However, mono-phenyl fulvene, obtained from Aldrich, gave no<br />
peak on the silicone column and infrared indicated extensive hydroxyl and carbonyl ab-<br />
sorptions. 1,3,5-hexatriene from Aldrich gave two major and two minor peaks on the<br />
squalane column. Infrared27 indicated largely the transisomer with only a small cis ab- )<br />
sorption at 818 ern-'. 1<br />
Fulvene was prepared by the method <strong>of</strong> Meuche. l7 Infrared (1662, 925,<br />
890, 765 cm-l) and ultraviolet (A1 = 242 mu, 4 2 = 360 mu) were in good agreement<br />
literature values: '5 '<br />
Reaction Conditions<br />
The equipment used in these experiments was presented in Figures 1 and<br />
2. Helium was bubbled through the benzene reservoir (55") at 100 ml/min. to carry 4.2<br />
~(0.054 moles) benzene through the corona reactor per hour. The reactor temperature<br />
was 45": 2". A total <strong>of</strong> 101 gm. (1.3 moles) benzene was passed through the 15 KV, 60<br />
cycle, corona discharge in 23.7 hours. A small helium purge was maintained throughout<br />
all sampling operations and necessary downtime to preclude oxidation.<br />
Product Isolation and Analysis !<br />
At the conclusion <strong>of</strong> the run, the reactor was partially filled with benzene<br />
and refluxed on a steam bath for 2 hours. ?he berizene.soluble material from the column<br />
was combined with the yellow brown solid which collected in the lower receiver. The<br />
insoluble material was swelled by bekene and generally loosened from the glass di-<br />
electric surface. This insoluble material was filtered <strong>of</strong>f, washed several times with<br />
chlor<strong>of</strong>orm and dried overnight at 70" in a vacuum oven. The reactor was cleaned<br />
with.a 5 per cent hydr<strong>of</strong>luoric acid solution to remove final traces <strong>of</strong> polymer after<br />
each run. Benzene insoluble polymer - 0.7 gm. yellow brown color, % C 86.95, %H<br />
7.17, C/H 1.01.<br />
The benzene containing all solid products from the reactor and pot<br />
residue was reduced to 30 ml. volume by vacuum distillation at 50". Considerable<br />
foaming was encountered, requiring careful control <strong>of</strong> the distillation. The 30 ml. Of<br />
I<br />
'<br />
I<br />
\<br />
\<br />
\<br />
\<br />
,<br />
\<br />
\<br />
i
- . .. . - . . . -<br />
457<br />
brown benzene solution was added to 300 ml. isooctane at room temperature. A yellow solid<br />
precipitated immediately and was collected by filtration. After several washings with hot<br />
methanol, the material was dried for 8 hours at 70° in a vacuum oven. Benzene soluble<br />
polymer - Fraction 1, 2.3 gm., M. W. 4360, %C 88.32, %H 7.18, C/H 1.02.<br />
The isooctane-methanol solution was reduced to 100 ml. volume (all<br />
methanol removed) by vacuum distillation at 60". The solution was a bkilliant yellow<br />
color at this point. On cooling to room temperature, a small proportion <strong>of</strong> a deep yellow<br />
polymeric solid precipitated and was collected by filtratlon. After washing with hot<br />
methanol, this yellow material was dried at 70Ofor 8 hours in a vacuum oven. Benzene<br />
soluble fraction 2, 0.2 gm., M.W. 1555, %C 87.26, %H 7.09, C/H 1.03.<br />
The isooctane-methanol soluble portion was reduced to 20 gm. by vacuum<br />
distillation and sufficient benzene added to insure solubility <strong>of</strong> ell components. The per<br />
cent solids was determined for this sample, being careful to.limit sublimation <strong>of</strong> biphenyl<br />
(about 2 hours at 70° in a vacuum oven). This sample contains the low molecular weight<br />
products and a yellow resinous polymer. Qualitative analysis for the low molecular weight<br />
components was accomplished by gas-liquid chromatography using the following liquid<br />
phases: silicone gum rubber, Carbowax 20 M, and Reoplex 400. Quantitative data were<br />
obtained on the silicone column (Figure 4) using calibration curves (peak height) prepare-d<br />
from appropriate standards. The concentration <strong>of</strong> the small peak eluted before<br />
biphenyl was estimated using the biphenyl calibration curve and the peak after'o-terphenyl<br />
was estimated using a commercial hydrogenated terphenyl mixture (Monsanto<br />
HB-40) as a reference. The F & M 500 gas-liquid chromatograph equipped with a<br />
standard thermoconductivity detector was used for these analyses. These low molecular<br />
weight products (biphenyl, 0-, m-, p-terphenyl.and phenylcycloalkenes, M. W. up<br />
to -250) amount to 0.72 gms. (See Table 3). Peak confirmation was provided by infrared<br />
and NMR analysis after fraction collection from the chromatographic separation.<br />
The concentration <strong>of</strong> the low molecular weight resinous polymer was<br />
determined by subtracting the total biphenyl fraction weight from the total isooctane-<br />
methanol soluble material. A sample <strong>of</strong> this material was placed in the vacuum oven<br />
at 100" unnl the GLC trace indicated neglipble biphenyl content and the molecular<br />
weight was determined. Benzene soluble fraction 3, 3.5 grn., M. W. 305, %C 87.23,<br />
%H 7.09, C/H 1.03. Prolonged heating at 150" in air gwes a hard, brittle yellow film.<br />
Infrared analysis indicates oxidation is involved in the drpng process.<br />
The benzene fraction was initially examined by GLC using 100 ft.<br />
support coated open tubular columns containing Carbowax 1540 poly (ethylene glycol )<br />
and squalane (Figure 3) as the liquid phase. Quantitative data were obtained at 45O<br />
on the squalane column with a helium flow <strong>of</strong>.3.0 ml/min. The Perkin Elmer Model<br />
2.80 gas-liquid chromatograph, equipped with a stream splitting device and hydrogen<br />
flame detector was used for these analyses. Peak identification was by comparison<br />
with commercial standards and the synthesized fulvene. Additionally, the beniene<br />
fraction was refluxed in pressure bottles with maleic anhydride, azobisieobutyronitrile,<br />
and ammoniacal cuprous chloride solution prior to chromatographic analysis to note<br />
peak changes due to adduct formation, polymerization or presence <strong>of</strong> acetylenic hydro-<br />
gen. Acetylene content was estimated from weight loss <strong>of</strong> the -7O"and -195" traps.<br />
The vapor space above those cold traps and the gas stream directly below the corona<br />
were analyzed by infrared using an 8 cm. gas cell. Although the study was not<br />
exhaustive, several samplings were made and only acetylene was detected.<br />
'
458<br />
h/lethylacetylene, allene and butadiene were not present in sufficient quantity to be detected.<br />
The yellow ,benzene from several runs ( 1000 mlt ) was fractionated and<br />
900 mi. distillate obtained from 79-80". The distillate was refluxed with maleic anhydride<br />
until colorless and the benzene removed at reduced pressure. Dilute sodium hydroxide<br />
solution was added and the resulting solution was extracted several times with chlor<strong>of</strong>orm.<br />
After acidification with dilute hydrochloric acid, the solution was extrayted with ether.<br />
The et1ie.r was removed after drying to give a white solid which, on recrystallization from<br />
chlor<strong>of</strong>orm and pet ether, gave the adduct 7-methylene-5-norbornene-2, 3-dicarboxylic<br />
acid \VI ich melted at 146°-1500. The infrared showed maxima at 1553, 875 and 710 cm-',<br />
(lit ("' 1555, 874, 712 cm-'). Fulvene was prepared in low yield (less than one per cent)<br />
from cyclopentadiene and formaldehyde with sodium ethoxide catalyst. l7 The adduct was<br />
prepared by adding maleic anhydride to the crude fulvene reaction product in Freon I13<br />
(b.p. 47.6O) and refluxing until colorless. After removal <strong>of</strong> the solvent, hydrolysis and<br />
recrystallization yielded the adduct, m. p. 147-150°, which gave properties identical with<br />
the fulvene adduct obtained from the corona discharge. Bryce-Smith obtained a melting<br />
range <strong>of</strong> 105-110° for this adduct which was likely a mixture <strong>of</strong> the ex0 and endo isomers.25<br />
Stille prepared the fulvene-maleic anhydride adducr with a melting point <strong>of</strong> 149-150" which<br />
may be a single isomer.12<br />
Through the courtesy <strong>of</strong> the Perkin Elmer Company at Norwalk, Con-<br />
necticut, the discharged benzene was analyzed by combination <strong>of</strong> gas-liquid chromatography<br />
and mass spectroscopy. The peak attributed to fulyene gave a parent mass <strong>of</strong> 78 and<br />
fragments at m/e 77, 52, 51, 50 and 39 in good agreement with the literature. I' Con-<br />
firmation <strong>of</strong> other assignments was obtained by this technique and the one unidentified<br />
component on the squalane column gave a parent mass <strong>of</strong> 78.<br />
Molecular weight and carbon-hydrogen determinanons were made by<br />
Schwarzkopf Microanalytical Laboratories, New York, New York. Molecular weight<br />
was determined in benzene by vapor pressure osmometry.<br />
LITERAT-URE CITED<br />
1 Present address: Tarrytown Technical Center, Union Carbide, Tarrytown, New York.<br />
2 G. Glockler and S. C. Lind, "The Electrochemistry <strong>of</strong> Gases and Other Delectrics,"<br />
John Wiley and Sons, New York (1939).<br />
3 Ozone Chemistry and Technology, Advances in Chemistry Series No. 21, American<br />
Chemical Society (1959).<br />
4 J. A. C<strong>of</strong>fman and W. A. Browne, Scientific American, 89, June (1965).<br />
5 M. Bertholot, Compt. rend., 82, 1357 (1876).<br />
6 S. M. Losanitsch, Bull, SOC. stun. Bucharest, 233 (1914).<br />
7 A. P. Davis, J. Phys. Chem., 35, 3330 (1931).<br />
8 V. Karapet<strong>of</strong>f, W. H. Trebler, E. G. Linder, and A. P. Davis, Research Reports,<br />
Detroit EdisOn Company, Cornel1 Univ. (1928-32).<br />
9 G. P. Brown, R. E. Rippere, Am. Chem. SOC., Dv. Petrol. Chem., Preprints 2,<br />
NO. 3, 149-54 (1957).<br />
10 H. SchUler, K. Prchal, and E. Kloppenburg, Z. Naturforsch., 15a, 308 (1960).<br />
11 A. Streitwieser, Jr., and H. R. Ward, J. Am. Chem. SOC., 84, 1065 (1962), 85,<br />
539 (1963).<br />
12 J. K. Stille, R. L. Sung, and J. Vander Kooi, J. Org. Chem., 30, 3116 (1965).<br />
13 U. S. I. Chemicals, Polyolefin News, Feb. (1964).
459<br />
14 G. J. Jam, J. Chem. Phys., 22, 751 (1954).<br />
15 J. Thiec and J. Wiemann, Bull. SOC. chim., France, 177 (1956).<br />
16 J. M. Blair and D. Bryce-Smith, Proc. Chem. SOC., 1287 (1957).<br />
17 D. Meuche, N. Neuenschwander, H. Schaltegger, and H. V. Schlunegger, Helv.<br />
Chim. Acta, 47, 1211 (1964).<br />
18 G. E. Gibson, N. Blake, and M. Kalm, J. Chem. Phys., 21, 1000 (1953).<br />
19 G. F. Woods and L. H. Schwartzman, J. Am. Chem. SOC., 70, 3384 (1948).<br />
20 E. E. Van Tamelen and S. P. Pappas, J. Am. Chem. SOC., 85, 3297 (1963).<br />
21 E. L. Eliel, J. W. McCoy, and C. C. Prlce, J. Org. Chem., 22, 1533 (1958).<br />
22 J. G. Powles, Polymer, 1, 219 (1960).<br />
23 W. N. Patrick and M. Burton, J. Am. Chem. SOC., 76, 2626 (1954).<br />
24 A. G. Davies and A. Wassermann, J. Polymer Sci., 4, 1887 (1966).<br />
25 H. J. F. Angus, J. M. Blair, and D. Bryce-Smlth, J. Chem. SOC., 2003 (1960).<br />
26 G. A. Mortimer and L. C. Arnold, J. Am. Chem. SOC., 84, 4986 (1962).<br />
27 J. C. H. Hwa, P. L. de Bonneville and H. J. Sims, J. Am. Chem. Soc., 82, 2538 (1960).
Introduction<br />
VAPOR PHASE DECOMPOSITION OF<br />
AROhIATIC 'HYDROCARBONS BY ELECTRIC DISCHARGE<br />
hlasayuki Kawahata<br />
Research and Development Center<br />
General Electric Company<br />
Schenectady, New York<br />
Decomposition <strong>of</strong> organic compounds in presence <strong>of</strong> electric dis-<br />
charge generally involves fragmentation and polymerization induced by<br />
inelastic collision with high energy electrons. When hydrogen is<br />
added, active hydrogen atoms produced by electric discharge also par-<br />
ticipate in the decomposition reactions. The reaction mechanisms are<br />
complex involving excited molecules, free radicals, and ions.<br />
In this laboratory hydrocracking <strong>of</strong> coal, coal volatiles and related<br />
materials by electric discharge in hydrogen was studied. It is<br />
postulated by Given1 that coal (vitrinite) molecules contain aromatic<br />
and hydroaromatic structures and probably fused aromatic ring nuclei<br />
linked together by methylene or ethylene groups forming hydroaromatic<br />
rings. Many <strong>of</strong> the replaceable hydrogens in the structure are substituted<br />
by hydroxyl or carbonyl groups. Short alkyl groups and<br />
alicyclic rings may also be attached as side chains. In pyrolysis<br />
at 500 - 6OO0C., dissociation <strong>of</strong> hydroxyl groups and dehydrogenation<br />
<strong>of</strong> naphthenic rings take place. These acts lead to formation <strong>of</strong> OH<br />
t<br />
and H radicals which in turn help to break the linkages between aromatic 1<br />
nuclei, forming smaller, partly aromatic volatile fragments, and at<br />
the same time leaving a more aromatic residual structure behind.<br />
On the other hand, high pressure hydrogenation <strong>of</strong> coal gives partial'<br />
or complete hydrogenation <strong>of</strong> the aromatic structure. When this is sub-<br />
sequently cracked, the light products tend to be aliphatic rather than<br />
aromatic hydrocarbons. It was thought that hydrocracking <strong>of</strong> coal by<br />
electric discharge might be somewhere between pyrolysis and high pressure<br />
hydrocracking. In the course <strong>of</strong> study it was thought important to test<br />
this hypothesis by subjecting several aromatic or hydroaromatic compounds<br />
to electric discharge in a hydrogen stream for better understanding <strong>of</strong><br />
the process. Organic compounds selected were m-cresol, d-methylnaphthalen<br />
tetrahydronaphthalene, decahydronaphthalene, and 9, 10-dihydrophenanthrend<br />
Experimental Apparatus and Procedures<br />
The apparatus consisted <strong>of</strong> a vaporizer, a preheater, a discharge<br />
reactor, and a product collecting system: The organic compound was fed<br />
into the vaporizer at a certain rate and vaporized. Hydrogen was also<br />
P. H. Given, presented Am. Chem. SOC., January, 1963 in<br />
Cincinnati, Ohio<br />
\<br />
\<br />
\<br />
'6 1<br />
1<br />
?<br />
1
461<br />
fed into the vaporizer at a certain flow rate and mixed with the organic<br />
vapor. The resultant mixture was then fed into the reactor through<br />
the preheater. The liquid products condensed in a water cooler were<br />
collected in a receiver and the gaseous products were collected in a<br />
liquid nitrogen trap. These products were analyzed by mass spectro-<br />
scope and vapor phase chromatograph employing a 6 ft. column <strong>of</strong> 10%<br />
silicone rubber, SE-30 and 60-80 Chromosorb P.<br />
I<br />
The discharge reactor was fabricated with quartz and was <strong>of</strong> a<br />
concentric tube design similar to an ozonizer. The inside <strong>of</strong> the inner<br />
barrier (40 mm OD and 38 mm ID) and the outside <strong>of</strong> the outer barrier<br />
(48 mm OD and 46 mm ID) were coated with conductive, transparent, tin<br />
oxide. The former was connected to the high voltage terminal and the<br />
latter to ground. In this arrangement the electric discharge was<br />
sustained in an annular space, 46 mm OD, 40 mm ID, and 200 mm long.<br />
The inside barrier tube was filled with stainless steel wool and a<br />
thermometer was placed in the center. This thermometer and three therm-<br />
istors attached to the outside electrode were used to determine the<br />
reactor temperature. The reactor was insulated with glass wool, and the<br />
reactor temperature was maintained at approximately 30OoC. for all the<br />
runs.<br />
The electric discharge power was supplied by Peeding the output<br />
<strong>of</strong> a 10,000 Hertz, 30 kilowatt inductor-alternator to the primary <strong>of</strong><br />
a 50 kilovolt transformer and, in turn, to a tuned circuit, to the<br />
reactor and to the high voltage instrumentation. The electric discharge<br />
power was determined by measuring the area <strong>of</strong> parallelogram on the<br />
oscilloscope2.<br />
When making the run, hydrogen was fed into the reactor at a definite<br />
flow rate and the electric discharge was applied to heat the reactor.<br />
When the reactor temperature was stabilized at about 3OO0C., the feed<br />
<strong>of</strong> the organic compound was started. The discharge power sustained for<br />
the reaction was in a range from 140 to 170 watts.<br />
Experimental Results and Discussions<br />
For cresol-hydrogen mistures, two runs were made using the empty<br />
reactor and three runs were made by filling the reactor space with<br />
porous or activated aluminum oxide grains. For all the runs, the reactor<br />
pressure maintained at 300 mm Hg. Experimental results, including<br />
product distribution, are listed in Table 1. The principal products<br />
were phenol, toluene, benzene, aliphatic hydrocarbons, carbon dioxide<br />
and water. Among the aliphatic hydrocarbons, acetylene was present in<br />
the largest amount and about 8% was unsaturates except for Run 5. In<br />
this run the concentration <strong>of</strong> unsaturates was 55 %. This is probably<br />
due to a higher hydrogen concentration in the feed causing somewhat<br />
T. C. Manley, Trans. Am. Electro Chem. SOC. 84 83, 1943
462<br />
more efficient hydrogenation. The bond energies <strong>of</strong> C6H5-CH3 and<br />
C H5-OH are 90 and 73 Kcal/mol. respectively. Despite this, it was<br />
otserved that, in the products‘. phenol was in higher concentration<br />
than toluene.<br />
Use <strong>of</strong> aluminum oxide packing in the discharge space was intended ,<br />
to investigate the possibility <strong>of</strong> increasing the energy yeild. Narrowing<br />
the gaseous discharge gap with dielectric packings may cpuse the follow- \<br />
ing two effects on the discharge: (1) increase <strong>of</strong> discharge current<br />
for the same discharge power dissipated, and (2) increase <strong>of</strong> gaseous<br />
space breakdown field strength, if the gap decreases beyond a certain<br />
l i m i t . The exact nature <strong>of</strong> the electric discharge employed in this study<br />
is still debatable.3,4<br />
However, it can be reasonably assumed that the )<br />
primary reaction rate <strong>of</strong> the organic vapor with either high energy<br />
electrons or active hydrogen atoms may be dependent on discharge current<br />
density and field strength, if the system pressure and partial pressure<br />
<strong>of</strong> the reactant are constant. In electric discharge product <strong>of</strong> ozone.<br />
an increase in ozone concentration after filling the discharge gap with<br />
various dielectric packings is also reported by Morinaga and Su2uki.S<br />
In this study for approximately the same concentration <strong>of</strong> cresol vapor,<br />
the use <strong>of</strong> aluminum oxide grains in the discharge space appeared to in-<br />
crease the energy yield somewhat, but not conclusively.<br />
In all the runs, the formation <strong>of</strong> brown solid films was observed<br />
on the reactor wall or on the surface <strong>of</strong> the grains. These solid films<br />
were not analyzed but they were insoluble in methylethyleketone or 1<br />
I<br />
toluene. It is presumed that they are indicating possibly highly cross-<br />
linked polymerized products derived from the cresol. The energy yield<br />
for film formation was estimated to be in a range from 30 to 50 g/KWH;<br />
considerably higher than that for the fragmentation products.<br />
Experimental results for the polycyclic compounds are summarized<br />
in Table 2. Methylnaphthalene vapor in hydrogen was tested under pressures<br />
<strong>of</strong> 760 and 74 mm Hg. The principal lighter products were aliphatic<br />
hydrocarbons, benzene and toluene. At the higher pressure the concentration<br />
<strong>of</strong> the lighter aliphatic hydrocarbons was in the order C2)c3>C4>C5. ~<br />
A t the lower pressure, however, this order was reversed. In both cases ,\<br />
about 32 - 35% were unsaturates. The energy yield was approximately<br />
doubled by lowering the pressure.<br />
For tetrahydronaphthalene, three experiments were made under diff<br />
erent pressures. Among the 1 ighter aliphatic hydrocarbons produced,<br />
the C2 fraction was present in the largest amount, in which ethylene<br />
was in highest concentration followed by acetylene and ethane. The<br />
)<br />
total percentage <strong>of</strong> unsaturates increased as the pressure decreased.<br />
This increase was essentially due to the increase in C2 and C un-<br />
7<br />
saturates. As observed for methylnaphthalene, the energy yeiqd at .:<br />
70 mm Hg was twice as high as that at 760 mm Hg. A considerably higher<br />
R. W. Lunt, Advanced Chem. Series, “Ozone Chemistry and Technology”,<br />
Am. Chem. SOC., p. 286 (1959) 1<br />
M. Suzuki and Y. Naito, Proc. Japan Academy, 2 469 (1959) \<br />
K. Morinaga and M. Suzuki, Bull. Chem. SOC., Japan 35 429 (1962)<br />
\<br />
‘1 I<br />
\<br />
j<br />
‘I<br />
4<br />
1<br />
I<br />
\
i<br />
453<br />
energy yield than for the other two runs was observed at the intermediate<br />
pressure, 300 mm Hg.<br />
1 760 mm Hg using 6.7% concentration in hydrogen. Another run was made<br />
When decahydronaphthalene was tested, one experiment was made under<br />
under 300 mm Hg using 67% concentration. In the latter run, a higher<br />
/energy yield was obtained. The product distribution was richer in the<br />
c2 and C3 fraction and also unsaturate concentration wab higher.<br />
'<br />
One experiment was made to test dihydrophenanthrene in hydrogen.<br />
The gaseous products were essentially in the C2 fraction; about 50%<br />
being unsaturated. Vapor phase chromatography <strong>of</strong> the liquid product<br />
showed two peaks. They were not identified but by the combined results<br />
<strong>of</strong> the VPC and mass spectroscope one <strong>of</strong> the peaks was tentatively<br />
I identified as butflbenzene. Biphenyl, which is a likely decomposition<br />
' product, was not found. For the polycyclic aromatic and hydroaromatic<br />
compounds tested in this study, the energy yield from electric discharge<br />
hydrocarcking for production <strong>of</strong> the lighter hydrocarbons was in the following<br />
order:<br />
/<br />
I<br />
I<br />
I The number <strong>of</strong> experiments are not sufficient to permit drawing a<br />
concrete relationship between energy yield and molecular structure.<br />
As observed in cresol runs, solid films were formed on the reactor W a l l ,<br />
but they were not analyzed.<br />
The resonance energies <strong>of</strong> benzene, naphthalene, and phenenthrene are<br />
39, 75, and 110 Kcal, respectively. It is reasonable to assume that<br />
' the condensed aromatic ring structure absorbs large amounts <strong>of</strong> energy<br />
and requires high energy for cracking. The radiation effect on various<br />
polycyclic aromatic compounds were studied by Weiss et a16. They dis-<br />
,\ cussed correlations between the radiation stability and various strut-<br />
$ 1 tural factors. These include resonance energy, electron affinity,<br />
I and ionization constant. Since there is a close similarity between<br />
radiation and electric discharge in principle, further information along<br />
this line would be helpful for a better understanding <strong>of</strong> the electric<br />
discharge hydrocracking process.<br />
b<br />
Elucidation <strong>of</strong> the reaction scheme in detail is beyond the scope<br />
/<strong>of</strong> the this study. However, it was indicated that in electric discharge<br />
hydrocracking <strong>of</strong> aromatic or hydroaromatic hydrocarbons, dissociation<br />
<strong>of</strong> the side chains and rupture <strong>of</strong> the rings are followed by secondary<br />
freactions involving the decomposed species; this leads to the formation<br />
<strong>of</strong> aromatic or aliphatic lighter compounds. Energy requirement to form<br />
these lighter compounds appear to be too high for practical applica-<br />
[ tions. The formation <strong>of</strong> the solid polymerized products, which takes<br />
\<br />
1<br />
I<br />
\<br />
v ' ~<br />
$<br />
J. Weiss, C. H. Collins, J. Sucker, and N. Carciello, Ind. Eng. Chem,<br />
Prod. Res. and Development, 3, 73 (1964)
464<br />
place in parallel with fragmentation, as seen for cresol runs, requires<br />
considerably less energy. Study on the formation <strong>of</strong> polymer films<br />
starting with various monomers and using the present discharge system<br />
presents an extremely interesting problem which is currently under<br />
investigation.<br />
This is a part <strong>of</strong> the work supported by the Office <strong>of</strong> Coal Research,<br />
The author is thankful fpr their generous\<br />
U. S. Department <strong>of</strong> Interior.<br />
support .<br />
1<br />
\<br />
\
Ldd<br />
ffl<br />
cc<br />
3 .I+<br />
X<br />
z<br />
\rl<br />
M O<br />
C<br />
- 0<br />
fflc<br />
e a<br />
0<br />
k<br />
&.2 u”<br />
vc,<br />
ldv<br />
c E\<br />
ld o z<br />
M k<br />
k 4 u<br />
0 u<br />
u)<br />
kM<br />
oc<br />
c, .I4<br />
O X<br />
ldv<br />
alld<br />
m a<br />
0<br />
2<br />
a<br />
X<br />
W<br />
In<br />
W<br />
0<br />
IC<br />
hl<br />
m<br />
0<br />
rl<br />
m<br />
hl<br />
In<br />
0<br />
0<br />
E<br />
In<br />
In<br />
(0<br />
hl<br />
In<br />
In<br />
In<br />
IC<br />
rl<br />
m<br />
In<br />
r(<br />
rl<br />
hl<br />
a3<br />
0<br />
0<br />
a0<br />
m<br />
m<br />
a<br />
al<br />
ld c,d<br />
m c ldc<br />
I n c<br />
U a b<br />
m c o<br />
w - M<br />
46 5<br />
rl<br />
0<br />
,ul<br />
al<br />
k<br />
V<br />
A<br />
y-c<br />
0<br />
u<br />
d<br />
k<br />
c,<br />
V<br />
al<br />
rl<br />
W
466<br />
P<br />
rl<br />
0,<br />
k<br />
7<br />
rnI<br />
rn<br />
al<br />
k<br />
a<br />
I<br />
hl<br />
m<br />
b<br />
m<br />
l-l<br />
I-I<br />
hl<br />
w<br />
hl<br />
m<br />
m<br />
hl<br />
(D<br />
(D<br />
0<br />
0<br />
(0<br />
t-<br />
In<br />
w<br />
v)<br />
m<br />
N<br />
0<br />
*<br />
Q,<br />
N<br />
*<br />
N<br />
0<br />
m<br />
N<br />
r(<br />
*<br />
b<br />
m<br />
ro<br />
w<br />
b<br />
hl<br />
l-l<br />
m<br />
hl<br />
rl<br />
b<br />
b<br />
In<br />
In<br />
rl<br />
0<br />
(D<br />
b<br />
In<br />
In<br />
m<br />
hl<br />
d<br />
m<br />
(D<br />
d<br />
m<br />
IC<br />
rl<br />
m<br />
m<br />
0<br />
0<br />
m<br />
m<br />
t-<br />
0<br />
Q,<br />
In<br />
In<br />
hl<br />
m<br />
hl<br />
*<br />
b<br />
I<br />
I<br />
0<br />
m<br />
0<br />
b<br />
w<br />
b<br />
Q,<br />
*<br />
rl<br />
CJ<br />
b<br />
0<br />
d<br />
0<br />
@a<br />
a0<br />
m<br />
*<br />
Q,<br />
m<br />
0<br />
W<br />
b<br />
b<br />
W<br />
0 4<br />
4 4<br />
W<br />
b<br />
b<br />
hl<br />
b<br />
b<br />
hl<br />
(D<br />
ua<br />
*<br />
9<br />
In<br />
0<br />
0<br />
m<br />
b<br />
(0<br />
(0<br />
*<br />
*<br />
rl<br />
t-<br />
rl<br />
@a<br />
m<br />
t-<br />
m<br />
N<br />
m<br />
Q,<br />
(D<br />
0<br />
0<br />
W<br />
b<br />
In<br />
*<br />
al<br />
k<br />
c<br />
V<br />
.d<br />
c<br />
3<br />
m<br />
c<br />
0<br />
.A<br />
c,<br />
V<br />
cd<br />
k<br />
w<br />
k<br />
0,<br />
.A<br />
><br />
cd<br />
0)<br />
c<br />
al<br />
c<br />
c,<br />
m<br />
cd<br />
3<br />
h<br />
$<br />
(D<br />
c<br />
7<br />
k<br />
I/)<br />
.A<br />
c<br />
c,<br />
k<br />
0<br />
w<br />
c<br />
0<br />
.A<br />
c,<br />
1<br />
P<br />
.A<br />
k<br />
c,<br />
rn<br />
.A<br />
5<br />
c,<br />
V<br />
7<br />
P<br />
0.<br />
kU<br />
aa<br />
><br />
al<br />
Ch<br />
W P<br />
+la<br />
oa,<br />
.A<br />
al'u<br />
04<br />
EIc,<br />
dfi<br />
dal<br />
dU<br />
alc'<br />
co<br />
bfi<br />
v<br />
nlc<br />
*<br />
P<br />
fi<br />
cd<br />
V<br />
.A<br />
c,<br />
d<br />
Em<br />
O V<br />
kfi<br />
41<br />
0<br />
v a<br />
E<br />
4 0<br />
hU<br />
u<br />
A V<br />
d .I4<br />
Oc,<br />
a d<br />
E<br />
rn wo<br />
.E O h<br />
0 (d<br />
.A M O<br />
c, Eh<br />
u dQ<br />
cd Y h<br />
k OX<br />
w d<br />
k<br />
Q, V<br />
0<br />
k<br />
U<br />
I<br />
rl a<br />
V h<br />
X<br />
fi<br />
.A<br />
m<br />
.a,<br />
c,<br />
cd<br />
k<br />
1<br />
c,<br />
cd<br />
m<br />
fi<br />
1<br />
'u<br />
0<br />
a,<br />
M<br />
cd<br />
c,<br />
fi<br />
0)<br />
0<br />
k<br />
a,<br />
a<br />
*<br />
*<br />
I<br />
a'<br />
I<br />
1
HYDROCARBONS AND CllRBON FROM A ROTATING ARC HEATER<br />
C. Hirayama and D. A. Maniero<br />
Westinghouse Electric Corporation, Pittsburgh, Pa.<br />
INTRODUCTION<br />
A brief description <strong>of</strong> the rotating arc heater utilized in this work,<br />
and some preliminary hydrocarbon processing data, were described a year<br />
ag0.l A more detailed description <strong>of</strong> the hbater, and some <strong>of</strong> the operating<br />
characteristics have also been presented within the past year.2 The<br />
arc heater consists essentially <strong>of</strong> water-cooled, toroidal electrodes, in<br />
which the arc is rapidly rotated as a result <strong>of</strong> interaction <strong>of</strong> the arc<br />
current with a magnetic field. The heater is a high current, low voltage<br />
device with a rating <strong>of</strong> 3 megawatts into the arc. Both alternating and<br />
direct current power operation are possible with this device.<br />
In contrast,<br />
most arc devices previously reported3 8pe <strong>of</strong> the long arc, high voltage<br />
type which are operated on d.c. only.<br />
We wish to report, herewith, some <strong>of</strong> the res.ilts obtained more recently<br />
in the pyrolysis <strong>of</strong> methane at atmospheric and at slightly elevated pressures.<br />
The feed gas was commercial grade methane <strong>of</strong> 96 per cent purity which<br />
was injected into the heater at ambient temperature. The flar rate was controlled<br />
by regulating the pressure drop across a sonic orifice.<br />
The arc heater was operated only on a.c. parer at electrode separations<br />
<strong>of</strong> 0.38, 0.75 and 1.0 inch. The arc power with methane ranged from 550 to<br />
2200 kilowatts, with thermal efficiencies between 25 and 76 per cent, depending<br />
on operating conditions. For operation at elevated chamber pressure,<br />
the nozzle was choked with a graphite plate which had an orifice <strong>of</strong> 0.5 or<br />
0.75 inch diameter in the center <strong>of</strong> the plate. The chamber pressure was as<br />
high as 100 psig.<br />
The heater was operated at two different field coil currents to determine<br />
arc rotation velocity on the degree <strong>of</strong> reaction.<br />
The products <strong>of</strong> the reaction were quenched and collected through a<br />
water-cooled copper probe, 118 or 1/4 inch I.D., inserted about an inch<br />
inside <strong>of</strong> the nozzle. The product from the probe was first passed through<br />
'<br />
a fiber filter (Purolater Co.) to separate the carbon, then collected at<br />
appropriate intervals in ga8 sapling tubes on a manifold, a8 previously<br />
described .1<br />
The gas analyses were made mas8 spectrometrically. A typical composition<br />
<strong>of</strong> a sam3le is shown in Table I; Table I1 shows the approldmate material<br />
balance based on the &/C ratios <strong>of</strong> the feed and product gases. The only<br />
variation with operating conditione is In the relative concentration <strong>of</strong><br />
the different species.
458<br />
TABLE I. COMPOSI'IION OF GASEOUS PRODUCT, MOU PERCENT<br />
~ ~ a c y 4 ~ ~ ~ c &<br />
77.68 1.27 0.09 5.81 13.58 1.01 0.12 0.32 0.14<br />
TABU 11. MATERIAL BALANCE OF PRODUCTS IN MOLE PERCENT<br />
c o c q cy4 sc&='ac<br />
73.06 1.19 0.08 5.46 12.77 0.95 0.11 0.30 0.13 15.00<br />
Electron micrographs were obtained on a number <strong>of</strong> the carbon samples<br />
by replicating an amyl acetate suspension on a carbon film. X-ray spectra<br />
were obtained by packing the soot into a disc-sarnple holder and irradiating<br />
with Cu K-a radiation. The sample packing and irradiation were kept as<br />
nearly identical as possible for all <strong>of</strong> the samples. The d-spacing and line<br />
intensity were compared against AUC graphite measured under identical conditione.<br />
RESULTS AM, DISCUSSION<br />
As shown in Table 11, the product consists <strong>of</strong> species which are expected<br />
from the high temperature pyrolysis <strong>of</strong> methane. The primary products are H2,<br />
C2H2, and carbon, with varying concentration <strong>of</strong> C&, depending on the degree<br />
<strong>of</strong> reaction. The diacetylene and benzene axe fonned from the polymerization<br />
<strong>of</strong> the acetylene; the methyl acetylene, although not intermediate in the<br />
conversion <strong>of</strong> to C2H2, is known to be a minor product found in the pyrolysis<br />
<strong>of</strong> diacetyleney The C,H, is an intermediate in the C'&+ C,H, reaction.<br />
There is a slight error in the material balance since the hydrogen content<br />
<strong>of</strong> the soot was not taken into account because <strong>of</strong> sampling difficulty. The<br />
soot usually contains about one percent <strong>of</strong> hydrogen, and the presence <strong>of</strong><br />
aromatic constituents is evident from the odor.<br />
Considering the difficulty <strong>of</strong> obtaining exact experimental parameters<br />
during an experiment, quite good correlation was obtained between the degree<br />
<strong>of</strong> reaction and arc enthalpy, whereas the correlation was poor with respect<br />
to the net enthalpy increase <strong>of</strong> the gas. Figure 1 shows the relative degree<br />
<strong>of</strong> methane pyrolysis as a function <strong>of</strong> the arc enthalpy. The curves show the<br />
comparison between runs made at 2500 and 1500 amperes field coil current.<br />
The former effects an arc rotation velocity approximately 66 percent greater<br />
than that at the 1500 amps field coil current. There is a significantly<br />
steeper slope at higher arc rotation velocity, this result probably arising<br />
from the greater degree <strong>of</strong> mixing.<br />
Note that the curyes saturate at high<br />
enthalpies, where the system is at equilibrium, or near equilibrium condition.<br />
A minimum just above the critical enthalpy (or initiation temperature).<br />
The electron micrographs <strong>of</strong> the soot, quenched at the heater nozzle,<br />
,500X magnification, Figure 2, shows spherical particles <strong>of</strong> less than<br />
100 at 2l to approximetely 5000 A diameter. The nature <strong>of</strong> the background is not<br />
knonw at present, but blow-ups <strong>of</strong> the photomicrographs to approximately<br />
153,OoOX suggests that the cloud consists <strong>of</strong> extremely fine, smokey carbon<br />
dust.
J<br />
i<br />
I<br />
i<br />
I<br />
469<br />
The d-spacings ob ained from the x-raa diffraction <strong>of</strong> the soot varies<br />
between 3.45 and 3.49 B ,ocompared to 3.35 A for pure, crystalline graphite.<br />
The lower value <strong>of</strong> 3.45 A was obtained on the carbon collected from the high<br />
pressure runs, where the residence times were up to seven times longer than<br />
the runs at atmospheric pressure. The degree <strong>of</strong> crystallinity <strong>of</strong> the soot,<br />
based on the x-ray intensity, varied Prom two to ten percent <strong>of</strong>:AUC graphite.<br />
The x-ray crystallinity, as suspected, is a function <strong>of</strong> the residence time.<br />
It was <strong>of</strong> interest to plot the major product compositions on a semilog<br />
plot, as a function <strong>of</strong> reciprocal arc enthalpy since the latter is proportional<br />
to the temperature in the gas. Figure 3 shows that the slopes<br />
for acetylene and hydrogen are approximately the same, whereas the slope for<br />
carbon is much steeper. The difference in slopes for carbon, as compared to<br />
CBH2 and H, clearly shows the different mechanisms for the formation <strong>of</strong> these<br />
species.
REFERENCES<br />
1. C. Hirayama and D. A. Maniero, "A New Rotating Arc Heater for Chem-<br />
ical Processing," Preprints, Div. <strong>of</strong> Fuel Chem., Am. Chem. SOC.<br />
Meeting, Pittsburgh, March 22-31, 1966, Vol. 10, No. 1, pp. 123-132.<br />
2.<br />
D. A. Maniero, P. F. Kienest, and C. Hirayama, Westinghodse Engineer<br />
- 26, 66 (1966). Also, same authors, paper presented at Electrochem.<br />
Sac. Eat. Meeting, Phila., Oct. 12, 1966.<br />
3. See, for example, S. A. Miller, "Acetylene, Its Properties, Manufacture<br />
and Uses." Vol. I, Academic Press, New York, 1965, pp. 391-405.<br />
4.<br />
K. C. Hou and H. B. Palmer, J. Phys. Chem. 69, 858 (1965).<br />
tj<br />
I
Arc Enthalpy ,-)<br />
- 2500 Amps Field Coil Current<br />
-- 1500 " 11 It I1<br />
Fig. 1. Methane conversion as functions <strong>of</strong> arc enthalpy and arc rotation<br />
Fig. 2. Electron micrographs <strong>of</strong> carbon at 25,500X
i<br />
472<br />
Reciprocal Arc Enthalpy ,-><br />
Fig. 3. Product composition as a function <strong>of</strong><br />
reciprocal aro enthalpy
473<br />
THE REARRANGEMENT OF METHANE AND METHYL CHLORIDE IN A MICROWAVE<br />
DISCHARGE<br />
I. INTRODUCTION'<br />
J. P. Wightman<br />
Virginia Polytechnic Institute, Blacksburg, Virginia<br />
N. J. Johnston<br />
NASA - Langley Research Center, Hampton, Virginia<br />
In a previous paper(1) the rearrangement <strong>of</strong> a series <strong>of</strong><br />
hydrocarbons including methane in a microwave discharge was reported.<br />
The work reported here is an investigation <strong>of</strong> the effect <strong>of</strong> the<br />
introduction <strong>of</strong> a functional group in methane on the nature <strong>of</strong> both<br />
the gaseous and solid products <strong>of</strong> rearrangement. Such measurements<br />
should ultimately support a model <strong>of</strong> the rearrangement mechanism<br />
in a microwave discharge. Methane and methyl chloride werc passed<br />
separately in the absence <strong>of</strong> a diluent through a microwave dis-<br />
charge.<br />
11. EXPERIMENTAL<br />
A. Materials<br />
Methane (ultrahigh purity grade) was obtained from the<br />
Matheson Co. and used without further purification. The following<br />
analysis was supplied with the methane: C02 - (5 ppm; O2 5 ppm;<br />
N - 19 ppm: C2H6 - 14 ppm: C3H8 - (5 ppm. Methyl chloride<br />
(gigh purity grade) was obtained from the Matheson Co. and used<br />
without further purification.<br />
B. Apparatus and Procedure<br />
The rearrangements were carried out in a high vacuum<br />
flow system. The power source for the microwave discharge was a<br />
Raytheon generator(Mode1 KV-1?4) and was operated at a power level<br />
corresponding to about 40 r.f. watts at 2450 Mc. The generator<br />
was connected to an air-cooled cavity(Raytheon - KV series) by a<br />
coaxial cable. Two procedures were used depending upon whether<br />
an analysis <strong>of</strong> the gaseous products was being made or whether solid<br />
film was being deposited for subsequent analysis. In the former<br />
case, the parent gas was passed through a variable leak valve<br />
(Veeco - VL). The pressure in a ty ical experimental run was 0.15<br />
torr at a mass flow rate <strong>of</strong> 1 x lo-' moles/min. and a linear flow<br />
rate <strong>of</strong> 20 cm./sec. In the latter case, the parent gas vas passed<br />
through a Teflon needle valve (Fischer-Porter) . The pressure in a<br />
typical experimental run was O.5Oitorr. Normal deposition time<br />
was 20 min.<br />
The gaseous rearrangement products <strong>of</strong> methane and methyl<br />
chloride were determined using a mass spectrometer(Associated<br />
Electronics Industries - MS-10). The sampling volume was located<br />
about 140 cm. downstream from the discharge. After establishment<br />
<strong>of</strong> a constant flow rate, the parent gas stream was sampled. An<br />
<strong>of</strong>f-on valve upstream from the discharge allowed the system to
474<br />
be pumped down without altering the setting <strong>of</strong> the variable leak.<br />
Between analyses pumpdown resulted in faster attainment <strong>of</strong> steady<br />
state concentrations <strong>of</strong> rearrangement products. Flow was again<br />
started and the discharge initiated with a Tesla coil. After<br />
establishment <strong>of</strong> a constant flow rate the discharged gas stream<br />
was sampled.<br />
The solid polyermic films were removed mechanically<br />
from the walls <strong>of</strong> the Pyrex tubing. The infrared spectra <strong>of</strong> the<br />
films <strong>of</strong> both gases were obtained using either a Perkin-hlmer<br />
421 or 621 spectrophotometer. The electron spin resonance spectra<br />
<strong>of</strong> the neat films from both gases were obtained on a Varian ESR 6<br />
spectrometer. Elemental analyses <strong>of</strong> both films were made by<br />
Gailbraith Labs.<br />
111. RESULTS<br />
’ A. Gaseous Products<br />
1. Mass spectra<br />
Selected peaks from the mass spectra <strong>of</strong> methane and<br />
methyl chloride obtained with the discharge on and <strong>of</strong>f are shewn<br />
in Figures la and lb. The most significant features ip the methane<br />
spectra(Figure la) were the incrTase in the m/e = 2 (H- ) peak and<br />
the decrease in the m/e = 15 (CH3) peak when methane W ~ S passed<br />
through the discharge. Trace quantities <strong>of</strong> higher molecular weight<br />
hydrocarbons were also noted. The most significant features <strong>of</strong><br />
the methyl+chloride spectra (Figure lb) were the increaae in the<br />
m/e - 2 (H2) peak, the appearance o$ the m/e = 30 (C2H6) peak and<br />
the decrease in the m/e = 50 (CH3C1 ) peak when methyl chloride<br />
was passed though the discharge. Further, HC1 was noted when<br />
the liquid nitrogen cooled trap downstream from the discharge<br />
was opened.<br />
B. Solid Films<br />
A solid film was observed to form in the discharge region<br />
when either methane or methyl chloride was passed through the dis-<br />
charge. Both films were characterized in several ways.<br />
1. Elemental analysis<br />
Empirical formulas for the solid films were established<br />
by elemental analysis. The formulas for the solid films formed<br />
form methane and methyl chloride were (CH1.5)x and (CHo.65Clo.045 1<br />
respectively .<br />
2. Infrared spectra<br />
The infrared spectrum <strong>of</strong> the film obtained from methane<br />
is shown in Figure 2 where a neat sample was used. Peaks ascribed<br />
to the film were noted in the regipn <strong>of</strong> the following frequencies5<br />
2900, 1700, 1450, 1380 and 880 cm . The assignments <strong>of</strong> these<br />
freauencies follow those given by Jesch et al(2). The band at 880<br />
cm hay be due to rocking vibrsfion <strong>of</strong> -CH in a multiple carbon<br />
atom chain. The band at 1290 cm is characgeristic <strong>of</strong> -CH deforma-<br />
3<br />
tion. The band at 1450 cm is character’stic <strong>of</strong> - CH2 symmetric<br />
scissors vibration. The band at 1700 cm-‘ was probably due gy<br />
carbonyl formed on exposure <strong>of</strong> the film to air. The 2900 cm band<br />
!
4<br />
i<br />
J<br />
1<br />
i<br />
475<br />
is due to C-H stretching vibration.<br />
The infrared spectrum <strong>of</strong> the film obtained from methyl<br />
chloride is shcyln in Figure 3 where a neat sample was used. The<br />
low intensity peaks were a characteristic <strong>of</strong> the methyl chloride<br />
films whether run as a neat sample or as a KBr pellet. Peaks<br />
ascribed to the film were noted in the region-pf the following<br />
frequencies: 2900, 1700, 1580, 1440 and 875 cm . The peak at<br />
1380 cm observed in the methane film- s absent in the methyl<br />
chloride film and a new peak at 1580 cm iis observed in tHe methyl<br />
chloride film.<br />
3. Electron spin resonance<br />
The electron spin resonance spectra <strong>of</strong> the films pro-<br />
duced from methane and methyl chloride are shown in Figures 4a<br />
and 4b. A significantly larger free spin concentration on the<br />
order <strong>of</strong> one thousand times greater was noted in the case <strong>of</strong> the<br />
methyl chloride film. No fine structure was observed in the case<br />
<strong>of</strong> the methyl chloride film. g-values for both films were<br />
essentially identical to the value for pitch(g = 2.000).<br />
4. Solubility studies<br />
An extensive series <strong>of</strong> liquids were tested as possible<br />
Solvents for the films prior to NMR measurements. Solubility <strong>of</strong><br />
the methane film was observed only in the case <strong>of</strong> hexamethyl-<br />
phosphoamide and concd. H2S04.<br />
IV. DISCUSSION<br />
Investigations <strong>of</strong> chlorinated hydrocarbons in a microwave<br />
discharge have not been reported in the literature previously.<br />
Swift et a1 (3) however have investigated the effect <strong>of</strong> an rf<br />
discharge on CC14 in the absence <strong>of</strong> a diluent. A number <strong>of</strong><br />
chlorinated gaseous products were reported in addition to a<br />
chlorinated polymer. The present work indicated a limited number<br />
<strong>of</strong> gaseous products. The results are consistent with the argument<br />
that molecules in a xicrowave discharge are subjected to a<br />
greater degree <strong>of</strong> fragmentation than in an rf discharge thereby<br />
limiting both the number aild ;he complexity <strong>of</strong> gaseous products.<br />
From the previous work <strong>of</strong> Vastola and Wightplan (1) the<br />
following postulate could be advanced: if a parent hydrocarbon<br />
has a hydrogen to carbon (H/C) ratio greater than about 1.6, a<br />
hydrogen saturated solid film will be produced on passage <strong>of</strong> the<br />
hydrocarbon through a microwave discharge in addition to hydrogen.<br />
Conversely, if a parent hydrocarbon has a (H/C) ratio less than about<br />
1.6 a hydrogen deficient film will be produced and no hydrogen<br />
will be observed.<br />
The present work is an attempt to test the validity <strong>of</strong><br />
this postulate if a functional group in introduced into the hydro-<br />
carbon molecule. Methyl chloride was chosen since it represents<br />
a straightfarward extension <strong>of</strong> the methane case. Methyl chloride<br />
has a (H/C) ratio <strong>of</strong> 3 and if the C1 is neglected, the formation<br />
<strong>of</strong> a hydrogen saturated film would be predicted. However, HC1<br />
was observed as a rearrangement product. Hence the C1 can not<br />
be neglected and yet assuming the limiting case <strong>of</strong> a 1:l corres-<br />
pondence between CH3C1 and HC1, the (H/C) ratio <strong>of</strong> the remaining<br />
fragment would be 2. Thus the formation <strong>of</strong> a hydrogen saturated<br />
film would still be predicted but which in fact was not observed.
476<br />
Instead a hydrogen deficient film was produced. The formation <strong>of</strong><br />
a hydrogen deficient film from a parent molecule which has a high<br />
enough (H/C) ratio to form a hydrogen saturated film can be attributed<br />
to two factors. In the first instance, C1 preferentially<br />
appears in the gas phase as is indicated by the low percentage <strong>of</strong><br />
C1 in the film, the absence <strong>of</strong> a significant C-C1 absorption peak<br />
in the infrared spectrum(Figure 3) and the presence <strong>of</strong> HC1 downstream<br />
from the discharge. The extension <strong>of</strong> this work to other<br />
functional groups is anticipated to determine if this is a general<br />
scheme. In the second instance, the tendency to form hydrogen in<br />
a microwave discharge is again noted in the case <strong>of</strong> methyl chloride<br />
as in the case <strong>of</strong> methane(Pigures la and lb). Apparently for<br />
hydrocarbons containing functional groups, the formation <strong>of</strong> hydrogen<br />
is favored even at the expense <strong>of</strong> the formation <strong>of</strong> a hydrogen<br />
deficient film. The formation <strong>of</strong> ethane observed as a product <strong>of</strong><br />
the methyl chloride discharge could be due to the recombination<br />
<strong>of</strong> methyl radicals produced in the discharge.<br />
Films formed in various types <strong>of</strong> electrical <strong>discharges</strong> have<br />
not been extensively characterized. The recent work <strong>of</strong> Jesch et a1<br />
(2) described the infrared analysis <strong>of</strong> a series <strong>of</strong> films produced<br />
from hydrocarbons in a glow discharge. Direct comparison between<br />
the present results and those <strong>of</strong> Jesch et a1 is not possible since<br />
two different types <strong>of</strong> electrical <strong>discharges</strong> were used. The type<br />
<strong>of</strong> discharge used dramatically alters the nature <strong>of</strong> both the gaseous<br />
and solid rearrangement products as indicated above<br />
The properties <strong>of</strong> the film produced from methyl chloride<br />
are similar to those reported previously(1) for the hydrogen<br />
deficient films formed from acetylene, benzene and naphthalene.<br />
The color <strong>of</strong> the methyl chloride f ilm was dark (brownish-black)<br />
characteristic <strong>of</strong> hydrogen deficient films in contrast to the light<br />
yellow methane film characteristic pf hydrogen saturated films.<br />
The high electron spin concentration <strong>of</strong> the methyl chloride film<br />
ibad also been oberved for the hydrogen deficient film from acetylene.<br />
The films produced from both methane and methyl chloride<br />
appear to be highly cross-linked as evidenced by the negligible<br />
solubility is liquids used as solvents for other polymeric systems.<br />
The methyl chloride film contains a greater number <strong>of</strong> free electrons<br />
than the methane film indicative <strong>of</strong> a significant number <strong>of</strong> unsaturated<br />
valences in the methyl chloride film. The methyl chloride film<br />
appears to be characterized by a greater degree <strong>of</strong> unsaturation<br />
than the methane film. Highly unsaturated polymeric systems have<br />
been found difficult to analyze by infrared spectroscopy* which<br />
has also been noted in the present results. Definitive NMR work<br />
would be helpful in elucidating the nature <strong>of</strong> these polymeric films.<br />
* private communication with Dr. Vernon Bell(NASA - Langley Research<br />
Center)<br />
Acknowledgements - The authors would like to acknowledge the help <strong>of</strong><br />
Dr. R. E. Dessy in obtaining the ESR spectra and the experimental<br />
assistance <strong>of</strong> Mr. R. 0. McGuffin‘.<br />
REFERENCES<br />
(1). F. J. Vastola and J. P. Wightman, J. Appl. Chem., 14, 69 (1964).<br />
(2). K. Jesch, J. E. Bloor and P. L. Kronick, J. Poly. Sci. A-1, 4,<br />
1487 (1966).<br />
(3). E. Swift, ‘r., R. L. Sung, J. Doyle and J. K. Stille, J. Org.<br />
Chem., a, 3114 (1965).
1<br />
I<br />
I<br />
/ 10000<br />
Y<br />
1<br />
-<br />
I<br />
before<br />
di sc harge<br />
I discharge<br />
during<br />
Fig.la. Selected<br />
mass .spectra o<br />
ih a microwave<br />
2 15<br />
MAS SIC HA RGE RATIO<br />
eaks from Fig.1b. Selected peaks from<br />
Pmethane mass spectra <strong>of</strong> methyl<br />
discharge. chloride in a discharge.<br />
n
i<br />
478
L<br />
t<br />
479
480<br />
FATTY ACIDS AND n-ALKANES IN GREEN<br />
RIVER OIL SHALE: CHANGES WITH DEPTH<br />
Dale L. Lawlor and W. E. Robinson<br />
Laramie Petroleum Research Center, Laramie, Wyoming<br />
INTRODUCTION<br />
Maqy papers have been published relating fatty acids to n-alkanes in ancient sedi-<br />
ments. Breger (1) postulated that n-alkanes may be formed in sediments by beta-<br />
keto acid decarboxylation. Jurg and Eisma (5) demonstrated in the laborator) th6:<br />
a homologous series <strong>of</strong> n-alkanes can be produced by heating the C22 fatty aci6 ir.<br />
the presence <strong>of</strong> bentonite. Cooper and Bray (3 suggested that odd-carbon-numbered<br />
n-alkanes and odd-carbon-numbered fatty acids may be produced from naturally occur-<br />
ring even-carbon-numbered fatty acids by a free-radical decarboxylation mech-plsin<br />
Mair (I) and Martin and coworkers (a) have discussed the relationship between fa-r,?<br />
acids and n-alkanes in petroleum genesis.<br />
If fatty acids are converted to n-alkanes in sediments, a distributional relation-<br />
ship would likely be apparent between the two compound classes. Such a relationship<br />
would be most relevant if both the acids and the alkanes were indigenous to each<br />
sample taken from one geological formation. In this respect the Green River Forma-<br />
tion is ideal for study <strong>of</strong> the relationship between fatty acids and n-alkanes. This<br />
formation represents 6 million years <strong>of</strong> accumulation <strong>of</strong> organic debris from a highly<br />
productive Eocene lake which existed approximately 50 million years ago. A 900-foot<br />
core representing 3 million years <strong>of</strong> the Green River Formation was sampled and the<br />
fatty acids were extracted and analyzed.<br />
An earlier paper (5) presented a correlation between the carbon number distribution<br />
<strong>of</strong> the fatty acids and the carbon number distribution <strong>of</strong> the n-alkanes in the<br />
Mahogany zone portion <strong>of</strong> the Formation. The present paper tests the postulate that<br />
n-alkanes are formed from fatty acids by maturation processes resulting from the<br />
time differential existing between the bottom and the top <strong>of</strong> the core. The results<br />
showed that both maturation and changes in organic source material explain the dif-<br />
ferences in samples taken from several stratigraphic positions in the formation.<br />
EXPERIMENTAL<br />
Ten oil-shale samples were taken from the Green River Formation, nine from a 900-<br />
foot core (Equity Oil Co., Sulfur Creek No. lo), and one from the Mahogany zone.<br />
The stratigraphic positions and the oil yields <strong>of</strong> the 10 samples are presented in<br />
table 1. The samples were the same as those used by Robinson and coworkers (2) in<br />
a study <strong>of</strong> the distribution <strong>of</strong> n-alkanes at different stratigraphic positions within<br />
the formation.<br />
The samples, ground to pass a 100 mesh screen, were treated with 10 percent hydrochloric<br />
acid to remove mineral carbonates and to convert salts <strong>of</strong> acids to free<br />
acids. Two-hundred grams <strong>of</strong> each <strong>of</strong> the acid-treated samples were refluxed with 400<br />
ml <strong>of</strong> 7 percent borontrifluoride in methyl alcohol for 6 hours. This reaction converted<br />
free and esterified acids to methyl esters. The reaction mixture was cooled,<br />
filtered, and washed with methyl alcohol until the washing was clear. The filtrate<br />
was transferred to a separatory funnel, water was added, and the solution wae<br />
extracted with successive portions <strong>of</strong> carbon tetrachloride until the solvent was<br />
colorless. The carbon tetrachloride solution <strong>of</strong> the methyl esters was dried OV(?<br />
night using anhydrous sodium sulfate. The fatty acid methyl esters were isolated<br />
from the carbon tetrachloride solution by urea adduction.<br />
I
TA?.LE 1, - StratigraphiL po:itio;. ar.d pyrolytic cil<br />
yields <strong>of</strong> the oil-shale samples<br />
Approximate pyrolytic<br />
Samp le Stratigraphic position, oil yield, gallons per<br />
Vamber feet from surface ton <strong>of</strong> shale<br />
0<br />
i<br />
)<br />
3<br />
- I.<br />
5<br />
6<br />
I<br />
6<br />
9<br />
93 0<br />
1036-105b<br />
1056 - 1081<br />
1152-11 ia<br />
123b-12-1<br />
1450-1L62<br />
1597-1628<br />
1668 -1696<br />
1786 -1825<br />
168G - 19 i-<br />
Fatty acid methyl esters were further purified by thin layer chromatography using<br />
silica gel The thin layer plate was developed with a mixture <strong>of</strong> n-hexane, ethyl<br />
ether. acd acetic acid (9O/lO,'lj. The adsorbcnt zone where esters occur was scraped<br />
from the plate and extracted with a solution <strong>of</strong> 10 percent methyl alcohol in carbon<br />
tetrachloride The solvent was remoLed from the methyl esters by evaporation at<br />
room temperature under a stream <strong>of</strong> nitrogeg" The carbon number distribution <strong>of</strong> these<br />
isolated fatty acid methyl esters was determined 5y mass spectrometry using the<br />
intensity parent peak as a measure <strong>of</strong> the quantity<br />
Carboxyl aqd ester coqtents were determined for the 10 shale samples by analytical<br />
procedurts deieloped by Fester and hsbixon (I).<br />
RESirLTS AbD DISCJSSIOK<br />
Thc fatty acid distribution with,depth (table 2), shows that the predominant acid at<br />
various stratigraphic depths within the formation is not constant. The sample number,<br />
percentage, acd chain length <strong>of</strong> the predominant acid <strong>of</strong> the samples are: No. 0, 16.2,<br />
C30; NO. 1, 14.1, C24; NO. 2, 21.7, c28; NO. 3, 9.8, c28; NO. 4, 16.6, C24; NO. 5,<br />
9.2, c28; No. 6, 12.8, C14; No. 7, 12.6, Clb; No. 8, 12.1, c28; and No. 9, 23.9, c28.<br />
The C28 acid is the predominant acid in five <strong>of</strong> the samples, and is among the three<br />
most abundant acids in all the other samples except samples 1 and 4 where it is pres-<br />
ent in small amounts. The C20 acid content in samples 5 through 9, despite being<br />
more abundant in nature than the C1g and e21 acids, is less than the C1g and C21<br />
acids in these samples. The c18 acid is quite low in samples 1 and 2 relative to<br />
the other eight samples. The C32 acid is present in large amounts only in sample<br />
Yo. 0. There appears to be no consistent relationship between the acids <strong>of</strong> one Sam-<br />
ple to that <strong>of</strong> another sample. This lack <strong>of</strong> relationship may bcdue to differences<br />
in source material or environmental changes rather than maturation changes.<br />
Significant differences are apparent in the .relative distribution <strong>of</strong> the fatty acids<br />
and the n-alkanes from published data (Q), shown in figures 1 and 2, where the data<br />
from four samples are plotted for comparative purposes. These particular samples<br />
were chosen because they represent three stratigraphic levels below the Mahogany zone<br />
sample approximately equi-distant from each other. If maturation <strong>of</strong> sediments causes<br />
decarboxylation <strong>of</strong> fatty acids to form n-alkanes, a relationship between the distribulion.<br />
<strong>of</strong> the two components would be expected. Except for sample No. 0, little.<br />
similarity is evident. Since the distributions correlate poorly, either the original<br />
postulate is 11r.true and n-alkanes are not produced from fatty acids having one more<br />
60<br />
35<br />
34<br />
42<br />
it8<br />
39<br />
35<br />
37<br />
28<br />
20
482<br />
TA5L,E 2. - Carbon number distributions <strong>of</strong> fatty acids<br />
Fcttv acids, wt percent <strong>of</strong> total<br />
Carbon Sample number<br />
Numb e r 0 1 2 3 h 5 6 7 8 9<br />
14<br />
15<br />
16<br />
17<br />
ia<br />
19<br />
20<br />
21<br />
22<br />
23 '<br />
24<br />
25<br />
26<br />
27<br />
28<br />
29<br />
30<br />
31<br />
32<br />
5.1 7.1<br />
3.0 3.4<br />
2.7 7.1<br />
2.0 4.0<br />
3.4 2.6<br />
2.4 3.4<br />
2.7 8.2<br />
2.4 7.1<br />
7.4 8.8<br />
4.7 4.7<br />
7.1 14.1<br />
3.7 7.6<br />
12.1 11.8<br />
2.7 2.6<br />
11.1 4.2<br />
2.7 1.3<br />
16.2 1.3<br />
1.7 0.4<br />
7.1 0.4<br />
2.8<br />
2.8<br />
2.4<br />
1.9<br />
2.1<br />
1.8<br />
1.9<br />
2.2<br />
3 .o<br />
4.4<br />
6.2<br />
5.0<br />
13.3<br />
8.2<br />
21.7<br />
5.1<br />
8.6<br />
2.5<br />
2.9<br />
6.3 :.3 8.0 12.8<br />
6.3 2.8 7.2 2.1<br />
5.6 4.0 5.8 6.4<br />
4.9 2.9 5.1 5.9<br />
4.2 4.2 5.i 8.0<br />
3.5 3.8 4.0 4.3<br />
2.8<br />
2.8<br />
L.5<br />
5.6<br />
2.9<br />
3.6<br />
4.2<br />
4.8<br />
4.9 9:; 5.8 11.6<br />
5.6 i2.5 5.8 3.8<br />
4.2 16.6 3.3 8.0<br />
3.5 8.3 1.5 3.8<br />
8.4 11.8 6.5 4.9<br />
6.3 3.7 8.0 2.7<br />
9.8 4.0 9.2 9.8<br />
4.2 1.2 4.3 1.5<br />
7.7 1.1 5.8 2.5<br />
4.2 0.3 4.0 0.8<br />
4.9 0.5 3.3 0.9<br />
12 .,6<br />
6.3<br />
7.8<br />
4.3<br />
7.4<br />
3.6<br />
2.9<br />
3.6<br />
8.3<br />
4.7<br />
6.3<br />
3.4<br />
5.9<br />
4.9<br />
8.5<br />
2.9<br />
3.2<br />
1.6<br />
1.6.<br />
11.4 6.5<br />
8.1 3.7<br />
9.4 5.4<br />
2.4 2.0<br />
7.4 6.1<br />
2.0 2.0<br />
2.0 1.3<br />
2.0 1.7<br />
10.7 13.0<br />
3.4 3.3<br />
8.7 12.0<br />
2.7 2.6<br />
4.1 5.2<br />
3.4 3.7<br />
12.1 23.9<br />
2.7 2.4<br />
2.7 2.8<br />
2.0 1.3<br />
2.0 1.1<br />
carbon atom or other factors are more significant. One factor to consider is that<br />
the fatty acids recovered from the lower portions <strong>of</strong> the core may not be representa-<br />
tive <strong>of</strong> the acids originally present since they may be residual acids from selective<br />
maturation.<br />
Groups <strong>of</strong> even numbered acids vary in abundance as shown graphically in figures 1<br />
and 2. Sample No. 0 has predominant acid peaks between c26 and C30, sample No. 4,<br />
between C22 and C26, sample No. 6, between C22 acd C28, and sample No. 9, between<br />
C22 and c28. The latter two samples show the greatest overall similarity.<br />
The carboxyl content <strong>of</strong> the shale generally decreases with depth, table 3, beginning<br />
with sample No. 0 having 23.6 mg <strong>of</strong> carboxyl per gm <strong>of</strong> organic carbon and ending<br />
with sample No. 9 having 4.8 mg <strong>of</strong> carboxyl per gm <strong>of</strong> organic carbon. This general<br />
decrease in carboxyl content with depth, and a corresponding increase in the n-alkane<br />
content, as shown by Robinson and coworkers (9, implies that the fatty acids may<br />
have been converted to n-alkanes by decarboxylation.<br />
The ester content <strong>of</strong> the samples, table 3, did not show a trend; this is in contrast<br />
to the trend shown by the acids. Random distributions found for the ester samples<br />
reflect more accurately changes in organic source material than do the acid<br />
distributions.<br />
Maturation changes are evident in the decreasing amount <strong>of</strong> carboxyl content with<br />
depth, the increasing amount <strong>of</strong> n-alkane content with depth, and the decreasing<br />
ratio <strong>of</strong> odd to even-carbon-numbered n-alkanes. Source material or environmental<br />
changes are evident in the variations in the amount and carbon number <strong>of</strong> the domi-<br />
nant fatty acids, and n-alkanes and the random distribution <strong>of</strong> ester content with<br />
depth.
1.<br />
2.<br />
3.<br />
4.<br />
5.<br />
TABLE 3. - Carbowl and esler contents <strong>of</strong> the<br />
10. shale samles<br />
Samp 1 e Carboxyl, Ester,<br />
Number mgf rn carbon mgfgm carbodf<br />
0 23.6 36.6<br />
1 18.3 22.2<br />
2 10.1 33.9<br />
.?I! -- --<br />
4 3.8 26l.7<br />
5 6 7 1; .q<br />
6 ~4.0 37.1<br />
7 4.7 26.0<br />
8 4.9 42.9<br />
9 k.8 29.3<br />
- 1' Ca,culated as COOH<br />
?/ 1ns.ifficient sample.<br />
CONCLUSIONS<br />
73e postulate that fatty acids from the organic debris in the Green River Formation<br />
may form n-alkanes <strong>of</strong> one less carbon atom was found to be subject to question.<br />
Relatable fatty acid and n-alkane distributions were shown to be preeent in the<br />
younger portions <strong>of</strong> the Formation but the older portions showed little relation.<br />
batbration and organic source material differences may account for the obeenved dis-<br />
tri5ut;ons <strong>of</strong> fatty acids in sections <strong>of</strong> Green River Formation oil shale. Evidence<br />
<strong>of</strong> matLration is found in the decreasing amount <strong>of</strong> odd-carbon-numbered n-alkanes and<br />
in the decreasing amount <strong>of</strong> carboxyl content with depth. Evidence <strong>of</strong> organic source<br />
material differences is found in the inconsistent distribution <strong>of</strong> the predominant<br />
fatty acids from sample to sample.<br />
The ester content is not related to the free-acid content, indicating different<br />
formative histories.<br />
REFERENCES<br />
Breger, I. A. Diagenesis <strong>of</strong> Metabolites and a Discussion <strong>of</strong> the Origin <strong>of</strong><br />
Petroleum Hydrocarbons. Geochim. et Cosmochim. Acta, v. 19, 1961, pp. 297-308.<br />
Cooper, J. E., and E. E. Bray. A Postulated Role <strong>of</strong> Fatty Acids in Petroleum<br />
Formation. Geochim. et Cosmochim. Acta, v. 27, 1963, pp. 1113-1127.<br />
Cumins, 3. J., and W. E. Robinson. Normal and Isoprenoid Hydrocarbons Isolated<br />
from Oil-Shale Bitumen. J. Chem. Eng. Data, v. 9, 1964, Pp. 304-301.<br />
Fester, J. I., and W. E. Robinson. Oxygen Functional Groups in Green River Oil<br />
Shale and Trona Acids. Ch. in "Advances in Cheeietry Series." American Chemical<br />
Society, Washington, D.C. (1966), pp. 22-31.<br />
.'urg, J. W ., and E. Eisma. Petroleum Nydrocarbons: Generation from Fatty Acide.<br />
Science, v 144, 196i, pp. 1451-1152.
6.<br />
7.<br />
a.<br />
9.<br />
484<br />
Lawlor, D. L., and W. E. Robinson. Division <strong>of</strong> Petroleum Chemistry, Am. Chem.<br />
SOC., Detroit, Mich., April 1965, Petroleum Division General Papers No. 1, 1965,<br />
pp. 5-9.<br />
Mair, B. J. Terpenoids, Fatty Acids and Alcohols as Source Materials for<br />
Petroleum Hydrocarbons. Geochim. et Cosmochim. Acta, v. 28, 1964, pp. 1303-1321<br />
Martin, R. L., J. C. Winters, and J. A. Williams. Distributions1 <strong>of</strong> n-Paraffins<br />
in Crude Oils and Their Implication to Origin <strong>of</strong> Petroleum. Nature, v. 199,<br />
1963, pp. 110-114.<br />
Robinson, W. E., J. J. Cumins, and G. U. Dinneen. Changes in Green River Oil<br />
Shale Paraffins with Depth. Geochim. et Cosmochim. Acta, v. 29, 1965, pp.<br />
249 - 258.<br />
h<br />
1<br />
c<br />
/<br />
‘I
i<br />
I<br />
I<br />
,<br />
r<br />
t<br />
\ ..<br />
4<br />
3<br />
I I I I I I 1 . 1 I<br />
13 17 21 25 29 13 17 21 25 29<br />
ALKANE CARBON NUMBER<br />
I<br />
'1 I<br />
F&ure 1. Caupiwison between fatty acld a d n-allme<br />
distributions, Sample Uos. 0 and 4<br />
.. .
' I ' Sample No. 6 '<br />
12<br />
IO<br />
8<br />
6<br />
z 4<br />
z<br />
4<br />
52<br />
I 1 I I<br />
4<br />
0/4 I: i2 /6 . i0 Ib I\ ;2 26 '<br />
v)<br />
n<br />
$14<br />
ACID CARBON NUMBER<br />
t24.7<br />
Sample No.6<br />
n-AI kaner<br />
Sample No. 9<br />
n-AI kanes<br />
13 17 21 25 29 13 17 21 25 29<br />
ALKANE CARBON NUMBER '<br />
Figure 2. m i s o n between fatty acid and n-alhue<br />
diatributions, sample Nos. 6 and 9
487<br />
ELECTRICAL PROPERTIES OF IODINE COXPLEXES OF ASPHALTENES<br />
Gustave A. Sill and Teh Fu Yen<br />
Mellon Institute I<br />
Pittsburgh, Pennsylvania 152 13<br />
INTRODUCTION<br />
Polynuclear aromatic hydrocarbons generally form charge-transfer<br />
complexes with halogens. Some <strong>of</strong> the fused aromatic hydrocarbons, e.g.,<br />
perylene, violanthrene, yield solid complexes exhibiting extremely good<br />
semiconduction (l,2,3) while others, e.g., coronene, show only fair to<br />
poor semiconduction (4).<br />
A number <strong>of</strong> charge-transfer complexes <strong>of</strong> aromatic<br />
containing polymers have been investigated for possible differences in<br />
electrical properties (5,6,30).<br />
A polymeric dielectric may be converted to a polymeric semi-<br />
conductor by increasing the aromaticity <strong>of</strong> the insulator, followed by<br />
complex formation with a halogen (7,8). The increase in aromaticity can<br />
be effected by radiation--e.g., cyclization <strong>of</strong> polyethylene and followed<br />
by dehydrogenation (7); or by heat--e. g., pyrolysis or graphitization to<br />
a pyropolymer (8).<br />
lhe resulting products when treated with iodine exhibit<br />
a wide range <strong>of</strong> interesting electrical properties.<br />
From a structural standpoint asphaltenes (9) are considered to<br />
consist <strong>of</strong> two-dimensioned fabrics <strong>of</strong> condensed aromatic rings, intermingled<br />
with short aliphatic chains and fused naphthenic ring systems (10).<br />
diffraction (11,12) and ESR investigations (13) have indicated that these<br />
aromatic oyotemo tend to form otacko <strong>of</strong> graphite-like lryrro rurroundrd<br />
by a disorganized zig-zag chain structure <strong>of</strong> saturated carbons.<br />
they may<br />
be considered as a highly associated "multipolymer" (14), the<br />
X-ray<br />
Morphologically
.<br />
488<br />
uiolecular weight <strong>of</strong> which can vary from a few thousand (unit or particle<br />
weight) to a few million (micelle weight) (IS).<br />
The aromatic centers,<br />
roughly 15; in diameter, are considcred to be pericondensed (~3,iB).<br />
has been demonstrated that asphaltics can form charge-transfer complexes,<br />
due to the presence <strong>of</strong> such aromatic systems (20).<br />
Most polynuclear aromatic compounds form w ell defined crystals,<br />
the iodine complexes <strong>of</strong> which are stable and stoichiometric in composition.<br />
Asphaltics are mesomorphic (17) owing to the raridom distribution and iso-<br />
tropic orientation <strong>of</strong> the structural units, and it is to be anticipated,<br />
therefore, that the conduction mechanism will be different from that <strong>of</strong><br />
the crystalline environment (due to diffusion and phonon processes) (16). 1<br />
\.<br />
Since there is a difference in the conduction mechanism between crystalline<br />
and amorphous aromatics, one would like to know whether the mesomorphic<br />
nature <strong>of</strong> asphaltics would retard or inhibit the conductivity, and if so )I<br />
to what extent. I<br />
The aims <strong>of</strong> this research were two-fold. The first was simply<br />
to observe where asphaltenes fall in the conductivity range and to<br />
determine the extent to which conductivity can be enhanced by iodine com-<br />
plex formation. The second was a more general study <strong>of</strong> the electrical<br />
properties <strong>of</strong> asphaltics as another approach to a better understanding<br />
<strong>of</strong> their structure. To the authors' best knowledge, there is no published<br />
work on the electrical characteristics <strong>of</strong> these materials. Iodine may<br />
be visualized as a tracer or indicator for condensed aromatic systems,<br />
even when buried in a matrix <strong>of</strong> paraffinic or cycloparaffinic material.<br />
It was thought, therefore, that iodine complex formation and its effect<br />
It<br />
I<br />
r
489<br />
on the ovcrall electrical properties <strong>of</strong> the asphaltene might yield inde-<br />
pendent information <strong>of</strong> the size anti distribution <strong>of</strong> the aromatic centers<br />
I<br />
in these multipolymers.<br />
Res 1 stance Eka sur cment s<br />
ESPERIPfEhTAL<br />
A General Radio type-1230b electrometer was used for specimens<br />
with resistance less than 1012 ohms (ambient ,temperature), and a Cary<br />
model 31 electrometer was used for specimens <strong>of</strong> higher resistance. In<br />
each case a glass vessel equipped with a ball joint and appropriate<br />
e1ec:rostatic snielding was coupled to the head <strong>of</strong> the electrometer (Fig. 1).<br />
specimen (approximately 1 x 0.5 x 0.1 cm) was pelleted with a Beckman KBr<br />
press at 7.09 x lo3 kg/cm2 between two pieces <strong>of</strong> 52 mesh platinum screen.<br />
The pellet was degassed for eight hours and the electrical measurements<br />
made in a vacuum <strong>of</strong> 5 x Torr. Using the high resistance leak method,<br />
a standard resistor served as calibrating reference (21); data were taken<br />
under conditions <strong>of</strong> both falling and rising temperature, a minimum <strong>of</strong> 30<br />
minutes being allowed for equilibration at 10" levels. Upon completion<br />
<strong>of</strong> the temperature dependence measurements, the physical dimensions <strong>of</strong><br />
the specimen block were obtained with a travelling microscope (1OX) with<br />
an x,y micrometer attachment (0.0001 cm precision).<br />
Preparation <strong>of</strong> Sample<br />
The asphaltene sample was prepared by our standard procedure (9).<br />
Two native asphaltenes were investigated, one from the Boscan crude oil<br />
from Venezuela (Sample VY), the other from the Baxterville crude from<br />
Each
Xississippi (Sample GS).<br />
Stock solutions in benzene <strong>of</strong> iodine and <strong>of</strong> the<br />
individual asphaltenes were made up in fixed concentration and samples<br />
I<br />
<strong>of</strong> varied composition prepared by mixing appropriate quantities <strong>of</strong> these<br />
stock solutions at room temperature and lyophylizing at reduced pressure<br />
to yield powdered solids with a homogeneous iodine distribution. These<br />
samTles were analyzed before and after the electrical measurements for<br />
$1 by ignition in oxygen, reduction with hydrazine sulfate, and poteniometric ,<br />
titration <strong>of</strong> the resulting iodide with &NO3 using a Beckman model K<br />
automatic titrator. Usually there was no observable loss <strong>of</strong> free iodine<br />
during electrical measurements; lO-l5$ loss <strong>of</strong> iodine was found after<br />
degassing. The iodine values used in the present work are those values<br />
obtained after completion <strong>of</strong> the electrical measurements for the entire<br />
specimen.<br />
Treatment <strong>of</strong> Data<br />
The resistance values along with the corresponding temperature<br />
data and dimensions <strong>of</strong> the specimen were key punched on IEiM cards and<br />
evaluated on an IBM 7090-1401 digital computer system. Given A, the area<br />
<strong>of</strong> the cross section, and L, the Length <strong>of</strong> the specimen, the resistivity,<br />
p, can be evaluated from the resistance, R, as follows:<br />
p = AR/L. The<br />
temperature dependence <strong>of</strong> the resistivity is then evaluated by the relationship:<br />
4<br />
PT = 00 exp (+kT), i<br />
where k is Boltzman's constant, E is the energy gap in eV and po is the<br />
resistivity extrapolated to ,$ = 0.<br />
By use <strong>of</strong> a California Computer Products %<br />
30-in. plotter (300 steps; l/5OO-in. per step) the temperature dependence<br />
data were fitted to straight lines (Fig. 2) as given by the equation<br />
\<br />
<<br />
\<br />
\.<br />
V<br />
\<br />
4<br />
3'<br />
I<br />
/<br />
3<br />
2 b<br />
V<br />
t<br />
491<br />
log p = log pJ-€E/(2kT In 10)<br />
From the digital output, p250c and E can be obtained. The applied voltage<br />
I<br />
was limited to values under LO V; in this region Ohm's law was followed.<br />
The temperature range examined was from ambient to 90°C.<br />
There is error involved in any single measurement <strong>of</strong> resistance,<br />
owing to systematic errors in the electrometer; errors also enter in the<br />
measurement <strong>of</strong> the dimensions <strong>of</strong> the specimen. That the results were not<br />
influenced by such systematic errors is evident from the two sets <strong>of</strong> data<br />
for two different preparations <strong>of</strong> an asphaltene (VY)-iodine complex, as<br />
given in Table I. The uncertainties in the per cent iodine and sample<br />
size may be judged from the variations in the independent measurements.<br />
Despite these variations, the resistivity at 2 5 O C and the energy gap are<br />
within ca. 5% <strong>of</strong> the mean values.<br />
Infrared Analysis<br />
Differential IR spectra were obtained from a scan <strong>of</strong> an iodine-<br />
containing asphaltene versus a reference asphaltene at equal asphaltene<br />
concentration in CSg (the iodine-containing sample is normalized to lo@$<br />
asphaltene for purposes <strong>of</strong> comparison) using a Beckman IR-12 instrument.<br />
A control scan <strong>of</strong> asphaltene in CS2 solution against itself also was made<br />
for each sample.<br />
X-ray Diffraction<br />
A Norelco x-ray diffractometer equipped with a CuxGL radiation<br />
source and a geiger tube detector was used to study the asphaltene-iodine<br />
system. In order to record the shift <strong>of</strong> the d-spacing due only to change<br />
in mass absorption coefficients, adamantane was added to an asphaltene-<br />
iodine complex (24% I) and to the original asphaltene.<br />
Strong (111) and<br />
(260) reflections due to the adamantane mixed with the VY asphaltene were
492<br />
found at 5.7 and 4.9& for the W asphaltene-iodine complex shifts were<br />
observed to 5.5 and 4.7A, respectively. The spacing is reproducible to<br />
I<br />
9. ai.<br />
Eleztron S?in Resonance<br />
ESR spectra were taken with a Varian V-4502 x-brand EPR spectrometer<br />
system in conjunction with a 12-inch magnet and a "Fieldial."<br />
intenslty observed was used as a guide for the spin concentration <strong>of</strong> the<br />
asphaltene (VY)-iodine complexes and native asphaltene (VY) (13).<br />
RESULTS<br />
The relative<br />
All asphaltene-iodine samples studied gave repeatable linear<br />
relations in the temperature range investigated as shown in Fig. 2.<br />
is no significant deviation from Ohm's law through the range 2.5 to 97 V<br />
as indicated by Fig. 3 in the temperature interval 313" to 372"K.<br />
insulator range.<br />
There<br />
The native asphaltenes (Points 1, Fig. 4) fall generally in the<br />
Upon the addition <strong>of</strong> iodine the resistivity f;Lls, 5 to<br />
- b, then increases sharply, to c, and finally drops, 5 to a, as iodine<br />
content rises. Both complexes appear to yield curves <strong>of</strong> similar shape.<br />
The gap energy values for the asphaltene-iodine complexes are plotted<br />
versus their iodine contents (Fig. 5). . The smallest energy gap measured<br />
in each case was -Q.5eVf but the absolute minimum is uncertain, These<br />
minima (points b) correspond to the sharp transitions <strong>of</strong> resistivity shown<br />
in Fig. 4..<br />
Scanning in the far IR region revealed no C-I stretching frequencies<br />
for those iodine complexes for which 0 was determined. However, the dif-<br />
ferential IR measured in the 7OO-l2OO cm-1 did show an additional band at
i'<br />
I<br />
Y<br />
'<br />
493<br />
..<br />
10E.G ~r,~-l {i;ig. 6) , and freshly ;,rc,,ar'cd asi'i~alecl-ic-iodine cosplexes in<br />
CSz also csiiibited an enhanccmcnr in absorprion 'in the regi<strong>of</strong>i <strong>of</strong> lOC0 to<br />
11 jO cn-1, w:IicIi is generally ascl-iilcu to complex iormation.<br />
Tile x-ray spectrograms in rile region 20 = 2 - 42" <strong>of</strong> the asphaltene'<br />
(mi)-iodine 'systca yielded in aiiior;),ious pattern with broad halos as shown<br />
. .<br />
in Xg. 7. For samples with an iodinc content <strong>of</strong> less tian 5 per cent,<br />
the j. ji band.stil1 appeared as ii siioulder.<br />
was fornad at a;oound 8.7a.<br />
In thc meantime a new band<br />
AT bigher'iodine conhenis (>IO$), the 3.5i<br />
band. has apparently disappeared and the 8.7x band became clearly visible.<br />
".<br />
liisrc is a general overall decrease <strong>of</strong> total intensity as $1 increases<br />
since the iodine itself absorbs an increasingly large fraction <strong>of</strong> the<br />
.. .<br />
diffracted x-rays. In the present case, the noise Level coupled with the<br />
intensity reduction has made the disappearance <strong>of</strong> the 3.51 shoulder dif-<br />
ficult KO detect.<br />
In gcnera1,the . . ESR,spectra obtained froqthe asphaltene-iodine<br />
samples indicated an incre,ase in free radical concentration with increase<br />
-in iodine content. The increase in relacive intensity for an asphaltene<br />
(Wj-iodine complex ,.containing 2@ I over the corresponding native asphal-<br />
. .<br />
tenes is about 3.8 fold.<br />
DISCUSSIOX'<br />
In all the samples studied, including both.native asphalcenes<br />
and their iodine. adducts, a negative temperature coefficient <strong>of</strong> resistivity<br />
was obtained.<br />
by the plots shown.in Fig. 2:<br />
The linearity <strong>of</strong> log D vs. reciprocal T .is substantiated<br />
The resistivity is inversely related to<br />
the concentration <strong>of</strong> the charge carriers (holes and electrons), but the<br />
fact that the number <strong>of</strong> charge-carriers increases exponentially with tempera-<br />
ture does not enable a choice to be made between electronic or ionic conduction<br />
"
494<br />
mechanisms. However, the voltade dependence for the current at a relatively<br />
low electric field is linear, indicating that Ohm's law is valid (Fig. 3).<br />
I<br />
This adherence to Ohm's law supports the belief that the conduction is<br />
electronic (19).<br />
The fact that the asphaltene-iodine sample exhibits (Fig. 6)<br />
strong enhancement <strong>of</strong> absorption near 1080 crn'l suggests that an iodine<br />
molecule forms a donor-acceptor complex with the aromatic portion <strong>of</strong> the<br />
asphaltenes. It is known that the iodine molecule forms such a charge-<br />
transfer complex with benzene and other alkylated benzenes, and that<br />
these complexes, in general, exhibit bands from 9 9 cm'l to 1200 cm'l<br />
(24,25).<br />
Arguing from analogy, it is plausible that the complex assumes<br />
the axial model (model A <strong>of</strong> Mulliken) while the acceptor molecule is sitting<br />
perpendicular to the plane. <strong>of</strong> the aromatics (26).<br />
Further, our failure<br />
to' locate any C-I stretching frequencies in the 400-600 cm-1 region sup-<br />
ports the view that iodination <strong>of</strong> the asphaltene samples did not occur'.<br />
crystalline.<br />
Xost charge-transfer complexes <strong>of</strong> iodine with aromatics are<br />
An exception to this is the violanthrene-iodine system,<br />
which x-ray diffraction indicates to be amorphous (2).<br />
X-ray results also indic'ate a low degree <strong>of</strong> order for the asphaltene-<br />
iodine Complexes. If the acceptors (I2) are homogeneously distributed in<br />
the host matrix (asphaltene), these systems may be considered analogous<br />
to the impurity or valence-controlled semiconductor systems. Disappearance<br />
<strong>of</strong> the 3.5i spacing <strong>of</strong> the (002) band means that the layered structure <strong>of</strong><br />
asphaltene must have been altered (Fig. 7).<br />
The 4.68, y-band, due to the<br />
saturated carbon in the structure, did not change in the complexing process.
d<br />
:- /<br />
I<br />
i'<br />
I<br />
495<br />
:"ne ~CIJ hznd A: S.?h thcn nay bo iuc to the cx;>ansion <strong>of</strong> the zironatic<br />
intcrplsnzr. distazia to aliow for coir,plexing by the iodine no;ecule.<br />
0<br />
thc case <strong>of</strong> the ;>eryicnc-iodinc systcns, Liie spacing found at 10.7A was<br />
interpreced (3) as the distance bctwecn perylene moleculcs when iodine<br />
moiccnlcs were sandwiched between tiic aromatic layers. The present ob-<br />
served value <strong>of</strong> &.?A can be viewed as the sua <strong>of</strong> the intcrplanar distance<br />
(5.5.:) and the iodine length '(4 x l.j$).<br />
linked layers resembles that oi an intercalation compound <strong>of</strong> graphite<br />
(Fig. 6).<br />
Tile picture <strong>of</strong> these inter-<br />
. .<br />
Beferring again to Fig. 4, the room temperature resistivities for<br />
the two nztive asphaltenes are seen to be in tne insulator range (>lo14 Ohm).<br />
Upon addition <strong>of</strong> the iodine, the resistivity decreases about six decades<br />
or a niLlion fold. This is essentially the same behavior as that observed<br />
for polymeric charge-transfer complexes such as poly(viny1pyridium TCNQ)<br />
ar.6 its derivatives.<br />
These polymeric salts are dependent upon the TCNQ con-<br />
centration wnicn at best increases the conductivity six decades. Here<br />
!lave to point out that it is not easy to prepare a polymeric charge-<br />
rransfer complex with good semiconduction.<br />
In<br />
Slough (5) nade a number <strong>of</strong><br />
polymeric complexes.from arosatic-containing polymers, with acceptors such<br />
GS cetracyanoechylene, chloranil, etc., and found the conductivities <strong>of</strong><br />
these conplexes were not measurably higher than those <strong>of</strong> the original poly-<br />
meric donors.<br />
These mesomorphic materials may lack the order <strong>of</strong> the K-<br />
systems needed to open a path for charge carriers.<br />
More likely, the<br />
aroGtic systems are too small to form stable charge-transfer complexes.
For pure polynuclcar arom.i;ic hydrocarbons, donor-acceptor com-<br />
Irlescs, cxpccially the iodine coinpicscs (2,3), exhibit increases in con-<br />
I ,<br />
ductivity <strong>of</strong> 12 to 16 decadcs when compared to the parent hydrocarbon.<br />
The enhancement in conduction by the addition <strong>of</strong> iodine can be illustrated<br />
by comparison <strong>of</strong> the complexes with a valence controlled semiconductor, a typical<br />
esample being nickel (11) oxide doped with lithium oxide (22).<br />
In Fig. 9 a com-<br />
parison is made between this system and the aromatic-iodine complexes. The<br />
trends in resistivity with the concentration <strong>of</strong> impurity are quite similar.<br />
- Asphaltenes contain fused ring aromatics, the peripheral hydrogens<br />
<strong>of</strong> which are substituted heavily by short chain alkyl groups (23).<br />
to the relatively large porportion <strong>of</strong> methyl groups (20$) and the large<br />
Owing<br />
average layer diameters (La - 1$), the asphaltic molecule can be viewed<br />
as a typical aromatic donor ( D); halogens such as iodine can behave as an<br />
acceptor (A). Through charge-transfer by overlapping <strong>of</strong> the molecular I<br />
!<br />
orbitals <strong>of</strong> the two moieties, the dative structure (D'A') should result<br />
in which asphaltene is the positive ion. \ ,<br />
Fig. 4 clearly indicates that the increase <strong>of</strong> conductivity follows<br />
two different paths, the first 5 to b, is the path followed for small in- <<br />
crements <strong>of</strong> iodine, terminating at k with fixed composition (VY, 15.5%;<br />
GS, lo.&); the other, c to rl, is for higher percentages <strong>of</strong> iodine.<br />
1<br />
Line & represents the transition state. The curve &may be extrapolated 1<br />
to a resistivity value <strong>of</strong> 5.8 x 106 ohm-cm, corresponding to that for pure<br />
12 (27)'. The same results were found (23) for violanthrene-iodine system with<br />
a minimum corresponding to a 2:l iodine-violanthrene molar ratio for the<br />
complex. Apparently in Fig. 4 corresponds to a stoichiometric ratio <strong>of</strong><br />
~<br />
I<br />
\ ,<br />
\<br />
\<br />
\<br />
a<br />
7<br />
1<br />
a<br />
\
497<br />
a stable complex where conductivity is at a maximum.<br />
It is assumed that<br />
for iodinc contents less than that corresponding to the minimum the amount is<br />
I<br />
insuiiisicnt to form the complex. At tile resistivity is highly sensitive<br />
to the number <strong>of</strong> iodinc molecules. When c is passed, excess iodine acts<br />
(is (in impurity in the stoichiornctric complex.<br />
reflecccd by the energy gap plot in Fig. 5. The same fixed composition<br />
(VY, 15.5';;; GS, 10.W;) is obtained in cither plot.<br />
the energy gap value is ca. 0.5cV suggesting favorable condicions for<br />
conduction.<br />
The transition at a is also<br />
At these minima,<br />
A number <strong>of</strong> aromatic-iodine complexes have been reported (2,3)<br />
and from their phase diagrams (either temperature or density vs. mole<br />
per cent <strong>of</strong> iodine) the complexes are found to be stoichiomecric (29).<br />
For example perylene-iodine can have 2:3 or l:3; pyranthrene-iodine i s<br />
1:2; violanthrene-iodine is 1:2; pyrene-iodine is 1:2. In all cases for<br />
peri-type aromatics the ratio <strong>of</strong> 12 to aromatic is higher than unity.<br />
Since the diameter <strong>of</strong> the aromatic system in asphaltics falls in the<br />
range 8-l$ (12), the system would be comparable in size to violanthrene<br />
or perylene.<br />
Assuming the composition at the transition '(q 15.5%; GS, lo.@)<br />
is stoichiometric, then for any given ratio <strong>of</strong> aromatic and iodine,<br />
the molecular weight <strong>of</strong> the asphaltene can be calculated. We have taken<br />
the liberty <strong>of</strong> calculating this weight for W and GS asphaltenes based<br />
on sample ratios <strong>of</strong> 1a:asphaltene <strong>of</strong> 2:1, 3:2 and 1:l. Since all layers<br />
contain the aromatic moieties and the sample is free <strong>of</strong> wax contamination,<br />
the molecular weight obtained is that <strong>of</strong> the unit sheet weight (weight <strong>of</strong> an<br />
average sheet containing both aromatic and saturated carbon atoms). These
-<br />
'-'. . ' . .<br />
values are listed in Table 11. Next, provided aromaticity is also known<br />
(fa for W, 0.35; for GS, O.5l), the disk weight (weight <strong>of</strong> aromatic<br />
carbon atoms in a single sheet) and the layer diameter also dan'be approxi-<br />
mated. These values are also listed in Table 11. Experimental values<br />
for the W and GS asphaltenes from a previous paper (15) are included: It i<br />
is <strong>of</strong> interest that the unit weight values obtained from GPC, mass spectrometry,<br />
x-ray diffraction and the electron microscopic measurements agree in general<br />
magnitude with the weights obtained by the present method.<br />
Deviations <strong>of</strong> the<br />
disk weight <strong>of</strong> GS calculated from resistivity from that obtained by mass<br />
spectrometry may by due to the polydispersity <strong>of</strong> the GS asphaltene (e.g.,<br />
Mw/MN for VY is 1.27; for GS, 1.74 (15)).<br />
From Table 11, the asphaltene-iodine complexes formed appear to<br />
correspond to an 12:asphaltene ratio <strong>of</strong> about l.5:l. This composition is<br />
J<br />
shown in model A <strong>of</strong> Fig. 8.<br />
Actually aromatic disks <strong>of</strong> the size present<br />
in asphaltenes should be able to accomodate more than one molecule <strong>of</strong> iodine.<br />
Finally the increase in the free spins, as demonstrated by the<br />
EPR spectra, may be indicative <strong>of</strong> an increase in carrier concentration (2).<br />
The nature <strong>of</strong> these charge carriers will have a strong bearing on the<br />
conduction mechanism and will be the subject <strong>of</strong> a separate investigation. . <<br />
ACKNOWLEDGMENT<br />
The authors thank Mr. Timothy R. Drury, Mellon Institute, Research<br />
Services, for his technical assistance; and Mrs. Marianne P. Hoy and<br />
Dr. Sidney S. Pollack, Mellon Institute, Research Services for the x-ray<br />
diffraction work.<br />
This work was sponeored by Gulf Research & Development Company<br />
as part <strong>of</strong> the research program <strong>of</strong> the Multiple Fellowship on Petroleum.<br />
. .<br />
, . I<br />
.. .<br />
.. . .<br />
. .<br />
.. .*. .i<br />
. .<br />
\<br />
i<br />
\\<br />
\<br />
i<br />
7<br />
(<br />
t
499<br />
(it Lnk>;t,icC,i: ii., ani k:taiiiatu, ii., Solid state i'iiysics, 2, ~3 (1561).<br />
(2) Schida, T., and Akclmatu, li., liull. Ciicm. SOC. (Japan), 2, 921 (1562).<br />
(;) ~c.li& , T., 9nd .q;aimtu, li., BULL. Clicm. ~oc. (Japan), 2, 1015 (1.961).<br />
(1;) Lab~s: X. X., Sciir, R. A., and Bosc, hl., "Yrocccdings <strong>of</strong> the Princeton<br />
';.;ivcrsicy Conr'crencc on Semiconductors in Molecular Solids,"<br />
?ri;i$.ccon, S. J., 1960.<br />
(j) Slau:.;ii, Id., Tyans. Faradzy SOC., 2, 2360 (1362).<br />
(6) Baith, S. .4., Vannikov, A. V., Grishina, A. D., and Sizhnil, S. V.,<br />
!). 'F.. \<br />
(9) The asphzlccne fraction <strong>of</strong> a crude oil or refinery residue is a brown-<br />
to-black, amorphous, powdery,. solid material, insoluble in n-<br />
;>i'i;tAI>e but soluble in bcnzene; see Ref. 11 for sample prepara-<br />
tion.<br />
(10) Yen, T. F., and Erdman, J. G., A. C. S., Div. Petroleum Chem.,<br />
PreTrints, 1, No. 1, 5 (1962).<br />
(11) Yen, T. I?., Erdman, J. G., and Pollack, S. S., Anal. Cnem., z, 1587<br />
(1561).<br />
I .<br />
,2) Yea, T. F., and Erdnan, J. G., in "Encyclopedia <strong>of</strong> X-rays and Gammarays"<br />
(G. L. ' Clark, Ed. ), Reinhold Publishing Co.,<br />
s. Y., 1963, p. 65.<br />
New York,<br />
(l>) Ycn, T. F., Erdman, J. G., and Saraceno, A. J., Anal. Chem., 3,<br />
694 (1952).<br />
(14) The WOYL "multipolper" refers: to a polymer with a very large number<br />
ijL different buildin; blocks, say ca. 100. Simpler structures<br />
can Dc referred to by more specific terms, as for example,<br />
"copolymer" (2 blocks) or "terpolymer" (3 blocks).
500<br />
(1;) Ten, T. 17.: a;1d,Dic;
'/<br />
925°C. x lO"2<br />
Sn.:>Lc SL>." "Iodine ..\ (cm) I. (cm) OLlrn-cn E (CVY<br />
23 i7.1 1.252, 0.0994 1.75c 1.742<br />
/ 25 16.6 1.266.L 0.1053 1.61, 1.825<br />
27 17.2 1.2592 0.1192 1.773 2. 007<br />
(a) Sane namers used in curves 95 Fig. 4; Sample Nos. 2j, 25, and 27 are from<br />
\ one ?reparatLon; Sample Nos. 24, 28, and 29 are fros an-<br />
d other preparation.<br />
i<br />
I<br />
c<br />
1'1<br />
\I
- ~<br />
Resistivity Czlculation<br />
2: 1<br />
j:2<br />
1: i<br />
x-ray DL ffrsct ionh<br />
(La)<br />
502<br />
27?Od k570 %9e 2330 1k.6f 22.6<br />
2080 34jO 727 1750 12.6 19.5,<br />
1360 2290 484 1170 10.3 16.0<br />
- - - 11.9 17.0<br />
E 1 ec t ron Xi cros cope" J<br />
.-<br />
(Particle weight) 3440 LO30 - - - -<br />
(a) Weight 0; a single sheet containing both aromatic and saturated carbon atoms<br />
(see 2. P. Dickie and T. F. Yen, A. C. S. , Div. Petroleum<br />
C'nem. , Preprints, Miami meeting, April, 1967).<br />
(b) Weight <strong>of</strong> aromatic carbon atoms in a single sheet.<br />
(c) Diameter <strong>of</strong> aronatic cluster; see T. F. Yen, J. G. Erdman, and S. S. Pollack,<br />
Anal. Chem. , u, 1587 (1961).<br />
(d) Calculated based on 254t'lR (LOO-t; where R is the ratio <strong>of</strong> 12 to asphaltene<br />
ana t is the %I corresponding to the transition in resistivity and<br />
gap energy.<br />
(e) Calculated from fa x (unit weight).<br />
(f) La = (2.62 CA);/', CA from disk vcight without contribution <strong>of</strong> hydrogens.<br />
(g) From pel permestion chromatography data on the native asphaltene, number average<br />
rdiecular weight. 1,<br />
(h) Expcrio.cn:ai dct; from J. P. Dickie and T. F. Yen, A. C. S., Div. Petrolen=<br />
C:ics. , Preprints, Miami rr,eet;ng, April, 1967.<br />
(i) J. P. DLciiic ar.d T. F. Yen. A. C. S., Div. Petroleum Cilem., Preprints,<br />
11, 39 (1%6).<br />
1<br />
?<br />
L<br />
-.<br />
\<br />
,<br />
,
503
504<br />
o '<br />
C<br />
0s 2<br />
0<br />
v<br />
x<br />
i<br />
8<br />
2<br />
0<br />
!<br />
P<br />
-<br />
a,<br />
0<br />
C<br />
0<br />
in<br />
.-<br />
In<br />
a,<br />
cc<br />
u-<br />
0<br />
al<br />
0<br />
C<br />
a,<br />
D<br />
C<br />
a,<br />
9<br />
n
505<br />
up”<br />
/<br />
U<br />
d<br />
L<br />
.-
506<br />
i
507<br />
c
d 601<br />
L<br />
P<br />
LL
I'<br />
i<br />
i<br />
C<br />
.. .<br />
i?<br />
2<br />
s
I I I I I I I I<br />
m W N<br />
W N 0 (D<br />
?<br />
0 2 0 2 0 0 0 0<br />
D<br />
N<br />
IO<br />
,
, 513<br />
THE ELECTRON SPIN RESONANCE SPECTRA OF CELLULOSE<br />
CHARS TREATED WITH HALOGENS*<br />
Evidence for Donor-Acceptor Complexes in Coals and Chars<br />
R.M. El<strong>of</strong>son and K.F. Schulz<br />
Research Counci I <strong>of</strong> AI berta<br />
Edmonton, Alberta<br />
INTRODUCTION<br />
Coals and low- and medium-temperature chars have been shown to be paramagnetic<br />
by bulk magnetic susceptibility measurementsl,2fr3 and by electron spin resonance studies4r5t6.<br />
If due care is taken to eliminate errors, reasonable agreement in the estimate <strong>of</strong> the number <strong>of</strong><br />
unpaired electrons present can be obtained'. While there is agreement on the existence <strong>of</strong><br />
paramagnetic centres in these materials, the origin and nature <strong>of</strong> these free radicals is not<br />
understood8,"10,'?,12. It is the purpose <strong>of</strong> this article to present new evidence to suggest that<br />
these unpaired electrons arise from donor-acceptor forces present in these materials.<br />
One way <strong>of</strong> studying the nature <strong>of</strong> free radicols is by observing the behavior <strong>of</strong> the<br />
electron spin resonance signals when the substrate is treated with adsorbed gases and other reagents.<br />
Early workers in the field found that poramagnetic gases such as oxygen affected only the spin-<br />
lattice relaxation times <strong>of</strong> chars prepared at from 300" to 5OOOC. For chars prepared in the 550'<br />
to 800" temperature range, however, reversible line broadening occurred with an apparent<br />
decrease in free radical concentration at higher oxygen pressures. Removal <strong>of</strong> oxygen restored<br />
the original signal, which indicated that the phenomenon was due to physical and not <strong>chemical</strong><br />
processes. Halogens were at first found to have no effect on coals and chars and the oxygen<br />
effect was attributed to spin broadening due to the paramagnetism <strong>of</strong> the oxygen molecules.<br />
Similar results to oxygen were obtained with nitric oxide.<br />
In 1963 Wynne-Jone~~~ and co-workers found that e.s.r. signals in chars obtained<br />
by heating polyvinylidene chloride (saran) were affected by adsorption <strong>of</strong> halogens. These<br />
workers noted that IBr, ICI, I, Br, and C12 caused partial or complete reduction in spin<br />
concentration at constant line width. The original signal was not restored by outgassing in vacuo<br />
at room temperature but could be partially restored by reheating the specimen at 100°C in vacuo.<br />
Since, as mentioned above, no halogen effect had been obtained on coals or more common chars,<br />
these authors attributed the susceptibility to halogens <strong>of</strong> these saran chars to a high degree <strong>of</strong><br />
porosity. Reinvestigation <strong>of</strong> the interaction <strong>of</strong> a series <strong>of</strong> cellulose chars with iodine, bromine<br />
and hydrogen iodide in this laboratory has shown pronounced effects on the electron spin resonance<br />
<strong>of</strong> some <strong>of</strong> these materials.<br />
EXPERIMENTAL<br />
&llulose chars were prepared by fint charring cellulose powder (Whatman Cellulose<br />
Powder, Standard Grade) in an autoclave at 195"-2OO0C under nitrogen. Subsequently this char<br />
* Contribution No. 360 from the Research Council <strong>of</strong> Alberta, Edmonton, Alberta.
514<br />
was heated under nitrogen in a Vycor tube to the desired temperature and held at temperature for<br />
one hour. Aliquots <strong>of</strong> the chars were then treated with a lawe.excess <strong>of</strong> 0.1 M solutions <strong>of</strong> iodine<br />
or bromine in carbon tetrachloride. After standing in the solutions for twenty-four hours, the<br />
samples were washed once with carbon tetrachloride and dried. Samples for e.s.r. measurements<br />
were ploced in 3 mm. 0.d. glass tubes and evacuated.<br />
Samples to be treated with hydrogen iodide were placed in open 3 mm. 0.d. tubes<br />
placed in the cavity <strong>of</strong> the spectrometer and purged with purified nitrogen. Gaious hydrogen<br />
iodide (Matheson) was passed directly over the sample in the cavity. Excess hydrogen iodide was<br />
removed after treatment by subsequent purging with nitrogen.<br />
Electron spin resonance measurements were performed with a Varian Model 4500<br />
electron spin resonance spectrometer fitted with a TE102 cavity and a 100 Kc modulation attachment.<br />
Saturation phenomena were avoided by working at 10 db attenuation and check for saturation were<br />
made. Line widths ond spin concentrations were measured from first derivative cutves, generally<br />
by direct inspection but in the case <strong>of</strong> very wide and very narrow signals resort was made to<br />
graphical integration. A standard 500" cellulose char, used for calibration, was standardized<br />
against DPPH, CuSO, and a Varian Standard char. The amount <strong>of</strong> sample was always less than<br />
10 mg. and asymmetric line shapes were not observed, indicating the absence <strong>of</strong> serious skin effects.<br />
RESULTS<br />
In Figures 1 and 2 are summarized the results <strong>of</strong> iodine and bromine treatment on line<br />
width and spin concentration as measured at room temperature. The untreated chars prepared at<br />
625" and 650"show a marked decrease in line width as compared with those prepared at higher<br />
or lower temperature -an effect attributed to exchange4. It is seen that both iodine and bromine<br />
remove this intense narrowing in the 625" and 650" chars. Neither has o significant effect on the<br />
300" to 500" chars but both broaden further the signal from the 700" char. The broadening due to<br />
bromine i s not as great as that due to iodine and for the 600" char, in particular, the broadening<br />
effect <strong>of</strong> the bromine is minimal. The effect on spin concentration shows that line broadening is<br />
accompanied by a considerable drop in concentration in both the bromine and iodine treated chars.<br />
Measurements made at 78°K showed that whereas the signals for the 300" to 500" chars<br />
were broadened somewhot, those from the iodine- and bromine-treated chars in the 600" to 700"<br />
range were narrowed. Washing the iodine-treated chars with cold 0.1 M sodium thiosulfate<br />
solution restored the original narrow signals. This showed that no <strong>chemical</strong> reaction had occurred<br />
to alter the signals.<br />
Treatment <strong>of</strong> a series <strong>of</strong> chars with hydrogen iodide gas for sixty hours resulted in<br />
reduction <strong>of</strong> the signal strength and broadening <strong>of</strong> the signals as shown in Table 1. The broadening<br />
for the 300°, 400" and 500" chars was moderate but that <strong>of</strong> the exchange narrowed chars (600"<br />
to 700") was extensive. Cold 0.1 M sodium thiosulfate produced no further change in the signals<br />
but refluxing for twenty-four hours with thiosulfate restored the original signals14. The restored<br />
signals <strong>of</strong> the 600", 625", 650° and 700' chars were sensitive to air whereas the restored signals<br />
<strong>of</strong> the 300", 400" and 500" chars were not. The e.s.r. signals <strong>of</strong> the untreated chars refluxed<br />
with 0.1 N Na, S, 0, were unchanged.
I<br />
./<br />
i<br />
i<br />
L \<br />
Y'<br />
J<br />
J<br />
/<br />
51-5<br />
TABLE I<br />
Effect <strong>of</strong> Hydrogen iodide on Free Radical Content <strong>of</strong> Chars<br />
Electron Spin Resonance Signals<br />
spins/g in vacua and width in gauss* 1<br />
Char<br />
Hi Treotment Na, S, 0,O. 1 M<br />
Temperature Untreated 60 hours Refluxed 24 hours<br />
300<br />
400<br />
500<br />
600<br />
2.1 x 1010<br />
4.9<br />
2.3x 1019<br />
6.1<br />
5.1 1019<br />
4.8<br />
1.4 x 10"O<br />
0.45<br />
2.0~ 1017<br />
7.9<br />
5.3x 10l8<br />
8.3<br />
3.3x 1019<br />
5.9<br />
1.3 1019<br />
19.1<br />
8.3x 1017<br />
5.76<br />
2.bX 1019<br />
6.3<br />
5.4x 1019<br />
4.1<br />
1.4 x 1020<br />
0.49<br />
625 1.3 x 10"' 1.7 1019 9.8 1019<br />
, 0.40 22.4 0.49<br />
650 1.0 x 1020 4 . 1019 ~ ~ 1.0 x 1020<br />
1.4 41 .O 1.2<br />
700 7.4x 1019 1;2 1019 7.2 1019<br />
1.4 43 2.0<br />
* Width measured between points <strong>of</strong> maximum slope.<br />
DISCUSSION<br />
Two aspects <strong>of</strong> these results must be considered -the interaction with the halogens<br />
which are typical electron occeptors and the interactions with hydrogen iodide. In agreement<br />
with Wynne -Jones and co-workers, we are <strong>of</strong> the opinion that iodine, and to a lesser extent<br />
bromine, can form donor-acceptor complexes with the chars and in so doing affect the e.s.r.<br />
signals. Presumably the iodine and bromine act as acceptors and aromatic ring systems <strong>of</strong> the<br />
char act as donors in these complexes. The low-temperature chars, 300" to 500°, having few<br />
aromatic ring systems as large or larger than four or five rings1', are not affected by iodine or<br />
bromine. The 600" char apparently has a high enough donor ability - low enough ionization<br />
potential -to interact with iodine but not sufficient to react definitely witti bromine, which is a<br />
much weaker acceptor. Both bromine and iodine readily form complexewith the 625", 650"<br />
and 700" chars. The fact that the signals <strong>of</strong> the halogen affected chars are narrowed on cooling<br />
to 79OK is also indicative <strong>of</strong> donor-acceptor interaction between halogens and aromatic moietiesle.<br />
It is possible that lack <strong>of</strong> porosity is sufficient explanation <strong>of</strong> the failure <strong>of</strong> the 300" to 500" chars<br />
to be affected by iodine',. However, the difference between iodine and bromine on the 600"<br />
char suggests that the electrophilicity and not the size <strong>of</strong> the adsorbed molecule is important.
The line broadening at 79OK <strong>of</strong> the 300°, 400' and 500' chars treated with halogens<br />
is not only indicative <strong>of</strong> no complex formation between halogen and the aromatic system but also<br />
suggests that the initial signals <strong>of</strong> these chars are due to complexes between quinonoid acceptors<br />
and carbocyclic donorsI6. This is further borne out by the effects <strong>of</strong> hydrogen iodide on these<br />
In order to illustrate the relative importance <strong>of</strong> charge-transfer complexes in these<br />
cham a plot has been made in Figure 3 <strong>of</strong> the number <strong>of</strong> free radicals vs. carbon content. There<br />
has also been plotted the reciprocal <strong>of</strong> crystallite size in no./g. vs. carbon content calculated<br />
from the work <strong>of</strong> Hirschl' and van Kre~elen~~ assuming that there is an average crystallite height<br />
<strong>of</strong> three graphitic layers and that there is no unordered material. It should be noted that the<br />
average height <strong>of</strong> the crystallites would be somewhat less than three in the low temperature cham<br />
and possibly somewhat greater than three in the higher temperature chars. In any event, it is<br />
seen that in the 600' to 650° range the number af crystallites clasely approximates the number <strong>of</strong><br />
free radicals, accounting for the results <strong>of</strong> Schuyera2. In fact these latter cham are apparently<br />
composed <strong>of</strong> little but charge transfer complexes in close proximity to one another which explains<br />
the intensive exchange narrowing <strong>of</strong> the e.s.r. signals <strong>of</strong> these materials. The decrease in free<br />
radicals at higher temperatures is at least in part due to the decrease in number <strong>of</strong> cr)+staIlites<br />
at higher charring temperatures.<br />
Extendiw the conclusions on cellulase chars to coal and related materials, suggests<br />
that e.s.r. signals in all the& materials result from donor-acceptor complexes which, while<br />
reaching a maximum <strong>of</strong> importance in anthracites and 600' to 70O0 chars, may still be <strong>of</strong><br />
significance in lower rank coals and humic acids. Such a conclusion find, suppolt from a number<br />
<strong>of</strong> other aspects <strong>of</strong> the behavior <strong>of</strong> coal and humic acids. Charge-transfer forces between coal<br />
and solvent can be used to explain the order <strong>of</strong> effectiveness <strong>of</strong> coal solvents. Thus catechol<br />
and anthracene oils, which am donor molecules, are very effective in extracting or dispersing<br />
coal OT humic materials. Charge-tmnsfer equilibria can also rationalize the failure <strong>of</strong> coal<br />
extmcts once dried to redissolve completely in the original solvent used for extraction. FIW<br />
,<br />
I<br />
I
z<br />
I<br />
51.7<br />
radicals in coals and chars are physically affected by oxygen, as indicated by line broadening <strong>of</strong><br />
the e.s.r. signal, but are stable to <strong>chemical</strong> attack. Such behavior could be explained as due<br />
either to spin-spin coupling <strong>of</strong> the oxygen molecule with the free electron <strong>of</strong> the charge-transfer<br />
complex or to formation <strong>of</strong> new donor-acceptor complexes between aromatic centres and oxygena4.<br />
The so-called molecular weight <strong>of</strong> humic acids and cool extracts should be re-examined<br />
in the light <strong>of</strong> the concept <strong>of</strong> donor-acceptor complexes. Thus molecular weight determinations<br />
<strong>of</strong> humic acids in catechola5, a substance conceivably capable <strong>of</strong> breaking up dbnor-acceptor<br />
complexes <strong>of</strong> humic acids, resulted in a low molecular weight estimate. Determinations on apparently<br />
similar material in sulfalane, a poor complexing agent, produced very much higher estimates <strong>of</strong><br />
molecular weight 6.<br />
finally, cools and related materials have a well-known broad absorption band at<br />
1600 cm -l in the infrared region which has defied interpretation. Donor-acceptor phenomena<br />
have been shown to produce strong and broad absorption in this regi~n"~~"~ in aromatic systems,<br />
as shown in Figure 4, and hence may be the cause <strong>of</strong> this band in coal and chars.<br />
1.<br />
2.<br />
3.<br />
4.<br />
5.<br />
6.<br />
7.<br />
8.<br />
9.<br />
10.<br />
11.<br />
12.<br />
REFERENCES<br />
Honda, H., and Ouchi, K., Rep. Resources Inst. Japan No. 1, 1952.<br />
Adamsan, A.F., and Blayden, H.E.,Proc. <strong>of</strong> the 3rd Conference on Carbon (1957),<br />
Pergammon Press, London, 1959, p. 147.<br />
Berkowitz, N., Cavell, P.A., and Elafson, R.M., Fuel 40, - 279 (1961).<br />
Ingram, D. J.E., and Bennett, J.E.P., Oil Mez. - 45, 545 (1954).<br />
Uebersfeld, J., Etienne, A., and Combrisson, J.,Nature - 174, 614 (1954).<br />
Winslow, F.H., Baker, W.O., and Yager, W.A.,J. Am. Chem. Soc. - 77, 4751<br />
(1 955).<br />
Akamatu, H., Mrozowski, S., and Wobschall, D.,Proc. <strong>of</strong> the 3rd Conference on<br />
Carbon (1957), Pergammon Press, London, 1959, p. 135.<br />
Singer, L.S., Spry, W. J., and Smith, W. H., Proc. <strong>of</strong> the 3rd Conference an Carbon<br />
(1957), Pergammon Press, London, 1959, p. 121.<br />
Ingram, D.J.E., Free Radicals as Studied by Electron Spin Resonance, Butterworths,<br />
London, 1958, p. 213.<br />
bbersfeld, J. and Erb, E., Proc. <strong>of</strong> the 3rd Conference on Carbon (1957), Pergammon<br />
Press, London, 1959, p. 103.<br />
Antonowicz, K., Proc. ,<strong>of</strong> the 5th Conference on Carbon (1961), Pergammon Press,<br />
London, 1963, p. 61.<br />
Mrozowski, S.,Proc. <strong>of</strong> the 4th Conference on.Carbon (1959), Pergammon Press,<br />
London, 1960, p. 271.
13.<br />
14.<br />
15.<br />
16.<br />
17.<br />
18.<br />
19.<br />
20.<br />
21.<br />
22.<br />
23.<br />
24.<br />
25.<br />
26.<br />
27.<br />
28.<br />
518<br />
Campbell, D., Jackson, C., and Wynne-Jones, W.F.K.,Nature - 199, 1090 (1963).<br />
Toland, W.G.,U.S. Patent 2,856,424 (1958).<br />
Hirsch, P.B., Proc. Roy. SOC. (London), - A226, 143 (1954).<br />
Base, M. , and Lobes, M.M., J. Am. Chem. SOC. - 83, 4505 (1961).<br />
Mitchell, H.K. and William, R.J.,J. Am. Chem. SOC. - 60, 2723 (1938).<br />
Deno, N.C., Freedman, N., Hodge, J.D., MacKay, F.P. and Saines, G., J. Am.<br />
Chem. Soc. - 84, 4713 (1962).<br />
Given, P.H. , and Peover, M.E., J. Chem. SOC. 394 (1960).<br />
Voet, A.C. and Teter, A.C., Amer. Ink Maker 38, No. 9, 94 (1960).<br />
Dulov, A.A., Slinkin, A.A., Rubinshtein, A.M., and Kotlyarevski, I.L.,lzvestia<br />
Akod. Nauk S.S.S.R. Seriya Khim. No. 11, p. 1910 (1963).<br />
Schuyer, J.,Brenn. Chemie. - 37, 74 (1956).<br />
van Krevelen, D.W. and Schuyer, J., Coal Science, Elsevier Publishing Co. ,<br />
London (1957), p. 176.<br />
Tsubomura, H., and Mulliken, R.S.,J. Am. Chem. SOC. 82, 5966 (1960).<br />
Polansky, T.S., and Kinney, C.R.,Fuel 1, 409 (1952).<br />
Wood, J.C., Moschopedis, S.E., and El<strong>of</strong>son, R.M.,Fuel - 40, 193 (1961).<br />
El<strong>of</strong>son, R.M., Anderson, D.H., Gutowsky, H.S., Sandin, R.B., and Schulz,<br />
K. F., J. Am. Chem. SOC. - 85, 2622 (1963).<br />
Sandin, R.B. , El<strong>of</strong>son, R.M. , and Schulz, K.F., J. Org. Chem. - 30, 1819 (1965).<br />
-
J 519<br />
i<br />
I<br />
\<br />
d<br />
i<br />
L<br />
300 400 500 600 700<br />
CHAR TEMPERATURE 'C.<br />
IO#'<br />
300 400 500 000 100<br />
CHAR TEMPERATUR€ 'C<br />
Effect <strong>of</strong> I, and Br, on line widths <strong>of</strong><br />
cellulose chars measured in vacuo at<br />
room temperature.<br />
Figure 2.<br />
rn no treatment<br />
A treated with 0.1 M I, in CCI,<br />
treated with 0.1 M Br, in CCI,<br />
Effect <strong>of</strong> , and Br, on spin concentrations<br />
in cellulose chars measured in vacuo at<br />
room tempera tu re<br />
m no treatment<br />
A treated with 0.1 M I, in CCI.<br />
treated with 0.1 M Br, in CCI
W<br />
L,<br />
Z<br />
6<br />
m<br />
(1:<br />
0<br />
6<br />
d j o 2 ' 1<br />
0.0<br />
g 0.2<br />
I I<br />
0.4<br />
I700<br />
I<br />
1400 1700<br />
I<br />
I400<br />
FREQUENCY (CM-I)<br />
6<br />
Figure 3.<br />
Comparison <strong>of</strong> the number <strong>of</strong> free radicals<br />
with the size <strong>of</strong> crystallites in cellulose chars.<br />
A.<br />
8.<br />
.number <strong>of</strong> free radicals per gram vs.<br />
Carbon content<br />
number <strong>of</strong> crystallites possible per gram<br />
vs. Carbon content<br />
Figure 4.<br />
The influence <strong>of</strong> charge transfer forces<br />
on infrared spectra.<br />
A. Tetrakis [p-dimethylaminophenyl]<br />
ethylene in fluarolube mull<br />
'<br />
B. Iodine complex <strong>of</strong> tetrakis<br />
[p-dimethylaminophenyl 1 ethylene<br />
in fluorolute mull<br />
t.<br />
I<br />
i'<br />
\
521<br />
INFRARED SPECTRA OF OXYGEN-18 LABELED CHARS<br />
R. A. Friedel<br />
Pittsburgh Coal Research Center, Bureau <strong>of</strong> Mines,<br />
U.S. Department <strong>of</strong> the Interior,Pittsburgh, Pa.<br />
R. A. Durie and Y. Shewchyk<br />
Commonwealth Scientific and Industrial Research Organization,<br />
Chatswood, New South Wales, Australia<br />
SUMMARY<br />
Chars <strong>of</strong> four 0-18 labeled compounds were prepared. Infrared spectra were<br />
examined for isotopic shifts. On the basis <strong>of</strong> a few published results on pure compounds<br />
the shifts for C=O18 at 1600 cm-l were expected to be -20 cm-l or more. The<br />
observed shifts are less than 5 cm-l; this result may indicate that chelated carbonyls<br />
are not involved. However, very strong hydrogen bond'ing as in chelated<br />
conjugated carbonyl structures may be responsible for the small shifts.<br />
labeled chelate is being prepared at the CSIRO laboratories in order to investigate<br />
this point.<br />
- Chars<br />
Introduction<br />
!<br />
An 0l8-<br />
Since the early work at the Bureau <strong>of</strong> Mines on carbohydrate char&/ the<br />
infrared spectra <strong>of</strong> chars ( 5 500" C) <strong>of</strong> various <strong>chemical</strong>s have been studied.m/<br />
Spectral investigations <strong>of</strong> structure <strong>of</strong> chars have included studies <strong>of</strong> the effect<br />
<strong>of</strong> elemental constitution; the effect <strong>of</strong> introducing oxygen into chars <strong>of</strong> hydrocarbons<br />
indicated that spectral absorption bands, particularly the intense 1600 cm-1<br />
band, were attributable to oxygenated structums.4'6II/<br />
Isotope-labeling <strong>of</strong> chars<br />
has been utilized; a successful study <strong>of</strong> deuterium labeling was carried out by comparing<br />
infrared spectra <strong>of</strong> chars <strong>of</strong> an aliphatic hydrocarbon and a corresponding<br />
deuterocarbon. From this study definite evidence was obtained for assignment <strong>of</strong><br />
the 730-910 cm-1 absorption bands to CH out-<strong>of</strong>-plane aromatic vibrations.=/<br />
further indications were found that the most intense band in char spectra, 1600 cm-i,<br />
was attributable to oxygenated structures.<br />
A more direct investigation <strong>of</strong> the specific structures responsible for the 1600<br />
cm'l band seemed desirable. Charring <strong>of</strong> compounds labeled with oxygen-18 was<br />
judged to be a direct method with good chances for success. Three structures were<br />
considered on the basis <strong>of</strong> their intense absorption, near 1600 an-1, to be possible<br />
sources <strong>of</strong> the 1600 cm-1 band in chars:<br />
(1) Carbonyl groups in ketones, quinones, or conju ated chelate structures<br />
should show a shift to low frequency in going from GOLg to C=018; (2) aromatic<br />
nuclei should produce no change in spectral frequency; (3) aromatic nuclei with<br />
enhanced intensities due to oxygenated substituents on the ring should not undergo<br />
any appreciable frequency shift in the 1600 cm-l region; a shift should occur in<br />
the C-0 stretch region at 1100 cm-1.<br />
Also
Coals<br />
522<br />
Studies <strong>of</strong> the 1600 cm-l band in char spectra may produce information on the<br />
origin <strong>of</strong> the same band in the spectra <strong>of</strong> coals. Spectral data from chars <strong>of</strong><br />
carbohydrates would be particularly applicable for comparison with coal spectra as<br />
carbohydrate chars produce spectra nearly identical to spectra <strong>of</strong> coals.u/ Unfortunately<br />
018-labeled carbohydrates are not available.<br />
Information on the intense 1600 cm-' band in coals has been obtained in studies<br />
<strong>of</strong> absorption intensities for many pure compounds in an effort to determine what<br />
structures could produce the 1600 cm-l absorption.j/ The principal type <strong>of</strong> struc-<br />
ture found to have sufficient intensity is the conjugated chelated carbonyl group.<br />
These groups can only produce the 1600 band if all or nearly all <strong>of</strong> the oxygen in<br />
the coal is in the form <strong>of</strong> hydroxyl chelated carbonyl groups.<br />
018-Labeled Compounds<br />
The minimum spectral shift in the infrared spectra for a labeled carbonyl<br />
group (C=018) relative to an unlabeled group (C=O16) is -40 cm-l, on the basis <strong>of</strong><br />
the simple ratio <strong>of</strong> the reduced masses. Hooke's law for the stretching vibration<br />
<strong>of</strong> ~=016 is: u16 = 2rrc J =, where<br />
p16 =<br />
mC + m016 ; u = frequency, cm-1; k = constant;<br />
%mol6<br />
For C=OL8 the same equation applies; dividing,<br />
'or ~ 1 = 8<br />
Jl.05 '<br />
For = 1600 cm-1, "18 = 1560 cm-li Au = -40 cm-l. If this 1600 cm-l band in a<br />
char or coal spectrum is due to a simple carbonyl structure, the corresponding<br />
labeled carbonyl group should produce an absorption band shifted to 1560 cm-l. If<br />
the stretching vibration ,<strong>of</strong> the carbonyl structure is complex the simple value obtained<br />
from the reduced mass calculation will differ somewhat from 40 crn-l. A<br />
ketone investi ated by Karabatsos?/ showed an appreciable deviation; 2,3-dimethyl-<br />
3-pentanone-018 shows a shift <strong>of</strong> 31 cm-l instead <strong>of</strong> the expected 40. This shift<br />
is in the neighborhood predicted by FrancisEI for a mixed vibration composed <strong>of</strong><br />
about 80 percent C=O stretch and 20 percent C-C stretch. With conjugation the<br />
shift does not change for the benzenoid carbonyl compounds:<br />
benzoates, benzophenone, and benzoquinone.<br />
Benzoic acid, two
523<br />
AV, cm-1<br />
Benzoic acid (monomer) -32- 111<br />
111<br />
Methylbenzoate -31-<br />
Cinnamyl nitrobenzoate<br />
Benzophenone<br />
p -Benzoquinone<br />
Benzoylchloride<br />
-25- 11/<br />
151<br />
Benzamide -24-<br />
The decreased shift for benzamide is attributable &greater participation <strong>of</strong><br />
other groups <strong>of</strong> the molecule in the carbonyl vibration.- Apparently the same<br />
explanation may be given for the decreased shift observed for benzoylchloride. It<br />
is surprising that the shifts observed indicate no particular increase due to con-<br />
jugation with an aromatic ring.<br />
(A large effect due to ring conjugation is observed<br />
by comparison <strong>of</strong> the spectra <strong>of</strong> nitromethane-018 and nitrobenzene-018.16,17/)<br />
Future investigations <strong>of</strong> more aliphatic ketones may produce different results.<br />
The only published indication <strong>of</strong> the effect <strong>of</strong> conjugated chelation is on<br />
benzoic acid dimer:<br />
AU, cm-1<br />
Benzoic acid dimer -20<br />
Benzoic acid monomer -32<br />
Thus hydrogen bonding is seen to produce a significant decrease in the OI8<br />
shift <strong>of</strong> a conjugated carbonyl system. It was anticipated that the spectra <strong>of</strong> chars<br />
having carbonyl groups enriched in 0l8 may shaw shifts for the 1600 cm-l band <strong>of</strong><br />
-20 to -30 cm-l. Such shifts in char spectra would.be measurable, even though absorption<br />
bands are broad.<br />
Experiment a 1<br />
On the initiation <strong>of</strong> this work only three usable 0l8-labeled organic compounds<br />
were commercially available. They were investigated in the following order:<br />
Linoleic acid, phenol, and benzoic acid; sodium phenate and sodium benzoate-018<br />
were also prepared and charred. First the unlabeled compounds were charred under<br />
various conditions in order to determine the most favorable condition for produc-<br />
ing intense 1600 cm-l bands.<br />
Temperatures required to eliminate the absorption <strong>of</strong><br />
monosubstituted benzenes in order to produce coal-like absorption bands in the 730-<br />
900 cm-1 aromatic band region were usually so high that other spectral details,<br />
including the 1600 cm-I band, were weakened.<br />
Samples <strong>of</strong> OI6 compounds were placed in a glass tube or a steel bomb, sealed<br />
or unsealed, in vacuo or under nitrogen. Amounts <strong>of</strong> sample used were 50-100 mg<br />
because <strong>of</strong> the scarcity <strong>of</strong> OI8 samples. Pyrolysis temperatures were varied from<br />
350" C to 620" C; times were varied from 1 to 58 hours.
L in ole i c a c id - 0'<br />
Two samples were investigated: (a) A sample <strong>of</strong> 22 percent 018-enriched acid<br />
(kindly given by Dr. A. Miko, <strong>of</strong> the Yeda Research and Development Co., Rehovoth,<br />
Israel), and (b) a commercial sample <strong>of</strong> 38 percent enriched acid. The best condi-<br />
tions found for obtaining 1600 cm-l bands were 400° C, 2 hours, under 1 atmosphere<br />
<strong>of</strong> nitrogen in a sealed pyrex tube. The 22 percent enriched sample was donated,<br />
with the warning that polymerization probably had occurred; it is ais0 possible<br />
that polymerization occurred in the 38 percent enriched sample. Yeda has taken<br />
linoleic acid-018 <strong>of</strong>f the market.<br />
Phenol-018, 86 percent<br />
Pyrolysis <strong>of</strong> phenol <strong>of</strong> 86 percent OI8 enrichment was carried out at tempera-<br />
tures near 550" C for about 18 hours in a sealed tube under nitrogen.<br />
Sodium phenate<br />
The 0I8-labeled phenate was not prepared because <strong>of</strong> insufficient phenol-018.<br />
Sodium phenate-016 was prepared from ordinary phenol and charred. Conditions were<br />
similar to those used for phenol.<br />
Benzoic acid-0I8, 95.6 percent<br />
Conditions were similar to those used for phenol.<br />
Sodium benzoate-0I8<br />
A nitrogen atmosphere and a temperature <strong>of</strong> 500' C were used. Pyrolysis times<br />
were about 24 hours.<br />
In all samples except the sodium phenate and sodium benzoate, char was ob-<br />
tained in the form <strong>of</strong> a thin film on the walls <strong>of</strong> the sealed tube. These films<br />
were sufficiently thin to give spectra <strong>of</strong> good quality directly; data obtained from<br />
such films was qualitative or semi-quantitative as thicknesses <strong>of</strong> the film varied<br />
considerably. In all cases spectra were also obtained by means <strong>of</strong> KC1 pellets<br />
which were better for quantitative absorption measurements.<br />
Spectra were determined first on gases produced. Then the samples were washed<br />
with hexane and/or benzene and the spectra <strong>of</strong> the soluble products were obtained.<br />
The benzene-insoluble char was then investigated.<br />
Ultimate analyses <strong>of</strong> all <strong>of</strong> the chars are given in table 1.<br />
Results and ,Discussion <strong>of</strong> Results<br />
Results obtained on the four substances investigated show that the spectral<br />
shifts are very small or negligible (table 2); no shift was definitely detectable<br />
for any <strong>of</strong> the chars. In figure 1 the spectra <strong>of</strong> chars from benzoic acid-0I6 and<br />
-0l8 are given; they are practically identical, including the positions <strong>of</strong> the<br />
two 1600 cm-l bands.<br />
1600 cm-I bands were obtained in all ,chars but in no case were they intense<br />
bands. This is a possible indication that,carbonyl groups may not have been in-<br />
volved. For linoleic acid and benzoic acid it is nearly certain that no oxygen-<br />
containing group could be an important contributor to the 1600 cm-l band, as the<br />
oxygen contents <strong>of</strong> the chars were found to be negligible. Hydrocarbon structures<br />
could have produced the weak bands found.
Y
526<br />
Table 1.- Ultimate analyses <strong>of</strong> chars<br />
c - . H ' " 0<br />
" ' State<br />
Table 2.- Negative spectral shifts <strong>of</strong> oxygen-18 labeled chars<br />
Spectral shift,<br />
cm-1 11<br />
Linoleic acid-0I8
The phenol chars also showed no shift from lbOU cm-', even though one char contains<br />
20 percent oxygen. It is not surprising if the pyrolysis <strong>of</strong> phenol at 550" C<br />
does not produce carbonyl groups. The 1600 cm-l band observed in the spectrum <strong>of</strong><br />
the phenol char must be due to aromatic absorption enhanced by oxygen-containing<br />
substituents on the aromatic rings (ethers and phenols).<br />
The char <strong>of</strong> sodium benzoate also shows a negligible shift. If was the most<br />
likely possibility for producing an isotope shift, as the elimination <strong>of</strong> oxygen<br />
encountered in the chars <strong>of</strong> linoleic and benzoic acid does not occur for the char<br />
<strong>of</strong> sodium benzoate. The sizeable percentage <strong>of</strong> oxygen present in the char, 4 per-<br />
cent, is likely to exist in the char as carbonyl groups <strong>of</strong> some sort; even so, no<br />
appreciable shift is observed.<br />
The lack <strong>of</strong> appreciable shift in the spectra <strong>of</strong> 018-labeled chars may be attributable<br />
to extremely strong association (chelation, or other). With the sizeable<br />
oxygen content <strong>of</strong> some <strong>of</strong> these chars there are presumably some carbonyl groups pre-<br />
sent.<br />
Unfortunately no O18-labeled pure compounds having very strong hydrogen-<br />
bonding have been investigated. As mentioned above, the spectrum <strong>of</strong> benzoic acid-<br />
OI8 shows a smaller s ift for the associated dimer acid (-20 cm-l) than for the free<br />
monomer (-32 cm-l).g7' These results are for a rather weakly associated acid dimer;<br />
the vibration <strong>of</strong> the C=O group in the dimer obviously involves participation <strong>of</strong><br />
other groups in the molecule. For a more strongly hydrogen-bonded chelate, as<br />
acetylacetone, the participation <strong>of</strong> other groups in the molecule would be greater<br />
and the shift probably would be less. On this basis the small shifts in chars are<br />
explainable. Certainly the negligible shift observed for chars does not exclude<br />
the presence <strong>of</strong> C=O groups in structures producing the 1600 cm-1 band. In order to<br />
investigate the effect <strong>of</strong> strong chelation on the C=OI8 shift preparation <strong>of</strong> dibeneo-<br />
ylmethane, H5Cg-C018-CH2-C018-C6H5, is being carried out at the CSIRO laboratories.<br />
Little or no e018 shift is expected. If an appreciable shift is observed for this<br />
compound it w ill be necessary to conclude that chars do not contain strongly chelated<br />
carbonyls.<br />
Further studies will be carried out on labeled chars as other Ol8-containing<br />
compounds become available. It is expected that prices <strong>of</strong> these compound will de-<br />
crease greatly, for cheaper methods <strong>of</strong> preparation are being deve1oped.d<br />
References cited<br />
1. Friedel, R. A., and M. G. Pelipetz. Infrared Spectra <strong>of</strong> Coal and Carbohydrate<br />
Chars. J. Opt. SOC. <strong>of</strong> Am., v. 43, No. 11, November 1953, pp. 1051-1052.<br />
2. Friedel, R. A., and J. A. Queiser. Infrared Analysis <strong>of</strong> Bituminous Coals and<br />
Other Carbonaceous Materials. Anal. Chem., v. 28, 1956, pp. 22-30.<br />
3. Friedman, S., R. Raymond, L. Reggel, I. Wender, and R. A. Friedel. Chemistry<br />
and Structure <strong>of</strong> Coal. Formation <strong>of</strong> Coallike Chars From Pure Organic Com-<br />
pounds. Abstracts, Am. Chem. SOC. meeting, Dallas, Texas, April 1956.<br />
4. Friedel, R. A. Spectra and the Constitution <strong>of</strong> Coal and Derivatives. Proc.<br />
4th Carbon Conf., Univ. <strong>of</strong> Buffalo, Pergamon Press, London, 1960, pp. 320-336.<br />
5. Brown, T. H., and Turkevich, J. A Study <strong>of</strong> the Low-Temperature Carbonization<br />
<strong>of</strong> d-Glucose Using Electron Magnetic Resonance and Infrared Absorption.<br />
Proc. 3rd Conf. on Carbon, Pergamon Press, London, 1959, pp. 113-120.
528<br />
6. Friedel, R. A., and H. Retc<strong>of</strong>sky. 'Spectral Studies <strong>of</strong> Coal. Proc. 5th Biennial<br />
Conf. on Carbon, The Pennsylvania State Univ., Pergamon Press, London, 1963,<br />
V. 2, pp. 149-165.<br />
7. Friedel, R. A. The Use <strong>of</strong> Infrared Spectra <strong>of</strong> Chars in Coal Structure Research.<br />
Applied Optics, v. 2, No. 11, Nov. 1963, pp. 1109-1111.<br />
8. Friedel, R. A., R. A. Durie, and Y. Shewchyk. The Origin <strong>of</strong> the 1600 Wavenurn- i<br />
ber Absorption dad in the Infr
529<br />
Recovery <strong>of</strong> Nitrogen Bases with<br />
Weak Acids<br />
by<br />
T. F. Johnson, P. X. Masciantonio<br />
and J. H. Schelllng<br />
Applied Research <strong>Laboratory</strong><br />
United States Steel Corporation<br />
Monroeville , Pennsylvanid<br />
Introduction<br />
The recovery <strong>of</strong> nitrogen bases is o'f particular interest. in<br />
<strong>chemical</strong> plants that operate in conjunction with coal-carbonization<br />
facilities.. . Traditionally, nitrogen bases' such as ammonia, pyridine, and<br />
quinoline have been recovered by processes that involve acid extraction<br />
followed by neutralization with caustic soda or lime. , 2, Such processes<br />
have inherent disadvantages with regard to excessive consumption,<strong>of</strong> acids<br />
and bases as well as concomitant waste-disposal problems'.<br />
Recently, several processes have been developed for nitrogenbase<br />
recovery by regenerative cyclic techniques. For example, recovery<br />
<strong>of</strong> ammonia from coke-oven gas and pyridine from coke-oven light oil have<br />
been reported.. * 5, These processes for recovery <strong>of</strong> nitrogen bases are<br />
characterized by the use <strong>of</strong> an aqueous acid solution for the extraction<br />
step followed by a high-temperature stripping, or springing, operation<br />
that liberates the nitrogen bases and simultaneously regenerates the acid<br />
extractant. The selection <strong>of</strong> an appropriate acid extractant for a<br />
. '<br />
particular nitrogen-base system is an important factor in designing or<br />
developing a regenerative process. Previously reported studies3 , ')<br />
have demonstrated that mqnoammonium acid phosphate is a suitable extractan,t<br />
for ammonia recovery, whereas either phosphoric acid or monopyridinium<br />
sulfate is adequate for pyridine recovery. However, the theory for<br />
selection <strong>of</strong> appropriate reagents for regenerative nitrogen-base recovery<br />
has not been developed and, consequently, the general applicability <strong>of</strong><br />
this recovery technique has not received significant attention. It is the<br />
object <strong>of</strong> this paper to demonstrate that readily available thermodynamic<br />
data can be used to determine the feasibility <strong>of</strong> a regenerative technique<br />
for recovery <strong>of</strong> partkular nitrogen bases:'The discussion is based on<br />
the behavior <strong>of</strong> particular acids during the extraction and springing<br />
operations. Data are also presented on the extraction and springing steps<br />
for recovery <strong>of</strong> pyridine and quinoline' from coal-tar fractions.<br />
. .<br />
Discussion. <strong>of</strong> the Regenerative Technique<br />
The general scheme for a regeneratlve recovery process for<br />
See References.<br />
I
egeneration. The system can be seen in Figure 1 as a generalized flow<br />
diagram. Usually, nitrogen bases are recovered from streams in which<br />
they are present in low concentrations (about 0.1 to 5%), such as coke-oven<br />
light-oil streams.<br />
Selection. <strong>of</strong> Reagents for Regenerative Systems<br />
The selection <strong>of</strong> an appropriate acidic extractant for a particular '<br />
nitrogen base 1s the initial problem encountered in developing a regenerative'<br />
process. Although such factors as solubility, volatility, decomposition, and<br />
side reactions must be- considered, the acid component essentially must be<br />
Free Hydronium Protonated Water<br />
Base Ion Base<br />
The temperature dependence <strong>of</strong> the equilibrium constant follows<br />
the expression<br />
where<br />
AH = the heat <strong>of</strong> reaction, and<br />
R = the gas constant.<br />
I<br />
I<br />
,<br />
\,<br />
(2) I<br />
It is desirable that the ratio (KT-/KT~) be less than unity for a \i<br />
regenerative system (AH, the heat <strong>of</strong> reaction, must be negative).<br />
I<br />
In an effort to develop suitable parameters for the selection <strong>of</strong> .<br />
reagents for regenerative systems, the ionization equilibria <strong>of</strong> acids and I<br />
bases have been examined. I<br />
In a discussion <strong>of</strong> acid-base systems, the following equations are 1<br />
applicable. _ _<br />
Acid extraction <strong>of</strong> a nitrogen base (B) from a hydrocarbon Stream'<br />
involves the equilibrium reaction<br />
(<br />
B + H~O+ = BH+ + H ~ O<br />
(3) '<br />
which can be represented by an equilibr<br />
um- constant<br />
I<br />
I<br />
!<br />
I<br />
1<br />
i
I<br />
1<br />
I<br />
i<br />
i<br />
I<br />
The ionization constant <strong>of</strong> the base involves the equll~brlum<br />
B + H20 = BtI+ + 011-<br />
expressed by an ionization constant<br />
5 31<br />
I<br />
Kb = lBHfl [OH-] (6)<br />
f,. I 13 I<br />
,/<br />
I<br />
,'<br />
By usina the lonlzati@n const
532<br />
and can be expressed in terms <strong>of</strong> Equation 8 to give<br />
Therefore<br />
[Bl =I?)<br />
[BH+]lI2<br />
I<br />
!<br />
I<br />
i<br />
(16) ' I<br />
Thus, for a given concentration [BH'] in the aqueous extract, the concentra- ~<br />
tion [B] <strong>of</strong> base is high for bases <strong>of</strong> low base strength (Kb).<br />
From Equations 12 and 17, the following generalizations can be<br />
made concerning the selection <strong>of</strong> an acid and.a base for recovery <strong>of</strong> nitrogen<br />
i<br />
bases by a regenerative process. To get good extraction, maximize [BH+) by<br />
using an acid with as large a value for Ka as is possible, consistent with<br />
other requirements <strong>of</strong> the process. To get good regeneration, maximize [B]<br />
(a base with a small Kb value should be involved).<br />
i<br />
Accordingly, suitabie systems appear to involve an acid extractant<br />
with a large 1<br />
Ka value and a nitrogen base with a small Kb value. Consistent;<br />
with the above, the ApK was examined as a useful guide in selecting compo.-<br />
1<br />
nents 'for regenerative systems, . ,<br />
where<br />
pK = -log K (19) 1<br />
ApK = (pKb -pKa) , and (20)<br />
ApK is expressed as the absolute value <strong>of</strong> (PKb - pKa). It is<br />
anticipated that the larger the ApK <strong>of</strong> a system, the better it w ill function!<br />
in a regenerative recovery process. Therefore it appears that recovery <strong>of</strong><br />
nitrogen bases is facilitated by combinations <strong>of</strong> relatively weak bases and<br />
relatively strong acids or relatively strong bases and weak acids. /<br />
\<br />
Systems for Recovery <strong>of</strong> Ammonia and Pyridine Bases<br />
\<br />
As mentioned above, several processes have previously been<br />
reported for recovery <strong>of</strong> ammonia and pyridine bases from coke-oven product<br />
streams. The data on ammonia and pyridine are consistent with the discussiol<br />
presented above, Tables I and 11. Experimentally it has been demonstrated I<br />
that, although ammonia can be recovered in a regenerative manner using<br />
'q<br />
monoammonium phosphate, neither phosphoric acid nor sulfuric acid can be<br />
(<br />
used (H2PO4- is suitable for NH3 recovery, whereas H3PO4, HS04-, and H2S04<br />
are not applicable). '.<br />
\<br />
It has also been shown that pyridine can be recovered successfull<br />
with either HjPOl or HSOq-; however, neither H2P04- nor H2S04 can be '6<br />
employed.<br />
i<br />
1<br />
k
1<br />
i 533<br />
1,<br />
I<br />
I<br />
The use <strong>of</strong> the parameter ,pK does not appear to fit the observed<br />
1 behavior <strong>of</strong> all the systems examined. However, the elementary ionlzation<br />
ionization equilibrium theory explains certain inconsistencies with ApK<br />
values. For example, in general, (1) systems in which both components have<br />
\ pK values greater than about 6 are not useful since interaction is too weak<br />
to effect extraction, (2) systems in which both componcnts,have pK values<br />
less than about 6 are not useful since interaction is too strong tor<br />
' dissociation <strong>of</strong> the acid-base salt during regeneration, and (3) when el ther<br />
' component is a strong acid or a strong base, the system is not applicable<br />
I because interaction is too strong for regeneration <strong>of</strong> the reagent.<br />
I<br />
I<br />
/<br />
Recovery <strong>of</strong> Pyridine and Quinoline from Coal-Tar Fractions<br />
A regenerative system for recovery <strong>of</strong> pyridine from coal-tar<br />
fractions has been examined on the basis <strong>of</strong> the above consideration ot ApK.<br />
Values <strong>of</strong> ApK for the pyridine - HS04- and pyridine - H3P04 systems are<br />
6.85 and 6.64, respectively, Table 11. Each system was examined to determine<br />
I<br />
the behavior <strong>of</strong> these acids in the extraction and regenerative steps. Data<br />
on the quinoline regenerative recovery system are presented for comparison.<br />
Ionization data on quinoline (Kb = 6.3 x pK = 9.87) suggest that<br />
'<br />
either HS04- or H3PO4 could be used as the sxtractant.<br />
The respective values<br />
<strong>of</strong> ApK are 7.95 and 3.22. Since HSO4- gives a larger value <strong>of</strong> ApK, the<br />
recovery <strong>of</strong> quinoline with quinolinium acid sulfate solution as the extractant<br />
was examined for comparison.<br />
Experimental<br />
Data on the extraction <strong>of</strong> pyridine and quinoline bases were<br />
obtained by using conventional laboratory equipment consisting <strong>of</strong> a stirred<br />
1-liter 3-neck flask equipped with a bottom outlet stopcock. Single-stage<br />
extraction tests were conducted over the temperature range 0 to 55 C. The<br />
data on the pyridine -,sulfuric acid system are presented in Table 111, and<br />
data on the pyridine - phosphoric acid and the quinoline - sulfuric acid<br />
systems appear in Tables IV and V, respectively. The extraction data<br />
indicate that the nitrogen bases can be concentrated in the aqueous phase<br />
by the extraction technique.<br />
To examine the vapor-liquid equilibrium data for the pyridine -<br />
phosphoric acid system, the pyridine - sulfuric acid system, and the<br />
quinoline - sulfuric acid system, solutions were prepared <strong>of</strong> various<br />
strengths. <strong>of</strong> acid and concentration <strong>of</strong> the respective nitrogen bases. Acid<br />
strengths covered the range 20 to 40 percent and base-to-acid mole ratios<br />
varied from 0.6 to 1.9.<br />
An Othmer equillbrium still was used to determine concentrations<br />
<strong>of</strong> pyridine and quinoline in the vapor and liquid phases for the various<br />
sample solutions.<br />
Discussion<br />
The Othmer equilibrium data indicate that pyridine concentration
534<br />
in the vapor increases with increasing concentration <strong>of</strong> acid (consistent wit<br />
Equation 17) and, at a given level <strong>of</strong> acid strength, with increasing pyri-<br />
dine-to-acid mole ratio, see Figures 2 and 3, Tables VI and VII.<br />
Although it was expected that quinoline should behave similar to<br />
pyridine, such was not the case. Quinoline was realized in the distillate i<br />
<strong>of</strong> the Othmer still; however, the quinoline concentration lis significantly<br />
lower than pyridine for a given mole ratio <strong>of</strong> base to acid. Two factors, ,~<br />
low vapor pressure and low solubility, cause problems in the quinoline I<br />
system. Because <strong>of</strong> the relatively poor solubility <strong>of</strong> quinoline in water ;<br />
-(0.08 g/100 ml at.25 C), recovery <strong>of</strong> quinoline as a distillate product<br />
depends on the limitations <strong>of</strong> a steam distillation system. Accordingly,<br />
the distillate composition is a function <strong>of</strong> the vapor pressure <strong>of</strong> quinoline ':<br />
at the boilihg point <strong>of</strong> the mixed liquid phase. Because <strong>of</strong> the low volatil-<br />
ity <strong>of</strong> quinoline, only about 2 weight percent quinoline is realized in the<br />
distillate during the regeneration step. Thus, typical Othmer equilibrium<br />
data are not realized for quinoline.<br />
Therefore, although quinoline can be recovered by a cyclic 1<br />
regenerative process, the system is not analogous to the pyridine system.<br />
The quinoline system appears suitable on the basis <strong>of</strong> ApK data, but other<br />
factors such as solubility and volatility must be considered for practical<br />
application.<br />
Summary<br />
A method for selecting reagents for recovery <strong>of</strong> weak bases from<br />
hydrocarbon streams has been developed, based on thermodynamic ionization<br />
equilibrium constants. An,acid can be selected for recovery <strong>of</strong> a given<br />
base according to the value <strong>of</strong> ApK calculated for the system. Experimental<br />
data have been presented on the pyridine - sulfuric acid system, the<br />
pyridine - phosphoric acid system, and the quinoline - sulfuric acid system<br />
and demonstrate the utility and limitations <strong>of</strong> the method.<br />
The technique should also be applicable to development <strong>of</strong> systems \<br />
When it is required to effectively remove nitrogen '<br />
for recovery <strong>of</strong> acids from effluent streams by extraction with selected<br />
nitrogen base reagents.<br />
impurities in conjunction with a subsequent refining operation on a hydrocarbon<br />
stream, a regenerative process may also be applicable; however,<br />
provision must be made for adequate removal <strong>of</strong> nitrogen bases by using a<br />
sufficient number <strong>of</strong> extraction stages.<br />
References<br />
1. H. H. Lowry, "Chemistry <strong>of</strong> Coal Utilization," Vol I1<br />
John Wiley 6 Sons, Inc., New York, 1945.<br />
2. P. J. Wilson and J. H. Wells, "Coal, Coke and Coal Chemicals,"<br />
McGraw Hill, N. Y., 1950.<br />
'I<br />
I
t<br />
'.<br />
I<br />
1 3.<br />
\ 4.<br />
I 5.<br />
/<br />
i<br />
\<br />
t<br />
J<br />
1,<br />
535.<br />
H. L. Stewart, U. S. Patent 2,410,906, November 12, 1946.<br />
R. D. Rice, U. S. Patent 3,024,090, March 6, 1962.<br />
G. D. Kharlampovich and R. I. Kudryashova, Coke and Chemistry (USSR),<br />
- 2, 29, (1964). I
NH3<br />
NH3<br />
NH3<br />
NH3<br />
Pyridine<br />
Pyridine<br />
Pyridine<br />
Pyridine<br />
536<br />
Table I<br />
Data on Regenerative Recovery Systems<br />
for Nitrogen Bases1) --<br />
Nitrogen Bases Extractant<br />
Ka-<br />
6.23 x<br />
7.52 10-33)<br />
1.20 x 10-22)<br />
-4)<br />
6.23 x<br />
7.52 x 10-<br />
1.20 x 10-Z2)<br />
33)<br />
-4)<br />
1) Handbook <strong>of</strong> Chemistry and Physics 44th Ed., Chemical Rubber<br />
Publishing Co., Cleveland, Ohio, 963.<br />
2) Ka for second hydronium-ion dissociation.<br />
3) Ka for first hydronium-ion dissociation.<br />
4) Completely dissociated.<br />
Sys terns<br />
NH3- (NH4) H2P04<br />
NH3-H3P04<br />
NH3-NH4HS04<br />
NH J-H~SO~ Pyridine- (PyH) H2P04<br />
Pyr idine-H 3P04<br />
Pyridine- (PyH) HS04<br />
Pyridine-H2S04<br />
* ApK Not applicable.<br />
' Table I1<br />
ApK Values for Acid-Base Systems<br />
Q!i<br />
2.46<br />
2.63<br />
2.83<br />
*<br />
1.55<br />
6.64<br />
6.85<br />
*<br />
1<br />
Kb-<br />
1.79 10-5<br />
1.79 x 10-5<br />
1.79 10-5<br />
1.79 10-5<br />
1.71 10-9<br />
1.71<br />
1.71<br />
1.71<br />
Suitability <strong>of</strong> System<br />
Yes<br />
No<br />
No<br />
No<br />
No<br />
Yes<br />
Yes<br />
No
537<br />
I Table 111<br />
E<br />
Extraction <strong>of</strong> Pyridine Bases from Lig$t Oil<br />
i With Pyridinium Sulfate Solution<br />
L I<br />
I<br />
I<br />
i<br />
I<br />
1,<br />
Test No.<br />
1<br />
2<br />
3.<br />
4<br />
Mole Ratio <strong>of</strong> Pyridine in Pyridine in<br />
Pyridine to H2E4 Washed Light Oil, % Extract, %<br />
1.68<br />
1.67<br />
1.63<br />
1.54<br />
0.15<br />
0.12<br />
0.08<br />
0.05<br />
~ * In each test, light oil containing 0.44 percent <strong>of</strong> P-bases was<br />
washed with a lean pyridinium sulfate solution (mole ratio 1.05)<br />
prepared by adding crude P-bases to a 30 weight percent sulfuric<br />
1 acid solution.<br />
\<br />
I<br />
i<br />
I<br />
Table IV<br />
Extraction <strong>of</strong> Pyridine Bases from Light Oil<br />
(30 Percent Phosphoric Acid at 55 C)<br />
I<br />
Mole Ratio <strong>of</strong> Pyridine in Pyridine in Pyridine in<br />
?est No. Pyridine to H3E4 Light Oil, % Washed Light Oil, %. Extract, %<br />
I<br />
I,) One-stage wash.<br />
!) Two-stage wash.<br />
I<br />
.\<br />
I'<br />
i<br />
i<br />
I<br />
t<br />
3<br />
1.0<br />
1.0<br />
0.70<br />
0.92<br />
0.95<br />
5.67<br />
2.10<br />
4.60<br />
4.60<br />
4.60<br />
0.44<br />
0.40<br />
0.24<br />
0.59<br />
0.58<br />
29<br />
29<br />
28<br />
27<br />
19.5<br />
19.2<br />
14.5<br />
18.1<br />
18.6
Mole Ratio <strong>of</strong><br />
Test No. Quinoline to H2s4<br />
1<br />
2<br />
538'<br />
Table V<br />
Extraction <strong>of</strong> Quinoline from a Tar Fraction<br />
With Sulfuric Acid1 8 2,<br />
I<br />
1.46 '<br />
1.41<br />
Quinoline in<br />
Washed Oil-, %<br />
1.59<br />
2.06<br />
1) Tar fraction contained 12.1 weig t percent quino<br />
2) Extractant contained 30 weight percent, H2S04.<br />
Table VI<br />
Quinoline in<br />
Acid Extract, % '<br />
me bases.<br />
Pyridine Concentrations in Vapor at Equilibrium With<br />
Refluxing H2E4 Solutions - Othmer Still Data<br />
36.7<br />
35.7<br />
<<br />
Initial H2SO4<br />
Concentration, Mole Ratio <strong>of</strong> Pyridine Content<br />
wt % Pyridine to H2E4 <strong>of</strong> Vapor, wt % \<br />
20.4<br />
19.9<br />
30.9<br />
30.4<br />
30.6<br />
40.3<br />
1.63<br />
1.95<br />
1.60<br />
1.81<br />
1.98<br />
1.60<br />
4.7<br />
21.1<br />
10.4<br />
23.8<br />
41.6<br />
21.3<br />
i<br />
I<br />
!<br />
\<br />
r<br />
'1
.. 539<br />
Table VI11<br />
Pyridine Conceqtrations in Vapor at Equilibrium With<br />
I<br />
Refluxing H3pO4 Solutions - Othmer Still Data<br />
Initial H3P04 Pyridine Content<br />
Concentration, Equivalent Ratio <strong>of</strong> <strong>of</strong> Vapor,<br />
wt % Pyridine to Acid wt %<br />
10.45<br />
10.44<br />
10.41<br />
19.88<br />
20.42<br />
20.40<br />
20.34<br />
31.20<br />
30.38<br />
31.08<br />
30.98<br />
31.06<br />
0.60<br />
0.80<br />
0.98.<br />
0.40<br />
a 0.60<br />
0.81<br />
0.97<br />
0.60<br />
0.80<br />
0.98<br />
0. 9ga)<br />
0.99b’<br />
a) Pyridine contained 23.4 percent picoline.<br />
b) Pyridine replaced with picoline.<br />
c) Analyzed as picoline.<br />
0.54<br />
2.08<br />
6.86<br />
0.0<br />
0.87<br />
4.72<br />
13.84<br />
1.11<br />
6.52<br />
21.46<br />
19.92<br />
21.65‘)
IXPROFJCTION<br />
D; M. Mason<br />
Institute <strong>of</strong> Gas Technology<br />
Chicago, Illinois<br />
Luminescence is the emission <strong>of</strong> noneguilibrium or nonthermal radiatio;<br />
as opposed to incandescence. A number <strong>of</strong> materials have been observed<br />
to l,mine?:ce !:.hen placed in a r'lame. This phenomenon, called candolurr.ineszence,<br />
cqst be distirguished from thermoluminescence - in which<br />
a xat?rial is excite? by radioactivity or other means but luminesces<br />
~nl:: 7.h2n rai~~c to a higher temperature - and from incandescence and *<br />
lurnirescxxe <strong>of</strong> the flame itself. The blue luminescence <strong>of</strong> an aerated<br />
!i:Cthtln.- flax:. is an example <strong>of</strong> the latter. Candoluminescence is <strong>of</strong><br />
- i nt;::r?st from two standpoints - that <strong>of</strong> obtaining a better understandin-<br />
cf flmes 2nd flme-solid interactions, and secondly the possibilit:;,<br />
though remot?, <strong>of</strong> developing a new.method <strong>of</strong> gaslighting. The<br />
wwl: reported here was supported in part by the American Gas Association.<br />
Cm5nliuninescence has been known and studied for many years. It<br />
vas probably first reported by Balmain in 1842 (3) in his description<br />
<strong>of</strong> th? material boron nitride, which he was the first to prepare.<br />
Apparently the phenomenon vas not recognized with other materials until<br />
it 1uas reportej by Donau in 1913 (5). The subject was reviewed by .<br />
L. T. Xinchin in 1938 (8) and by E.C.W. Smith in 1941 (11).<br />
The materials that luminesce in flames share the characteristics<br />
<strong>of</strong> phospiizrs generally; that is, they are comprised <strong>of</strong> a colorless<br />
crystalline host or matrix material into which a small concentration<br />
<strong>of</strong> foreign atoms or ions, called the activator, is incorporated. The<br />
radiation is typically continuous and extends over only a small wave-<br />
length range, thus having a definite color. The color is usually<br />
characteristic <strong>of</strong>' the activating ion and can also be obtained with<br />
2tkr means <strong>of</strong> excitstion, such as ultraviolet light or cathode rays.<br />
?>:citation by a hydrogen flame has been used in most studies <strong>of</strong><br />
candolminescence, although Tiede and Buescher reported that, in ad- \<br />
dition to hydrogen, flames <strong>of</strong> ethyl alcohol, hydrogen sulfide, and<br />
carbon disulfide excited the luminescence <strong>of</strong> boron nitride (14). EXCI- 1<br />
tation by the flame <strong>of</strong> city gas has also been reported (9).<br />
The existence <strong>of</strong> the phenomenon at relatively low temperatures has<br />
not been seriously questioned. The temperature range here is from<br />
the temperature attained when a flame impinges on a thin film <strong>of</strong> phosnhor<br />
spread on a cool metal support up to a bright red heat. This<br />
upper limit is based on the observation <strong>of</strong> Tiede and Buescher (14)<br />
that blue luminescence occurred when a flame touched boron nitride in<br />
a carbon boat heated electrically to redness.<br />
E. L. Nichols (10) claimed that at much higher temperatures he had<br />
rsbserved light outputs from materials heated in an oxyhydrogen flame<br />
that :.rere several times greater than the light output from a blackbody<br />
at thc- same temperature. Apparently this work was inspired by the<br />
,<br />
\<br />
1<br />
I<br />
L
limelight which was produced bG4keating a cylinder <strong>of</strong> lime with an<br />
oxyhydrogen flame. The fact that a fresh portion <strong>of</strong> the lime had to<br />
be brought to the flame from time to time was interpreted to mean<br />
that somethm other than thema1 radiation was inmlved (8).<br />
Neunhoeffer (3) reports that Lhe natural chalks from which the lime<br />
was Prepared contain rare earths. If this were known to Nichols, it<br />
would no doubt have suggested to him that the rare earths were activators<br />
in a luminescence process. However, the work <strong>of</strong> later investigators<br />
indicated that the limelight was similar to the light <strong>of</strong> the<br />
Welsbach mantle in that both are only excited thermalky.<br />
Nichols applied his "Dhosphors" on ceramic adjacent to a uraniumoxide-coated<br />
8rea. and heated both areas evenly with an oxyhydrogen<br />
flame. Observations were made <strong>of</strong> both areas with an optical pyrometer;<br />
the uranium oxide vas taken to behave essentially as a blackbody and<br />
to have the same temperature as the companion material. Many mixtures<br />
Of colorless oxides uith small amounts <strong>of</strong> rare earths, or with some<br />
other elements that were also regarded as activators, had a greater<br />
emittance than uranium oxide. E.C.W. Smith (11) repeated some <strong>of</strong><br />
Nichols's experiments and obtained similar emittance readings. He<br />
then proceeded to determine or approximate the actual temperature <strong>of</strong><br />
the two coatings - a matter <strong>of</strong> some difficulty. By means <strong>of</strong> thermocouples<br />
made <strong>of</strong> extremely fine wires, he was able to show lwge differences<br />
in the temperatures <strong>of</strong> the two coatings - differences suffi- *<br />
cient to conclude that at incandescence temperatures there was no<br />
emission other than thermal. This conclusion has been confirmed by<br />
Sokolov and his eo-workers (12), (13).<br />
In hydrogen flames, at least, recombination <strong>of</strong> hydrogen atoms is I<br />
generally accepted as the major source <strong>of</strong> energy for the low-temperature<br />
excitation (1) , (7); earlier Donau (5) and Nichols (10 considered<br />
an oxidation-reduction mechanism, and Neunhoef fer (9 1 thought<br />
that electrons in the flame were responsible. Smith, and Arthur and<br />
Townend (l), (2) investigated this question with phosphors composed<br />
<strong>of</strong> calci1.m oxide activated with manganese, antimony, or bismuth. They<br />
found that:<br />
1. Activated oxides that luminesce in hydrogen flames also luminesce<br />
when subjected to the action <strong>of</strong> hydrogen atoms produced in an electrical<br />
discharge. When subjected to the latter action and heated<br />
electrically, the color <strong>of</strong> the luminescence approached the color<br />
produced in the flame. Presumably, the temperature <strong>of</strong> the heated<br />
oxides approached the temperature <strong>of</strong> oxides in the flame.<br />
2. The intensity <strong>of</strong> hydrogen flame luminescence increases when the<br />
hydrogen is burned under reduced pressure. This effect was presumed<br />
to be caused by a decrease in the number <strong>of</strong> three-body<br />
collisions. These collisions remove hydrogen atoms by recombination.<br />
3. The luminescence is strongest in hydrogen, erratic and very faint<br />
in town gas, and absent in carbon monoxide flames. It is also<br />
absent in methane and ethylene flames tested at pressures from<br />
atmospheric to 10 em Hg.<br />
4. The luminescence is extinguished when small amounts <strong>of</strong> hydrocarbon<br />
gases or vapors are added to the hydrogen.
-4:c~rciiny to recent studies o ;i42 flame mechanisms, hydrogen atoms are<br />
m>zs?Iit i_n the methane-air flame as well as in the hydrogen flame (15).<br />
This it aypna!-eC! reasonable to search for phosphors that candoluminesce<br />
in th3 :?,t!isnL7-air flame. This search and the study <strong>of</strong> the characteristl-s<br />
<strong>of</strong> this phmonenon were the objectives <strong>of</strong> our investigation.<br />
?.!-I; c pial s<br />
R-rc zarthc: vere obtaineci as ?..?.'?:I pure oxides from Lindsay i<br />
Ci?c.:-:iccl Pivision <strong>of</strong> Amrican lotash and Chemical Corporation. Other<br />
zhmi. :?-ls vert rcsearch grade.<br />
Ca1ci.w OxiSe Phosphors<br />
I Calcium and rare .earth nitrate solutions were prepared and mixed<br />
in the required amounts. The mixed nitrate solution was slowly poured<br />
intc hot mionium carbonate solution with stirring. The precipitate<br />
vas Cilt:?md, mshed vitli hot ammonium carbonate solution, dried at<br />
13C)°C, and ignited at 900°C for 30 minutes. -The pure rare earth oxides<br />
:wr? prcpared by precipitation from the nitrate in the same manner. !<br />
Yttrium Europium Tunastate - (YO. ~Euo. I)PO~WO~ - and Gadolinium<br />
Europ.i.um Molybdate - ( Gdo. &uo. 1 >Os. Mooa<br />
T!ie required amounts <strong>of</strong> the oxides (tungstic acid in the case <strong>of</strong><br />
tjqsten) wme mixed, heated in a platinum crucible at 1000°C for 2<br />
hours, :-round with mortar and pestle, and reheated at 1000°C for 2<br />
hours. The X-ray patterns <strong>of</strong> the two-products showed that in both a<br />
new phase had been .formed.<br />
Screcninp; Tests<br />
I<br />
Mounting strips about 2 cm wide and 10 em long were made <strong>of</strong> insulating<br />
fire brick and <strong>of</strong> copper sheet. Phosphor powder, thick enough to hide '<br />
the surface <strong>of</strong> the strip, was pressed on to it with a spatula. The '<br />
nearl:i vertical face <strong>of</strong> the strip was observed as it was passed through<br />
a methane-air flame (bunsen burner) and a hydrogen diffusion flame in<br />
a dark room. (<br />
Spectra<br />
Thrne different methods <strong>of</strong> mounting the phosphors were used in<br />
obtainiw the spectra. When we wished to observe the effect <strong>of</strong> temperature,<br />
the phosphor was applied as a paste to a silicon carbide rod.<br />
Pastes made with monomethyl ester <strong>of</strong> ethylene glycol seemed to adhere<br />
after drying somewhat better than those made with other liquids. The<br />
silicon carbide rod, about 6 mm in diameter, was held between two<br />
vater-cooled electrical terminals to allow the electrical current<br />
passing between the terminals to heat the silicon carbide rod and the<br />
phosphor.<br />
iYhen hydrogen was used as a fuel, the phosphor could be coated dry<br />
on the fritted disk <strong>of</strong> a Gas-dispersion tube. The fritted disk was<br />
operated as a porous-plat€ burner (Figure 1) without p-imary air.<br />
Knoiin ai-,o~ints <strong>of</strong> other gases could be added to the hydrogen.<br />
I<br />
I<br />
\<br />
I<br />
,(<br />
I<br />
1<br />
I<br />
b
I<br />
I<br />
1<br />
543 - ,<br />
The phosphor ijas coated on a water-cooled eo r plate when a<br />
methane-air flame was to be used. The plate was 1 in. square x l/$ in.<br />
thick wit!i trro l/k-in. copper tubes soldered to the back for cooling<br />
water. Plating the face with silver was found to be necessary to prevent<br />
slow Poisoiling <strong>of</strong> the phosphor by the copper. The phosphor vas<br />
settled onto the plate from a water suspension. The plate was mounted<br />
facing upwards at about 45' to the horizontal and the flame <strong>of</strong> a<br />
1 <strong>National</strong> Welding Equipment Go. Type-3A blowpipe was d3,rected downward<br />
jonto it.<br />
I A Beckmail DK-2 spectrophotometer with an IP 28 photomultiplier was<br />
used to record spectra. The backplate <strong>of</strong> the lamp housing was removed<br />
to al1o:s direct entrance <strong>of</strong> the radiation from the excited phosphor<br />
into the spectrophotometer.<br />
A slit opening <strong>of</strong> 0.4 mm without an aux-<br />
iliary light-gathering system gave sufficient enera. Varying light<br />
intensity Prom the flickering <strong>of</strong> the flame was troublesome and made it<br />
necessary to use the highest time constant and the slowest recording<br />
rete <strong>of</strong> the instrument. The porous-plate burner gave the steadiest<br />
radiation.<br />
RZSULTS<br />
Screening tests to discover phosphors that candoluminesce in methaneair<br />
flames or in hydrogen flames were run on a number <strong>of</strong> commercial<br />
phosphors and on others prepared in our laboratories. The latter were<br />
principally rare earths - pure and in calcium oxide. The phosphors<br />
were spread on fire-brick slabs and on copper strips - the latter to<br />
hold down the temperature <strong>of</strong> the phosphor at least momentarily - and ,<br />
tested by impi,agement in both hydrogen and methane-air flames. Sev-<br />
, eral phosphors that candolumenesce in both flames irere found. Detailed<br />
results are shown in Tables 1 and 2. These results should be regarded<br />
as tentatlve rather than conclusive.<br />
A few additional phosphors not listed in the tables were tested.<br />
' Three General Electric silver-activated zinc sulfides (NOS. 118-2-3,<br />
118-2-11, and 118-3-1) showed emission in the blue. Their spectra,<br />
abtained with a hydrogen diffusion flame on a fritted glass disk,<br />
showed that the emission was the band spectrum <strong>of</strong> S2 and that no cando-<br />
)luminescence was present (6). The S2 was undoubtedly formed by decom-<br />
position <strong>of</strong> the phosphor.<br />
Zinc sulfide activated by silver and cop er<br />
showed both candoluminescence and the band spectrum <strong>of</strong> S2 (Figure 27.<br />
A boron nitride sample from Carborundum' Co., Electronics Division<br />
showed weak, green luminescence in hydrogen and methane flames. Two<br />
recently developed rare earth phosphors, (YO. s EUO. 1)203-W03 and<br />
( Gdo. eEuo. 2) 203- MOOS were prepared and tested (4) . Both luminesced red<br />
in hydrogen and metharle flames on the fire-brick strip.<br />
,<br />
Light emission in some <strong>of</strong> the screening tests may have had a source<br />
other than candoluminescence. The emission from magnesium germanate<br />
,was probably thermal. This is indicated by the color <strong>of</strong> the emission<br />
(vhite to yellowish white), by the absence <strong>of</strong> a maximum <strong>of</strong> emission<br />
intensity vith increasing temperature <strong>of</strong> the phosphor, and by equal<br />
mission \?hen the phosphor is heated to the same temperature with or<br />
eithout the flame. For this test, the phosphor was mounted on the<br />
electrically neated silicon carbide rod, and temperature equivalence<br />
'was indicated by the emission <strong>of</strong> the phosphor at 2.25 microns. Light<br />
J<br />
!<br />
I
544<br />
Table 1. LIGHT EMISSION OF Y"TRIUM OXIDE AND RARE EARTHS (Pure and<br />
in CaO) IN HYDROGEN AND METHANE-AIR FLAMES<br />
Element<br />
Yttrium<br />
hnthanm.<br />
Neodymium<br />
Samarium<br />
GadOlinlum<br />
Dyspmsium<br />
Holmium<br />
Erbium<br />
Ytterbium<br />
- Fure oxide.<br />
-<br />
Atomic<br />
No. a<br />
39 Y<br />
57 la<br />
59 Pr<br />
60 Nd<br />
62 9m<br />
64 cd<br />
66 Dy<br />
67 H@<br />
68 Er<br />
70 Yb<br />
Metal<br />
Conc<br />
in<br />
CaO.<br />
vtF<br />
0.2<br />
1.0<br />
Po'<br />
0.2<br />
1.0<br />
PO<br />
. 0.2'<br />
1.0<br />
PO<br />
0.2<br />
1.0<br />
Po<br />
0.2<br />
1.0<br />
PO<br />
0.2<br />
1.0<br />
PO.<br />
0.2<br />
1.0<br />
PO<br />
0.2<br />
1.0<br />
Po<br />
0.2<br />
1.0<br />
Po<br />
0.2<br />
1.0<br />
PO<br />
ht msslon<br />
3ith €L LU W-<br />
On<br />
cu<br />
Trace<br />
Trace<br />
Weak<br />
Trace<br />
Trace<br />
Weak<br />
Trace<br />
Trace<br />
Nil<br />
Trace<br />
Trace<br />
Nil<br />
Trace<br />
Trace<br />
Nil<br />
Trace<br />
Trace<br />
Strong<br />
Trace<br />
Trace<br />
Nil<br />
Trace<br />
Trace<br />
Nil<br />
Trace<br />
Trace<br />
Nil<br />
Trace<br />
Trace<br />
Nil<br />
On<br />
Fire<br />
?M&<br />
Weak<br />
Trace<br />
Trace<br />
Weak<br />
Weak<br />
Trace<br />
Stmng<br />
Strong<br />
Trace<br />
Trace<br />
Weak<br />
Trace<br />
Trace<br />
Trace<br />
Nil<br />
Trace<br />
Weak<br />
Strong<br />
Trace<br />
Weak<br />
Nil<br />
Trace<br />
Weak<br />
Trace<br />
Trace<br />
Weak<br />
Trace<br />
Trace<br />
Trace<br />
Nil<br />
on<br />
cu<br />
Nil<br />
Nil<br />
Trace<br />
Nil<br />
Nil<br />
Nil<br />
Nil<br />
Trace<br />
Nil .<br />
Nil<br />
Nil<br />
Nil<br />
Trace<br />
Nil<br />
Nil<br />
Nil<br />
Nil<br />
Trace<br />
Nil<br />
Nil<br />
Nil<br />
Nil<br />
Nil<br />
Nil<br />
Nil<br />
Nil<br />
Nll<br />
Nil<br />
Nil<br />
Nil<br />
- r<br />
on<br />
Fire<br />
m<br />
Nil<br />
3:<br />
Nil<br />
Nil<br />
Nil<br />
Strong<br />
strong<br />
Nil<br />
Nil<br />
Nil<br />
NiI<br />
Trace<br />
Trace<br />
Nil<br />
Nil<br />
Nil<br />
Weak<br />
Nil<br />
Nil<br />
Nil<br />
Nil<br />
Nil<br />
Trace<br />
Nil<br />
Nil<br />
Nil<br />
Nil<br />
Trace<br />
Nil<br />
Color<br />
Violet !<br />
I<br />
Violet<br />
Green<br />
and<br />
Orange<br />
Lavender 4<br />
lavender<br />
Green<br />
-and<br />
Orange I<br />
Red<br />
Red I<br />
Red<br />
Violet<br />
Lavender<br />
Red<br />
Oee<br />
orange<br />
--<br />
Violet<br />
Violet<br />
Red<br />
Lavender<br />
Blue<br />
--<br />
Aqua to Blue<br />
Aqua<br />
Red<br />
Blue<br />
White<br />
Red<br />
Lavender<br />
Red<br />
--<br />
I<br />
!<br />
I<br />
i<br />
I<br />
1
\<br />
'i<br />
2<br />
w<br />
8<br />
?N4"?? I? IC9<br />
t-OO+r(N 0 A N<br />
545
emission in a few cases may be by decomposition <strong>of</strong> the phosphor,<br />
es vas shom in the case <strong>of</strong> the silver-activated zinc sulfides.<br />
The manganese-activated zinc phosphate (Sylvania No. 151) phosphor<br />
appeared to give the most intense luminescence emission in methane-air<br />
flames <strong>of</strong> my tested, and, accordingly, its light emission was investi-<br />
-<br />
aated further. \Then the phosphor was coated on a fritted disk and the<br />
Eisk used as e porous-plate burner, a steady emission was obtained with<br />
a hyerocen dif'fusion f'lame. However, no emission could be obtained in<br />
this manner vhm the fritted-disk burner was fed with Qure methane or<br />
vith a --thane-air mixture. Only when the phosphor was brought into a<br />
i3dnsnn flair.? -;:as the light emission observed. We do not know the cause<br />
sf this effsct. A greater concentration <strong>of</strong> hydrogen atoms in the hydro-<br />
play a. role. The high diffusivity and other properties<br />
hz't zause its flame to burn closer to the solid surface<br />
rL or greate? importance.<br />
Sssctre recor2ed with the Beclanan DK-2 spectrophoto.meter were used<br />
to' zonpare the light emission <strong>of</strong> this phosphor in the hydrogen flame<br />
vith its emission in the methane-air flame. The phosphor was coated<br />
on 2 silicon carbide rod for this experiment. No significant differ- ,<br />
ence other than that <strong>of</strong> the emission from the flame itself was observed<br />
('izurc, 3 ) . I<br />
T!ie effect <strong>of</strong> temperature on the light emission <strong>of</strong> the phosphor was<br />
i nvasti.?at?5 by electrically heating the silicon carbide rod. With a<br />
nethanerair flame impinging .on the phosphor-coated rod, and with the<br />
spectrophotometer focused on the rod near one <strong>of</strong> its water-cooled ends,<br />
the Li&t emission increased to a maximum with an increase in electrical .<br />
hGatin5, then decreased to near extinction with further heating. This<br />
agrees with the observation <strong>of</strong> Neunhoeffer (9) that the hydrogen flame.<br />
candoluminescence <strong>of</strong> calcium oxide impregnated with various activators '<br />
eshibit s temperature maxima.<br />
When'the phosphor was heated to a still higher temperature, thermal<br />
emission appeared. When the emission <strong>of</strong> the phosphor was corrected<br />
'i<br />
for the emission <strong>of</strong> the flame, no essential difference in emission at<br />
high temperatures with and without the flame was evident. These experi- .'<br />
ments indicated that the low-temperature light emission experienced<br />
with this. phosphor is candoluminescence, but that candoluminescence is<br />
not involved in the high-temperature emission.<br />
I<br />
This phosphor was'the only one <strong>of</strong> several tested that luminesced in<br />
E carbon monoxide flame. The question <strong>of</strong> excitation in this flame by<br />
high-energ;; species other than hydrogen atoms was not pursued.<br />
Spectra <strong>of</strong> the luminesccnces from several phosphors excited by<br />
hydrogen burning on the surface <strong>of</strong> a sintered glass disk were obtained, '<br />
a& the effect <strong>of</strong> the addition <strong>of</strong> small amounts <strong>of</strong> carbon monoxide and<br />
methan? to the fuel was observed. The.phosphors were calcium silicate '<br />
aztivatec with lead and manganese (Sylvania No. 290), calcium oxide ,<br />
activated vith ytterbium, calcium oxide activated with praseodymium,<br />
and salcim oxide activated with samarium. The spectra obtained from<br />
the f5rst three <strong>of</strong> these phosphors when they are excited with pure<br />
hj-d?Ggen burning on a fritted disk are shown in Figure 4. The Sylvania 1,<br />
KG. 24.9 phosphor emits with a broad peak in the green. Praseodymiumecti:retc.c<br />
cal2im oxide shows two peaks - one at 595 millimicrons /<br />
(p110:.1) and a weaker one at 490 millimicrons (blue-green). Ytterbium- 1<br />
i
I<br />
i<br />
t<br />
\<br />
I/'<br />
t<br />
I<br />
activated calcium oxide also sh&J two peaks - one at 375 millimicrons,<br />
which is in the ultraviolet. Samarium-activated calcium oxide also<br />
emits with a similar peak in the ultraviolet and another at 570 ,<br />
mllllmicrons (yellow), as shown in Figure 5.<br />
The effect <strong>of</strong> the addition <strong>of</strong> small amounts (1 to 10 volume percent)<br />
Of carbon monoxide or methane to the hydrogen burning on the fritted<br />
disk was different for each <strong>of</strong> these phosphors. Luminescence <strong>of</strong> the<br />
Sylvania No. 290 phosphor was substanitally unchanged with addition <strong>of</strong><br />
as much as 105 <strong>of</strong> the carbon monoxide or methane. Lwipescence <strong>of</strong> the<br />
Praseodymium-activated calcium oxide was not greatly affected by addition<br />
<strong>of</strong> 1" <strong>of</strong> the carbon monoxide or methane, but decreased rapidly<br />
with greater amounts.<br />
obtained with 62 carbon monoxide or 846 methane. The luminescence <strong>of</strong><br />
the ytterbium-activated calcium oxide was even more strongly quenched<br />
by the addition <strong>of</strong> carbon monoxide or methane. With these phosphors,<br />
only the intensity <strong>of</strong> the luminescence was affected.<br />
With samarium-activated calcium oxide, the spectral distribution <strong>of</strong><br />
the luminescence was also affected, as shown in Figure 5. Upon the<br />
addition <strong>of</strong> carbon monoxide, emission by this phosphor in both wavelength<br />
regions decreased in intensity, and, at about 3% carbon monoxide,<br />
emission in the 375-millimicron region had virtually disappeared. The<br />
570-millimicron peak, however, was replaced by two narrower emission<br />
bands with peaks at about 560 and 595 millimicrons. With further addition<br />
<strong>of</strong> carbon monoxide the intensity <strong>of</strong> this emission increased to a<br />
maximum several times more intense than the original peak emission,<br />
then decreased. These effects were not observed upon the addition <strong>of</strong><br />
methane to the flame. The variation <strong>of</strong> the intensity <strong>of</strong> the luminescence<br />
Fn the two cases is shown in Figure 6.<br />
At a late stage <strong>of</strong> the work, phosphors were deposited by settling<br />
from a water suspension onto a water-cooled copper plate. A few spectra<br />
were obtained in this way with methane-air excitation.<br />
Ehhancement <strong>of</strong> the luminescence from the manganese-activated zinc<br />
silicate (Sylvania No. 161) by the cooling <strong>of</strong> the copper plate was very<br />
pronounced. (Figure 7 ) .<br />
ACKNOWLEDGI4ENT<br />
Only about 10% <strong>of</strong> the original intensity was<br />
The author wishes to thank the American Gas Association for their<br />
support and for permission to publish these results. John Hasenberg<br />
did most <strong>of</strong> the experimental work. Peter Mrdjin identified the S2<br />
bands in the spectra <strong>of</strong> the zinc sulfide phosphors.<br />
. .<br />
. .
LITERATUR2 CITED<br />
i.<br />
C.<br />
-.<br />
.' *<br />
il.<br />
5.<br />
'2 .<br />
7.<br />
8.<br />
9.<br />
10.<br />
11.<br />
12.<br />
13.<br />
14.<br />
543<br />
Arthr, J. R. and Townend, D.T.A., "Fundamental Aspects 0;<br />
Sombustion - Some Reactions <strong>of</strong> Atomic Hydrogen in Flames,<br />
J. Inst. FLel 2-0, 44-51, 61 (1953) July.<br />
t rtnur, J., R. and Ttwnend, D.T.A., "Some Reactions <strong>of</strong> Atomic<br />
Hydrogen in Flames, in U.S. Natl. Bur. Standards, Circ. 523,<br />
"Znergy Transfer in Hot Gases, ' I 99-110. Washington, D.C. :<br />
.;ovt. Print. Office, 1954. I \<br />
Bslrrain, W. H., ,"Observations on the Formation <strong>of</strong> CEmpounds <strong>of</strong><br />
Boron ana Sllicon With Nitrogen and Certain Metals, Phil. Mag. 2l,<br />
2-3-7, (1342).<br />
Borchardt, H. J., "Rare Earth Phosphors, J. Chem. Phys. 2, 1251<br />
(-333) March 1.<br />
Pcnau, J., "New Kind <strong>of</strong> Luminescence <strong>of</strong> Alkaline Earth Preparation,<br />
Especially Calcium Preparations Which Contain Bismuth or Manganese<br />
Brought Out by the Hy$rogen Flame; as Well as the Detection <strong>of</strong> .<br />
Traces <strong>of</strong> the Latter, Monatsh. 2, 949-55 (1913).<br />
Gaydon, A. G., The Spectroscopy <strong>of</strong> Flames, 244. New York: John<br />
liiley, 1957.<br />
Gorban, A. N. and Sokolov, V. A., "On the Question <strong>of</strong> the Physico-<br />
Chemical Nature <strong>of</strong> Candoluminescence,'" Opt. Spectr. (U. S. S. R. )<br />
Eixlish Trans;. 1, 1C:-65 (1959) August.<br />
Minchin, L. T., "Luminescence <strong>of</strong> Oxides Under Flame Excitation, I'<br />
Trans. Farada SOC. 35, 11163-70 (1939) ; "Luminescence <strong>of</strong> Substances<br />
'Under Flame Gcitation, Trans. Faraday SOC. 2, 505-06 (1940).<br />
Neunhoeffer, O., "The Hydrogen Flame as a Source <strong>of</strong> Electrons, 'I<br />
Z. Physik. Chem. x, 179-85 (1951).<br />
Nichols, E. L., Howes, H. L. and Wilber, D. T., "CF)thodo-lumines-<br />
cence and the Luminescence <strong>of</strong> Incandescent Solids, Carnepie Inst.<br />
Pubi. No. 348 (1928).<br />
Smith, E. C. W. . "The Radiation From Solid Substances Under Flame<br />
Sokolov. V. A., "Candoluminescence From the Viewpoint <strong>of</strong> the<br />
Vavilox-Wiedemann Criterion and <strong>of</strong> Contemporary Physico<strong>chemical</strong><br />
Ideas, Izv. Akad. Nauk. U.S.S.R., Ser. Fiz. - 26, 514-17 (1962).<br />
Sokolov, V. A., Grozina, I. S. and $orban, A. N., "The Nature <strong>of</strong><br />
Candoluminescence <strong>of</strong> Calcium Oxide, Izv. Tomskogo Polite-. Inst.<br />
42, 253-56 (1958).<br />
Tiede, E. and Buescher, F., "Inorganic Luminescent Phenomena. 11.<br />
Luminous Boron Ni5ride (Balmain's Aethogen and the Flame Excitation 1<br />
<strong>of</strong> Luminescence), & a, 2206-14 (1920).<br />
<<br />
'<br />
i<br />
I
I<br />
15. ',Jise, H. and Rosser, W. ,,A., "Homogeneous and Heterogeneous Heactions<br />
<strong>of</strong> Fh~e Intermediates, in Ninth Symposium (International ) On<br />
Cmbustion, 733-42. New York: Academic Press, 19b3.
co, CH,,OR 1<br />
-<br />
OTHER ADDITIVE<br />
FRITTED<br />
1 GLASS<br />
+ DISK<br />
\LIGHT BEAM<br />
\ SPECTROPHOTOMETER<br />
L-KE-1<br />
' PHOSPHOR<br />
BURNER<br />
DETAIL 1-4121<br />
:-'L :.:?? 1. SCHEATIC REPRESENTATION OF THE, POEicJUS BURNER<br />
CPPAR?TUS AWR LIGHT ENISSIOIi STUDIES<br />
?:pire 2. Ei41SSION SPECTRUM OF ZnS: Cu:Ag EXCITED<br />
' EY A HYDROGEN DIFFUSION FLAME
B 549<br />
15- '-Jise, H. and Rosser, W. llA., "Homogeneous and Heterogeneous Reactions<br />
Of Flame Intermediates, in Ninth Symposium (International) on<br />
Canbustion, 733-42. New York: Academic Press, 1963.<br />
I
FRIT 'TED<br />
CO, CH,.OR 4<br />
___<br />
OTHER ADDITIVE<br />
i<br />
@)<br />
FUEL<br />
\ PHOSPHOR<br />
BURNER<br />
DETAIL A-b?Zl<br />
Z..;.:T? 1. SCHIGiATIC REPRESENTATION OF THE PGROUS BURNER<br />
."-PPAR3.TUS FOR LIGW E31ISSIGTJ STUDIES<br />
?:;Lire 7. Ei4ISSION SPECTRUM OF ZnS: Cu:Ag EXCITED<br />
' BY 4. HYDROGEN DIFFUSION F W
I<br />
0<br />
I I I I 1 I I I I 0 R<br />
2gDOc- 0 8 8 : . F l O 0 N - 0 0<br />
(3SNOdS3U 1133010Hd ljOj 03133kJU03Nn)<br />
NOllVlClVU 03111W3 30 AlISN31NI 3A11Wl3U<br />
8<br />
P<br />
0<br />
al IC)<br />
0<br />
a R
340 350 300 400 4 50 500 600 700 800 900<br />
WAVELENGTH, MILLIMICRONS 1 e.’<br />
Figure 4. EMISSION SPECTRA OF PHOSPHORS EXCITED BY A<br />
HYDROGEN DIFFUSION FLAME<br />
\<br />
300 400 450<br />
A -A<br />
WAVELENGTH, MILLIMICRONS * .e<br />
Figure 5. EFFWT OF CARBON MONOXIDE ON THE EMISSION<br />
SPECTRUM OF SAMARIUM-ACTIVATEE CALCIUM OXIDE
1<br />
2.2<br />
2.0<br />
0.8<br />
0.6<br />
0.4<br />
553<br />
0 2 4 . 6 8 IO<br />
CO OR CH, CONTENT, VOLUME K A-4771<br />
Figure 6. EFFECT OF FUEL COMPOSITION ON THE CANDOLUMINESCENCE<br />
OF SAMARIUM-ACTIVATED CALCIUM OXIDE<br />
. .
0<br />
554<br />
WITH COOLING<br />
(PLATE AT 40°C)<br />
COOLING<br />
4500 5000 6000<br />
WAVELENGTH, A A-4973<br />
Figure 7. THE EFFECT OF TEMPFBATUFE ON THE E3:ISSION SPECTRUM<br />
OF SYLVANIA NO. 161 PHOSPHOR EXCITED BY<br />
A METHANE-AIR FLcuvlE<br />
i<br />
I<br />
1<br />
1<br />
‘I<br />
I<br />
\<br />
I 1<br />
1<br />
\
555<br />
A RAPID, SIMBLE METhOD FOR THE DETERNINATION<br />
OF THE THERMAL CONDUCTIVITY OF SOLIDS<br />
Neal D. Smith<br />
Fay Fun<br />
Robert M. Visokey<br />
UNITED STATES STEEL CORPORATION<br />
Research Center I<br />
I' Monroeville, Pennsylvania<br />
I<br />
I<br />
I<br />
The rational design <strong>of</strong> equipment such as shaft coolers, heaters,<br />
I and rotary kilns for the heating and cooling <strong>of</strong> solids requires that<br />
the thermal properties <strong>of</strong> the solids be known. Thermal conductivity is<br />
one <strong>of</strong> these properties that to measure necessitates elaborate equipment<br />
I and tiqe-consuming techniques.<br />
A rapid, simple method has been developed €or determining the<br />
thermal conductivity <strong>of</strong> solids.' The solids can be either porous or non-<br />
porous and <strong>of</strong> either high or low conductivity. If high-conductivity<br />
, materials are tested, then both the thermal conductivity and heat capa-<br />
I city caq be simultaneously measured by the method.<br />
The procedure involves preparing a cylindrical briquette <strong>of</strong> the<br />
.test solid th'at has a thermocouple located in the center. This bril<br />
quette is heated to a constant temperature after which it is suspended<br />
' in an open-end glass tube and cooled by a known flow <strong>of</strong> nitrogen or any<br />
other nonreactive gas. The thermal conductivity is then computed from<br />
(.a digital computer comparison <strong>of</strong> the cooling curves for the test solid<br />
' versus a reference solid <strong>of</strong> known thermal properties and similar size<br />
1 that has undergone the same heating and cooling cycle. The method was<br />
validated by using the known thermal properties <strong>of</strong> lead, aluminum, and<br />
silver and computing the theoretical cooling curves. The theoretical<br />
, curves were in close agreement with the experimentally measured cooling<br />
curves for these materials.<br />
' marized as follows.<br />
Theory<br />
I The mathematical.basis for determining thermal conductivity by<br />
the described method is discussed in a paper by Newman') and is sum-<br />
Consider a cylindrical briquette as shown in Figure<br />
( 1. The differential equation for unsteady state heat transfer by conduction<br />
in the x-direction is (see nomenclature for definition <strong>of</strong> the vari-<br />
1 .<br />
1 For a briquette <strong>of</strong> thickness Za, the central plane being at x = 0 and<br />
) assuming:<br />
i<br />
' 1) uniform temperature at the start <strong>of</strong> cooling <strong>of</strong> the initially hot<br />
1 briquette
5 56<br />
then t = to when e = 0<br />
2) the final temperature <strong>of</strong> the briquette will be the temperature <strong>of</strong><br />
the surroundings : I<br />
therefore t = t, when e = *go<br />
3) there is no heat flow across the central plane because <strong>of</strong> symmetry: {<br />
I<br />
consequently -k (2;) - = 0 at x 0<br />
The heat balance on the briquette surface is made by equating heattrans-<br />
ferred to the surface by conduction with heat transferred from the surfac<br />
by convect&,on.. In differential form, the heat balance is: '<br />
-k dt = h (t - t,) at x = +a<br />
. (a4 %<br />
Newman') showed that the solution to Equations (1) through<br />
in terms <strong>of</strong> a dimensionless temperature ratio Yx is:<br />
ma<br />
(I+$ ma2 +ma) cospn<br />
where An= and<br />
pn are defined as the first, second, third, etc., roots <strong>of</strong><br />
cendental equation:<br />
Pn TAN pn - I /ma = 0<br />
The surface to solid thermal resistahce ratio, ma,<br />
ma = k/ha<br />
and Xa is defined as: xq = ae/a2<br />
where the thermal diffusivity is: a = k/p Cp<br />
(5)<br />
(5) expressed<br />
the trans-<br />
Similarily , considering radial heat transfer , the .radial briquettl<br />
heat balance is<br />
The initial condition equation is:<br />
toto WHEN' 8=0<br />
The final temperature equation is:<br />
t= 1, WHEN 8. -<br />
The boundary condition equations are:<br />
-k (g)=O AT r=O<br />
(7)<br />
(12) i
557<br />
and -k (%)= h (t-ts) AT f =R<br />
Solving Equations (11) through (15) gives:<br />
and Pn ar,e the first, second, third, etc., roots <strong>of</strong> the equation:<br />
and . .<br />
PnJI(fin) - I/mr Jo(Pn) =.O<br />
The surface to solid thermal resistance ratio, mr is<br />
I x, = a8/R2<br />
~ and<br />
is :<br />
(18)<br />
frIr = k/hR (19 1<br />
The complete differential equation for the case shown in Fig. 1<br />
the solution to Equation (21) is:<br />
, eqn. 22 becomes: . .<br />
I<br />
If the center temperature defined at r = 0, x = 0 is tc, then<br />
tC’+S<br />
Yc= - = Yr<br />
to ’tS<br />
Y,<br />
where y,<br />
1<br />
and YX are evaluated at r = 0 and x = 0.<br />
i The preceding mathematical analysis shows that the rate <strong>of</strong> cooling,<br />
lor change in center temperature for a cyclindrical briquette is a function<br />
I <strong>of</strong>, time (8 ) , density ( e) , thermal conductivity (k) , the surface heat<br />
) transfer coefficient (h) , specific heat (C,) and the briquette dimensions<br />
I as expressed by Equation (23).<br />
The experimental technique can now be described in terms <strong>of</strong> th;<br />
~p~vious discussion. If the change in center temperature with time is Rsa-<br />
,sued experimentally for a material <strong>of</strong> known thermal and physical prop-<br />
‘erties (standard briquette) , the surface heat transfer coefficient can be<br />
calculated from Equation (23), since it is the only unknown.<br />
(20)
5 58<br />
The surface heat transfer coefficient (h) is a function <strong>of</strong> the<br />
flow rate <strong>of</strong> the cooling gas and the geometry and size <strong>of</strong> the briquette.<br />
It is independent <strong>of</strong> all other physical, thermal, or <strong>chemical</strong> properties<br />
<strong>of</strong> the briquette. Therefore, any other briquette having similar dimensions<br />
and cooled at the same flow rate will have the same value for (h).<br />
Once (h) has been determined using the standard briquette, the<br />
thermal conductivity <strong>of</strong> any test material can be, determiped from Equa-<br />
. tion (23) since all other variables are known.<br />
A computor program has been written which through an iterative<br />
process determines the best value <strong>of</strong> (h) which makesthe calculated<br />
values for the dimensionless temperature ratio equal to the experimental<br />
values obtained when the standard briquette is cooled.<br />
With (h) deter&ned, another, computor program is run for the test<br />
specimen. Thermal conductivity is now the unknown variable and through<br />
another iterative scheme, the best value for (k) that makes the calcu-<br />
lated and experimental values for the temperature ratios equal is found.<br />
The input data for both programs consist <strong>of</strong> density, specific<br />
heat, time, briquette dimensions, and several experimental values for<br />
the temperature ratio. The output from the first: program (standard) is<br />
the best value for (h). Using this value for (h), the second program<br />
used to determine the k value €or any test material. If a highly conduc-<br />
tive material is tested, then it is possible to determine its heat capa-<br />
city since the solid thermal resistance will be small compared to the<br />
surface thermal resistance. A transient heat balance can be written for<br />
. the test solid cooling in a stream.<strong>of</strong> coolant gas.<br />
VpCp dt = hA (t - ts)<br />
de<br />
In the above equation, t = tc since the thermal gradient in the solid is<br />
'neglected. Integrating Equation (24) and using the dimensionless temperature<br />
ratio, Yc gives:<br />
Yc = exp(hA/pCpV) e (25)<br />
Thus, if the internal solid thermal resistance is neglible, a plot <strong>of</strong><br />
the experimental Yc versus e data on semilog paper should be linear as<br />
shown by Equation (25). The heat capacity, Cp, can be calculated from<br />
the slope <strong>of</strong> the line for Yc versus e since (h) is the same as for the<br />
standard briquette and the density, p, and total surface area, A, for<br />
the test material are also known.<br />
Materials and Experimental Work<br />
A primary advantage <strong>of</strong> the transient technique for determining<br />
thermal conductivities is the ease and swiftness with which the experi-<br />
ment can be conducted.<br />
In so far assunple preparation is concerned, any solid that Can<br />
be briquetted, cast, or fabricated around a centrally located rigid<br />
c
559<br />
thermocouple (x = 0; r = 0) may ue tested. Finished test sazple ciriin-<br />
ders should be approximately one inch in diameter, and one-haif inch ir.<br />
height; however, other dimensions can be used.<br />
Experimental Apparatus<br />
The experimental apparatus (see Figure 2) consists simply <strong>of</strong> a<br />
3-inch diameter glass tube approximately 3 feet in length. One end <strong>of</strong><br />
, the tube .is completely stoppered except for a one-half iAch circular<br />
opening through which the coolant gas flows. The other end <strong>of</strong> the tube<br />
I is open to the atmosphere. A small electric furnace is Lised to heat<br />
1 the briquette, and an automatic single point temperature recorder con-<br />
nected to the embedded thermocouple is used to measure the center temp-<br />
erature <strong>of</strong> the briquette.<br />
Experimental Procedure<br />
The experimental procedure is the same for both the standard and<br />
test briquettes. Either the standard (aluminum was chosen since its<br />
thermal properties are well established), or the test briquette is con-<br />
nected to the temperature recorder by way <strong>of</strong> the thermocouple lea&.<br />
The briquette is heated until the center temperature has reached a con-<br />
stant , predetermined value. The briquette is then quickly removed from<br />
1 the furnace and suspended in the cooling tube with the cooling gas flowing<br />
at a constant rate. The briquette is usually cooled to the temperature<br />
<strong>of</strong> the cooling gas within 20 minutes.<br />
Data Processing<br />
I<br />
i For the standard briquette , the experimental dimensionless temp-<br />
erature ratio versus time data points for the standard briquette along<br />
with the known thermal properties are used to calculate the surface co-<br />
t efficient, h, in the following manner. A digital computer program is<br />
’ written to compute Yc from Equations (6) through (231. By iteration<br />
. and assuming various values <strong>of</strong> (h), the computed values <strong>of</strong> Yc can be<br />
! made to converge on each <strong>of</strong> selected experimental Yc versus e data<br />
i points. Thus, for a selected data point, the best experimental (h) is<br />
that which when used in Equations (8) and (19) results in equal values<br />
for the computed and experimental Yc values.<br />
\<br />
t For low conductivity test materials, the same method is used to<br />
determine the best experimental value <strong>of</strong> k by using the h determined for<br />
\ the standard and the other properties <strong>of</strong> the test material. If the test<br />
1 material is a good conductor as discussed in the theory section, then<br />
t<br />
experience has shown that h should be computed from the experimental<br />
cooling curve and then this value is used tommpute k by the same method<br />
as for low conductivity test materials.
Discussion and Results<br />
560<br />
Three briquettes <strong>of</strong> aluminum, lead and silver were made to<br />
test the validity <strong>of</strong> the experimental technique since their thermal<br />
properties were available from the literature as shown in Table I.<br />
Sintered, dense hematite (Fe 0 ) and a briquette <strong>of</strong> porous carbon<br />
2.3 I<br />
made from a partially devolatized coal were used as test materials<br />
For these materials, all properties except the thermal Conductivities<br />
shown in Table I were previously measured. Cooling curves for each<br />
briquette were measured for a nitrogen flow rate <strong>of</strong> 0.9 scfm. Sur-<br />
face heat transfer coefficients for lead, silver and alumixC<br />
- vere<br />
calculated by the method discussed in the data processing section.<br />
For these materials , the llterature conductivity values were used to<br />
calculate the surface coefficient. Table I shows that the calculated<br />
or experiment@ h values for each metal are nearly identical.<br />
This result is consistent with the theoretical basis <strong>of</strong> the experiment<br />
and may be considered as establishing the validity <strong>of</strong> the method.<br />
Also as additional evidence, aluminum was choosen as the standard and<br />
k values for lead and silver were calculated using the h value for aluminum.<br />
Table I shows that the calculated or experimental k values were<br />
within 0.5 percent <strong>of</strong> the literature values. The conductivities for<br />
hematite and porous carbon were calculated using aluminum as the standard.<br />
Figure 3 shows the experimental data points with the solid lines ,<br />
calculated from the theory. Note that the line for the carbon is curved<br />
whereas those for the metals and hematite are linear. As discuss- '<br />
ed previously, a linear cooling curve is obtained if the surface to<br />
solid thermal resistance ratios are relatively large.<br />
Note that for the<br />
metals, lead which has the lowest conductivity and thermal diffusivity<br />
cooled the fastest. This result is explained by examination <strong>of</strong> eqn.<br />
(25) which shows that for similar gas flows and briquette dimensions,<br />
the rate <strong>of</strong> cooling for different materials is determined by the heat<br />
content, pC . It can be seen in Table I that the heat content for lead<br />
is the lowegt <strong>of</strong> all metals tested.<br />
. Summary<br />
A rapid, simple method for determining thermal conductivity for a solid<br />
has been developed. The solid can be either porous or non-porous and <strong>of</strong><br />
either high or low conductivity. If high conductivity materials are<br />
tested, then both conductivity and heat capacity can be simultaneously<br />
measured from one cooling experiment. The method was validated by using<br />
the known thermal properties <strong>of</strong> lead, aluminum, and silver and the<br />
experimental cooling curves in a comparsion with the computed results.<br />
References<br />
1- Newman, A. B., Industrial and Engineering Chemistry, Vol. 28, 1936, i<br />
pp. 545-548<br />
. .<br />
_. .-. - . . . .<br />
. .,<br />
. .<br />
I<br />
1<br />
i<br />
1<br />
I<br />
1<br />
1
1<br />
I<br />
A<br />
An<br />
''P<br />
'H<br />
I<br />
'h<br />
I<br />
k<br />
ma<br />
"r<br />
Q<br />
R<br />
r<br />
' S<br />
t<br />
/X<br />
tC<br />
tS<br />
xo<br />
I xr<br />
! yx<br />
yr<br />
( a<br />
I<br />
1 8<br />
I<br />
!<br />
J<br />
\<br />
*I<br />
e<br />
-<br />
551<br />
Nomenclature<br />
Half height <strong>of</strong> briquette; ft<br />
Area; ft2<br />
Coefficient in infinite series solution for temperature dis cri-<br />
bution in Briquette<br />
Specific heat; BTU/lb OF<br />
Overall heat transfer coefficient; BTU/hr ft2 OF<br />
Surface heat transfer coefficient; BTU/hr ft2 .OF<br />
Thermal conductivity; BTU/hr ft2 O F / f t<br />
Axial surface resistance; dimensionless<br />
Radial surface resistance; dimensionless<br />
Heat flux; BTU/hr<br />
Maximum radius <strong>of</strong> briquette; ft<br />
Radius <strong>of</strong> briquette; ft<br />
Half width <strong>of</strong> infinite plate; ft<br />
Temperature; OF<br />
Temperature at center <strong>of</strong> briquette; OF<br />
Initial temperature <strong>of</strong> briquette; OF<br />
Temperature <strong>of</strong> cooling gas; OF<br />
Distance <strong>of</strong> direction; ft<br />
- - a8 Dimensionless time parameter for axial component<br />
02<br />
= a8 Dimensionless time parameter for radial component<br />
r2<br />
= Symbol for temperature ratio, axial component; dimensionless<br />
= Symbol for temperature ratio, radial component; dimensionless<br />
=(k/eCp) Thermal diffusivity; ft2/hr<br />
= Time; minutes or hours<br />
= Density; wft3
__- ._<br />
562<br />
Table I<br />
THERMAL AND PHYSICAL PROPERTIES<br />
Aluminum Silver Lead<br />
a .01842 .02059 .01842<br />
R<br />
P<br />
cP<br />
PCP<br />
h (experimental)<br />
k (experimental)<br />
k (literature)<br />
a (experimental)<br />
.04208 .04210<br />
168.50 655.20<br />
.2273 -0578<br />
38.30 39.31<br />
5.58 5.70<br />
240.3<br />
121.7 240.0<br />
3.178* 6.113<br />
* Average <strong>of</strong> literature sources<br />
.04ii7<br />
707.43<br />
.0306<br />
21.65<br />
5.60<br />
18.99<br />
19.00<br />
.8770<br />
Hematite<br />
.01842<br />
.04208<br />
306 .OO<br />
-2090<br />
63.95<br />
5.58<br />
12.10<br />
none<br />
.1892<br />
.01958<br />
.Of121 1<br />
75.0<br />
.2360 I<br />
17.70<br />
5.58<br />
.0307<br />
none<br />
-00173<br />
/<br />
I
X<br />
563<br />
STANDARD CYLINDRICAL BRIQUETTE<br />
Figure 1
ln<br />
4<br />
I<br />
0<br />
4J<br />
v<br />
\<br />
1.0<br />
0.8<br />
0.6<br />
L<br />
-<br />
0.4 -<br />
0.3 -<br />
0.2 -<br />
-<br />
0.1<br />
-<br />
.08 -<br />
-<br />
f<br />
L M<br />
:ooling<br />
Tube LL<br />
Legend<br />
564<br />
1 Thermocouple<br />
I jl-. -2<br />
t<br />
rique tte<br />
hl<br />
EXPERIMENTAL APPARATUS<br />
0- Lead<br />
0- Silver<br />
0- Aluminum<br />
A- Porous Carbon Briquette '<br />
* - Sintered hematite<br />
Zlectri c<br />
Furnace<br />
I I I I I I I I I I<br />
I<br />
0 1<br />
2 3 4 5<br />
Time, 0, minutes<br />
, Theoretical Cooling Curves and Experimental<br />
Points for five Materials<br />
T - L<br />
c:::<br />
* I<br />
.<br />
stop<br />
Clock<br />
Figure 2<br />
Figuze 3