

Ab Initio Study of Silyloxonium Ions
Ab Initio Study of Silyloxonium Ions
Ab Initio Study of Silyloxonium Ions

Create successful ePaper yourself
Turn your PDF publications into a flip-book with our unique Google optimized e-Paper software.
<strong>Ab</strong> <strong>Initio</strong> <strong>Study</strong> <strong>of</strong> <strong>Silyloxonium</strong> <strong>Ions</strong> Organometallics, Vol. 16, No. 26, 1997 5947<br />
Table 5. Calculated Proton Affinities (6-31G*) <strong>of</strong> Model Oxygen Bases (Eq 13) (kcal/mol) a<br />
base ∆E0 e (HF) ∆E0 e (MP2) ∆E0 e (MP3) ∆E0 e (MP4) ∆E298 b ∆H298 c ∆S (cal/(mol K)) ∆G298 c<br />
4a -189.6 -189.2 -191.8 -191.1 5.8 -185.3 -27.9 -177.0<br />
5a -190.7 -187.8 -190.4 -189.3 5.6 -183.7 -14.1 -179.5<br />
6a -192.8 -192.6 194.7 194.4 5.7 -188.7 -36.1 -178.0<br />
7a -203.4 -200.5 -203.4 -201.9 5.9 -196.0 -23.2 -189.1<br />
8a d -199.3 -195.2 -197.1 196.5 6.5 -189.9 -21.8 -183.4<br />
9a d -197.9 -194.6 -197.1 196.1 6.1 -190.0 -21.8 -183.5<br />
a S(H + ) ) 26.039 cal/(mol K). 35 b ∆E298 ) E298 vib + ∆E298 rot + ∆E298 trans + ∆(PV); ∆E298 vib scaled by 0.893. 8 c Based on the MP4/6-31G*/<br />
/6-31G* energies. d HF, MP2, and MP3 values taken from ref 16b.<br />
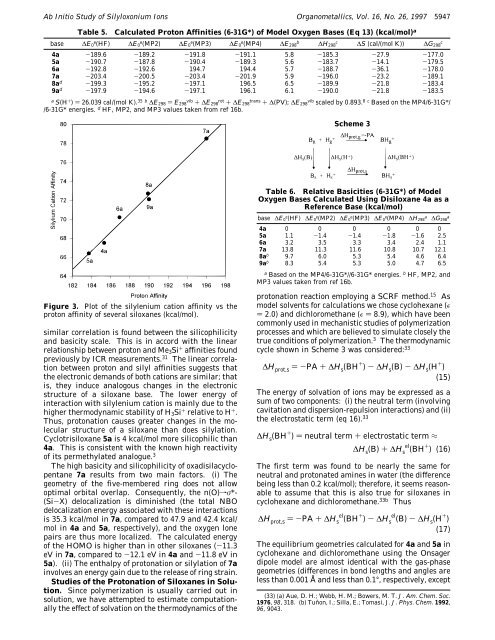
Figure 3. Plot <strong>of</strong> the silylenium cation affinity vs the<br />
proton affinity <strong>of</strong> several siloxanes (kcal/mol).<br />
similar correlation is found between the silicophilicity<br />
and basicity scale. This is in accord with the linear<br />
relationship between proton and Me3Si + affinities found<br />
previously by ICR measurements. 31 The linear correlation<br />
between proton and silyl affinities suggests that<br />
the electronic demands <strong>of</strong> both cations are similar; that<br />
is, they induce analogous changes in the electronic<br />
structure <strong>of</strong> a siloxane base. The lower energy <strong>of</strong><br />
interaction with silylenium cation is mainly due to the<br />
higher thermodynamic stability <strong>of</strong> H3Si + relative to H + .<br />
Thus, protonation causes greater changes in the molecular<br />
structure <strong>of</strong> a siloxane than does silylation.<br />
Cyclotrisiloxane 5a is 4 kcal/mol more silicophilic than<br />
4a. This is consistent with the known high reactivity<br />
<strong>of</strong> its permethylated analogue. 3<br />
The high basicity and silicophilicity <strong>of</strong> oxadisilacyclopentane<br />
7a results from two main factors. (i) The<br />
geometry <strong>of</strong> the five-membered ring does not allow<br />
optimal orbital overlap. Consequently, the n(O)fσ*-<br />
(Si-X) delocalization is diminished (the total NBO<br />
delocalization energy associated with these interactions<br />
is 35.3 kcal/mol in 7a, compared to 47.9 and 42.4 kcal/<br />
mol in 4a and 5a, respectively), and the oxygen lone<br />
pairs are thus more localized. The calculated energy<br />
<strong>of</strong> the HOMO is higher than in other siloxanes (-11.3<br />
eV in 7a, compared to -12.1 eV in 4a and -11.8 eV in<br />
5a). (ii) The enthalpy <strong>of</strong> protonation or silylation <strong>of</strong> 7a<br />
involves an energy gain due to the release <strong>of</strong> ring strain.<br />
Studies <strong>of</strong> the Protonation <strong>of</strong> Siloxanes in Solution.<br />
Since polymerization is usually carried out in<br />
solution, we have attempted to estimate computationally<br />
the effect <strong>of</strong> solvation on the thermodynamics <strong>of</strong> the<br />
Scheme 3<br />
Table 6. Relative Basicities (6-31G*) <strong>of</strong> Model<br />
Oxygen Bases Calculated Using Disiloxane 4a as a<br />
Reference Base (kcal/mol)<br />
base ∆E0 e (HF) ∆E0 e (MP2) ∆E0 e (MP3) ∆E0 e (MP4) ∆H298 a ∆G298 a<br />
4a 0 0 0 0 0 0<br />
5a 1.1 -1.4 -1.4 -1.8 -1.6 2.5<br />
6a 3.2 3.5 3.3 3.4 2.4 1.1<br />
7a 13.8 11.3 11.6 10.8 10.7 12.1<br />
8a b 9.7 6.0 5.3 5.4 4.6 6.4<br />
9a b 8.3 5.4 5.3 5.0 4.7 6.5<br />
a Based on the MP4/6-31G*//6-31G* energies. b HF, MP2, and<br />
MP3 values taken from ref 16b.<br />
protonation reaction employing a SCRF method. 15 As<br />
model solvents for calculations we chose cyclohexane (ɛ<br />
) 2.0) and dichloromethane (ɛ ) 8.9), which have been<br />
commonly used in mechanistic studies <strong>of</strong> polymerization<br />
processes and which are believed to simulate closely the<br />
true conditions <strong>of</strong> polymerization. 3 The thermodynamic<br />
cycle shown in Scheme 3 was considered: 33<br />
∆H prot,s )-PA + ∆H s (BH + ) - ∆H s (B) - ∆H s (H + )<br />
(15)<br />
The energy <strong>of</strong> solvation <strong>of</strong> ions may be expressed as a<br />
sum <strong>of</strong> two components: (i) the neutral term (involving<br />
cavitation and dispersion-repulsion interactions) and (ii)<br />
the electrostatic term (eq 16). 33<br />
∆Hs (BH + ) ) neutral term + electrostatic term ≈<br />
el +<br />
∆Hs (B) + ∆Hs (BH ) (16)<br />
The first term was found to be nearly the same for<br />
neutral and protonated amines in water (the difference<br />
being less than 0.2 kcal/mol); therefore, it seems reasonable<br />
to assume that this is also true for siloxanes in<br />
cyclohexane and dichloromethane. 33b Thus<br />
el + el<br />
∆Hprot,s )-PA + ∆Hs (BH ) - ∆Hs (B) - ∆Hs (H + )<br />
(17)<br />
The equilibrium geometries calculated for 4a and 5a in<br />
cyclohexane and dichloromethane using the Onsager<br />
dipole model are almost identical with the gas-phase<br />
geometries (differences in bond lengths and angles are<br />
less than 0.001 Å and less than 0.1°, respectively, except<br />
(33) (a) Aue, D. H.; Webb, H. M.; Bowers, M. T. J. Am. Chem. Soc.<br />
1976, 98, 318. (b) Tuñon, I.; Silla, E.; Tomasi, J. J. Phys. Chem. 1992,<br />
96, 9043.










