Programm „WinXPOW“
Programm „WinXPOW“
Programm „WinXPOW“

Erfolgreiche ePaper selbst erstellen
Machen Sie aus Ihren PDF Publikationen ein blätterbares Flipbook mit unserer einzigartigen Google optimierten e-Paper Software.
Auswertung von Röntgenpulvermessungen (STOE StadiP):<br />
<strong>Programm</strong> <strong>„WinXPOW“</strong><br />
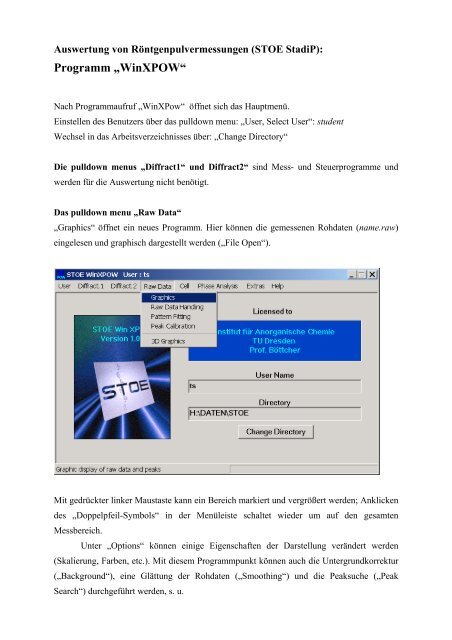
Nach <strong>Programm</strong>aufruf „WinXPow“ öffnet sich das Hauptmenü.<br />
Einstellen des Benutzers über das pulldown menu: „User, Select User“: student<br />
Wechsel in das Arbeitsverzeichnisses über: „Change Directory“<br />
Die pulldown menus „Diffract1“ und Diffract2“ sind Mess- und Steuerprogramme und<br />
werden für die Auswertung nicht benötigt.<br />
Das pulldown menu „Raw Data“<br />
„Graphics“ öffnet ein neues <strong>Programm</strong>. Hier können die gemessenen Rohdaten (name.raw)<br />
eingelesen und graphisch dargestellt werden („File Open“).<br />
Mit gedrückter linker Maustaste kann ein Bereich markiert und vergrößert werden; Anklicken<br />
des „Doppelpfeil-Symbols“ in der Menüleiste schaltet wieder um auf den gesamten<br />
Messbereich.<br />
Unter „Options“ können einige Eigenschaften der Darstellung verändert werden<br />
(Skalierung, Farben, etc.). Mit diesem <strong>Programm</strong>punkt können auch die Untergrundkorrektur<br />
(„Background“), eine Glättung der Rohdaten („Smoothing“) und die Peaksuche („Peak<br />
Search“) durchgeführt werden, s. u.
Mit dem <strong>Programm</strong> „Graphics“ können auch mehrere Messungen in einem Bild dargestellt<br />
werden. Zunächst Öffnen der gemessenen Daten name.raw mit „file, open“. Durch<br />
Rechtsklick in der Graphik erscheint ein Menü u. a. mit den Optionen „add raw data“. Hier<br />
kann der nächste einzulesende file, z. B name2.raw, ausgewählt werden. Zur besseren<br />
Darstellung sollte in der oberen Leiste die Optionen „R“ (= relative Intensitäten) und „L“ (=<br />
lineare Intensitäten) gewählt werden. Durch erneutes Rechtsklicken können auch Messkurven<br />
editiert, bzw. wieder gelöscht werden. Nach der gleichen Methode können auch Messdaten<br />
und Reflexdaten (zur Erstellung des Reflexdatenfiles s. u.) in einer Graphik dargestellt<br />
werden.<br />
Die Option „Smoothing“ benötigt zwei Startwerte für die Glättung der Messpunkte:<br />
die Halbwertsbreite der Reflexe und die Anzahl der Glättungspunkte (max. 25). Die<br />
geglätteten Daten können als rsm-file (name.rsm) im gleichen Format wie die Rohdaten<br />
gespeichert werden. Mit „Inc.Smo.“ bzw. „Dec.Smo.“ lässt dich die Glättungsfunktion<br />
variieren.<br />
Die Option „Background“ ermöglicht eine Untergrundkorrektur. Es wird eine<br />
Funktion mit mehreren Stützpunkten in den Untergrund gelegt. Diese Stützpunkte (rote<br />
Punkte) können manuell editiert werden. Doppelklick links erzeugt einen neuen Punkt, Klick<br />
rechts löscht einen Punkt. Mit gedrückter linker Maustaste lassen sich die Punkte verschieben.
Achtung Bug: Das <strong>Programm</strong> setzt in der Regel den letzten Punkt der Funktion zu hoch an,<br />
dadurch wird an der Stelle zu viel Untergrund abgezogen.<br />
Mit „Subtract BG“ wird der Untergrund abgezogen, die korrigierten Daten können als rmbfile<br />
(name.rmb) im gleichen Format wie die Rohdaten gespeichert werden.<br />
Für die Reflexsuche (Peak Search) müssen die Startparameter „Estimated Halfwidth“ (=<br />
geschätzte Halbwertsbreite der zu suchenden Reflexe), „Siginificance Level“ (= um welchen<br />
Faktor ist die Intensität des zu suchenden Peaks höher als die Intensität des Untergrundes) und<br />
„Intensity Limit“ (= gemessene Intensität, die ein zu suchender Peak mindestens aufweisen<br />
muß) angegeben werden. Eine Subtraktion von Spikes oder Alpha2-Peaks ist auf Grund der<br />
Messanordnung nicht notwendig. Die mit diesen Parametern gefundenen Peaks (dargestellt<br />
durch blaue Linien) lassen sich manuell editieren. Doppelklick mit der linken Maustaste<br />
erzeugt an dieser Stelle einen neuen Peak; klick mit der rechten Maustaste auf eine blau Linie<br />
löscht den Peak. Mit gedrückter linker Maustaste lässt sich die blaue Linie verschieben. Die<br />
Ergebnisse der Peaksuche können mit „Write Peakfile“ in einen ASCII-file abgespeichert<br />
werden (name.pks).<br />
Das <strong>Programm</strong> „Raw Data Handling“ wird in diesem Praktikum nicht benötigt.<br />
Mit dem <strong>Programm</strong> „Pattern Fitting“ lassen sich eine Anpassung von Rohdaten (bzw.<br />
geglätteten und/oder untergrundkorrigierten Daten) mit den durch „Peak Search“ gefundenen<br />
Reflexen durchführen. Das <strong>Programm</strong> benötigt also zwei Eingabefiles einen Peakfile<br />
(name.pks) und einen Datenfile (name.raw, name.rsm, oder name.rmb). Mit „Pattern Fitting“<br />
können Reflexlagen und -intensitäten nochmal manuell verändert werden, sowie eine<br />
Untergrundkorrektur durchgeführt werden (alles im pulldown menu „Operations“). Unter<br />
„Operations“ können dann auch durch eine lineare und nicht-lineare Regression die<br />
Übereinstimmung von gefundenen Reflexen und gemessenen Daten überprüft und aneinander<br />
angepasst werden. Die Differenz zwischen beiden wird jeweils im unteren Bildausschnitt<br />
graphisch dargestellt. Die angepassten Peaklagen und -intensitäten können in einen neuen<br />
Peakfile (name.pft; ASCII-Format) gespeichert werden.<br />
Die <strong>Programm</strong>e „Peak Calibration“ und „3D Graphics“ werden in diesem Praktikum nicht<br />
benötigt.
Das pulldown menu „Cell“<br />
Mit dem <strong>Programm</strong> „Index & Refine“ können die Pulverdaten indiziert werden, d. h. jedem<br />
Reflex wird ein Zahlentripel hkl zugeordnet. Mit diesen Informationen kann dann eine<br />
Elementarzelle berechnet werden. Da diese Indizierungen nur in wenigen Fällen eindeutige<br />
Ergebnisse liefern, ist hier etwas Fingerspitzengefühl bei der Einstellung der Startparameter<br />
und Durchhaltevermögen bei Auswahl und Durchführung gefragt.<br />
Das <strong>Programm</strong> benötigt als Eingabe entweder den Reflexfile (name.pks) oder den durch<br />
„Pattern Fitting“ veränderten Reflexfile (name.pft).<br />
Das Indizieren kann mit einem der drei <strong>Programm</strong>e „TREOR (Werners Algorithmus)“, „ITO<br />
(Vissers Algorithmus)“ oder „DICVOL (Louers Algorithmus)“ durchgeführt werden. Für alle<br />
kubischen, tetragonalen, hexagonalen und orthorhombischen Probleme empfiehlt sich das<br />
<strong>Programm</strong> „TREOR“.<br />
Vor der Indizierung müssen einige Startparameter eingegeben werden. Diese sind für<br />
TREOR z. B.:<br />
max. # of Peaks: Zahl sollte größer sein, als die gefundenen Reflexe im Reflexfile
max. cell edge, max. cell volume: nicht zu klein, sonst wird die „richtige“ Zelle nicht<br />
gefunden<br />
2Theta window: Fenster, in dem das <strong>Programm</strong> die Reflexlagen verschieben darf, um sie<br />
an eine Zelle anzupassen. Kritische Größe, wird sie zu klein gewählt passen Reflexe<br />
möglicherweise nicht in eine gegebene Indizierung, bei zu großem Wert ist oft keine<br />
eindeutige Zuordnung mehr möglich.<br />
unindexed lines: Anzahl der Linien die nicht in die Indizierung passen müssen<br />
peak intensity limit: Reflexe mit Intensitäten unterhalb dieses Schwellenwertes werden bei<br />
der Indizierung nicht berücksichtigt<br />
required FOM: Gütewert für die Anpassung, je höher der FOM desto besser die<br />
Übereinstimmung (FOM = figure of merit)<br />
crystal system: Kristallsystem in dem die Indizierung gestartet wird, wird keine ordentliche<br />
Übereinstimmung erzielt, wird in niedersymmetrischen Kristallsystemen weiter gesucht.<br />
<strong>Programm</strong>start (für TREOR) mit „W“ oder „Index, Run Werner“ in der Menüleiste. Die<br />
gefundenen Elementarzellen werden in einem Fenster ausgegeben. Im unteren Bereich des<br />
Fensters sieht man die graphische Anpassung mitlaufen.<br />
Achtung: Die angegebenen Lösungen sind stets nur mehr oder weniger gute Möglichkeiten<br />
der Indizierung!<br />
Durch Anklicken (rote Markierung) einer bestimmten Lösung wird ein neues Fenster<br />
geöffnet. Hier sind die berechneten Gitterparameter, die Anzahl der nicht indizierten Reflexe<br />
(sollte möglichst Null oder nahe bei Null sein) die hkl-Werte der Reflexe und die<br />
Abweichungen der Reflexlagen (2Θobs-2Θcalc) aufgelistet.<br />
Mit Anklicken von „R“ oder „Refine“ in der Menüleiste wird für die rot markierte Lösung<br />
eine Fehlerquadratminimierung durchgeführt. Dazu wird wieder ein Fenster eingeblendet, in<br />
dem Startwerte (Kristallsystem, Bravais Typ, Gitterparameter, etc.) eingegeben werden<br />
können/müssen. Kritische Größen sind hier das „2Theta Window“ (s. o.) und „2Theta Zero“<br />
das eine mögliche Fehljustierung des Nullpunktes des Diffraktometers berücksichtigt, die<br />
Option „Refine 2Theta Zero“ sollte eingeschaltet werden.<br />
Als Ergebnis erscheint wieder ein Fenster mit Startwerten und Ergebnissen der Verfeinerung,<br />
der Anzahl der nicht indizierten Reflexe, den Indizes und Abweichungen der Reflexlagen für<br />
jeden Reflex, sowie einem FOM der wiederum möglichst groß sein sollte. Diese Ergebnisse<br />
können in den peak-file (name.pks, name.pft) abgespeichert werden.
Mit dem <strong>Programm</strong> „Theoretical Pattern“ können Röntgenbeugungsdiagramme simuliert<br />
werden. Dazu sind zwingend mindestens folgende Parameter nötig: Formel, Raumgruppe,<br />
Gitterparameter, und Besetzung der kristallographischen Lagen (daraus ergeben sich u. a.<br />
weitere Angaben). Diese Daten können aus Datenbanken (z. B. aus der ICSD),<br />
Nachschlagewerken oder der Originalliteratur entnommen werden.<br />
Mit „Setup, Crystal Parameters“ werden zunächst die kristallographischen Abgaben: Formel<br />
(Achtung: jeweils ein Leerzeichen zwischen den Elementsymbolen bzw. stöch.<br />
Koeffizienten), Z, Raumgruppe, und Gitterparameter eingegeben. Als Wellenlänge wird Cu<br />
eingestellt, der zu berechnende Diffraktogrammauschnitt (2Theta min, max) wird den<br />
gemessenen Daten angepasst. Danach zeigt das <strong>Programm</strong> zunächst alle möglichen<br />
Reflexlagen an.<br />
Mit „Setup, Atom List“ werden nun die Atome an die entsprechende Stelle in der<br />
Elementarzelle platziert:<br />
New Atom: name = eindeutige Atombezeichnung (z. B. Na1 oder Cl2), Type = Element<br />
gemäß Liste rechts, x/a, y/b, z/c = relative Atomkoordinaten, Occupancy = Besetzung ( in der<br />
Regel 1), U11 = therm. Auslenkungsparameter (in der Regel 0.01 – 0.02).<br />
Für jedes neue Atom muss diese Eingabemaske mir „New Atom“ wieder neu gestartet<br />
werden.<br />
Nach Eingabe aller Atome Maske schließen und speichern („File, Save As“). Die Daten<br />
werden in einem ASCII-file name.tin gespeichert. Erst beim eneuten Aufruf dieses<br />
gespeicherten files wird das berechnete Pulverdiffraktogramm angezeigt (Bug).<br />
Mit „Setup, Pattern Parameters“ kann das berechnete Diffraktogramm nun den<br />
Messbedingungen noch angepasst werden. Wichtig für die Messungen am STOE STADI P:<br />
Geometry: Transmission, Monochromator: Germanium, keine alpha2-Peaks. Weiterhin kann<br />
man die Profilfunktion variieren, die Halbwertsbreite der zu berechnenden Reflexe, die max.<br />
Intensität, und einen gemitttelten Absorptionskoeffizienten (Mu*T) eintragen.<br />
Das <strong>Programm</strong> „Theoretical Pattern“ speichert insgesamt vier files: name.tin = die<br />
Eingabedaten, name.pth = die Reflexliste, name.lth = Reflexliste mit zusätzlichen kristallographischen<br />
Angaben, und name.rth = Datenfile zur graphischen Darstellung des<br />
Diffraktogramms.<br />
Berechnete und gemessene Datenfiles können im <strong>Programm</strong> „Graphics“ (pulldown<br />
menu: Raw Data) in einem Bild dargestellt werden. Z. B. so: zunächst Öffnen der gemessenen<br />
Daten name.raw mit „File, Open“. Durch Rechtsklick in der Graphik erscheint ein Menü u. a.<br />
mit den Optionen „Add Raw Data“. Hier kann der berechnete file name.rth ausgewählt<br />
werden. Zur besseren Darstellung sollte in der oberen Leiste die Option „R“ (= relative<br />
Intensitäten) gewählt werden.
Das pulldown menu „Phase Analysis“ :<br />
Das <strong>Programm</strong> „Search/Match“ dient zur Substanzidentifizierung durch Vergleich mit den in<br />
der Datenbank „Powder Diffraction File“ (PDF) gespeicherten Reflexdaten. Das gemessene<br />
Pulverdiffraktogramm kann entweder als graphischer Messdatenfile (name.raw, name.rmb)<br />
oder als Peakfile (name.pks, name.pft) eingelesen werden.<br />
Unter „Select“ kann zunächst der Teilbereich der Datenbank ausgewählt werden, der für die<br />
Auswertung relevant ist (z. B. gesamte Datenbank, Anorganische Substanzen, Minerale,<br />
Legierungen/Metalle). Dann können unter „Elements“ beliebige Kombinationen aus den<br />
Elementen des PSE vorgegeben werden. Der Farbcode für die Markierungen versteht sich wie<br />
folgt. rot: alle rot markierten Elemente müssen vorkommen; gelb: von den gelb markierten<br />
Elementen muss mindestens eins enthalten sein; blau: diese Elemente dürfen nicht enthalten<br />
sein; grau: diese Elemente dürfen enthalten sein, müssen aber nicht. Vor dem Suchen<br />
erscheint möglicherweise eine weitere Abfragebox, in der die Anzahl der Elemente<br />
anzugeben ist.<br />
Beispiel 1: Sie suchen nach allen Verbindungen, die nur aus Tl und Te bestehen. Markieren<br />
Sie Tl und Te rot, alle anderen Elemente blau. Diese Eingabe ist eindeutig, es gibt keine<br />
weitere Abfrageoption.<br />
Beispiel 2: Sie wissen, dass ihr Substanz ein Phosphat eines Alkalimetalls ist: markieren Sie P<br />
und O rot, Li, Na, K, Rb, Cs gelb. Es erscheint die Abfragebox für die Anzahl der in der<br />
Verbindung erhaltenen Elemente. Markieren Sie „min. 3, max. 3“ für eine ternäre<br />
Verbindung.
Bei völlig unbekannter Zusammensetzung der gemessenen Substanz kann nach einem<br />
anderen Kriterium gesucht werden, nämlich nach den Lagen (d-Werten oder 2Theta-Werten)<br />
der starken Reflexe. Diese werden Sie in die dann erscheinende Abfragebox eingegeben.<br />
Die Ergebnisse der Suche können mit „View“ in verschiedenen Formaten (z. B. mit Namen,<br />
mit d-Werten, oder 2theta-Werten) auf dem Bildschirm ausgegeben werden. Einzelne Treffer<br />
können mit linkem Mausklick markiert werden. Unter „View, Text Window“ kann der<br />
komplette Eintrag eines markierten Treffers, die so genannte PDF-Karteikarte angezeigt (und<br />
exportiert) werden. Auf dieser Karteikarte befinden sich alle gespeicherten Informationen zu<br />
dieser Substanz: alle relevanten Messdaten, die kristallographischen Angaben soweit bekannt,<br />
die Originalpublikation und die Autoren, der so genannte Hanawalt-Eintrag, und das<br />
wichtigste zuletzt, eine Tabelle mit d-Werten, 2Theta-Werten Intensitäten und hkl-Werten der<br />
gemessenen Reflexe. Zur Substanzidentifizierung müssen Sie diese Liste mit Ihren<br />
gemessenen Daten vergleichen (s. u.). Für eine positive Identifizierung einer Substanz muss<br />
die Lage aller Reflexe exakt übereinstimmen. Die Reflexintensitäten können durch<br />
verschiedene Effekte verfälscht werden und müssen daher nicht exakt passen. Jedoch ist die<br />
Intensitätsabfolge einer Substanz ebenfalls charakteristisch. Der Vergleich einer Messung mit<br />
einem oder mehreren Datenbankeinträgen geschieht zweckmäßigerweise im<br />
„GraphWindow“. Hier können die Abweichungen und Übereinstimmungen zwischen den<br />
gemessenen Rohdaten bzw. den gefundenen Peaks (grün) mit den Reflexlagen und -<br />
intensitäten der Datenbankeinträge (rot) für das gesamte Diffraktogramm überprüft werden.
Mit den Schalter in der Kopfzeile kann zwischen den einzelnen Treffern der<br />
Datenbankrecherche hin- und hergeblättert werden. Es können auch einzelne Einträge<br />
markiert („select“) oder verschiedene Bereiche aus dem Diffraktogramm vergrößert<br />
dargestellt werden (ziehen mit gedrückter linker Maustaste).<br />
Der Inhalt des „GraphWindow“ (Messdaten und Datenbankeinträge) kann ausgedruckt und<br />
exportiert werden.<br />
Das <strong>Programm</strong> „Crystallinity“ wird im Praktikum nicht benötigt.<br />
Das pulldown menu „Extras“ wird in diesem Praktikum nicht benötigt.