Dokument anzeigen
Dokument anzeigen
Dokument anzeigen
- Keine Tags gefunden...

Erfolgreiche ePaper selbst erstellen
Machen Sie aus Ihren PDF Publikationen ein blätterbares Flipbook mit unserer einzigartigen Google optimierten e-Paper Software.
Weisse Substanz von 0 bis 20MRT bei StoffwechselstörungenM. Warmuth-MetzAbteilung für NeuroradiologieUniversität Würzburg
Normale Myelinisation• beginnt im 5. Embryonalmonat• Fortschreiten von:– kaudal nach kranial– dorsal nach ventral– zentral nach peripher
Erkrankungen der weißen Substanz• Dysmyelinisationfehlerhafte Bildung oder Erhaltungvon Myelin• DemyelinisationVerlust von vormals normalemMyelin
Einteilung nach der Ätiologie• LysosomaleErkrankungen– MetachromatischeLeukodystrophie– M. Krabbe– M. Niemann-Pick– M. Tay-Sachs– Fukosidose– Mukopolysacharidose– Neuronale Zeroidlipofuszinose• PeroxisomaleErkrankungen– Zellweger Syndrom– M. Refsum– NeonataleAdrenoleukodystrophie– X-linkedAdrenoleukodystrophie• Aminosäurestörungen– M. Canavan– Phenylketonurie– Harnstoffzyklustörungen (z.B. OTC)– Azidurien• Mitochondriale Störungen– M. Leigh– MELAS– MERRF– Kearns Sayre Syndrom
Betroffenen Strukturen• Leukodystrophien– MetachromatischeLeukodystrophie– M. Canavan– M. Pelizaeus-Merzbacher– M. Alexander– Adrenoleukodystrophied – M. Krabbe– Aminosäurestoffwechsel-störungen• Überwiegend graueSubstanz– M. Tay-Sachs– NeuronaleZeroidlipofuszinose– Glykogenspeicherkrank-heiten• Graue und weiße Substanz– Mitochondriale Enzephalopathien– Peroxisomale ErkrankungenZellweger Syndrom,neonatale Adrenoleukodystrophie• Stammganglien-erkrankungenk– Chorea Huntington– M. Hallervorden-Spatz– M. Fahr– M. Wilson
Diagnostischer Wegweiser I• Ausgeprägter Mangel• Infarkte und infarkt-an Myelinähnliche Symptome– M. Canavan– M. Pelizaeus-Merzbacher• Makrozephalie– M. Canavan– M. Alexander– Mukopolysacharidosen(Hunter, Hurler)• Frontale Betonung– M. Alexander– AtypischeAdrenleukodystrophied – MitochondrialeEnzephalopathien– Harnstoffzyklusstörungen• Okzipitale Betonung– Adrenoleukodystrophie• Auffällig geschlängelteGefäße– Menkes Kinky Hair Disease
Diagnostischer Wegweiser II• Kraniozervikale oder zervikale • Tigroid id PatternEnge– M. Pelizaeus-Merzbacher– Hurler Syndrom– Cockayn Syndrom– L2-HydroxyglutarazidurieHd lt i• Verknüpfung mit• Ausparung periventrikulärerMuskeldystrophienRäume– Fukuyama Syndrom– L2-Hydroxyglutarazidurie– Myotonie• Weite Sylvische Fissur oder• Fettspeicherung im Kleinhirnsubdurale Ergüsse– Zerebrotendinöse Xanthomatose – Glutarazidurie• Hirnstammläsionen– ALD, AMN– M. Alexander• Stammganglienverkalkungen– Cockayn Syndrom– Mitochondriale i Erkrankungenk– Phosphatstoffwechselstörungen
Metachromatische Leukodystrophie MLDVererbungbetroffenKlinikTodUrsacheLaborDiagnoseTherapieautosomal rezessivKleinkinder (infantile), Kinder undJugendliche (juvenile) und Erwachsenen (adult variant)Hypotonie mit Gehschwierigkeiten,Polyneuropathie, Paresen, Kleinhirnsymptome,Verhaltensstörungen bis zur Psychose (adult)5 bis 15 Jahre ab Symptombeginn oder späterDefekt der Arylsulfatase A in LysosomenLiquoreiweiß kann erhöht sein, Sulfatitausscheidung im UrinerhöhtAktivitätsbestimmung der Arylsulfatase A inLeukozyten oder Fibroblastenbl evtl. Knochenmarktransplantation
Metachromatische Leukodystrophie MLD
Metachromatische Leukodystrophie MLD• bilaterale symmetrische Entmarkungen• Verbindung über den Balken• U-Fasern erhalten• keine KM-Aufnahme• einfacher Nachweis
M. Pelizaeus-MerzbacherVererbungbetroffenKlinikTodLaborDiagnoseTherapiex-chromosomal rezessivkonnatal (Typ Seitelberger),frühkindlich oder infantilHypotonie, unsystematische Störungder Augenmotilität, Stridor, EntwicklungsundWachstumsverzögerung,Krampfanfälle, kleiner Kopfje nach Typ im Kindes- oder Jungendalterkeine AuffälligkeitGenuntersuchung (Proteolipidprotein)auffällige Oligodendrozyten (Pathologie)keine
M. Pelizaeus-Merzbacher
M. Pelizaeus-Merzbacher• kleiner Kopf• kein Myelin• Tigroid Pattern
M. CanavanVererbungautosomal rezessivbetroffen Kleinkinder, KinderKlinikik Hypotonie, schlechtes ht Trinken,Erblindung, MakrozephalieTod zwischen 4 und über 10 J.Ursachen Mangel an Aspartoazyklase(spaltet N-Azetylaspartat inAspartat und Azetat)LaborN-Azethylaspartat im Serum, Urinund Liquor erhöhtDiagnose NAA in MRS erhöht, Liquor, SerumTherapiekeine
M. Canavan
• großer Kopf• kein MyelinM. Canavan• typischer Aspartatnachweis im Liquor undMRS
X-linked AdrenoleukodystrophieVererbung x-chromosomal rezessivbetroffen Knaben (5-9J.), Erwachsene AdrenomyloneuropathieAMN ca. 40J., AdrenoleukomyeloneuropathieALMN), Konduktorinnen (leichte Symptome)Klinik langsam progrediente Verhaltensstörungen, Gang-störungen, Sehverlust, Hörstörungen, Demenz,Krampfanfälle etc., M. AddisonTod ?UrsacheAbbau der sehr langkettigen Fettsäuren gestörtLabor Liquoreiweiß erhöht, NebenniereninsuffizienzDiagnose hohe Spiegel der sehr langkettigen Fettsäuren(VLCFA) im Serum oder ZellenTherapie Diät mit Restriktion der gesättigten Fettsäuren undZusatz von ungesättigten Fettsäuren, Gentherapiemit Transplantation von Knochenmark oderTransplantation von Oligodendrozyten
X-linked Adrenoleukodystrophie
X-linked Adrenoleukodystrophie
X-linked Adrenomyeloneuropathie
X-linked Adrenoleukodystrophie• nur Jungen• okzipitoparietale Entmarkung• Balkenbeteiligung• KM-Aufnahme im akuten Stadium• Verhaltensauffälligkeiten, Sehstörungen• Verwandte, Konduktorinnen
M. AlexanderVererbung sporadische Neumutationen, nicht familiärbetroffen keine Geschlechtsbevorzugung; angeboren(selten), infantil (2J.),Erwachsenenform (selten)Klinik großer Kopf, Neurologie variabelTod je früher betroffen, desto eherUrsache Mutation im GFAP (saures Gliafaser Protein)GenLabor keineDiagnoseMutatationsnachweis, t ti i Histologiei(Rosenthalfasern)Therapiekeine
M. AlexanderV d Knaap AJNR 2001; 22
M. Alexander
M. Alexander
M. Alexander• Großer Kopf bei schwer betroffenen Patienten• Diagnostische Kriterien (2001; M v d Knaap):– Extensive Entmarkungen, frontal betont– Periventrikuläre Säume (T1 hyper; T2 hypo)– Stammganglienbeteiligung– Hirnstammläsionen– KM-Aufnahme (mindestens eine der folgendenStrukturen): periventrikulär, frontal, Chiasma, Fornix,Stammganglien, g Thalamus, Nucl. Dentatus,Kleinhrinrinde, Hirnstamm– 4 von 5 müssen erfüllt sein, aber auch Verdacht beiweniger
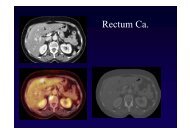
Neuronale ZeroidlipofuszinoseVererbung autosomal rezessiv und machmal autosomaldominant (adult)betroffen Kleinkinder (Santavouri), Kinder (Jansky-Bielschowsky), Jugendliche (Spielmeyer-Vogt,Batten), Erwachsenen (Kuf, Böhme)Klinikpsychomotorische Verschlechterung,Krampfanfälle, Ataxie, Hypotonie,Sprachstörungen, Sehverlust, DemenzTodje nach Typ zwischen 15 und bis zu 40 JahrenUrsache unklare Störung mit Speicherung von Lipofuszin(Zeroid)Diagnose Nachweis der doppelbrechendenSpeicherprodukte in Rektum-, Haut- oderSchleimhautbiopsieTherapie keine
Neuronale Zeroidlipofuszinose
M. Kufs
Neuronale Zeroidlipofuszinose• Bei jeder altersinadäquaten Demenz sollteman an eine e Neuronale ae Ceroidlipofuszinoseod osedenken, da die Diagnostik problemlos ist,wenn auch keine Therapie zur Verfügungsteht• überwiegend kortikale Atrophie• je jünger desto häufiger zusätzlichEntmarkungen
Ornithintranskarbamylasemangel (OTC)• Häufigster Harnstoffzyklusdefektf kt• Vererbung X-chromosomal rezessiv• betroffenNeonatal bis Kleinkindesalter• Klinik Episodische Lethargie und Koma,Hirndruckzeichen, hohe Mortalität in der akutenPhase• Tod ?• Ursache Ornithintranskarbamylase kondensiertKarbamylphosphat und Ornithin zu Citrullin• Labor Hyperammonämie, respiratorische Alkalose• Diagnose Enzymdefektnachweis in Leberzellen• Therapie Proteinarme Diät, ausreichende Ernährung,Zufuhr essentieller Aminosäuren, zukünftigGentherapie, Lebertransplantation
Ornithintranskarbamylasemangel (OTC)
Acidurien• Störung des Aminosäureabbaus• Ahornsirup Krankheit• Ketothiolase-Defizienz• Propionazidurie• Methylmalonazidurie• Glutarazidurie Typ I• Sulfit-Oxidase-Defizienz• Molybdän-Kofaktor-Defizienz
Methylmalon-Acidurie
Stammganglienläsionen• Aminoazidurien• mitochondriale Erkrankungen• Störungen des Energiestoffwechsels
Menkes SyndromVererbung x-chromosomal rezessivbetroffen KleinkinderKlinikik Gedeihstörung, Krampfanfälle, fäll Verlust vonFähigkeiten, auffällig brüchige, schüttereHaare, Pigmentstörungen, g subduraleBlutungen oder ErgüsseTod innerhalb weniger MonateLabor Coeruloplasmin im Serum vermindertUrsache verminderte Kupferaufnahme aus der Darm,verminderte Aktivität der kupferabhängigenEnzyme z.B. ZytochromoxidaseDiagnoseverminderte Coeruloplasminspiegel im SerumTherapie bei Früherkennung Substitution möglich
Menkes Syndrom
MELASVererbungdas Genom der Mitochondrien i wird ausschließlich h von derMutter vererbtbetroffen ErwachseneKlinikik myopathische h Störungen, Kleinwüchsigkeit, i it Seh- undHörstörungen, Stroke-like-Episoden erst im Verlauf, abhängigvon Defekten evtl. KrampfanfälleTod ?Ursache Störungen der Atemkette durch Mutationen desMitochondriengenoms. Da die Mitochondrien bei jederZellteilung zufällig verteilt werden, resultieren völligunterschiedliche und nicht vorhersehbareMitochondrienpopulationen in verschiedenen ZellenLabor Laktat in Serum und Liquor erhöhtDiagnose Störungsnachweis von Atemkettenenzymen in Muskelzellenund Fibroblasten, MutationsnachweisTherapieGabe von Co-Faktoren
MELAS
MELAS und MRS
MELAS und MRS
nicht klassifizierteMitochondrienstörung
Leigh-SyndromVererbung entweder autosomal rezessiv oder maternalbetroffen Kleinkinder und KinderKlinikAtemstörungen, Augensymptome, Hypotonie,Pyramidenbahnzeichen, Schwäche, Lethargie,Koma, evtl. Status epilepticus, je später derBeginn, desto milder der VerlaufTod innerhalb von Monaten oder JahrenUrsache Stoffwechselstörung im Energiehaushalt derMitochondrien, besonders Mangel an Cytochrom COxidase und PyruvatkinaseLaborLaktat und Pyruvat im Liquor und Serum erhöhtDiagnose Nachweis des Defekts in der Atmungskette vonMuskelzellen oder FibroblastenTherapie Gabe von Co-Faktoren und Vitaminen, L-Carnithin
Leigh-Syndrom
Pyruvatdehydrogenase Mangel
L2-HydroxyglutarazidurieVererbung autosomal rezessivbetroffen Kinder und JugendlicheKlinikRetardierung, zerebelläre Symptome, Entwicklungeiner Demenz, meist Krampfanfälle, häufigextrapyramidale Störungen, überdurchschnittl.Belastung mit HirntumorenTod ?Labor erhöhte Ausscheidung vonL2-Hydroxyglutarsäure im Urin, erhöhteWerte in Serum und LiquorUrsache unbekannt, vielleicht defekteL2-hydroxyglutardehydrongenase,hd lt dhdDiagnose typisches MRT, LaborTherapiekeine
L2-Hydroxyglutarazidurie
Zerebrotendinöse XanthomatoseVererbung:betroffen:Klinik:Tod:Ursache:Labor:Diagnose:Therapie:autosomal dominantErwachseneBorderline-Intelligenz, Sehnenxanthome, Katarakt,Kleinhirnzeichen, Krampfanfälle, Para- undTetraparese, Hinterstrangsymptome, Demenz,vorzeitige Arteriosklerose4. bis 6. DekadeStörung der mitochondrialen 27-Hydroxylase(Störung der Gallensäurebildung, durchRückkopplung vermehrte Cholesterol undCholestanolproduktion und – speicherung)Serumcholesterol normal, Serumcholestanol starkerhöht, Liquoreiweiß erhöht, abnormeGallensäureprodukte im UrinFehlen der 27 Hydroxylaseaktivität in FibroblastenUnterbrechung des Rückkopplungskreises mitInhibitoren der Cholesterinsynthese undChenodesoxycholsäure
Zerebrotendinöse Xanthomatose
Myotone Muskeldystrophie
Vielen Dank für IhreAufmerksamkeit
Fragen• Bei welchen Stoffwechselstöungen machteine e MR-Spektroskopie op e Sinn?• Defekte oder Läsionen in denStammganglien weisen auf welcheStoffwechselstörungen hin?• Welche Stoffwechselstörungen führenhäufig zur Makrozephalie?