EG-Leitfaden der Guten Herstellungspraxis für Arzneimittel zur ...
EG-Leitfaden der Guten Herstellungspraxis für Arzneimittel zur ...
EG-Leitfaden der Guten Herstellungspraxis für Arzneimittel zur ...

Sie wollen auch ein ePaper? Erhöhen Sie die Reichweite Ihrer Titel.
YUMPU macht aus Druck-PDFs automatisch weboptimierte ePaper, die Google liebt.
Pharmaceutical Legislation<br />
Volume 4<br />
EU Guidelines to<br />
Good Manufacturing Practice<br />
Medicinal Products for Human and Veterinary Use<br />
Introduction<br />
Commission Directive 2003/94/EC, of 8 October 2003, laying down the principles and<br />
guidelines of good manufacturing practice in respect of medicinal products for human use<br />
and investigational medicinal products for human use<br />
Replacement of Commission Directive 91/356/EC of 13 June 1991 to cover good<br />
manufacturing practice of investigational medicinal products.<br />
Commission Directive 91/412/EEC of 23 July 1991 laying down the principles and guidelines<br />
of good manufacturing practice for veterinary medicinal products.<br />
<strong>EG</strong>-<strong>Leitfaden</strong> <strong>der</strong> <strong>Guten</strong> <strong>Herstellungspraxis</strong> <strong>für</strong> <strong>Arzneimittel</strong><br />
<strong>zur</strong> Anwendung an Mensch o<strong>der</strong> Tier<br />
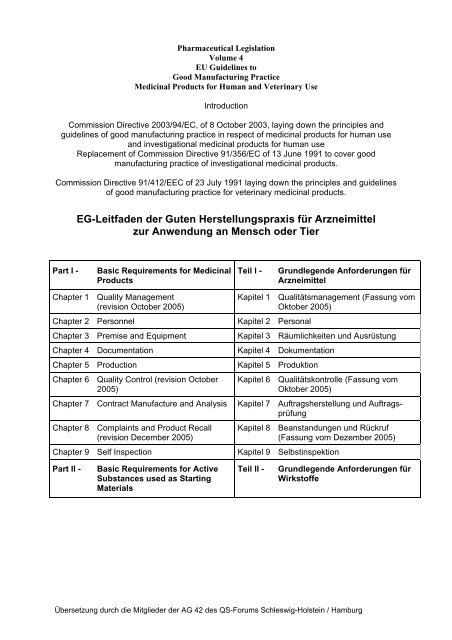
Part I - Basic Requirements for Medicinal<br />
Products<br />
Chapter 1 Quality Management<br />
(revision October 2005)<br />
Chapter 2 Personnel Kapitel 2 Personal<br />
Teil I - Grundlegende Anfor<strong>der</strong>ungen <strong>für</strong><br />
<strong>Arzneimittel</strong><br />
Kapitel 1 Qualitätsmanagement (Fassung vom<br />
Oktober 2005)<br />
Chapter 3 Premise and Equipment Kapitel 3 Räumlichkeiten und Ausrüstung<br />
Chapter 4 Documentation Kapitel 4 Dokumentation<br />
Chapter 5 Production Kapitel 5 Produktion<br />
Chapter 6 Quality Control (revision October<br />
2005)<br />
Kapitel 6 Qualitätskontrolle (Fassung vom<br />
Oktober 2005)<br />
Chapter 7 Contract Manufacture and Analysis Kapitel 7 Auftragsherstellung und Auftragsprüfung<br />
Chapter 8 Complaints and Product Recall<br />
(revision December 2005)<br />
Kapitel 8 Beanstandungen und Rückruf<br />
(Fassung vom Dezember 2005)<br />
Chapter 9 Self Inspection Kapitel 9 Selbstinspektion<br />
Part II - Basic Requirements for Active<br />
Substances used as Starting<br />
Materials<br />
Teil II - Grundlegende Anfor<strong>der</strong>ungen <strong>für</strong><br />
Wirkstoffe<br />
Übersetzung durch die Mitglie<strong>der</strong> <strong>der</strong> AG 42 des QS-Forums Schleswig-Holstein / Hamburg
ANNEXES Annexe<br />
Annex 1 Manufacture of Sterile Medicinal Products<br />
(May 2003)<br />
Manufacture of Steril Medicinal Products<br />
(revision February 2008)<br />
The revised annex should be implemented<br />
by 1 March 2009 except for the provisions<br />
on capping of freeze-dried vials,<br />
which should be implemented by 1 March<br />
2010.<br />
Annex 2 Manufacture of Biological Medicinal<br />
Products for Human Use<br />
Annex 3 Manufacture of Radio Pharmaceuticals<br />
GMP Annex 3 “Manufacture of Radiopharmaceuticals”<br />
of the GMP Guide has<br />
been revised (The revised annex should<br />
be implemented by 01 March 2009)<br />
Annex 4 Manufacture of Veterinary Medicinal<br />
Products other than Immunological<br />
Veterinary Medicinal Products<br />
Annex 5 Manufacture of Immunological Veterinary<br />
Medicinal Products<br />
Herstellung steriler <strong>Arzneimittel</strong> (Stand<br />
vom Mai 2003)<br />
Herstellung steriler <strong>Arzneimittel</strong> (Fassung<br />
vom Februar 2008)<br />
Die Neufassung ist bis zum 01. März<br />
2009 zu implementieren, ausgenommen<br />
die Regelungen zum Verschluss von Vials<br />
mit gefriergetrocknetem Inhalt, die zum<br />
01. März 2010 zu implementieren sind.<br />
Herstellung biologischer <strong>Arzneimittel</strong> <strong>zur</strong><br />
Anwendung am Menschen<br />
Herstellung radioaktiver <strong>Arzneimittel</strong><br />
Der Annex 3 wurde neu gefasst (Die<br />
Neufassung ist bis zum 01. März 2009 zu<br />
implementieren)<br />
Herstellung von <strong>Arzneimittel</strong>n <strong>zur</strong> Anwendung<br />
am Tier ausschließlich immunologischer<br />
<strong>Arzneimittel</strong> <strong>für</strong> Tiere<br />
Herstellung immunologischer <strong>Arzneimittel</strong><br />
<strong>für</strong> Tiere<br />
Annex 6 Manufacture of Medicinal Gases Herstellung medizinischer Gase<br />
Annex 7 Manufacture of Herbal Medicinal Products Herstellung pflanzlicher <strong>Arzneimittel</strong><br />
Annex 8 Sampling of Starting and Packaging<br />
Materials<br />
Annex 9 Manufacture of Liquids, Creams and<br />
Ointments<br />
Annex 10 Manufacture of Pressurised Metered<br />
Dose Aerosol Preparations for Inhalation<br />
Probenahme von Ausgangsstoffen und<br />
Verpackungsmaterialien<br />
Herstellung von Liquida, Cremes und<br />
Salben<br />
Herstellung von Dosieraerosolen <strong>zur</strong> Inhalation<br />
Annex 11 Computerised Systems Computergestützte Systeme<br />
Annex 12 Use of Ionising Radiation in the<br />
Manufacture of Medicinal Products<br />
Annex 13 Manufacture of Investigational Medicinal<br />
Products<br />
Annex 14 Manufacture of Products <strong>der</strong>ived from<br />
Human Blood or Human Plasma<br />
Verwendung ionisieren<strong>der</strong> Strahlen bei<br />
<strong>der</strong> Herstellung von <strong>Arzneimittel</strong>n<br />
Herstellung klinischer Prüfpräparate<br />
Herstellung von Produkten aus<br />
menschlichem Blut o<strong>der</strong> menschlichem<br />
Plasma<br />
Annex 15 Qualification and validation (July 2001) Qualifizierung und Validierung (Juli 2001)<br />
Annex 16 Certification by a Qualified person and<br />
Batch Release (July 2001)<br />
Zertifizierung einer Charge durch eine<br />
Sachkundige Person und Inverkehrbringen<br />
(Juli 2001)<br />
Annex 17 Parametric Release (July 2001) Parametrische Freigabe (Juli 2001)<br />
Annex 18 Good manufacturing practice for active <strong>EG</strong>-<strong>Leitfaden</strong> einer <strong>Guten</strong> Herstellungs-<br />
Übersetzung durch die Mitglie<strong>der</strong> <strong>der</strong> AG 42 des QS-Forums Schleswig-Holstein / Hamburg
pharmaceutical ingredients<br />
requirements for active substances used<br />
as starting materials from October 2005<br />
covered un<strong>der</strong> part II<br />
Annex 19 Reference and Retention Samples<br />
(December 2005)<br />
Annex 20 Quality Risk Management<br />
(February 2008)<br />
Glossary Glossar<br />
praxis <strong>für</strong> Wirkstoffe<br />
Die Anfor<strong>der</strong>ungen an Wirkstoffe finden<br />
sich seit Oktober 2005 in Teil II<br />
Referenz- und Rückstellmuster<br />
(Dezember 2005)<br />
Qualitätsrisikomanagement<br />
(Februar 2008)<br />
Übersetzung durch die Mitglie<strong>der</strong> <strong>der</strong> AG 42 des QS-Forums Schleswig-Holstein / Hamburg
Part I<br />
Chapter 1 Quality Management<br />
Teil 1<br />
Kapitel 1 Qualitätsmanagement<br />
Document History Dokumentenhistorie<br />
Revision to include new Chapter on Product<br />
Quality Review; October 2005<br />
Date of revised version coming into operation,<br />
indicating that a first Product Quality Review will<br />
be expected in 2006 for a minimum review<br />
period of at least 6 months. Subsequent reports<br />
Should cover a full 12 months’ period; 01<br />
January 2006<br />
Principle Grundsätze<br />
The hol<strong>der</strong> of a Manufacturing Authorisation<br />
must manufacture medicinal products so as to<br />
ensure that they are fit for their intended use,<br />
comply with the requirements of the Marketing<br />
Authorisation and do not place patients at risk<br />
due to inadequate safety, quality or efficacy. The<br />
attainment of this quality objective is the responsibility<br />
of senior management and requires the<br />
participation and commitment by staff in many<br />
different departments and at all levels within the<br />
company, by the company’s suppliers and by<br />
the distributors. To achieve the quality objective<br />
in a reliable manner there must be a<br />
comprehensively designed and correctly<br />
implemented system of Quality Assurance<br />
incorporating Good Manufacturing Practice and<br />
thus Quality Control. It should be fully<br />
documented and its effectiveness monitored. All<br />
parts of the Quality Assurance system should be<br />
adequately resourced with competent<br />
personnel, and suitable and sufficient premises,<br />
equipment and facilities. There are additional<br />
legal responsibilities for the hol<strong>der</strong> of the<br />
Manufacturing Authorisation and for the<br />
Qualified Person(s).<br />
Überarbeitete Fassung <strong>zur</strong> Aufnahme des neuen<br />
Kapitels über Produktqualitätsüberprüfung;<br />
Oktober 2005<br />
Datum des Inkrafttretens <strong>der</strong> überarbeiteten<br />
Fassung 01. Januar 2006. Für das Jahr 2006<br />
wird eine erste Produktqualitätsüberprüfung <strong>für</strong><br />
einen Zeitraum von mindestens 6 Monaten erwartet.<br />
Folgeberichte haben einen Zeitraum von<br />
12 Monaten abzudecken.<br />
Der Inhaber einer Herstellungserlaubnis muss<br />
<strong>Arzneimittel</strong> so herstellen 1 , dass ihre Eignung<br />
<strong>für</strong> den vorgesehenen Gebrauch gewährleistet<br />
ist, sie den im Rahmen <strong>der</strong> Zulassung spezifizierten<br />
Anfor<strong>der</strong>ungen entsprechen und sie die<br />
Patienten keiner Gefahr wegen un<strong>zur</strong>eichen<strong>der</strong><br />
Sicherheit, Qualität o<strong>der</strong> Wirksamkeit aussetzen.<br />
Für die Erreichung dieses Qualitätszieles<br />
ist die Geschäftsleitung eines Unternehmens<br />
verantwortlich; dies erfor<strong>der</strong>t die Beteiligung und<br />
Verpflichtung <strong>der</strong> Mitarbeiter in diversen Abteilungen<br />
und auf allen Ebenen des Unternehmens<br />
sowie <strong>der</strong> Lieferanten und Vertriebsunternehmen.<br />
Um dieses Qualitätsziel zuverlässig zu erreichen,<br />
muss das Unternehmen über ein umfassend<br />
konzipiertes und korrekt implementiertes<br />
System <strong>der</strong> Qualitätssicherung verfügen,<br />
das die Gute <strong>Herstellungspraxis</strong> und insoweit<br />
auch die Qualitätskontrolle beinhaltet. Dieses<br />
System ist vollständig zu dokumentieren und<br />
seine Funktionstüchtigkeit zu überwachen. Alle<br />
vom Qualitätssicherungssystem umfassten Bereiche<br />
sind angemessen mit kompetentem Personal<br />
sowie mit geeigneten und ausreichenden<br />
Räumlichkeiten und Ausrüstungen auszustatten.<br />
Für den Inhaber <strong>der</strong> Herstellungserlaubnis und<br />
<strong>für</strong> die Sachkundige(n) Person(en) bestehen zusätzliche<br />
rechtliche Verpflichtungen.<br />
1 Herstellung im Sinne des <strong>Leitfaden</strong>s bedeutet abweichend vom § 4 Abs. 14 AMG: „Alle Arbeitsgänge wie<br />
Beschaffung von Material und Produkten, Produktion, Qualitätskontrolle, Zertifizieren zum Inverkehrbringen,<br />
Lagerung und Vertrieb von <strong>Arzneimittel</strong>n und die dazugehörigen Kontrollen.“<br />
Übersetzung durch die Mitglie<strong>der</strong> <strong>der</strong> AG 42 des QS-Forums Schleswig-Holstein / Hamburg
1.1 The basic concepts of Quality Assurance,<br />
Good Manufacturing Practice and Quality<br />
Control are inter-related. They are described<br />
here in or<strong>der</strong> to emphasise their relationships<br />
and their fundamental importance to the production<br />
and control of medicinal products.<br />
Quality Assurance Qualitätssicherung<br />
1.2 Quality Assurance is a wide ranging concept<br />
which covers all matters which individually<br />
or collectively influence the quality of a product.<br />
It is the total sum of the organised arrangements<br />
made with the object of ensuring that medicinal<br />
products are of the quality required for their intended<br />
use. Quality Assurance therefore incorporates<br />
Good Manufacturing Practice plus other<br />
factors outside the scope of this Guide. The<br />
system of Quality Assurance appropriate for the<br />
manufacture of medicinal products should ensure<br />
that:<br />
(i) medicinal products are designed and developed<br />
in a way that takes account of the requirements<br />
of Good Manufacturing Practice and<br />
Good Laboratory Practice;<br />
(ii) production and control operations are clearly<br />
specified and Good Manufacturing Practice<br />
adopted;<br />
(iii) managerial responsibilities are clearly specified;<br />
(iv) arrangements are made for the<br />
manufacture, supply and use of the correct<br />
starting and packaging materials;<br />
(v) all necessary controls on intermediate<br />
products, and any other in-process controls and<br />
validations are carried out;<br />
(vi) the finished product is correctly processed<br />
and checked, according to the defined procedures;<br />
(vii) medicinal products are not sold or supplied<br />
before a Qualified Person has certified that each<br />
production batch has been produced and controlled<br />
in accordance with the requirements of<br />
the Marketing Authorisation and any other regulations<br />
relevant to the production, control and<br />
release of medicinal products;<br />
1.1 Die Grundkonzepte <strong>der</strong> Qualitätssicherung,<br />
<strong>der</strong> <strong>Guten</strong> <strong>Herstellungspraxis</strong> und <strong>der</strong><br />
Qualitätskontrolle sind miteinan<strong>der</strong> verflochten.<br />
Sie werden im Folgenden beschrieben, um ihre<br />
Verflechtung und grundlegende Bedeutung <strong>für</strong><br />
die Produktion und Prüfung von <strong>Arzneimittel</strong>n zu<br />
unterstreichen.<br />
1.2 Qualitätssicherung ist ein weitreichendes<br />
Konzept, das alles umfasst, was im Einzelnen<br />
o<strong>der</strong> im Zusammenwirken die Qualität eines<br />
Produktes beeinflusst. Dieses stellt die Gesamtheit<br />
aller vorgesehenen Maßnahmen dar, die<br />
getroffen werden, um sicherzustellen, dass <strong>Arzneimittel</strong><br />
die <strong>für</strong> den beabsichtigten Gebrauch<br />
erfor<strong>der</strong>liche Qualität aufweisen. Qualitätssicherung<br />
umfasst daher die Gute <strong>Herstellungspraxis</strong><br />
sowie weitere Faktoren, die über den Rahmen<br />
dieses <strong>Leitfaden</strong>s hinausgehen. Durch ein <strong>für</strong><br />
die Herstellung von <strong>Arzneimittel</strong>n geeignetes<br />
Qualitätssicherungssystem ist sicherzustellen,<br />
dass:<br />
(i) <strong>Arzneimittel</strong> unter Berücksichtigung <strong>der</strong> Anfor<strong>der</strong>ungen<br />
<strong>der</strong> <strong>Guten</strong> <strong>Herstellungspraxis</strong> und<br />
<strong>der</strong> <strong>Guten</strong> Laborpraxis konzipiert und entwickelt<br />
werden<br />
(ii) Produktions- und Prüfverfahren klar spezifiziert<br />
sind und die Gute <strong>Herstellungspraxis</strong> berücksichtigen<br />
(iii) Verantwortungsbereiche auf <strong>der</strong> Leitungsebene<br />
eindeutig festgelegt sind<br />
(iv) Vereinbarungen <strong>für</strong> die Herstellung, die Lieferung<br />
und den Einsatz <strong>der</strong> richtigen Ausgangsstoffe<br />
und Verpackungsmaterialien getroffen<br />
sind<br />
(v) alle notwendigen Prüfungen <strong>der</strong> Zwischenprodukte<br />
sowie alle weiteren Inprozesskontrollen<br />
und Validierungen durchgeführt werden<br />
(vi) das Fertigprodukt nach den festgelegten<br />
Verfahren ordnungsgemäß produziert und geprüft<br />
wird<br />
(vii) <strong>Arzneimittel</strong> nicht verkauft o<strong>der</strong> abgegeben<br />
werden, bevor eine Sachkundige Person die<br />
Produktion und Prüfung je<strong>der</strong> Herstellungscharge<br />
in Übereinstimmung mit den in <strong>der</strong> Zulassung<br />
festgelegten Anfor<strong>der</strong>ungen und allen an<strong>der</strong>en<br />
<strong>für</strong> die Produktion, Prüfung und das Inverkehrbringen<br />
von <strong>Arzneimittel</strong>n relevanten Vorschriften<br />
zertifiziert hat<br />
Übersetzung durch die Mitglie<strong>der</strong> <strong>der</strong> AG 42 des QS-Forums Schleswig-Holstein / Hamburg
(viii) satisfactory arrangements exist to ensure,<br />
as far as possible, that the medicinal products<br />
are stored, distributed and subsequently<br />
handled so that quality is maintained throughout<br />
their shelf life;<br />
(ix) there is a procedure for Self-Inspection<br />
and/or quality audit which regularly appraises<br />
the effectiveness and applicability of the Quality<br />
Assurance system.<br />
Good Manufacturing Practice for Medicinal<br />
Products (GMP)<br />
1.3 Good Manufacturing Practice is that part<br />
of Quality Assurance which ensures that<br />
products are consistently produced and<br />
controlled to the quality standards appropriate to<br />
their intended use and as required by the<br />
Marketing Authorisation or product specification.<br />
Good Manufacturing Practice is concerned with<br />
both production and quality control. The basic<br />
requirements of GMP are that:<br />
(i) all manufacturing processes are clearly defined,<br />
systematically reviewed in the light of experience<br />
and shown to be capable of consistently<br />
manufacturing medicinal products of<br />
the required quality and complying with their<br />
specifications;<br />
(ii) critical steps of manufacturing processes and<br />
significant changes to the process are validated;<br />
(iii) all necessary facilities for GMP are provided<br />
including:<br />
(iv) appropriately qualified and trained<br />
personnel;<br />
(v) adequate premises and space;<br />
(vi) suitable equipment and services;<br />
(vii) correct materials, containers and labels;<br />
(viii) approved procedures and instructions;<br />
(ix) suitable storage and transport;<br />
(x) instructions and procedures are written in an<br />
instructional form in clear and unambiguous language,<br />
specifically applicable to the facilities<br />
(viii) ausreichende Vorkehrungen bestehen, um<br />
so weit wie möglich sicherzustellen, dass die<br />
<strong>Arzneimittel</strong> so gelagert, vertrieben und anschließend<br />
gehandhabt werden, dass die Qualität<br />
während ihrer Laufzeit erhalten bleibt<br />
(ix) ein Verfahren <strong>der</strong> Selbstinspektion und/o<strong>der</strong><br />
Qualitätsaudit <strong>zur</strong> regelmäßigen Bewertung <strong>der</strong><br />
Wirksamkeit und Eignung des Qualitätssicherungssystems<br />
eingeführt ist.<br />
Gute <strong>Herstellungspraxis</strong> <strong>für</strong> <strong>Arzneimittel</strong><br />
(GMP)<br />
1.3 Die Gute <strong>Herstellungspraxis</strong> ist <strong>der</strong> Teil<br />
<strong>der</strong> Qualitätssicherung, <strong>der</strong> gewährleistet, dass<br />
die Produkte gleichbleibend nach den Qualitätsanfor<strong>der</strong>ungen<br />
produziert und geprüft werden,<br />
die <strong>der</strong> beabsichtigten Verwendung und den Zulassungsanfor<strong>der</strong>ungen<br />
o<strong>der</strong> <strong>der</strong> Produktspezifikation<br />
entsprechen. Die Gute <strong>Herstellungspraxis</strong><br />
betrifft sowohl die Produktion als auch die<br />
Qualitätskontrolle. Die grundlegenden Anfor<strong>der</strong>ungen<br />
<strong>der</strong> <strong>Guten</strong> <strong>Herstellungspraxis</strong> sind folgende:<br />
(i) Alle Vorgänge <strong>der</strong> Herstellung sind eindeutig<br />
definiert, werden unter Berücksichtigung <strong>der</strong><br />
vorliegenden Erfahrungen systematisch überprüft<br />
und sind nachweislich geeignet, kontinuierlich<br />
<strong>Arzneimittel</strong> zu erzeugen, die die gefor<strong>der</strong>te<br />
Qualität aufweisen und damit ihren Spezifikationen<br />
entsprechen;<br />
(ii) Kritische Schritte des Produktionsprozesses<br />
und alle wesentlichen Prozessän<strong>der</strong>ungen sind<br />
validiert;<br />
(iii) Alle <strong>für</strong> die Gute <strong>Herstellungspraxis</strong> erfor<strong>der</strong>lichen<br />
Voraussetzungen sind erfüllt, insbeson<strong>der</strong>e:<br />
(iv) Geeignetes qualifiziertes und geschultes<br />
Personal<br />
(v) Geeignete und ausreichend große Räume<br />
(vi) Geeignete Ausrüstung und <strong>der</strong>en technische<br />
Betreuung<br />
(vii) Richtige Ausgangsmaterialien, Behälter und<br />
Kennzeichnungen<br />
(viii) Genehmigte Verfahrens- und sonstige Anweisungen<br />
(ix) Geeignete Möglichkeiten <strong>für</strong> Lagerung und<br />
Transport;<br />
(x) Anweisungen und Verfahrensanweisungen<br />
sind als verbindliche Vorgaben schriftlich in<br />
klarer und eindeutiger Sprache verfasst, sie<br />
Übersetzung durch die Mitglie<strong>der</strong> <strong>der</strong> AG 42 des QS-Forums Schleswig-Holstein / Hamburg
provided;<br />
(xi) operators are trained to carry out procedures<br />
correctly;<br />
(xii) records are made, manually and/or by recording<br />
instruments, during manufacture which<br />
demonstrate that all the steps required by the<br />
defined procedures and instructions were in fact<br />
taken and that the quantity and quality of the<br />
product was as expected. Any significant deviations<br />
are fully recorded and investigated;<br />
(xiii) records of manufacture including<br />
distribution which enable the complete history of<br />
a batch to be traced, are retained in a<br />
comprehensible and accessible form;<br />
(xiv) the distribution (wholesaling) of the<br />
products minimises any risk to their quality;<br />
(xv) a system is available to recall any batch of<br />
product, from sale or supply;<br />
(xvi) complaints about marketed products are<br />
examined, the causes of quality defects investigated<br />
and appropriate measures taken in respect<br />
of the defective products and to prevent<br />
reoccurrence.<br />
Quality Control Qualitätskontrolle<br />
1.4 Quality Control is that part of Good Manufacturing<br />
Practice which is concerned with<br />
sampling, specifications and testing, and with<br />
the organisation, documentation and release<br />
procedures which ensure that the necessary<br />
and relevant tests are actually carried out and<br />
that materials are not released for use, nor<br />
products released for sale or supply, until their<br />
quality has been judged to be satisfactory.<br />
The basic requirements of Quality Control are<br />
that:<br />
(i) adequate facilities, trained personnel and approved<br />
procedures are available for sampling,<br />
gelten speziell <strong>für</strong> die entsprechende Betriebsstätte;<br />
(xi) Das ausführende Personal ist geschult, um<br />
die Arbeiten korrekt durchführen zu können;<br />
(xii) Während des Produktionsablaufs sind<br />
handschriftlich o<strong>der</strong> maschinell Aufzeichnungen<br />
zu erstellen, die belegen, dass alle Schritte, die<br />
nach den vorgegebenen Verfahren und Anweisungen<br />
erfor<strong>der</strong>lich sind, auch tatsächlich durchgeführt<br />
wurden. Es ist zu belegen, dass Qualität<br />
und Quantität des Produktes den Erwartungen<br />
entsprechen. Alle wesentlichen Abweichungen<br />
wurden untersucht und sind vollständig protokolliert;<br />
(xiii) Alle Aufzeichnungen über die Herstellung<br />
einschließlich <strong>der</strong> Abgabe an An<strong>der</strong>e müssen<br />
verständlich und leicht zugänglich sein. Sie<br />
müssen es ermöglichen, die vollständige Entstehungsgeschichte<br />
einer Charge <strong>zur</strong>ückverfolgen<br />
zu können;<br />
(xiv) Der Vertrieb <strong>der</strong> Produkte (Großhandel) erfolgt<br />
unter Bedingungen, die die Qualitätsrisiken<br />
minimieren:<br />
(xv) Es besteht ein System, das ermöglicht, jede<br />
Charge nach Auslieferung <strong>zur</strong>ück<strong>zur</strong>ufen;<br />
(xvi) Beanstandungen, die bereits in Verkehr gebrachte<br />
Chargen betreffen, sind zu überprüfen.<br />
Die Ursachen von Qualitätsfehlern sind zu ermitteln<br />
und die notwendigen Maßnahmen zu ergreifen.<br />
Vorkehrungen sind zu treffen, um ein erneutes<br />
Auftreten solcher Fehler in Zukunft zu<br />
vermeiden.<br />
1.4 Qualitätskontrolle ist <strong>der</strong> Teil <strong>der</strong> <strong>Guten</strong><br />
<strong>Herstellungspraxis</strong>, <strong>der</strong> sich mit Probenahme,<br />
Spezifikationen und Prüfungen sowie Organisations-,<br />
Dokumentations- und Freigabeverfahren<br />
2 befasst, mit denen gewährleistet wird, dass<br />
die jeweils notwendigen und relevanten Prüfungen<br />
tatsächlich durchgeführt werden und dass<br />
sowohl die benötigten Materialien freigegeben<br />
als auch die Fertigprodukte erst dann in den<br />
Verkehr gebracht werden, wenn ihre Qualität als<br />
angemessen beurteilt wurde.<br />
Die grundlegenden Anfor<strong>der</strong>ungen an die Qualitätskontrolle<br />
sind:<br />
(i) Geeignete Einrichtungen, geschultes Personal<br />
und genehmigte Verfahrensanweisungen<br />
2<br />
Mit o. g. „Freigabeverfahren“ ist we<strong>der</strong> das Verfahren gemäß § 16 AMWHV noch die Zertifizierung nach<br />
Annex 16 gemeint<br />
Übersetzung durch die Mitglie<strong>der</strong> <strong>der</strong> AG 42 des QS-Forums Schleswig-Holstein / Hamburg
inspecting and testing starting materials, packaging<br />
materials, intermediate, bulk, and finished<br />
products, and where appropriate for monitoring<br />
environmental conditions for GMP purposes;<br />
(ii) samples of starting materials, packaging materials,<br />
intermediate products, bulk products and<br />
finished products are taken by personnel and by<br />
methods approved by Quality Control;<br />
(iii) test methods are validated;<br />
(iv) records are made, manually and/or by recording<br />
instruments, which demonstrate that all<br />
the required sampling, inspecting and testing<br />
procedures were actually carried out. Any deviations<br />
are fully recorded and investigated;<br />
(v) the finished products contain active ingredients<br />
complying with the qualitative and quantitative<br />
composition of the Marketing Authorisation,<br />
are of the purity required, and are enclosed<br />
within their proper containers and correctly labelled;<br />
(vi) records are made of the results of inspection<br />
and that testing of materials, intermediate, bulk,<br />
and finished products is formally assessed<br />
against specification. Product assessment includes<br />
a review and evaluation of relevant production<br />
documentation and an assessment of<br />
deviations from specified procedures;<br />
(vii) no batch of product is released for sale or<br />
supply prior to certification by a Qualified Person<br />
that it is in accordance with the requirements of<br />
the Marketing Authorisation;<br />
(viii) sufficient reference samples of starting<br />
materials and products are retained to permit<br />
future examination of the product if necessary<br />
and that the product is retained in its final pack<br />
unless exceptionally large packs are produced.<br />
sind verfügbar <strong>für</strong> die Probenahme und Prüfung<br />
von Ausgangsstoffen, Verpackungsmaterialien,<br />
Zwischenprodukten, Bulkware sowie Fertigprodukten<br />
und, soweit dies die Gute <strong>Herstellungspraxis</strong><br />
erfor<strong>der</strong>t, <strong>für</strong> die Umgebungsbedingungen;<br />
(ii) Proben von Ausgangsstoffen, Verpackungsmaterialien,<br />
Zwischenprodukten, Bulkware und<br />
Fertigprodukten werden durch Personen und<br />
nach Methoden entnommen, die von <strong>der</strong> Qualitätskontrolle<br />
genehmigt wurden;<br />
(iii) Die Prüfmethoden sind validiert;<br />
(iv) Es werden handschriftlich und/o<strong>der</strong> maschinell<br />
Aufzeichnungen geführt, die belegen, dass<br />
alle erfor<strong>der</strong>lichen Probenahmen, Kontroll- und<br />
Prüfverfahren tatsächlich durchgeführt wurden.<br />
Jede Abweichung wird vollständig aufgezeichnet<br />
und untersucht;<br />
(v) Die Fertigprodukte enthalten Wirkstoffe, die<br />
qualitativ und quantitativ <strong>der</strong> zugelassenen Zusammensetzung<br />
entsprechen, weisen die erfor<strong>der</strong>liche<br />
Reinheit auf, befinden sich in den richtigen<br />
Behältnissen und sind ordnungsgemäß gekennzeichnet;<br />
(vi) Über die Prüfung <strong>der</strong> Materialien, Zwischenprodukte<br />
und Bulkware sowie <strong>der</strong> Fertigprodukte<br />
werden Aufzeichnungen (Protokolle) erstellt; die<br />
Ergebnisse werden mit den Anfor<strong>der</strong>ungen <strong>der</strong><br />
Spezifikation verglichen. Zur Produktbewertung<br />
gehören die Überprüfung und Beurteilung <strong>der</strong><br />
jeweiligen Produktionsdokumentation 3 und eine<br />
Bewertung eventueller Abweichungen von den<br />
festgelegten Verfahren;<br />
(vii) Eine Produktcharge darf erst in den Verkehr<br />
gebracht werden, wenn zuvor eine Sachkundige<br />
Person die Übereinstimmung mit den in <strong>der</strong> Zulassung<br />
festgelegten Anfor<strong>der</strong>ungen zertifiziert<br />
hat;<br />
(viii) Rückstellmuster von Ausgangsstoffen und<br />
Produkten werden in ausreichen<strong>der</strong> Menge aufbewahrt,<br />
um ggf. erneut untersucht werden zu<br />
können. Das Fertigprodukt ist in seiner endgültigen<br />
Verpackung aufzubewahren, es sei denn,<br />
es handelt sich um außergewöhnlich große<br />
Packungen. 4<br />
3 Herstellungsprotokoll gemäß § 13 Abs. 7 AMWHV<br />
4 Annex 19 unterscheidet zwischen reference samples (Referenzmuster <strong>zur</strong> Wie<strong>der</strong>holung <strong>der</strong> analytischen<br />
Prüfungen) und retention samples (Rückstellmuster als Anschauungsmuster von Fertigprodukten)<br />
Übersetzung durch die Mitglie<strong>der</strong> <strong>der</strong> AG 42 des QS-Forums Schleswig-Holstein / Hamburg
Product Quality Review Produktqualitätsüberprüfung<br />
1.5. Regular periodic or rolling quality reviews<br />
of all licensed medicinal products, including export<br />
only products, should be conducted with the<br />
objective of verifying the consistency of the<br />
existing process, the appropriateness of current<br />
specifications for both starting materials and<br />
finished product to highlight any trends and to<br />
identify product and process improvements.<br />
Such reviews should normally be conducted and<br />
documented annually, taking into account previous<br />
reviews, and should include at least:<br />
(i) A review of starting materials and packaging<br />
materials used for the product, especially those<br />
from new sources.<br />
(ii) A review of critical in-process controls and<br />
finished product results.<br />
(iii) A review of all batches that failed to meet<br />
established specification(s) and their investigation.<br />
(iv) A review of all significant deviations or nonconformances,<br />
their related investigations, and<br />
the effectiveness of resultant corrective and preventative<br />
actions taken.<br />
(v) A review of all changes carried out to the<br />
processes or analytical methods.<br />
(vi) A review of Marketing Authorisation variations<br />
submitted/granted/refused, including those<br />
for third country (export only) dossiers.<br />
(vii) A review of the results of the stability monitoring<br />
programme and any adverse trends.<br />
(viii) A review of all quality-related returns, complaints<br />
and recalls and the investigations performed<br />
at the time.<br />
(ix) A review of adequacy of any other previous<br />
product process or equipment corrective<br />
actions.<br />
(x) For new marketing authorisations and variations<br />
to marketing authorisations, a review of<br />
post-marketing commitments.<br />
(xi) The qualification status of relevant equip-<br />
1.5 Es sind in festgelegten Intervallen o<strong>der</strong><br />
kontinuierlich wie<strong>der</strong>kehrend Produktqualitätsüberprüfungen<br />
aller zugelassenen <strong>Arzneimittel</strong><br />
sowie <strong>der</strong> nur <strong>für</strong> den Export bestimmten Produkte<br />
mit dem Ziel durchzuführen, die Konstanz<br />
des vorhandenen Prozesses und die Angemessenheit<br />
<strong>der</strong> aktuellen Spezifikationen sowohl <strong>für</strong><br />
die Ausgangsstoffe als auch <strong>für</strong> das Fertigprodukt<br />
zu belegen, um Trends aufzuzeigen sowie<br />
Verbesserungsmöglichkeiten <strong>für</strong> Produkte und<br />
Abläufe zu erkennen. Solche Überprüfungen<br />
werden üblicherweise jährlich und unter Berücksichtigung<br />
vorhergehen<strong>der</strong> Überprüfungen<br />
durchgeführt und dokumentiert. Sie haben mindestens<br />
zu beinhalten:<br />
Eine Zusammenstellung und Bewertung <strong>der</strong><br />
Daten<br />
(i) <strong>der</strong> <strong>für</strong> das Produkt eingesetzten Ausgangsstoffe<br />
und Verpackungsmaterialien, beson<strong>der</strong>s<br />
von solchen, die von neuen Lieferanten bezogen<br />
wurden<br />
(ii) kritischer Inprozesskontrollen und <strong>der</strong> Ergebnisse<br />
von Fertigproduktprüfungen<br />
(iii) aller Chargen, die den festgelegten Spezifikationen<br />
nicht entsprachen und <strong>der</strong> dazugehörigen<br />
Untersuchungen<br />
(iv) aller signifikanten Abweichungen o<strong>der</strong> OoS-<br />
Ergebnisse, <strong>der</strong> dazugehörigen Untersuchungen<br />
und <strong>der</strong> Effektivität daraus resultieren<strong>der</strong><br />
Korrektur- und Präventivmaßnahmen (CAPA)<br />
(v) eine Überprüfung und Bewertung aller durchgeführten<br />
Än<strong>der</strong>ungen am Prozess o<strong>der</strong> den<br />
Analysenverfahren<br />
(vi) eine Überprüfung und Bewertung <strong>der</strong> eingereichten/genehmigten/abgelehnten<br />
Än<strong>der</strong>ungen<br />
an <strong>der</strong> Zulassung - das gleiche gilt <strong>für</strong> nicht zugelassene<br />
<strong>Arzneimittel</strong>, die ausschließlich <strong>für</strong><br />
den Export in Drittlän<strong>der</strong> bestimmt sind<br />
(vii) eine Überprüfung und Bewertung aller Ergebnisse<br />
<strong>der</strong> Stabilitätsprüfung und etwaiger<br />
Trends<br />
(viii) eine Überprüfung und Bewertung aller qualitätsbezogenen<br />
Rücklieferungen, Beanstandungen<br />
und Rückrufe und <strong>der</strong> zum jeweiligen Zeitpunkt<br />
durchgeführten Untersuchungen<br />
(ix) eine Überprüfung und Bewertung <strong>der</strong> Angemessenheit<br />
aller früheren Korrekturmaßnahmen<br />
an Herstellungsprozess o<strong>der</strong> Ausrüstung<br />
(x) bei neuen Zulassungen o<strong>der</strong> bei Zulassungsän<strong>der</strong>ungen<br />
die Überprüfung <strong>der</strong> im Rahmen<br />
<strong>der</strong> Zulassung erteilten Auflagen<br />
(xi) den Qualifizierungsstatus erfor<strong>der</strong>licher Aus-<br />
Übersetzung durch die Mitglie<strong>der</strong> <strong>der</strong> AG 42 des QS-Forums Schleswig-Holstein / Hamburg
ment and utilities, e.g. HVAC, water, compressed<br />
gases, etc.<br />
(xii) A review of Technical Agreements to ensure<br />
that they are up to date. The manufacturer and<br />
marketing authorisation hol<strong>der</strong>, where different,<br />
should evaluate the results of this review and an<br />
assessment should be made whether corrective<br />
and preventative action or any revalidation<br />
should be un<strong>der</strong>taken. Reasons for such corrective<br />
actions should be documented. Agreed corrective<br />
and preventative actions should be completed<br />
in a timely and effective manner. There<br />
should be management procedures for the ongoing<br />
management and review of these actions<br />
and the effectiveness of these procedures verified<br />
during selfinspection. Quality reviews may<br />
be grouped by product type, e.g. solid dosage<br />
forms, liquid dosage forms, sterile products, etc.<br />
where scientifically justified. Where the marketing<br />
authorisation hol<strong>der</strong> is not the manufacturer,<br />
there should be a technical agreement in place<br />
between the various parties that defines their<br />
respective responsibilities in producing the quality<br />
review. The Qualified Person responsible for<br />
final batch certification together with the marketing<br />
authorisation hol<strong>der</strong> should ensure that the<br />
quality review is performed in a timely manner<br />
and is accurate.<br />
CHAPTER 2 PERSONNEL Kapitel 2 Personal<br />
Principle Grundsätze<br />
The establishment and maintenance of a satisfactory<br />
system of quality assurance and the correct<br />
manufacture of medicinal products relies<br />
upon people. For this reason there must be<br />
sufficient qualified personnel to carry out all the<br />
tasks which are the responsibility of the<br />
manufacturer. Individual responsibilities should<br />
be clearly un<strong>der</strong>stood by the individuals and<br />
recorded. All personnel should be aware of the<br />
principles of Good Manufacturing Practice that<br />
affect them and receive initial and continuing<br />
training, including hygiene instructions, relevant<br />
to their needs.<br />
5<br />
6<br />
„Unverzüglich“ im Sinne von „ohne schuldhaftes Zögern“<br />
„Zertifizierung“ entspricht <strong>der</strong> Freigabe im Sinne des § 16 <strong>der</strong> AMWHV<br />
rüstung und Betriebsmittel, z. B. Heizung / Belüftung<br />
/ Klimatisierung, Wasser, Druckgase etc.<br />
(xii) Ferner ist eine Überprüfung und Bewertung<br />
<strong>der</strong> Verantwortungsabgrenzungsverträge durchzuführen,<br />
um sicherzustellen, dass sie auf aktuellem<br />
Stand sind. Der Hersteller und, sofern<br />
nicht identisch, <strong>der</strong> Zulassungsinhaber haben<br />
die Ergebnisse dieser Überprüfung zu bewerten<br />
und eine Einschätzung abzugeben, ob korrigierende<br />
und/o<strong>der</strong> vorbeugende Maßnahmen erfor<strong>der</strong>lich<br />
sind o<strong>der</strong> es einer Revalidierung bedarf.<br />
Die Begründungen <strong>für</strong> solche Korrekturmaßnahmen<br />
sind zu dokumentieren. Sind Korrekturmaßnahmen<br />
beschlossen, sind sie unverzüglich<br />
5 und in wirksamer Weise umzusetzen.<br />
Verfahrensabläufe <strong>für</strong> die verantwortliche Leitungsebene<br />
<strong>zur</strong> Überwachung <strong>der</strong> Maßnahmen<br />
müssen vorhanden sein. Ihre Wirksamkeit ist im<br />
Rahmen von Selbstinspektionen nachzuweisen.<br />
Qualitätsüberprüfungen können nach Arzneiformen<br />
zusammengefasst werden, z. B. feste<br />
o<strong>der</strong> flüssige Darreichungsformen, Sterilprodukte,<br />
etc. soweit dies wissenschaftlich gerechtfertigt<br />
ist. Falls <strong>der</strong> Zulassungsinhaber nicht <strong>der</strong><br />
Hersteller des Produktes ist, sind Verantwortungsabgrenzungsverträge<br />
zwischen allen<br />
beteiligten Parteien abzuschließen. Darin sind<br />
die jeweiligen Verantwortlichkeiten <strong>für</strong> die<br />
Durchführung <strong>der</strong> Qualitätsüberprüfung und<br />
-bewertung festzulegen. Die Sachkundige<br />
Person, die <strong>für</strong> die Zertifizierung 6 <strong>der</strong> Chargen<br />
verantwortlich ist, hat zusammen mit dem Zulassungsinhaber<br />
sicherzustellen, dass die Qualitätsüberprüfung<br />
und Bewertung korrekt und in<br />
angemessener Zeit durchgeführt wird.<br />
Aufbau und Erhaltung eines angemessenen<br />
Qualitätssicherungssystems und die ordnungsgemäße<br />
Herstellung von <strong>Arzneimittel</strong>n sind im<br />
wesentlichen abhängig vom Personal. Daher<br />
muss qualifiziertes Personal in ausreichen<strong>der</strong><br />
Zahl vorhanden sein, um alle in <strong>der</strong> Verantwortung<br />
des Herstellers liegenden Aufgaben ausführen<br />
zu können. Die jeweiligen Verantwortungsbereiche<br />
sind schriftlich nie<strong>der</strong>zulegen. Die<br />
Beteiligten müssen sich ihrer Verantwortung bewusst<br />
sein. Alle Mitarbeiter haben mit den sie<br />
betreffenden Grundsätzen <strong>der</strong> <strong>Guten</strong> <strong>Herstellungspraxis</strong><br />
vertraut zu sein.<br />
Übersetzung durch die Mitglie<strong>der</strong> <strong>der</strong> AG 42 des QS-Forums Schleswig-Holstein / Hamburg
Schulungen sind zu Beginn <strong>der</strong> Tätigkeit und<br />
fortlaufend durchzuführen; dazu gehören auch<br />
die jeweils notwendigen Hygieneunterweisungen.<br />
General Allgemeine Anfor<strong>der</strong>ungen<br />
2.1 The manufacturer should have an adequate<br />
number of personnel with the necessary<br />
qualifications and practical experience. The responsibilities<br />
placed on any one individual<br />
should not be so extensive as to present any<br />
risk to quality.<br />
2.2 The manufacturer must have an organisation<br />
chart. People in responsible positions<br />
should have specific duties recorded in written<br />
job descriptions and adequate authority to carry<br />
out their responsibilities. Their duties may be<br />
delegated to designated deputies of a<br />
satisfactory qualification level. There should be<br />
no gaps or unexplained overlaps in the<br />
responsibilities of those personnel concerned<br />
with the application of Good Manufacturing<br />
Practice.<br />
2.1 Der Hersteller hat über Personal in ausreichen<strong>der</strong><br />
Zahl und mit <strong>der</strong> erfor<strong>der</strong>lichen Qualifikation<br />
und praktischen Erfahrung zu verfügen.<br />
Die dem Einzelnen zugewiesenen Verantwortungsbereiche<br />
dürfen nicht so umfangreich sein,<br />
dass sich daraus Risiken <strong>für</strong> die Qualität ergeben.<br />
2.2 Der Hersteller muss über ein Organigramm<br />
verfügen. Für Mitarbeiter in verantwortlicher<br />
Stellung sind <strong>der</strong>en Aufgaben in Stellenbeschreibungen<br />
festzulegen. Diesen Mitarbeitern<br />
sind die entsprechenden Vollmachten zu<br />
erteilen, damit sie ihrer Verantwortung gerecht<br />
werden können. Aufgaben können auch auf<br />
hier<strong>für</strong> benannte, ausreichend qualifizierte Vertreter<br />
übertragen werden. Zwischen den Verantwortungsbereichen<br />
des mit <strong>der</strong> Umsetzung <strong>der</strong><br />
<strong>Guten</strong> <strong>Herstellungspraxis</strong> befassten Personals<br />
dürfen keine Lücken o<strong>der</strong> unbegründeten Überschneidungen<br />
bestehen.<br />
Key Personnel Personal in Schlüsselstellung<br />
2.3 Key Personnel include the head of Production,<br />
the head of Quality Control, and if at<br />
least one of these persons is not responsible for<br />
the duties described in Article 51 of Directive<br />
2001/83/EC, the Qualified Person(s) designated<br />
for the purpose. Normally key posts should be<br />
occupied by full-time personnel. The heads of<br />
Production and Quality Control must be independent<br />
from each other. In large organisations,<br />
it may be necessary to delegate some of the<br />
functions listed in 2.5, 2.6 and 2.7.<br />
2.4 The duties of the Qualified Person(s) are<br />
fully described in Article 51 of Directive<br />
2001/83/EC 10 , and can be summarised as<br />
follows:<br />
a) for medicinal products manufactured within<br />
the European Community, a Qualified Person<br />
must ensure that each batch has been produced<br />
and tested/checked in accordance with the di-<br />
7<br />
2.3 Zum Personal in Schlüsselstellung 7 gehören<br />
<strong>der</strong> Leiter <strong>der</strong> Herstellung, <strong>der</strong> Leiter <strong>der</strong><br />
Qualitätskontrolle sowie, wenn nicht mindestens<br />
einer <strong>der</strong> beiden Genannten <strong>für</strong> die in Art. 51 <strong>der</strong><br />
Direktive 2001/83/<strong>EG</strong> 8 beschriebenen Aufgaben<br />
verantwortlich ist, die hier<strong>für</strong> zuständige(n)<br />
Sachkundige(n) Person(en). Schlüsselstellungen<br />
sind üblicherweise mit Vollzeitbeschäftigten<br />
zu besetzen. Der Leiter <strong>der</strong> Herstellung und <strong>der</strong><br />
Leiter <strong>der</strong> Qualitätskontrolle müssen voneinan<strong>der</strong><br />
unabhängig sein. In großen Unternehmen<br />
kann es notwendig sein, einige <strong>der</strong> unter 2.5,<br />
2.6 und 2.7 genannten Aufgaben zu delegieren.<br />
2.4 Die Pflichten einer/<strong>der</strong> Sachkundigen Person(en)<br />
sind in Artikel 51 <strong>der</strong> Direktive<br />
2001/83/<strong>EG</strong> 8 umfassend beschrieben und können<br />
folgen<strong>der</strong>maßen zusammengefasst werden:<br />
a) <strong>für</strong> die innerhalb <strong>der</strong> Europäischen Gemeinschaft<br />
hergestellten <strong>Arzneimittel</strong> muss eine<br />
Sachkundige Person sicherstellen, dass jede<br />
Charge in Übereinstimmung mit den gesetz-<br />
In Deutschland gehören dazu auch <strong>der</strong> Stufenplanbeauftragte und <strong>der</strong> Informationsbeauftragte<br />
8 Für Tierarzneimittel gilt Artikel 55 <strong>der</strong> Direktive 2001/82/<strong>EG</strong>.<br />
Übersetzung durch die Mitglie<strong>der</strong> <strong>der</strong> AG 42 des QS-Forums Schleswig-Holstein / Hamburg
ectives and the marketing authorisation 9 ;<br />
(b) for medicinal products manufactured outside<br />
the European Community, a Qualified Person<br />
must ensure that each imported batch has un<strong>der</strong>gone,<br />
in the importing country, the testing<br />
specified in paragraph 1 (b) of Article 51;<br />
(c) a Qualified Person must certify in a register<br />
or equivalent document, as operations are<br />
carried out and before any release, that each<br />
production batch satisfies the provisions of<br />
Article 51. The persons responsible for these<br />
duties must meet the qualification requirements<br />
laid down in Article 49 of the same Directive,<br />
they shall be permanently and continuously at<br />
the disposal of the hol<strong>der</strong> of the Manufacturing<br />
Authorisation to carry out their responsibilities.<br />
Their responsibilities may be delegated, but only<br />
to other Qualified Person(s).<br />
2.5 The head of the Production Department<br />
generally has the following responsibilities:<br />
i. to ensure that products are produced and<br />
stored according to the appropriate documentation<br />
in or<strong>der</strong> to obtain the required quality;<br />
ii. to approve the instructions relating to production<br />
operations and to ensure their strict implementation;<br />
iii. to ensure that the production records are<br />
evaluated and signed by an authorised person<br />
before they are sent to the Quality Control Department;<br />
iv. to check the maintenance of his department,<br />
premises and equipment;<br />
v. to ensure that the appropriate validations are<br />
done;<br />
lichen Bestimmungen und den For<strong>der</strong>ungen <strong>der</strong><br />
Zulassung produziert und geprüft wurde 9 ;<br />
b) <strong>für</strong> außerhalb <strong>der</strong> Europäischen Gemeinschaft<br />
hergestellte <strong>Arzneimittel</strong> muss eine Sachkundige<br />
Person sicherstellen, dass jede importierte<br />
Charge im Einfuhrland den in Artikel 51<br />
Abs. 1(b) 10 aufgeführten Prüfungen unterzogen<br />
wurde;<br />
c) eine Sachkundige Person muss vor jedem<br />
Inverkehrbringen in einem Register o<strong>der</strong> einem<br />
gleichwertigen Dokument zertifizieren, dass jede<br />
produzierte (Fertigprodukt-) Charge den Anfor<strong>der</strong>ungen<br />
des Artikels 51 8 genügt. Das Register<br />
o<strong>der</strong> vergleichbare Dokument ist auf aktuellem<br />
Stand zu halten. Die <strong>für</strong> diese Pflichten verantwortlichen<br />
Personen müssen die in Artikel 49<br />
<strong>der</strong> o. a. Direktive 11 festgelegte Qualifikation erfüllen<br />
und müssen dem Inhaber <strong>der</strong> Herstellungserlaubnis<br />
ständig <strong>zur</strong> Verfügung stehen,<br />
um ihren Verpflichtungen nachkommen zu können.<br />
Ihre Pflichten können delegiert werden, jedoch<br />
nur an (eine) an<strong>der</strong>e Sachkundige<br />
Person(en).<br />
2.5 Der Leiter <strong>der</strong> Herstellung hat im Allgemeinen<br />
folgende Verantwortlichkeiten:<br />
(i) Sicherstellung, dass die Produkte gemäß den<br />
entsprechenden Anweisungen produziert und<br />
gelagert werden, um die gefor<strong>der</strong>te Qualität zu<br />
erhalten<br />
(ii) Genehmigung <strong>der</strong> Anweisungen <strong>für</strong> die Produktionsvorgänge<br />
und Sicherstellung, dass diese<br />
genau eingehalten werden<br />
(iii) Sicherstellung, dass die Herstellungsprotokolle<br />
von einer befugten Person 12 überprüft und<br />
unterschrieben werden, bevor sie an die Abteilung<br />
<strong>für</strong> Qualitätskontrolle weitergegeben werden<br />
(iv) Kontrolle <strong>der</strong> Wartung, <strong>der</strong> Räumlichkeiten<br />
und <strong>der</strong> Ausrüstung seiner Abteilung<br />
(v) Sicherstellung, dass die notwendigen Validierungen<br />
durchgeführt werden<br />
9 According to Directive 75/319/EEC (now codified Directive 2001/83/EC) and the Ruling (Case 247/81) of the<br />
Court of Justice of the European Commununities, medicinal products which have been properly controlled in<br />
the EU by a Qualified Person do not have to be recontrolled or rechecked in any other Member State of the<br />
Community.<br />
Gemäß <strong>der</strong> Direktive 75/319/EWG (jetzt kodifizierte Direktive 2001/83/<strong>EG</strong>) und <strong>der</strong> Entscheidung des<br />
Europäischen Gerichtshofs (Fall 247/81) brauchen <strong>Arzneimittel</strong>, die in <strong>der</strong> <strong>EG</strong> durch eine Sachkundige Person<br />
ordnungsgemäß zertifiziert worden sind, in keinem an<strong>der</strong>en Mitgliedstaat <strong>der</strong> Gemeinschaften nochmals<br />
zertifiziert o<strong>der</strong> nachgeprüft zu werden.<br />
10<br />
<strong>für</strong> Tierarzneimittel Artikel 55 Abs. 1(b) <strong>der</strong> Direktive 2001/82/<strong>EG</strong><br />
11<br />
Für die Herstellung von Tierarzneimitteln siehe Direktive 2001/82/<strong>EG</strong>, Artikel 53<br />
12<br />
Nach § 12 AMWHV ist dies <strong>der</strong> Leiter <strong>der</strong> Herstellung.<br />
Übersetzung durch die Mitglie<strong>der</strong> <strong>der</strong> AG 42 des QS-Forums Schleswig-Holstein / Hamburg
vi. to ensure that the required initial and continuing<br />
training of his department personnel is<br />
carried out and adapted according to need.<br />
2.6 The head of the Quality Control Department<br />
generally has the following responsibilities:<br />
i. to approve or reject, as he sees fit, starting<br />
materials, packaging materials, and intermediate,<br />
bulk and finished products;<br />
ii. to evaluate batch records;<br />
iii. to ensure that all necessary testing is carried<br />
out;<br />
iv. to approve specifications, sampling instructions,<br />
test methods and other Quality Control<br />
procedures;<br />
v. to approve and monitor any contract analysts;<br />
vi. to check the maintenance of his department,<br />
premises and equipment;<br />
vii. to ensure that the appropriate validations are<br />
done;<br />
viii. to ensure that the required initial and continuing<br />
training of his department personnel is<br />
carried out and adapted according to need.<br />
Other duties of the Quality Control Department<br />
are summarised in Chapter 6.<br />
2.7 The heads of Production and Quality<br />
Control generally have some shared, or jointly<br />
exercised, responsibilities relating to quality.<br />
These may include, subject to any national<br />
regulations:<br />
— the authorisation of written procedures and<br />
other documents, including amendments;<br />
— the monitoring and control of the manufacturing<br />
environment;<br />
— plant hygiene;<br />
— process validation;<br />
— training;<br />
— the approval and monitoring of suppliers of<br />
materials;<br />
— the approval and monitoring of contract<br />
(vi) Sicherstellung, dass die erfor<strong>der</strong>liche anfängliche<br />
und fortlaufende Schulung des Personals<br />
seiner Abteilung durchgeführt und entsprechend<br />
den jeweiligen Erfor<strong>der</strong>nissen angepasst<br />
wird.<br />
2.6 Der Leiter <strong>der</strong> Qualitätskontrolle hat im<br />
Allgemeinen folgende Verantwortlichkeiten:<br />
(i) Billigung o<strong>der</strong>, falls er es <strong>für</strong> erfor<strong>der</strong>lich hält,<br />
Zurückweisung von Ausgangsstoffen, Verpackungsmaterialien,<br />
Zwischenprodukten, Bulkware<br />
und Fertigprodukten<br />
(ii) Überprüfung <strong>der</strong> Chargenprotokolle<br />
(iii) Sicherstellung, dass alle erfor<strong>der</strong>lichen Prüfungen<br />
durchgeführt werden<br />
(iv) Genehmigung von Spezifikationen, Anweisungen<br />
<strong>zur</strong> Probenahme, Prüfmethoden und<br />
an<strong>der</strong>en Verfahren <strong>zur</strong> Qualitätskontrolle<br />
(v) Genehmigung und Überwachung von Auftragslaboratorien<br />
(vi) Kontrolle <strong>der</strong> Wartung, <strong>der</strong> Räumlichkeiten<br />
und <strong>der</strong> Ausrüstung seiner Abteilung<br />
(vii) Sicherstellung, dass die notwendigen Validierungen<br />
durchgeführt werden<br />
(viii) Sicherstellung, dass die erfor<strong>der</strong>liche anfängliche<br />
und fortlaufende Schulung des Personals<br />
seiner Abteilung durchgeführt und entsprechend<br />
den jeweiligen Erfor<strong>der</strong>nissen angepasst<br />
wird. Weitere Pflichten <strong>der</strong> Abteilung <strong>für</strong><br />
Qualitätskontrolle sind in Kapitel 6 zusammengefasst.<br />
2.7 Der Leiter <strong>der</strong> Herstellung und <strong>der</strong> Leiter<br />
<strong>der</strong> Qualitätskontrolle teilen im Allgemeinen einige<br />
die Qualität betreffenden Verantwortungsbereiche<br />
untereinan<strong>der</strong> auf o<strong>der</strong> üben die Verantwortung<br />
gemeinsam aus. Je nach nationalen<br />
Regelungen können dies sein:<br />
— Genehmigung schriftlicher Verfahrensanweisungen<br />
und an<strong>der</strong>er Vorgaben einschließlich Ergänzungen<br />
— Überwachung und Kontrolle <strong>der</strong> Umgebungsbedingungen<br />
bei <strong>der</strong> Herstellung<br />
— Betriebshygiene<br />
— Validierung von Verfahren<br />
— Schulung<br />
— Genehmigung und Überwachung von Lieferanten<br />
— Genehmigung und Überwachung <strong>der</strong> Auf-<br />
Übersetzung durch die Mitglie<strong>der</strong> <strong>der</strong> AG 42 des QS-Forums Schleswig-Holstein / Hamburg
manufacturers;<br />
— the designation and monitoring of storage<br />
conditions for materials and products;<br />
— the retention of records;<br />
— the monitoring of compliance with the requirements<br />
of Good Manufacturing Practice;<br />
— the inspection, investigation, and taking of<br />
samples, in or<strong>der</strong> to monitor factors which may<br />
affect product quality.<br />
Training Schulung<br />
2.8 The manufacturer should provide training<br />
for all the personnel whose duties take them into<br />
production areas or into control laboratories (including<br />
the technical, maintenance and cleaning<br />
personnel), and for other personnel whose activities<br />
could affect the quality of the product.<br />
2.9 Besides the basic training on the theory<br />
and practice of Good Manufacturing Practice,<br />
newly recruited personnel should receive<br />
training appropriate to the duties assigned to<br />
them. Continuing training should also be given,<br />
and its practical effectiveness should be<br />
periodically assessed. Training programmes<br />
should be available, approved by either the<br />
head of Production or the head of Quality<br />
Control, as appropriate. Training records should<br />
be kept.<br />
2.10 Personnel working in areas where contamination<br />
is a hazard, e.g. clean areas or areas<br />
where highly active, toxic, infectious or sensitising<br />
materials are handled, should be given specific<br />
training.<br />
2.11 Visitors or untrained personnel should,<br />
preferably, not be taken into the production and<br />
quality control areas. If this is unavoidable, they<br />
should be given information in advance, particularly<br />
about personal hygiene and the prescribed<br />
protective clothing. They should be closely supervised.<br />
2.12 The concept of Quality Assurance and all<br />
the measures capable of improving its un<strong>der</strong>standing<br />
and implementation should be fully discussed<br />
during the training sessions.<br />
Personnel Hygiene Personalhygiene<br />
2.13 Detailed hygiene programmes should be<br />
established and adapted to the different needs<br />
tragshersteller<br />
— Festlegung und Überwachung <strong>der</strong> Lagerungsbedingungen<br />
<strong>für</strong> Materialien und Produkte<br />
— Aufbewahrung von Protokollen<br />
— Überwachung <strong>der</strong> Einhaltung <strong>der</strong> Anfor<strong>der</strong>ungen<br />
<strong>der</strong> <strong>Guten</strong> <strong>Herstellungspraxis</strong><br />
— Überprüfung, Untersuchung und Entnahme<br />
von Proben <strong>zur</strong> Überwachung von Faktoren, die<br />
die Produktqualität beeinflussen können.<br />
2.8 Der Hersteller hat <strong>für</strong> die Schulung aller<br />
Personen zu sorgen, die Aufgaben in den Produktionsbereichen<br />
o<strong>der</strong> in Kontrolllaboratorien<br />
zu erfüllen haben (einschließlich des technischen,<br />
des Wartungs- und Reinigungspersonals).<br />
Auch an<strong>der</strong>es Personal, dessen Tätigkeit<br />
die Produktqualität beeinflussen könnte, ist zu<br />
schulen.<br />
2.9 Neben <strong>der</strong> theoretischen und praktischen<br />
Basisschulung in Guter <strong>Herstellungspraxis</strong> sind<br />
neu eingestellte Personen entsprechend den<br />
ihnen jeweils zugewiesenen Aufgaben zu schulen.<br />
Darüber hinaus ist eine fortlaufende Schulung<br />
durchzuführen und <strong>der</strong>en erfolgreiche Umsetzung<br />
in die Praxis regelmäßig zu überprüfen.<br />
Schulungsprogramme, je nach Inhalt vom Leiter<br />
<strong>der</strong> Herstellung o<strong>der</strong> vom Leiter <strong>der</strong> Qualitätskontrolle<br />
genehmigt, sind erfor<strong>der</strong>lich. Die Schulungsprotokolle<br />
sind aufzubewahren.<br />
2.10 Personal, das in Bereichen mit beson<strong>der</strong>en<br />
Kontaminationsrisiken arbeitet (z. B. in Reinräumen<br />
o<strong>der</strong> in Bereichen, in denen mit hochaktiven,<br />
toxischen, infektiösen o<strong>der</strong> sensibilisierenden<br />
Stoffen umgegangen wird), ist speziell<br />
zu schulen.<br />
2.11 Besucher o<strong>der</strong> ungeschultes Personal<br />
sollen möglichst keine Produktions- und Qualitätskontrollbereiche<br />
betreten. Ist dies jedoch unumgänglich,<br />
sind sie vorher insbeson<strong>der</strong>e über<br />
Personalhygiene und die vorgeschriebene<br />
Schutzkleidung zu informieren. Sie sind streng<br />
zu beaufsichtigen.<br />
2.12 Im Rahmen <strong>der</strong> Schulung sind das Konzept<br />
<strong>der</strong> Qualitätssicherung und alle Maßnahmen,<br />
die dessen Verständnis und Anwendung<br />
verbessern können, ausführlich darzulegen.<br />
2.13 Detaillierte Hygieneprogramme – den unterschiedlichen<br />
Erfor<strong>der</strong>nissen im Betrieb ange-<br />
Übersetzung durch die Mitglie<strong>der</strong> <strong>der</strong> AG 42 des QS-Forums Schleswig-Holstein / Hamburg
within the factory. They should include procedures<br />
relating to the health, hygiene practices<br />
and clothing of personnel. These procedures<br />
should be un<strong>der</strong>stood and followed in a very<br />
strict way by every person whose duties take<br />
him into the production and control areas.<br />
Hygiene programmes should be promoted by<br />
management and widely discussed during<br />
training sessions.<br />
2.14 All personnel should receive medical examination<br />
upon recruitment. It must be the<br />
manufacturer’s responsibility that there are instructions<br />
ensuring that health conditions that<br />
can be of relevance to the quality of products<br />
come to the manufacturer’s knowledge. After the<br />
first medical examination, examinations should<br />
be carried out when necessary for the work and<br />
personal health.<br />
2.15 Steps should be taken to ensure as far as<br />
is practicable that no person affected by an infectious<br />
disease or having open lesions on the<br />
exposed surface of the body is engaged in the<br />
manufacture of medicinal products.<br />
2.16 Every person entering the manufacturing<br />
areas should wear protective garments appropriate<br />
to the operations to be carried out.<br />
2.17 Eating, drinking, chewing or smoking, or<br />
the storage of food, drink, smoking materials or<br />
personal medication in the production and<br />
storage areas should be prohibited. In general,<br />
any unhygienic practice within the<br />
manufacturing areas or in any other area where<br />
the product might be adversely affected, should<br />
be forbidden.<br />
2.18 Direct contact should be avoided between<br />
the operator’s hands and the exposed product<br />
as well as with any part of the equipment that<br />
comes into contact with the products.<br />
2.19 Personnel should be instructed to use the<br />
hand-washing facilities.<br />
2.20 Any specific requirements for the manufacture<br />
of special groups of products, for example<br />
sterile preparations, are covered in the annexes.<br />
passt – sind erfor<strong>der</strong>lich. Sie beinhalten Vorschriften<br />
<strong>zur</strong> Gesundheit, über hygienisches<br />
Verhalten und <strong>zur</strong> Schutzkleidung des Personals.<br />
Diese Vorschriften müssen von jedem, <strong>der</strong><br />
bei <strong>der</strong> Durchführung seiner Aufgaben Produktions-<br />
und Qualitätskontrollbereiche betritt, verstanden<br />
sein und sehr genau befolgt werden.<br />
Hygieneprogramme sind von den Führungskräften<br />
aktiv zu unterstützen und im Rahmen <strong>der</strong><br />
Schulung eingehend zu erläutern.<br />
2.14 Je<strong>der</strong> Mitarbeiter ist vor Aufnahme <strong>der</strong><br />
Tätigkeit ärztlich zu untersuchen. Der Inhaber<br />
<strong>der</strong> Herstellungserlaubnis ist da<strong>für</strong> verantwortlich,<br />
dass Anweisungen vorhanden sind, mit<br />
denen sichergestellt wird, dass ihm Än<strong>der</strong>ungen<br />
des Gesundheitszustandes des Personals, die<br />
von Bedeutung <strong>für</strong> die Produktqualität sein<br />
könnten, gemeldet werden. Nach <strong>der</strong> Erstuntersuchung<br />
sind Folgeuntersuchungen durchzuführen,<br />
wenn dies aus betrieblichen o<strong>der</strong> gesundheitlichen<br />
Gründen erfor<strong>der</strong>lich ist.<br />
2.15 Soweit praktisch möglich, sind Vorkehrungen<br />
zu treffen, die sicherstellen, dass in <strong>der</strong> <strong>Arzneimittel</strong>herstellung<br />
niemand beschäftigt wird,<br />
<strong>der</strong> an einer ansteckenden Krankheit leidet o<strong>der</strong><br />
offene Verletzungen an unbedeckten Körperstellen<br />
aufweist.<br />
2.16 Jede Person, die Produktionsbereiche betritt,<br />
hat eine den jeweils auszuführenden Arbeiten<br />
angepasste Schutzkleidung zu tragen.<br />
2.17 Essen, Trinken, Kaugummikauen o<strong>der</strong><br />
Rauchen sowie die Aufbewahrung von Speisen,<br />
Getränken, Tabakerzeugnissen o<strong>der</strong> <strong>Arzneimittel</strong>n<br />
<strong>für</strong> den persönlichen Gebrauch sind in den<br />
Produktions- und Lagerbereichen zu verbieten.<br />
Allgemein ist jedes unhygienische Verhalten in<br />
jedem Bereich, in dem das Produkt beeinträchtigt<br />
werden könnte, zu verbieten.<br />
2.18 Der direkte Kontakt zwischen den Händen<br />
eines Beschäftigten und dem offenen Produkt ist<br />
ebenso zu vermeiden wie <strong>der</strong> direkte Kontakt<br />
mit irgendeinem Ausrüstungsteil, das mit den<br />
Produkten in Berührung kommt.<br />
2.19 Das Personal ist anzuhalten, die Handwaschgelegenheiten<br />
zu benutzen.<br />
2.20 Spezielle Anfor<strong>der</strong>ungen bei <strong>der</strong> Herstellung<br />
beson<strong>der</strong>er Produktgruppen, wie z. B. steriler<br />
Zubereitungen, werden in den Anhängen<br />
abgehandelt.<br />
Übersetzung durch die Mitglie<strong>der</strong> <strong>der</strong> AG 42 des QS-Forums Schleswig-Holstein / Hamburg
CHAPTER 3 PREMISES AND EQUIPMENT Kapitel 3 Räumlichkeiten und Ausrüstung<br />
Principle Grundsätze<br />
Premises and equipment must be located, designed,<br />
constructed, adapted and maintained to<br />
suit the operations to be carried out. Their layout<br />
and design must aim to minimise the risk of errors<br />
and permit effective cleaning and maintenance<br />
in or<strong>der</strong> to avoid crosscontamination,<br />
build up of dust or dirt and, in general, any<br />
adverse effect on the quality of products.<br />
Räumlichkeiten und Ausrüstung müssen so gelegen<br />
bzw. angeordnet, ausgelegt, konstruiert,<br />
angepasst und instand gehalten sein, dass sie<br />
<strong>für</strong> die vorgesehenen Arbeitsgänge geeignet<br />
sind. Ihre Anordnung und Auslegung müssen<br />
darauf ausgerichtet sein, das Risiko von Fehlern<br />
zu minimieren und eine gründliche Reinigung<br />
und Wartung zu erlauben, um Kreuzkontamination,<br />
Staub- o<strong>der</strong> Schmutzansammlungen und<br />
ganz allgemein jeden die Qualität des Produktes<br />
beeinträchtigenden Einfluss zu vermeiden.<br />
Premises Betriebstätte / Räumlichkeiten<br />
General Allgemeine Anfor<strong>der</strong>ungen<br />
3.1 Premises should be situated in an environment<br />
which, when consi<strong>der</strong>ed together with<br />
measures to protect the manufacture, presents<br />
minimal risk of causing contamination of materials<br />
or products.<br />
3.2 Premises should be carefully maintained,<br />
ensuring that repair and maintenance operations<br />
do not present any hazard to the quality of products.<br />
They should be cleaned and, where applicable,<br />
disinfected according to detailed written<br />
procedures.<br />
3.3 Lighting, temperature, humidity and ventilation<br />
should be appropriate and such that they<br />
do not adversely affect, directly or indirectly,<br />
either the medicinal products during their manufacture<br />
and storage, or the accurate functioning<br />
of equipment.<br />
3.4 Premises should be designed and<br />
equipped so as to afford maximum protection<br />
against the entry of insects or other animals.<br />
3.5 Steps should be taken in or<strong>der</strong> to prevent<br />
the entry of unauthorised people. Production,<br />
storage and quality control areas should not be<br />
used as a right of way by personnel who do not<br />
work in them.<br />
Production Area Produktionsbereiche<br />
3.6 In or<strong>der</strong> to minimise the risk of a serious<br />
medical hazard due to cross-contamination,<br />
dedicated and self contained facilities must be<br />
available for the production of particular medici-<br />
3.1 Die Betriebsstätte sollte in einer Umgebung<br />
gelegen sein, <strong>der</strong>en Kontaminationsrisiko<br />
<strong>für</strong> Materialien o<strong>der</strong> Produkte unter Berücksichtigung<br />
<strong>der</strong> Schutzmaßnahmen bei <strong>der</strong> Produktion<br />
möglichst gering ist.<br />
3.2 Die Räumlichkeiten sind sorgfältig instandzuhalten;<br />
Reparatur- und Wartungsarbeiten<br />
dürfen die Qualität <strong>der</strong> Produkte nicht gefährden.<br />
Die Räumlichkeiten sind nach detaillierten,<br />
schriftlich festgelegten Verfahren zu reinigen<br />
und, falls notwendig, zu desinfizieren.<br />
3.3 Beleuchtung, Temperatur, Luftfeuchtigkeit<br />
und Belüftung müssen geeignet und so beschaffen<br />
sein, dass sie we<strong>der</strong> direkt noch indirekt die<br />
<strong>Arzneimittel</strong> während <strong>der</strong> Produktion und Lagerung<br />
o<strong>der</strong> das einwandfreie Funktionieren <strong>der</strong><br />
Ausrüstung beeinträchtigen.<br />
3.4 Die Räumlichkeiten sind so auszulegen<br />
und auszustatten, dass <strong>der</strong> größtmögliche<br />
Schutz gegen das Eindringen von Insekten o<strong>der</strong><br />
an<strong>der</strong>en Tieren gewährleistet ist.<br />
3.5 Es sind Vorkehrungen gegen den Zutritt<br />
Unbefugter zu treffen. Produktions-, Lager- und<br />
Qualitätskontrollbereiche sind von dort nicht<br />
beschäftigtem Personal nicht als Durchgang zu<br />
benutzen.<br />
3.6 Um das Risiko einer ernsten Gesundheitsgefährdung<br />
durch Kreuzkontamination zu<br />
minimieren, müssen <strong>für</strong> die Produktion bestimmter<br />
<strong>Arzneimittel</strong>, wie solchen mit hochsensibili-<br />
Übersetzung durch die Mitglie<strong>der</strong> <strong>der</strong> AG 42 des QS-Forums Schleswig-Holstein / Hamburg
nal products, such as highly sensitising<br />
materials (e.g. penicillins) or biological<br />
preparations (e.g. from live micro-organisms).<br />
The production of certain additional products,<br />
such as certain antibiotics, certain hormones,<br />
certain cytotoxics, certain highly active drugs<br />
and non-medicinal products should not be<br />
conducted in the same facilities. For those<br />
products, in exceptional cases, the principle of<br />
campaign working in the same facilities can be<br />
accepted provided that specific precautions are<br />
taken and the necessary validations are made.<br />
The manufacture of technical poisons, such as<br />
pesticides and herbicides, should not be allowed<br />
in premises used for the manufacture of<br />
medicinal products.<br />
3.7 Premises should preferably be laid out in<br />
such a way as to allow the production to take<br />
place in areas connected in a logical or<strong>der</strong> corresponding<br />
to the sequence of the operations<br />
and to the requisite cleanliness levels.<br />
3.8 The adequacy of the working and inprocess<br />
storage space should permit the or<strong>der</strong>ly<br />
and logical positioning of equipment and materials<br />
so as to minimise the risk of confusion between<br />
different medicinal products or their components,<br />
to avoid cross-contamination and to<br />
minimise the risk of omission or wrong application<br />
of any of the manufacturing or control steps.<br />
3.9 Where starting and primary packaging<br />
materials, intermediate or bulk products are exposed<br />
to the environment, interior surfaces<br />
(walls, floors and ceilings) should be smooth,<br />
free from cracks and open joints, and should not<br />
shed particulate matter and should permit easy<br />
and effective cleaning and, if necessary, disinfection.<br />
3.10 Pipework, light fittings, ventilation points<br />
and other services should be designed and sited<br />
to avoid the creation of recesses which are difficult<br />
to clean. As far as possible, for maintenance<br />
purposes, they should be accessible from outside<br />
the manufacturing areas.<br />
3.11 Drains should be of adequate size, and<br />
have trapped gullies. Open channels should be<br />
avoided where possible, but if necessary, they<br />
sierenden Stoffen (z. B. Penicilline) o<strong>der</strong> biologischen<br />
Zubereitungen (z. B. aus lebenden<br />
Mikroorganismen), fest zugeordnete und in sich<br />
geschlossene Räume/Einrichtungen <strong>zur</strong> Verfügung<br />
stehen. Die Produktion weiterer Produkte<br />
wie bestimmter Antibiotika, Hormone, Zytostatika,<br />
hochwirksamer <strong>Arzneimittel</strong> sowie von Nicht-<br />
<strong>Arzneimittel</strong>n darf nicht in den gleichen Räumen/<br />
Einrichtungen erfolgen. Für diese Produkte kann<br />
jedoch in Ausnahmefällen das Prinzip <strong>der</strong> Produktion<br />
in Kampagnen in den gleichen Räumen/<br />
Einrichtungen unter <strong>der</strong> Voraussetzung akzeptiert<br />
werden, dass spezielle Vorsichtsmaßnahmen<br />
getroffen werden und die notwendigen Validierungen<br />
durchgeführt sind. Die Herstellung<br />
technischer Gifte, wie Pestizide und Herbizide,<br />
ist nicht in Räumlichkeiten gestattet, die <strong>zur</strong> Produktion<br />
von <strong>Arzneimittel</strong>n genutzt werden.<br />
3.7 Die Räumlichkeiten sind möglichst so anzuordnen,<br />
dass die Produktion in logisch aufeinan<strong>der</strong><br />
folgenden Schritten erfolgen kann, entsprechend<br />
<strong>der</strong> Reihenfolge <strong>der</strong> Arbeitsgänge<br />
und den erfor<strong>der</strong>lichen Reinheitsklassen.<br />
3.8 Arbeitsflächen und Zwischenproduktlager<br />
im Produktionsbereich sind so zu bemessen,<br />
dass diese die ordnungsgemäße und prozessorientierte<br />
Aufstellung <strong>der</strong> Ausrüstung und Bereitstellung<br />
<strong>der</strong> Materialien ermöglichen, um das<br />
Risiko von Verwechslungen unterschiedlicher<br />
<strong>Arzneimittel</strong> o<strong>der</strong> ihrer Bestandteile zu minimieren,<br />
Kreuzkontamination zu vermeiden und um<br />
die Gefahr zu verringern, irgendeinen Produktions-<br />
o<strong>der</strong> Kontrollschritt auszulassen o<strong>der</strong><br />
falsch anzuwenden.<br />
3.9 Wo Ausgangsstoffe und primäre Verpackungsmaterialien,<br />
Zwischenprodukte o<strong>der</strong><br />
Bulkware Umgebungseinflüssen ausgesetzt<br />
sind, haben die Innenflächen (Wände, Fußböden,<br />
Decken) glatt und frei von Rissen und<br />
offenen Fugen zu sein, sollen keine Partikel abgeben<br />
und müssen sich leicht und gründlich<br />
reinigen und, wenn nötig, desinfizieren lassen.<br />
3.10 Rohrleitungen, Beleuchtungskörper, Belüftungseinrichtungen<br />
und an<strong>der</strong>e Versorgungsanlagen<br />
sind so zu konstruieren und anzubringen,<br />
dass keine schwer zu reinigenden Stellen<br />
entstehen. Es ist, soweit möglich, anzustreben,<br />
zu Wartungszwecken <strong>für</strong> die o. g. Ausrüstung<br />
separate Zugänge, außerhalb <strong>der</strong> Produktionsbereiche<br />
gelegen, zu haben.<br />
3.11 Abflüsse sind ihrer Benutzung entsprechend<br />
zu dimensionieren und jeweils mit Rückstauklappe<br />
zu versehen. Offene Abflüsse sind,<br />
Übersetzung durch die Mitglie<strong>der</strong> <strong>der</strong> AG 42 des QS-Forums Schleswig-Holstein / Hamburg
should be shallow to facilitate cleaning anddisinfection.<br />
3.12 Production areas should be effectively<br />
ventilated, with air control facilities (including<br />
temperature and, where necessary, humidity<br />
and filtration) appropriate both to the products<br />
handled, to the operations un<strong>der</strong>taken within<br />
them and to the external environment.<br />
3.13 Weighing of starting materials usually<br />
should be carried out in a separate weighing<br />
room designed for that use.<br />
3.14 In cases where dust is generated (e.g.<br />
during sampling, weighing, mixing and processing<br />
operations, packaging of dry products), specific<br />
provisions should be taken to avoid crosscontamination<br />
and facilitate cleaning.<br />
3.15 Premises for the packaging of medicinal<br />
products should be specifically designed and<br />
laid out so as to avoid mix-ups or crosscontamination.<br />
3.16 Production areas should be well lit, particularly<br />
where visual on-line controls are carried<br />
out.<br />
3.17 In-process controls may be carried out<br />
within the production area provided they do not<br />
carry any risk for the production.<br />
Storage Areas Lagerbereiche<br />
3.18 Storage areas should be of sufficient<br />
capacity to allow or<strong>der</strong>ly storage of the various<br />
categories of materials and products: starting<br />
and packaging materials, intermediate, bulk and<br />
finished products, products in quarantine, released,<br />
rejected, returned or recalled.<br />
3.19 Storage areas should be designed or<br />
adapted to ensure good storage conditions. In<br />
particular, they should be clean and dry and<br />
maintained within acceptable temperature limits.<br />
Where special storage conditions are required<br />
(e.g. temperature, humidity) theseshould be provided,<br />
checked and monitored.<br />
3.20 Receiving and dispatch bays should protect<br />
materials and products from the weather.<br />
wenn irgend möglich, zu vermeiden. Wenn sie<br />
dennoch erfor<strong>der</strong>lich sind, dürfen sie nur so tief<br />
sein, dass Reinigung und Desinfektion möglich<br />
sind.<br />
3.12 Produktionsbereiche sind wirkungsvoll zu<br />
belüften. Die Belüftungssysteme (einschließlich<br />
Temperatur- und, wo notwendig, mit Luftfeuchtigkeits-<br />
und Filterkontrollsystemen) müssen den<br />
darin gehandhabten Produkten, den durchgeführten<br />
Arbeitsgängen und <strong>der</strong> äußeren Umgebung<br />
angemessen sein.<br />
3.13 Das Abwiegen von Ausgangsstoffen ist<br />
üblicherweise in einem separaten, <strong>für</strong> diesen<br />
Zweck ausgelegten Wägeraum durchzuführen.<br />
3.14 An Stellen, an denen Staub entstehen<br />
kann (z. B. bei <strong>der</strong> Probenahme, beim Abwiegen,<br />
Mischen und Verarbeiten, beim Abpacken<br />
trockener Produkte), sind beson<strong>der</strong>e Maßnahmen<br />
zu ergreifen, um Kreuzkontamination zu<br />
vermeiden und die Reinigung zu erleichtern.<br />
3.15 Räumlichkeiten, in denen <strong>Arzneimittel</strong> verpackt<br />
werden, sind so auszulegen, dass Verwechslungen<br />
und Kreuzkontamination vermieden<br />
werden.<br />
3.16 Produktionsbereiche sind gut auszuleuchten,<br />
beson<strong>der</strong>s dort, wo prozessbegleitend visuelle<br />
Kontrollen durchgeführt werden.<br />
3.17 Inprozesskontrollen können innerhalb des<br />
Produktionsbereichs durchgeführt werden, vorausgesetzt,<br />
dass sie kein Risiko <strong>für</strong> die Produktion<br />
darstellen.<br />
3.18 Lagerräume sind ausreichend groß auszulegen,<br />
um eine ordnungsgemäße Lagerung<br />
<strong>der</strong> unterschiedlichen Materialien und Produkte<br />
zu erlauben: Ausgangsstoffe und Verpackungsmaterialien,<br />
Zwischenprodukte, Bulkware und<br />
Fertigprodukte, in Quarantäne befindliche, freigegebene,<br />
<strong>zur</strong>ückgewiesene, <strong>zur</strong>ückgegebene<br />
o<strong>der</strong> <strong>zur</strong>ückgerufene Produkte.<br />
3.19 Die Lagerräume sind so auszulegen o<strong>der</strong><br />
anzupassen, dass gute Lagerungsbedingungen<br />
gewährleistet sind, insbeson<strong>der</strong>e sollen sie sauber<br />
und trocken sein und angemessene Temperaturbereiche<br />
aufweisen. Sind beson<strong>der</strong>e Lagerungsbedingungen<br />
(z. B. hinsichtlich Temperatur,<br />
Luftfeuchtigkeit) erfor<strong>der</strong>lich, sind diese zu<br />
schaffen, zu überprüfen und zu überwachen.<br />
3.20 Im Bereich <strong>der</strong> La<strong>der</strong>ampen (Annahme<br />
und Versand) sind die Materialien und Produkte<br />
Übersetzung durch die Mitglie<strong>der</strong> <strong>der</strong> AG 42 des QS-Forums Schleswig-Holstein / Hamburg
Reception areas should be designed and<br />
equipped to allow containers of incoming materials<br />
to be cleaned where necessary before<br />
storage.<br />
3.21 Where quarantine status is ensured by<br />
storage in separate areas, these areas must be<br />
clearly marked and their access restricted to<br />
authorised personnel. Any system replacing the<br />
physical quarantine should give equivalent security.<br />
3.22 There should normally be a separate<br />
sampling area for starting materials. If sampling<br />
is performed in the storage area, it should be<br />
conducted in such a way as to prevent contamination<br />
or cross-contamination.<br />
3.23 Segregated areas should be provided for<br />
the storage of rejected, recalled or returned<br />
materials or products.<br />
3.24 Highly active materials or products should<br />
be stored in safe and secure areas.<br />
3.25 Printed packaging materials are consi<strong>der</strong>ed<br />
critical to the conformity of the medicinal<br />
product and special attention should be paid to<br />
the safe and secure storage of these materials.<br />
vor Witterungseinflüssen zu schützen. Annahmebereiche<br />
sind so auszulegen und auszustatten,<br />
dass Behälter mit eingehenden Materialien<br />
erfor<strong>der</strong>lichenfalls vor <strong>der</strong> Einlagerung gereinigt<br />
werden können.<br />
3.21 Soll <strong>der</strong> Quarantänestatus durch Lagerung<br />
in abgetrennten Bereichen gewährleistet<br />
werden, sind diese Bereiche deutlich zu kennzeichnen,<br />
<strong>der</strong> Zutritt ist auf Befugte zu beschränken.<br />
Jedes System, das die o. g. Quarantänelagerung<br />
ersetzt, muss die gleiche Sicherheit<br />
bieten.<br />
3.22 Üblicherweise hat die Probenahme von<br />
Ausgangsstoffen in einem abgetrennten Bereich<br />
zu erfolgen. Wird die Probenahme im Lagerbereich<br />
durchgeführt, hat dies in einer Weise zu<br />
erfolgen, die eine Verunreinigung o<strong>der</strong><br />
Kreuzkontamination verhin<strong>der</strong>t.<br />
3.23 Es sind separate Bereiche vorzusehen,<br />
um <strong>zur</strong>ückgewiesene, <strong>zur</strong>ückgerufene o<strong>der</strong> <strong>zur</strong>ückgegebene<br />
Materialien o<strong>der</strong> Produkte zu lagern.<br />
3.24 Hochaktive Materialien o<strong>der</strong> Produkte<br />
sind in sicheren und geschützten Bereichen zu<br />
lagern.<br />
3.25 Bedruckte Verpackungsmaterialien werden<br />
als kritisch <strong>für</strong> die Qualität eines <strong>Arzneimittel</strong>s<br />
angesehen, beson<strong>der</strong>e Aufmerksamkeit ist<br />
daher auf eine sichere und geschützte Lagerung<br />
dieser Materialien zu richten.<br />
Quality Control Areas Qualitätskontrollbereiche<br />
3.26 Normally, Quality Control laboratories<br />
should be separated from production areas. This<br />
is particularly important for laboratories for the<br />
control of biologicals, microbiologicals and<br />
radioisotopes, which should also be separated<br />
from each other.<br />
3.27 Control laboratories should be designed<br />
to suit the operations to be carried out in them.<br />
Sufficient space should be given to avoid mixups<br />
and cross-contamination. There should be<br />
adequate suitable storage space for samples<br />
and records.<br />
3.28 Separate rooms may be necessary to<br />
protect sensitive instruments from vibration,<br />
electrical interference, humidity, etc.<br />
3.26 Üblicherweise sind Kontrolllaboratorien<br />
von den Produktionsbereichen abzutrennen.<br />
Dies ist insbeson<strong>der</strong>e von Bedeutung bei Laboratorien<br />
<strong>zur</strong> Kontrolle von biologischen o<strong>der</strong> mikrobiologischen<br />
Produkten sowie von Radiopharmaka.<br />
Solche Laboratorien sind zudem<br />
noch voneinan<strong>der</strong> zu trennen.<br />
3.27 Kontrolllaboratorien sind so auszulegen,<br />
dass sie <strong>für</strong> die vorgesehenen Arbeiten geeignet<br />
sind. Sie müssen ausreichend groß sein, damit<br />
Verwechslungen und Kreuzkontamination (<strong>der</strong><br />
Proben) vermieden werden. Für die Aufbewahrung<br />
von Proben und Protokollen ist hinreichend<br />
Platz vorzusehen.<br />
3.28 Separate Räume können notwendig sein,<br />
um empfindliche Instrumente vor Erschütterungen,<br />
elektrischen Störeinwirkungen, Feuchtigkeit<br />
usw. zu schützen.<br />
3.29 Special requirements are needed in labo- 3.29 In Laboratorien, in denen mit beson<strong>der</strong>en<br />
Übersetzung durch die Mitglie<strong>der</strong> <strong>der</strong> AG 42 des QS-Forums Schleswig-Holstein / Hamburg
atories handling particular substances, such as<br />
biological or radioactive samples.<br />
Ancillary Areas Nebenbereiche<br />
3.30 Rest and refreshment rooms should be<br />
separate from other areas.<br />
3.31 Facilities for changing clothes, and for<br />
washing and toilet purposes should be easily<br />
accessible and appropriate for the number of<br />
users. Toilets should not directly communicate<br />
with production or storage areas.<br />
3.32 Maintenance workshops should as far as<br />
possible be separated from production areas.<br />
Whenever parts and tools are stored in the production<br />
area, they should be kept in rooms or<br />
lockers reserved for that use.<br />
3.33 Animal houses should be well isolated<br />
from other areas, with separate entrance<br />
(animal access) and air handling facilities.<br />
Equipment Ausrüstung<br />
3.34 Manufacturing equipment should be designed,<br />
located and maintained to suit its intended<br />
purpose.<br />
3.35 Repair and maintenance operations<br />
should not present any hazard to the quality of<br />
the products.<br />
3.36 Manufacturing equipment should be designed<br />
so that it can be easily and thoroughly<br />
cleaned. It should be cleaned according to detailed<br />
and written procedures and stored only in<br />
a clean and dry condition.<br />
3.37 Washing and cleaning equipment should<br />
be chosen and used in or<strong>der</strong> not to be a source<br />
of contamination.<br />
3.38 Equipment should be installed in such a<br />
way as to prevent any risk of error or of contamination.<br />
3.39 Production equipment should not present<br />
any hazard to the products. The parts of the production<br />
equipment that come into contact with<br />
the product must not be reactive, additive or absorptive<br />
to such an extent that it will affect the<br />
quality of the product and thus present any<br />
hazard.<br />
3.40 Balances and measuring equipment of an<br />
appropriate range and precision should be avail-<br />
Substanzen wie biologischen o<strong>der</strong> radioaktiven<br />
Proben umgegangen wird, sind spezielle Anfor<strong>der</strong>ungen<br />
zu erfüllen.<br />
3.30 Aufenthalts- und Pausenräume sind von<br />
an<strong>der</strong>en Bereichen zu trennen.<br />
3.31 Umkleide- und Waschräume sowie Toiletten<br />
sollen leicht erreichbar und <strong>der</strong> Benutzerzahl<br />
angemessen sein. Toiletten sollen nicht in direkter<br />
Verbindung mit Produktions- o<strong>der</strong> Lagerräumen<br />
stehen.<br />
3.32 Werkstätten sollen, soweit möglich, von<br />
Produktionsbereichen getrennt sein. Wenn Ersatzteile<br />
und Werkzeuge im Produktionsbereich<br />
gelagert werden, sind sie in da<strong>für</strong> vorgesehenen<br />
Räumen o<strong>der</strong> Schränken aufzubewahren.<br />
3.33 Räume, in denen Tiere gehalten werden,<br />
sind von den an<strong>der</strong>en Bereichen gut zu isolieren<br />
(separater Eingang (Tierzugang) und separate<br />
Belüftungsanlagen).<br />
3.34 Die Produktionsausrüstung ist so auszulegen,<br />
anzuordnen und zu warten, dass sie <strong>für</strong><br />
den vorgesehenen Zweck geeignet ist.<br />
3.35 Reparatur- und Wartungsarbeiten dürfen<br />
die Qualität <strong>der</strong> Produkte keinesfalls gefährden.<br />
3.36 Die Produktionsausrüstung ist so auszulegen,<br />
dass sie sich leicht und gründlich reinigen<br />
lässt. Sie ist nach detaillierten schriftlichen Verfahren<br />
zu reinigen; vor Standzeiten ist die Ausrüstung<br />
zu reinigen und zu trocknen.<br />
3.37 Die zum Waschen und Reinigen verwendete<br />
Ausrüstung ist so auszuwählen und einzusetzen,<br />
dass sie selbst keine Quelle <strong>der</strong> Verunreinigung<br />
darstellt.<br />
3.38 Die Ausrüstung ist so zu installieren, dass<br />
keine Gefahr eines Fehlers o<strong>der</strong> einer Verunreinigung<br />
besteht.<br />
3.39 Gefahren <strong>für</strong> die Produkte durch die Produktionsausrüstung<br />
sind zu vermeiden. Kein mit<br />
dem Produkt in Berührung kommendes Ausrüstungsteil<br />
darf mit diesem so in Wechselwirkung<br />
treten, dass die Produktqualität beeinträchtigt<br />
wird und damit ein Risiko entsteht<br />
(egal, ob reaktiv, additiv o<strong>der</strong> absorptiv).<br />
3.40 Für Produktions- und Kontrollzwecke sind<br />
Waagen und Messgeräte mit geeigneten Mess-<br />
Übersetzung durch die Mitglie<strong>der</strong> <strong>der</strong> AG 42 des QS-Forums Schleswig-Holstein / Hamburg
able for production and control operations. bereichen und <strong>der</strong> jeweils erfor<strong>der</strong>lichen Empfindlichkeit<br />
vorzuhalten.<br />
3.41 Measuring, weighing, recording and control<br />
equipment should be calibrated and checked<br />
at defined intervals by appropriate methods.<br />
Adequate records of such tests should be maintained.<br />
3.42 Fixed pipework should be clearly labelled<br />
to indicate the contents and, where applicable,<br />
the direction of flow.<br />
3.43 Distilled, deionized and, where appropriate,<br />
other water pipes should be sanitised according<br />
to written procedures that detail the action<br />
limits for microbiological contamination and<br />
the measures to be taken.<br />
3.44 Defective equipment should, if possible,<br />
be removed from production and quality control<br />
areas, or at least be clearly labelled as<br />
defective.<br />
3.41 Die Mess-, Wäge-, Aufzeichnungs- und<br />
Kontrollausrüstung ist zu kalibrieren und in bestimmten<br />
Abständen mit geeigneten Methoden<br />
zu überprüfen. Hierüber sind aussagekräftige<br />
Aufzeichnungen aufzubewahren.<br />
3.42 Fest installierte Rohrleitungen sind deutlich<br />
mit <strong>der</strong> Angabe des Inhalts und, wo angezeigt,<br />
mit <strong>der</strong> Fließrichtung zu kennzeichnen.<br />
3.43 Leitungen <strong>für</strong> destilliertes und demineralisiertes<br />
Wasser und, wo angezeigt, an<strong>der</strong>e Wasserleitungen<br />
sind bei Bedarf zu desinfizieren;<br />
dies hat nach schriftlich festgelegten Vorgaben<br />
zu erfolgen, in denen Aktionsgrenzen (Überschreitung<br />
<strong>der</strong> zulässigen KBE) definiert und die<br />
dann zu treffenden Maßnahmen beschrieben<br />
sind.<br />
3.44 Schadhafte Ausrüstung ist, wenn möglich,<br />
aus Produktions- und Qualitätskontrollbereichen<br />
zu entfernen o<strong>der</strong> zumindest deutlich als schadhaft<br />
zu kennzeichnen.<br />
CHAPTER 4 DOCUMENTATION Kapitel 4 Dokumentation<br />
Principle Grundsätze<br />
Good documentation constitutes an essential<br />
part of the quality assurance system. Clearly<br />
written documentation prevents errors from<br />
spoken communication and permits tracing of<br />
batch history. Specifications, Manufacturing<br />
Formulae and instructions, procedures, and<br />
records must be free from errors and available<br />
in writing. The legibility of documents is of paramount<br />
importance.<br />
Eine gute Dokumentation 13 ist ein wesentlicher<br />
Teil des Qualitätssicherungssystems. Eindeutige<br />
Unterlagen verhin<strong>der</strong>n Irrtümer aus mündlicher<br />
Kommunikation und erlauben die Rückverfolgung<br />
des Werdegangs einer Charge. Spezifikationen,<br />
Rezepturen, Herstellungsanweisungen,<br />
sonstige Anweisungen und Protokolle müssen<br />
fehlerfrei sein und in Schriftform vorliegen. Die<br />
Lesbarkeit <strong>der</strong> Unterlagen ist äußerst wichtig.<br />
General Allgemeine Anfor<strong>der</strong>ungen<br />
4.1 Specifications describe in detail the requirements<br />
with which the products or materials<br />
used or obtained during manufacture have to<br />
conform. They serve as a basis for quality<br />
evaluation. Manufacturing Formulae, Processing<br />
and Packaging Instructions state all the starting<br />
materials used and lay down all processing and<br />
packaging operations. Procedures give directions<br />
for performing certain operations e.g.<br />
cleaning, clothing, environmental control, sampling,<br />
testing, equipment operation. Records<br />
provide a history of each batch of product, including<br />
its distribution, and also of all other rele-<br />
4.1 Spezifikationen beschreiben detailliert die<br />
Anfor<strong>der</strong>ungen, denen jedes Produkt o<strong>der</strong> Material,<br />
das bei <strong>der</strong> Produktion eingesetzt o<strong>der</strong> erhalten<br />
wird, entsprechen muss. Sie dienen als<br />
Grundlage <strong>der</strong> Qualitätsbewertung. Rezepturen,<br />
Herstellungsanweisungen (Bulkware und Fertigprodukt)<br />
bestimmen alle eingesetzten Ausgangsmaterialien<br />
und legen alle Produktionseinschließlich<br />
<strong>der</strong> Verpackungsvorgänge fest.<br />
Verfahrensanweisungen enthalten Vorgaben <strong>für</strong><br />
die Durchführung bestimmter Vorgänge wie Reinigung,<br />
Bekleidung, Umgebungskontrollen,<br />
Probenahme, Prüfung und den Betrieb von Ge-<br />
13 Dokumentation im Sinne dieses <strong>Leitfaden</strong>s umfasst sowohl Vorgaben (z. B. Anweisungen, Spezifikationen)<br />
als auch Nachweise (z. B. Protokolle, Zertifkate).<br />
Übersetzung durch die Mitglie<strong>der</strong> <strong>der</strong> AG 42 des QS-Forums Schleswig-Holstein / Hamburg
vant circumstances pertinent to the quality of the<br />
final product.<br />
4.2 Documents should be designed,<br />
prepared, reviewed and distributed with care.<br />
They should comply with the relevant parts of<br />
the manufacturing and marketing authorisation<br />
dossiers.<br />
4.3 Documents should be approved, signed<br />
and dated by appropriate and authorised persons.<br />
4.4 Documents should have unambiguous<br />
contents; title, nature and purpose should be<br />
clearly stated. They should be laid out in an<br />
or<strong>der</strong>ly fashion and be easy to check. Reproduced<br />
documents should be clear and legible.<br />
The reproduction of working documents from<br />
master documents must not allow any error to<br />
be introduced through the reproduction process.<br />
4.5 Documents should be regularly reviewed<br />
and kept up-to-date. When a document has<br />
been revised, systems should be operated to<br />
prevent inadvertent use of superseded<br />
documents.<br />
4.6 Documents should not be handwritten;<br />
although, where documents require the entry of<br />
data, these entries may be made in clear, legible,<br />
indelible handwriting. Sufficient space<br />
should be provided for such entries.<br />
4.7 Any alteration made to the entry on a<br />
document should be signed and dated; the alteration<br />
should permit the reading of the original<br />
information. Where appropriate, the reason for<br />
the alteration should be recorded.<br />
4.8 The records should be made or<br />
completed at the time each action is taken and<br />
in such a way that all significant activities<br />
concerning the manufacture of medicinal<br />
products are traceable. They should be retained<br />
for at least one year after the expiry date of the<br />
finished product.<br />
4.9 Data may be recorded by electronic data<br />
processing systems, photographic or other reliable<br />
means, but detailed procedures relating to<br />
the system in use should be available and the<br />
accuracy of the records should be checked. If<br />
documentation is handled by electronic data<br />
processing methods, only authorised persons<br />
should be able to enter or modify data in the<br />
räten. Protokolle dokumentieren den Werdegang<br />
einer jeden Charge, <strong>der</strong>en Vertrieb, sowie<br />
alle an<strong>der</strong>en Sachverhalte, die <strong>für</strong> die Qualität<br />
des Fertigproduktes von Belang sind.<br />
4.2 Vorgabedokumente sind sorgfältig zu konzipieren,<br />
zu erstellen, zu überprüfen und zu verteilen.<br />
Sie haben den Vorgaben <strong>der</strong> Herstellungserlaubnis<br />
und den For<strong>der</strong>ungen <strong>der</strong> Zulassung<br />
zu entsprechen.<br />
4.3 Die Vorgabedokumente sind von qualifizierten<br />
und dazu befugten Personen zu genehmigen<br />
und mit Datumsangabe zu unterzeichnen.<br />
4.4 Der Inhalt <strong>der</strong> Vorgabedokumente muss<br />
eindeutig sein; Titel, Sinn und Zweck sind eindeutig<br />
anzugeben. Die Vorgabedokumente sind<br />
übersichtlich zu gestalten, sie sollen leicht zu<br />
überprüfen sein. Vervielfältigte Dokumente sollen<br />
klar und gut lesbar sein. Werden Vervielfältigungen<br />
<strong>der</strong> Originalunterlagen als Arbeitsunterlagen<br />
erstellt, darf dies nicht zu Fehlern führen.<br />
4.5 Die Vorgaben sind regelmäßig zu überprüfen<br />
und auf aktuellem Stand zu halten. Wenn<br />
ein Dokument überarbeitet wurde, muss die versehentliche<br />
Verwendung <strong>der</strong> veralteten Fassung<br />
durch geeignete Maßnahmen verhin<strong>der</strong>t werden.<br />
4.6 Vorgabedokumente sollen nicht handgeschrieben<br />
sein. Wenn jedoch Daten manuell eingetragen<br />
werden müssen, kann dies in klarer,<br />
lesbarer und nicht zu entfernen<strong>der</strong> Handschrift<br />
gemacht werden. Für solche Eintragungen ist<br />
ausreichend Platz vorzusehen.<br />
4.7 Jede Än<strong>der</strong>ung einer Eintragung in einem<br />
Dokument ist mit Datum versehen abzuzeichnen.<br />
Trotz Än<strong>der</strong>ung muss die ursprüngliche Information<br />
lesbar bleiben. Soweit erfor<strong>der</strong>lich, ist<br />
<strong>der</strong> Grund <strong>für</strong> die Än<strong>der</strong>ung anzugeben.<br />
4.8 Protokolle sind zeitnah zum jeweiligen<br />
Vorgang und so anzufertigen o<strong>der</strong> zu vervollständigen,<br />
dass sich alle wichtigen, die Herstellung<br />
<strong>der</strong> <strong>Arzneimittel</strong> betreffenden Tätigkeiten<br />
<strong>zur</strong>ückverfolgen lassen. Die Protokolle sind mindestens<br />
ein Jahr über die Laufzeit des Fertigproduktes<br />
hinaus aufzubewahren.<br />
4.9 Daten können über DV-Systeme, fotografisch<br />
o<strong>der</strong> auf an<strong>der</strong>e zuverlässige Weise aufgezeichnet<br />
werden. Es müssen jedoch detaillierte<br />
Verfahrensanweisungen bezüglich des verwendeten<br />
Systems <strong>zur</strong> Verfügung stehen. Die Richtigkeit<br />
<strong>der</strong> Aufzeichnungen ist zu überprüfen. Bei<br />
einer Dokumentation mittels DV dürfen nur dazu<br />
befugte Personen Daten eingeben o<strong>der</strong> än<strong>der</strong>n<br />
Übersetzung durch die Mitglie<strong>der</strong> <strong>der</strong> AG 42 des QS-Forums Schleswig-Holstein / Hamburg
computer and there should be a record of<br />
changes and deletions; access should be restricted<br />
by passwords or other means and the<br />
result of entry of critical data should be independently<br />
checked. Batch records electronically<br />
stored should be protected by back-up transfer<br />
on magnetic tape, microfilm, paper or other<br />
means. It is particularly important that the data<br />
are readily available throughout the period of<br />
retention.<br />
können. Än<strong>der</strong>ungen und Löschungen sind zu<br />
protokollieren. Der Zugang zum System ist<br />
durch Passwörter o<strong>der</strong> auf an<strong>der</strong>e Weise zu<br />
schützen. Die Eingabe kritischer Daten ist von<br />
einer zweiten Person zu überprüfen. Elektronisch<br />
gespeicherte Chargenprotokolle sind<br />
durch zusätzliche Übertragung auf Magnetband,<br />
Mikrofilm, Papier o<strong>der</strong> auf an<strong>der</strong>e Weise zu<br />
sichern. Es ist beson<strong>der</strong>s wichtig, dass die Daten,<br />
solange sie gespeichert werden, kurzfristig<br />
verfügbar sind.<br />
Documents required Erfor<strong>der</strong>liche Vorgabedokumente<br />
Specifications Spezifikationen<br />
4.10 There should be appropriately authorised<br />
and dated specifications for starting and packaging<br />
materials, and finished products; where<br />
appropriate, they should be also available for<br />
intermediate or bulk products.<br />
Specifications for starting and packaging materials<br />
4.11 Specifications for starting and primary or<br />
printed packaging materials should include, if<br />
applicable:<br />
a) a description of the materials, including:<br />
— the designated name and the internal code<br />
reference;<br />
— the reference, if any, to a pharmacopoeial<br />
monograph;<br />
— the approved suppliers and, if possible, the<br />
original producer of the products;<br />
— a specimen of printed materials;<br />
b) directions for sampling and testing or reference<br />
to procedures;<br />
c) qualitative and quantitative requirements with<br />
acceptance limits;<br />
d) storage conditions and precautions;<br />
e) the maximum period of storage before re-examination.<br />
Specifications for intermediate and bulk<br />
products<br />
4.10 Erfor<strong>der</strong>lich sind von <strong>der</strong> hier<strong>für</strong> verantwortlichen<br />
Person genehmigte und datierte Spezifikationen<br />
<strong>für</strong> Ausgangsstoffe, Verpackungsmaterialien<br />
und Fertigprodukte. Soweit zutreffend,<br />
müssen auch Spezifikationen <strong>für</strong> Zwischen-<br />
o<strong>der</strong> Endprodukte (Bulkware) <strong>zur</strong> Verfügung<br />
stehen.<br />
Spezifikationen <strong>für</strong> Ausgangsstoffe und Verpackungsmaterialien<br />
4.11 Die Spezifikationen <strong>für</strong> Ausgangsstoffe<br />
und primäre o<strong>der</strong> bedruckte Verpackungsmaterialien<br />
haben (soweit zutreffend) zu umfassen:<br />
a) eine Beschreibung <strong>der</strong> Materialien mit:<br />
— <strong>der</strong> festgelegten Bezeichnung und <strong>der</strong> internen<br />
Artikelnummer<br />
— sofern vorhanden, einer Bezugnahme auf<br />
eine Arzneibuchmonographie<br />
— <strong>der</strong> Angabe <strong>der</strong> zugelassenen Lieferanten<br />
und, wenn möglich, <strong>der</strong> Originalhersteller <strong>der</strong><br />
Produkte<br />
— einem Muster <strong>der</strong> bedruckten Verpackungsmaterialien<br />
b) Anweisungen <strong>zur</strong> Probenahme und <strong>zur</strong> Prüfung<br />
o<strong>der</strong> einen Hinweis auf entsprechende Verfahrensanweisungen<br />
c) qualitative und quantitative Anfor<strong>der</strong>ungen<br />
einschließlich <strong>der</strong> zulässigen Grenzwerte<br />
d) Lagerungsbedingungen und etwaige Vorsichtsmaßnahmen<br />
e) die maximale Lagerdauer bis zu einer erneuten<br />
Überprüfung.<br />
Spezifikationen <strong>für</strong> Zwischen- und Endprodukte<br />
(Bulkware)<br />
Übersetzung durch die Mitglie<strong>der</strong> <strong>der</strong> AG 42 des QS-Forums Schleswig-Holstein / Hamburg
4.12 Specifications for intermediate and bulk<br />
products should be available if these are purchased<br />
or dispatched, or if data obtained from<br />
intermediate products are used for the evaluation<br />
of the finished product. The specifications<br />
should be similar to specifications for starting<br />
materials or for finished products, as<br />
appropriate.<br />
4.12 Spezifikationen <strong>für</strong> Zwischen- und Endprodukte<br />
(Bulkware) müssen vorhanden sein,<br />
wenn diese als solche bezogen o<strong>der</strong> vertrieben<br />
o<strong>der</strong> Daten von Zwischenprodukten <strong>für</strong> die Bewertung<br />
des Fertigprodukts herangezogen werden.<br />
Die Spezifikationen haben denen <strong>für</strong> Ausgangsstoffe<br />
bzw. denen <strong>für</strong> Fertigprodukte zu<br />
entsprechen.<br />
Specifications for finished products Spezifikationen <strong>für</strong> Fertigprodukte<br />
4.13 Specifications for finished products should<br />
include:<br />
a) the designated name of the product and the<br />
code reference where applicable;<br />
b) the formula or a reference to;<br />
c) a description of the pharmaceutical form and<br />
package details;<br />
d) directions for sampling and testing or a reference<br />
to procedures;<br />
e) the qualitative and quantitative requirements,<br />
with the acceptance limits;<br />
f) the storage conditions and any special handling<br />
precautions, where applicable;<br />
g) the shelf-life.<br />
Manufacturing Formula and Processing Instructions<br />
Formally authorised Manufacturing Formula and<br />
Processing Instructions should exist for each<br />
product and batch size to be manufactured.<br />
They are often combined in one document.<br />
4.14 The Manufacturing Formula should include:<br />
a) the name of the product, with a product reference<br />
code relating to its specification;<br />
b) a description of the pharmaceutical form,<br />
strength of the product and batch size;<br />
c) a list of all starting materials to be used, with<br />
the amount of each, described using the designated<br />
name and a reference which is unique to<br />
that material; mention should be made of any<br />
substance that may disappear in the course of<br />
processing;<br />
d) a statement of the expected final yield with<br />
the acceptable limits, and of relevant<br />
intermediate yields, where applicable.<br />
4.13 Die Spezifikationen <strong>für</strong> Fertigprodukte beinhalten:<br />
a) den festgesetzten Produktnamen und, sofern<br />
zutreffend, die interne Artikelnummer<br />
b) die Rezeptur o<strong>der</strong> eine Bezugnahme darauf<br />
c) eine Beschreibung <strong>der</strong> Darreichungsform und<br />
<strong>der</strong> Einzelheiten <strong>der</strong> Verpackung<br />
d) Anweisungen <strong>für</strong> die Probenahme und Prüfung<br />
o<strong>der</strong> einen Verweis auf entsprechende Verfahrensanweisungen<br />
e) die qualitativen und quantitativen Anfor<strong>der</strong>ungen<br />
einschließlich <strong>der</strong> zulässigen Grenzwerte<br />
f) die Lagerungsbedingungen und etwaige Vorsichtsmaßnahmen<br />
g) die Laufzeit.<br />
Rezeptur und Herstellungsanweisungen<br />
Für alle herzustellenden Produkte und jede<br />
Chargengröße sind ordnungsgemäß genehmigte<br />
Rezepturen und Herstellungsanweisungen<br />
erfor<strong>der</strong>lich. Sie werden häufig in einem Dokument<br />
zusammengefasst.<br />
4.14 Die Rezepturen haben zu enthalten:<br />
a) den Produktnamen mit Artikelnummer, die auf<br />
die Spezifikation des Produkts verweist<br />
b) eine Beschreibung <strong>der</strong> Darreichungsform, die<br />
Stärke des <strong>Arzneimittel</strong>s und die Chargengröße<br />
c) eine Auflistung aller einzusetzenden Ausgangsstoffe<br />
mit den jeweiligen Mengen, bezeichnet<br />
mit den festgelegten Namen und eindeutigen<br />
Artikelnummern; ebenso ist jede Substanz<br />
aufzuführen, die im Endprodukt nicht mehr<br />
enthalten ist;<br />
d) Angaben <strong>zur</strong> erwarteten Endausbeute mit<br />
den zulässigen Grenzwerten und, soweit zutreffend,<br />
Ausbeuteangaben auf relevanten Zwischenstufen.<br />
Übersetzung durch die Mitglie<strong>der</strong> <strong>der</strong> AG 42 des QS-Forums Schleswig-Holstein / Hamburg
4.15 The Processing Instructions should include:<br />
a) a statement of the processing location and<br />
the principal equipment to be used;<br />
b) the methods, or reference to the methods, to<br />
be used for preparing the critical equipment (e.g.<br />
cleaning, assembling, calibrating, sterilising);<br />
c) detailed stepwise processing instructions (e.g.<br />
checks on materials, pre-treatments, sequence<br />
for adding materials, mixing times, temperatures);<br />
d) the instructions for any in-process controls<br />
with their limits;<br />
e) where necessary, the requirements for bulk<br />
storage of the products; including the container,<br />
labelling and special storage conditions where<br />
applicable;<br />
f) any special precautions to be observed.<br />
4.15 Die Herstellungsanweisung (Bulkware)<br />
hat zu enthalten:<br />
a) Angaben <strong>zur</strong> Produktionsstätte und <strong>der</strong><br />
grundsätzlich zu verwendenden Ausrüstung<br />
b) die Methoden o<strong>der</strong> einen Verweis auf die<br />
Methoden, nach denen kritische Teile <strong>der</strong> Ausrüstung<br />
vorzubereiten sind (z. B. Reinigung,<br />
Montage, Kalibrierung, Sterilisation)<br />
c) detaillierte schrittweise Verfahrensanweisungen<br />
(z. B. Materialkontrollen, Vorbehandlungen,<br />
Reihenfolge <strong>der</strong> Materialzugabe, Mischzeiten,<br />
Temperaturen)<br />
d) Anweisungen <strong>für</strong> alle Inprozesskontrollen einschließlich<br />
<strong>der</strong> zulässigen Grenzwerte<br />
e) erfor<strong>der</strong>lichenfalls Anfor<strong>der</strong>ungen an die Lagerung<br />
<strong>der</strong> Bulkware, einschließlich <strong>der</strong> Behältnisse,<br />
<strong>der</strong> Kennzeichnung und, soweit zutreffend,<br />
spezieller Lagerungsbedingungen<br />
f) alle beson<strong>der</strong>en Vorsichtsmaßnahmen, die zu<br />
beachten sind.<br />
Packaging Instructions Herstellungsanweisung (Fertigprodukt)<br />
4.16 There should be formally authorised<br />
Packaging Instructions for each product, pack<br />
size and type. These should normally include, or<br />
have a reference to, the following:<br />
a) name of the product;<br />
b) description of its pharmaceutical form, and<br />
strength where applicable;<br />
c) the pack size expressed in terms of the number,<br />
weight or volume of the product in the final<br />
container;<br />
d) a complete list of all the packaging materials<br />
required for a standard batch size, including<br />
quantities, sizes and types, with the code or reference<br />
number relating to the specifications of<br />
each packaging material;<br />
e) where appropriate, an example or reproduction<br />
of the relevant printed packaging materials,<br />
and specimens indicating where to apply batch<br />
number references, and shelf life of the product;<br />
f) special precautions to be observed, including<br />
a careful examination of the area and equipment<br />
in or<strong>der</strong> to ascertain the line clearance before<br />
operations begin;<br />
4.16 Für jedes Produkt, jede Packungsgröße<br />
und jeden Packungstyp sind ordnungsgemäß<br />
genehmigte Herstellungsanweisungen (Fertigprodukt)<br />
erfor<strong>der</strong>lich. Diese haben in <strong>der</strong> Regel<br />
folgende Angaben o<strong>der</strong> Querverweise hierauf zu<br />
enthalten:<br />
a) Name des Produkts<br />
b) Beschreibung <strong>der</strong> Darreichungsform und soweit<br />
zutreffend <strong>der</strong> Stärke<br />
c) die Packungsgröße, ausgedrückt in Stückzahl,<br />
Masse o<strong>der</strong> Füllvolumen des Produktes im<br />
Endbehältnis<br />
d) eine vollständige Auflistung aller <strong>für</strong> eine<br />
Standardchargengröße erfor<strong>der</strong>lichen Verpackungsmaterialien<br />
nach Art, Größe und Menge,<br />
mit Angabe <strong>der</strong> Codierung o<strong>der</strong> Artikelnummer,<br />
die sich auf die Spezifikation des jeweiligen<br />
Verpackungsmaterials bezieht<br />
e) soweit angezeigt ein Muster o<strong>der</strong> eine Kopie<br />
<strong>der</strong> betreffenden bedruckten Verpackungsmaterialien<br />
sowie Muster, die erkennen lassen, wo<br />
Chargenbezeichnung und Verfalldatum des Produkts<br />
zu platzieren sind<br />
f) beson<strong>der</strong>e Vorsichtsmaßnahmen, die zu beachten<br />
sind, einschließlich einer sorgfältigen<br />
Überprüfung des Verpackungsbereichs und <strong>der</strong><br />
Ausrüstung, um die vollständige Räumung <strong>der</strong><br />
Übersetzung durch die Mitglie<strong>der</strong> <strong>der</strong> AG 42 des QS-Forums Schleswig-Holstein / Hamburg
g) a description of the packaging operation, including<br />
any significant subsidiary operations,<br />
and equipment to be used;<br />
h) details of in-process controls with instructions<br />
for sampling and acceptance limits.<br />
Anlage vor Beginn des neuen Verpackungsvorganges<br />
sicherzustellen<br />
g) eine Beschreibung <strong>der</strong> Verpackungsvorgänge<br />
mit allen wichtigen Nebenarbeiten und <strong>der</strong> einzusetzenden<br />
Ausrüstung<br />
h) Einzelheiten zu Inprozesskontrollen mit Hinweisen<br />
<strong>für</strong> die Probenahme und Angabe <strong>der</strong> zulässigen<br />
Grenzwerte.<br />
Batch Processing Records Herstellungsprotokolle (Bulkware)<br />
4.17 A Batch Processing Record should be<br />
kept for each batch processed. It should be<br />
based on the relevant parts of the currently approved<br />
Manufacturing Formula and Processing<br />
Instructions. The method of preparation of such<br />
records should be designed to avoid transcription<br />
errors. The record should carry the number<br />
of the batch being manufactured. Before any<br />
processing begins, there should be recorded<br />
checks that the equipment and work station are<br />
clear of previous products, documents or materials<br />
not required for the planned process, and<br />
that equipment is clean and suitable for use.<br />
During processing, the following information<br />
should be recorded at the time each action is<br />
taken and, after completion, the record should<br />
be dated and signed in agreement by the person<br />
responsible for the processing operations:<br />
a) the name of the product;<br />
b) dates and times of commencement, of significant<br />
intermediate stages and of completion of<br />
production;<br />
c) name of the person responsible for each<br />
stage of production;<br />
d) initials of the operator of different significant<br />
steps of production and, where appropriate, of<br />
the person who checked each of these operations<br />
(e.g. weighing);<br />
e) the batch number and/or analytical control<br />
number as well as the quantities of each starting<br />
material actually weighed (including the batch<br />
number and amount of any recovered or reprocessed<br />
material added);<br />
f) any relevant processing operation or event<br />
and major equipment used;<br />
g) a record of the in-process controls and the<br />
initials of the person(s) carrying them out, and<br />
4.17 Für jede gefertigte Charge ist ein Herstellungsprotokoll<br />
anzufertigen. Es basiert auf den<br />
wesentlichen Teilen <strong>der</strong> gültigen Rezeptur und<br />
<strong>der</strong> Herstellungsanweisung. Das Erstellen <strong>der</strong><br />
Protokolle hat so zu erfolgen, dass Übertragungsfehler<br />
vermieden werden. Das Protokoll<br />
hat die Chargenkennzeichnung <strong>der</strong> zu fertigenden<br />
Charge zu tragen. Vor Produktionsbeginn<br />
ist zu überprüfen, dass die Ausrüstung und <strong>der</strong><br />
Arbeitsbereich frei sind von allen vorherigen, <strong>für</strong><br />
den nächsten Vorgang nicht erfor<strong>der</strong>lichen Produkten,<br />
Unterlagen o<strong>der</strong> Materialien und die<br />
Ausrüstung sauber und einsatzbereit ist; dies ist<br />
zu dokumentieren. Während <strong>der</strong> Produktion sind<br />
jeweils zum Zeitpunkt <strong>der</strong> entsprechenden Arbeitsabläufe<br />
die folgenden Informationen aufzuzeichnen<br />
und nach Beendigung des Vorganges<br />
von <strong>der</strong> <strong>für</strong> die Herstellung verantwortlichen Person<br />
mit Datumsangabe zu unterzeichnen:<br />
a) Name des Produkts<br />
b) Daten und Zeiten des Produktionsbeginns,<br />
wichtiger Zwischenstufen und des Produktionsendes<br />
c) Name <strong>der</strong> <strong>für</strong> die jeweilige Produktionsstufe<br />
verantwortlichen Person<br />
d) Namenszeichen des jeweiligen Mitarbeiters,<br />
<strong>der</strong> den entsprechenden, signifikanten Produktionsschritt<br />
durchführte und, soweit zutreffend,<br />
Namenszeichen <strong>der</strong> Person, die diese Arbeitsgänge<br />
überprüft hat (z. B. die Einwaage)<br />
e) die Chargenkennzeichnung und/o<strong>der</strong> die<br />
Analysenkontrollnummer sowie die tatsächlich<br />
eingewogene Menge jedes Ausgangsstoffs (einschließlich<br />
Chargenkennzeichnung und Menge<br />
wie<strong>der</strong>gewonnener o<strong>der</strong> umgearbeiteter Materialien)<br />
f) je<strong>der</strong> relevante Produktionsvorgang, die wichtigste<br />
verwendete Ausrüstung sowie jedes beson<strong>der</strong>e<br />
Vorkommnis<br />
g) Aufzeichnungen über die Inprozesskontrollen<br />
sowie die erhaltenen Ergebnisse und das/die<br />
Übersetzung durch die Mitglie<strong>der</strong> <strong>der</strong> AG 42 des QS-Forums Schleswig-Holstein / Hamburg
the results obtained;<br />
h) the product yield obtained at different and<br />
pertinent stages of manufacture;<br />
i) notes on special problems including details,<br />
with signed authorisation for any deviation from<br />
the Manufacturing Formula and Processing Instructions.<br />
Namenszeichen <strong>der</strong> Person(en), die die Kontrollen<br />
ausgeführt hat/haben<br />
h) die Ausbeute auf den jeweils relevanten Stufen<br />
i) Angaben zu beson<strong>der</strong>en Vorkommnissen, einschließlich<br />
Einzelheiten zu je<strong>der</strong> Abweichung<br />
von <strong>der</strong> Rezeptur o<strong>der</strong> Herstellungsanweisung,<br />
mit Unterschrift <strong>der</strong> Person, die die Abweichung<br />
genehmigt hat.<br />
Batch Packaging Records Herstellungsprotokoll (Verpackung)<br />
4.18 A Batch Packaging Record should be kept<br />
for each batch or part batch processed. It should<br />
be based on the relevant parts of the Packaging<br />
Instructions and the method of preparation of<br />
such records should be designed to avoid transcription<br />
errors. The record should carry the<br />
batch number and the quantity of bulk product to<br />
be packed, as well as the batch number and the<br />
planned quantity of finished product that will be<br />
obtained. Before any packaging operation begins,<br />
there should be recorded checks that the<br />
equipment and work station are clear of<br />
previous products, documents or materials not<br />
required for the planned packaging operations,<br />
and that equipment is clean and suitable for use.<br />
The following information should be entered at<br />
the time each action is taken and, after<br />
completion, the record should be dated and<br />
signed in agreement by the person(s)<br />
responsible for the packaging operations:<br />
a) the name of the product;<br />
b) the date(s) and times of the packaging operations;<br />
c) the name of the responsible person carrying<br />
out the packaging operation;<br />
d) the initials of the operators of the different<br />
significant steps;<br />
e) records of checks for identity and conformity<br />
with the packaging instructions including the results<br />
of in-process controls;<br />
f) details of the packaging operations carried<br />
out, including references to equipment and the<br />
packaging lines used;<br />
g) whenever possible, samples of printed packaging<br />
materials used, including specimens of<br />
the batch coding, expiry dating and any<br />
4.18 Für jede Charge o<strong>der</strong> Teilcharge ist ein<br />
Herstellungsprotokoll (Verpackung) zu erstellen.<br />
Es hat auf den wesentlichen Teilen <strong>der</strong> Herstellungsanweisung<br />
zu beruhen. Das Erstellen solcher<br />
Protokolle hat so zu erfolgen, dass Übertragungsfehler<br />
vermieden werden. Im Protokoll<br />
sind die Chargenbezeichnung und die Menge<br />
<strong>der</strong> zu verpackenden Bulkware sowie die Chargenbezeichnung<br />
des Fertigprodukts und die geplante<br />
Stückzahl anzugeben. Vor Verpackungsbeginn<br />
ist zu überprüfen, dass die Ausrüstung<br />
und <strong>der</strong> Arbeitsbereich frei sind von allen vorherigen,<br />
<strong>für</strong> den nächsten Vorgang nicht erfor<strong>der</strong>lichen<br />
Produkten, Unterlagen o<strong>der</strong> Materialien<br />
und die Ausrüstung sauber und einsatzbereit ist;<br />
dies ist zu dokumentieren. Folgende Informationen<br />
sind jeweils zum Zeitpunkt <strong>der</strong> entsprechenden<br />
Arbeitsgänge aufzuzeichnen und nach<br />
<strong>der</strong> Beendigung des Verpackungsvorganges<br />
von <strong>der</strong> <strong>für</strong> die Herstellung verantwortlichen Person<br />
mit Datumsangabe zu unterzeichnen:<br />
a) Name des Produkts<br />
b) Daten und Zeiten <strong>der</strong> Verpackungsvorgänge<br />
c) Name <strong>der</strong> <strong>für</strong> den Verpackungsvorgang verantwortlichen<br />
Person<br />
d) Namenszeichen <strong>der</strong> Mitarbeiter, die die verschiedenen<br />
signifikanten Verpackungsschritte<br />
durchführten<br />
e) Aufzeichnungen über Identitätskontrollen und<br />
Überprüfung auf Übereinstimmung mit <strong>der</strong> Herstellungsanweisung<br />
(Verpackung), einschließlich<br />
<strong>der</strong> Ergebnisse von Inprozesskontrollen<br />
f) Einzelheiten zu den durchgeführten Verpackungsvorgängen,<br />
einschließlich Hinweisen<br />
auf die verwendete Ausrüstung und die eingesetzten<br />
Verpackungslinien<br />
g) wann immer möglich, Proben <strong>der</strong> verwendeten<br />
bedruckten Verpackungsmaterialien, einschließlich<br />
Mustern mit <strong>der</strong> Chargenbezeich-<br />
Übersetzung durch die Mitglie<strong>der</strong> <strong>der</strong> AG 42 des QS-Forums Schleswig-Holstein / Hamburg
additional overprinting;<br />
h) notes on any special problems or unusual<br />
events including details, with signed authorisation<br />
for any deviation from the Manufacturing<br />
Formula and Processing Instructions;<br />
i) the quantities and reference number or<br />
identification of all printed packaging materials<br />
and bulk product issued, used, destroyed or<br />
returned to stock and the quantities of obtained<br />
product, in or<strong>der</strong> to provide for an adequate<br />
reconciliation.<br />
nung, dem aufgedruckten Verfalldatum und<br />
an<strong>der</strong>en zusätzlichen Aufdrucken<br />
h) detaillierte Angaben zu speziellen Problemen<br />
o<strong>der</strong> beson<strong>der</strong>en Vorkommnissen, mit Genehmigungsvermerk<br />
(Datum/Unterschrift) bei je<strong>der</strong><br />
Abweichung von <strong>der</strong> Stückliste o<strong>der</strong> <strong>der</strong> Herstellungsanweisung<br />
(Verpackung)<br />
i) die Mengen und die Referenznummern o<strong>der</strong><br />
an<strong>der</strong>e Angaben <strong>zur</strong> Identifizierung 14 aller bereitgestellten,<br />
verwendeten, vernichteten o<strong>der</strong><br />
ins Lager <strong>zur</strong>ückgegebenen bedruckten Verpackungsmaterialien<br />
o<strong>der</strong> <strong>der</strong> Bulkware sowie<br />
<strong>der</strong> Menge des erhaltenen Fertigproduktes, um<br />
eine aussagekräftige Bilanzierung zu ermöglichen.<br />
Procedures and records Verfahrensanweisungen und Protokolle<br />
Receipt Wareneingang<br />
4.19 There should be written procedures and<br />
records for the receipt of each delivery of each<br />
starting and primary and printed packaging material.<br />
4.20 The records of the receipts should<br />
include:<br />
a) the name of the material on the delivery note<br />
and the containers;<br />
b) the "in-house" name and/or code of material<br />
(if different from a);<br />
c) date of receipt;<br />
d) supplier’s name and, if possible, manufacturer’s<br />
name;<br />
e) manufacturer’s batch or reference number;<br />
f) total quantity, and number of containers received;<br />
g) the batch number assigned after receipt;<br />
h) any relevant comment (e.g. state of the containers).<br />
4.21 There should be written procedures for<br />
the internal labelling, quarantine and storage of<br />
starting materials, packaging materials and<br />
other materials, as appropriate.<br />
4.19 Schriftliche Verfahrensanweisungen und<br />
Protokolle <strong>für</strong> die Annahme je<strong>der</strong> Lieferung<br />
eines jeden Ausgangsstoffs und jedes primären<br />
und/o<strong>der</strong> bedruckten Verpackungsmaterials<br />
müssen vorhanden sein.<br />
4.20 Die Wareneingangsprotokolle enthalten:<br />
a) den Namen des Materials auf dem Lieferschein<br />
und den Behältnissen<br />
b) den firmenintern gebräuchlichen Namen<br />
und/o<strong>der</strong> Materialcode (wenn dieser sich von<br />
Buchstabe a unterscheidet)<br />
c) das Datum des Wareneingangs<br />
d) den Namen des Lieferanten und, wenn möglich,<br />
des Herstellers<br />
e) die Chargenkennzeichnung o<strong>der</strong> Referenznummer<br />
des Herstellers<br />
f) die Gesamtmenge und die Anzahl <strong>der</strong> erhaltenen<br />
Behältnisse<br />
g) die <strong>der</strong> Lieferung nach dem Eingang ggf. zugewiesene<br />
firmeninterne Chargenkennzeichnung<br />
h) beson<strong>der</strong>e Bemerkungen (z. B. zum Zustand<br />
<strong>der</strong> Behältnisse).<br />
4.21 Schriftliche Verfahrensanweisungen <strong>für</strong><br />
die interne Kennzeichnung, die Quarantäne und<br />
die Lagerung <strong>der</strong> Ausgangsstoffe, <strong>der</strong> Verpackungsmaterialien<br />
und, soweit zutreffend, an-<br />
14<br />
Referenznummer o<strong>der</strong> an<strong>der</strong>e Angaben <strong>zur</strong> Identifizierung: vorzugsweise ist hier die Chargennummer des<br />
Materials zu verwenden.<br />
Übersetzung durch die Mitglie<strong>der</strong> <strong>der</strong> AG 42 des QS-Forums Schleswig-Holstein / Hamburg
Sampling Probenahme<br />
4.22 There should be written procedures for<br />
sampling, which include the person(s)<br />
authorised to take samples, the methods and<br />
equipment to be used, the amounts to be taken<br />
and any precautions to be observed to avoid<br />
contamination of the material or any<br />
deterioration in its quality (see Chapter 6, item<br />
13).<br />
Testing Prüfung<br />
4.23 There should be written procedures for<br />
testing materials and products at different<br />
stages of manufacture, describing the methods<br />
and equipment to be used. The tests performed<br />
should be recorded (see Chapter 6, item 17).<br />
Other Sonstiges<br />
4.24 Written release and rejection procedures<br />
should be available for materials and products,<br />
and in particular for the release for sale of the<br />
finished product by the Qualified Person(s) in<br />
accordance with the requirements of Article 51<br />
of Directive 2001/83/EC.<br />
4.25 Records should be maintained of the distribution<br />
of each batch of a product in or<strong>der</strong> to<br />
facilitate the recall of the batch if necessary (see<br />
Chapter 8).<br />
4.26 There should be written procedures and<br />
the associated records of actions taken or conclusions<br />
reached, where appropriate, for:<br />
— validation;<br />
— equipment assembly and calibration;<br />
— maintenance, cleaning and sanitation;<br />
— personnel matters including training, clothing,<br />
hygiene;<br />
— environmental monitoring;<br />
— pest control;<br />
<strong>der</strong>er Materialien haben vorzuliegen.<br />
4.22 Es haben schriftliche Verfahrensanweisungen<br />
<strong>für</strong> die Probenahme vorzuliegen, die Angaben<br />
enthalten über die <strong>zur</strong> Probenahme befugte(n)<br />
Person(en), die Methoden <strong>der</strong> Probenahme<br />
und die einzusetzende Ausrüstung, die<br />
zu entnehmenden Probenmengen und alle Vorsichtsmaßnahmen,<br />
die zu beachten sind, um<br />
eine Verunreinigung des Materials o<strong>der</strong> sonstige<br />
Qualitätsmin<strong>der</strong>ung zu vermeiden (siehe Nummer<br />
6.13).<br />
4.23 Für die Prüfung von Materialien und Produkten<br />
auf den verschiedenen Produktionsstufen<br />
haben schriftliche Verfahrensanweisungen<br />
vorzuliegen, in denen die Prüfmethoden<br />
und die einzusetzende Ausrüstung angegeben<br />
sind. Die durchgeführten Prüfungen sind zu protokollieren<br />
(siehe Nummer 6.17).<br />
4.24 Es haben schriftliche Verfahrensanweisungen<br />
<strong>für</strong> die Freigabe und Zurückweisung von<br />
Materialien und Produkten <strong>zur</strong> Verfügung zu stehen.<br />
In Übereinstimmung mit den Anfor<strong>der</strong>ungen<br />
in Artikel 51 <strong>der</strong> Direktive 2001/83/<strong>EG</strong> 8 gilt<br />
dies beson<strong>der</strong>s <strong>für</strong> die durch die Sachkundige(n)<br />
Person(en) erteilte Freigabe des Fertigprodukts<br />
zum Verkauf. 15<br />
4.25 Über den Vertrieb einer jeden Charge eines<br />
Produkts sind Aufzeichnungen aufzubewahren,<br />
um erfor<strong>der</strong>lichenfalls Rückrufe zu ermöglichen<br />
(siehe Kapitel 8).<br />
4.26 Es haben schriftliche Verfahrensanweisungen<br />
und die zugehörigen Protokolle über<br />
durchgeführte Maßnahmen o<strong>der</strong>, soweit angebracht,<br />
über getroffene Schlussfolgerungen vorzuliegen<br />
<strong>für</strong>:<br />
— Validierungen<br />
— Montage und Kalibrierung <strong>der</strong> Ausrüstung<br />
— Wartung, Reinigung und Desinfektion<br />
— personalbezogene Belange, einschließlich<br />
Schulung, Kleidung und Hygiene<br />
— Umgebungskontrollen<br />
— Schädlingsbekämpfungsmaßnahmen<br />
15 Der Annex 16 definiert als Schlüsselbegriffe „Confirmation“, „Certification“ und „Release“.<br />
Übersetzung durch die Mitglie<strong>der</strong> <strong>der</strong> AG 42 des QS-Forums Schleswig-Holstein / Hamburg
— complaints;<br />
— recalls;<br />
— returns.<br />
4.27 Clear operating procedures should be<br />
available for major items of manufacturing and<br />
test equipment.<br />
4.28 Log books should be kept for major or<br />
critical equipment recording, as appropriate, any<br />
validations, calibrations, maintenance, cleaning<br />
or repair operations, including the dates and<br />
identity of people who carried these operations<br />
out.<br />
4.29 Log books should also record in chronological<br />
or<strong>der</strong> the use of major or critical equipment<br />
and the areas where the products have<br />
been processed.<br />
— Beanstandungen<br />
— Rückrufe<br />
— Warenrückgaben.<br />
CHAPTER 5 PRODUCTION Kapitel 5 Produktion<br />
Principle Grundsätze<br />
Production operations must follow clearly<br />
defined procedures; they must comply with the<br />
principles of Good Manufacturing Practice in<br />
or<strong>der</strong> to obtain products of the requisite quality<br />
and be in accordance with the relevant<br />
manufacturing and marketing authorisations.<br />
4.27 Für die wichtigsten Teile <strong>der</strong> Produktionsund<br />
Prüfausrüstung haben klare Bedienungsanweisungen<br />
<strong>zur</strong> Verfügung zu stehen.<br />
4.28 Für wichtige o<strong>der</strong> kritische Ausrüstungsteile<br />
sind Logbücher zu führen, in denen, soweit<br />
angebracht, alle Validierungen, Kalibrierungen,<br />
Wartungen, Reinigungs- o<strong>der</strong> Reparaturarbeiten<br />
vermerkt werden. Datum und Namen <strong>der</strong> Personen,<br />
die diese Tätigkeiten ausgeführt haben,<br />
sind anzugeben.<br />
4.29 In Logbüchern sind die Benutzung <strong>der</strong><br />
wichtigsten o<strong>der</strong> kritischen Ausrüstungsteile und<br />
die Arbeitsbereiche, in denen die Produkte hergestellt<br />
wurden, chronologisch aufzuzeichnen. 16<br />
Produktionsprozesse haben nach klar definierten<br />
Verfahren zu erfolgen; sie haben den Grundsätzen<br />
<strong>der</strong> <strong>Guten</strong> <strong>Herstellungspraxis</strong> zu entsprechen,<br />
um zu Produkten zu gelangen, die die erfor<strong>der</strong>liche<br />
Qualität aufweisen und mit den gültigen<br />
Herstellungserlaubnissen und Zulassungen<br />
übereinstimmen.<br />
General Allgemeine Anfor<strong>der</strong>ungen<br />
5.1 Production should be performed and supervised<br />
by competent people.<br />
5.2 All handling of materials and products,<br />
such as receipt and quarantine, sampling,<br />
storage, labelling, dispensing, processing, packaging<br />
and distribution should be done in accordance<br />
with written procedures or instructions<br />
and, where necessary, recorded.<br />
5.3 All incoming materials should be checked<br />
to ensure that the consignment corresponds to<br />
the or<strong>der</strong>. Containers should be cleaned where<br />
necessary and labelled with the prescribed data.<br />
5.4 Damage to containers and any other<br />
problem which might adversely affect the quality<br />
of a material should be investigated, recorded<br />
5.1 Die Produktion ist von qualifiziertem Personal<br />
durchzuführen und zu überwachen.<br />
5.2 Je<strong>der</strong> Umgang mit Materialien und Produkten,<br />
wie z. B. Wareneingang und Quarantäne,<br />
Probenahme, Lagerung, Kennzeichnung,<br />
Bereitstellung, Verarbeitung, Verpackung und<br />
Vertrieb, ist in Übereinstimmung mit schriftlich<br />
festgelegten Verfahren o<strong>der</strong> Anweisungen<br />
durchzuführen und – soweit erfor<strong>der</strong>lich – zu<br />
protokollieren.<br />
5.3 Alle eingehenden Materialien sind zu<br />
überprüfen, um sicherzustellen, dass die Lieferung<br />
<strong>der</strong> Bestellung entspricht. Behältnisse sind<br />
erfor<strong>der</strong>lichenfalls zu reinigen und mit den vorgeschriebenen<br />
Angaben zu kennzeichnen.<br />
5.4 Schäden an Behältnissen und alle an<strong>der</strong>en<br />
Probleme, die die Qualität beeinträchtigen<br />
könnten, sind zu untersuchen, zu protokollieren<br />
16 Angaben zum Vorprodukt sollten im Herstellungsprotokoll nach AMWHV aufgeführt sein.<br />
Übersetzung durch die Mitglie<strong>der</strong> <strong>der</strong> AG 42 des QS-Forums Schleswig-Holstein / Hamburg
and reported to the Quality Control Department. und <strong>der</strong> Qualitätskontrollabteilung zu melden.<br />
5.5 Incoming materials and finished products<br />
should be physically or administratively quarantined<br />
immediately after receipt or processing,<br />
until they have been released for use or distribution.<br />
5.6 Intermediate and bulk products purchased<br />
as such should be handled on receipt as though<br />
they were starting materials.<br />
5.7 All materials and products should be<br />
stored un<strong>der</strong> the appropriate conditions established<br />
by the manufacturer and in an or<strong>der</strong>ly<br />
fashion to permit batch segregation and stock<br />
rotation.<br />
5.8 Checks on yields, and reconciliation of<br />
quantities, should be carried out as necessary to<br />
ensure that there are no discrepancies outside<br />
acceptable limits.<br />
5.9 Operations on different products should<br />
not be carried out simultaneously or consecutively<br />
in the same room unless there is no<br />
risk of mix-up or cross-contamination.<br />
5.10 At every stage of processing, products<br />
and materials should be protected from<br />
microbial and other contamination.<br />
5.11 When working with dry materials and<br />
products, special precautions should be taken to<br />
prevent the generation and dissemination of<br />
dust. This applies particularly to the handling of<br />
highly active or sensitising materials.<br />
5.12 At all times during processing, all materials,<br />
bulk containers, major items of equipment<br />
and where appropriate rooms used should be<br />
labelled or otherwise identified with an indication<br />
of the product or material being processed, its<br />
strength (where applicable) and batch number.<br />
Where applicable, this indication should also<br />
mention the stage of production.<br />
5.13 Labels applied to containers, equipment<br />
or premises should be clear, unambiguous and<br />
in the company’s agreed format. It is often<br />
helpful in addition to the wording on the labels to<br />
use colours to indicate status (for example,<br />
quarantined, accepted, rejected, clean, ...).<br />
5.5 Eingehende Materialien und Fertigprodukte<br />
sind unverzüglich nach Eingang o<strong>der</strong> Produktionsende<br />
bis zu ihrer Freigabe <strong>für</strong> eine Verarbeitung<br />
o<strong>der</strong> einer Freigabe <strong>für</strong> das Inverkehrbringen<br />
durch getrennte Lagerung o<strong>der</strong> durch<br />
geeignete organisatorische Maßnahmen in den<br />
Quarantänestatus zu überführen.<br />
5.6 Zwischenprodukte und Bulkware, die als<br />
solche gekauft werden, sind bei <strong>der</strong> Annahme<br />
wie Ausgangsstoffe zu behandeln.<br />
5.7 Alle Materialien und Produkte sind unter<br />
geeigneten, vom Hersteller festgelegten Bedingungen<br />
übersichtlich zu lagern, um eine Trennung<br />
nach Chargen und einen geordneten Umschlag<br />
des Lagerbestands zu ermöglichen.<br />
5.8 Überprüfungen <strong>der</strong> Ausbeuten und die<br />
Bilanzierung <strong>der</strong> Mengen sind notwendigerweise<br />
durchzuführen, um sicherzustellen, dass keine<br />
über die zulässigen Grenzen hinausgehenden<br />
Diskrepanzen auftreten.<br />
5.9 Die Bearbeitung unterschiedlicher Produkte<br />
ist nicht gleichzeitig o<strong>der</strong> nacheinan<strong>der</strong> in<br />
demselben Raum durchzuführen, es sei denn,<br />
es besteht keine Gefahr <strong>der</strong> Verwechslung o<strong>der</strong><br />
Kreuzkontamination.<br />
5.10 Auf je<strong>der</strong> Verarbeitungsstufe sind Produkte<br />
und Materialien vor mikrobieller und an<strong>der</strong>er<br />
Verunreinigung zu schützen.<br />
5.11 Beim Arbeiten mit trockenen Ausgangsmaterialien<br />
und Produkten sind beson<strong>der</strong>e Vorkehrungen<br />
zu treffen, um eine Staubbildung und<br />
–ausbreitung zu verhüten. Dies gilt beson<strong>der</strong>s<br />
<strong>für</strong> den Umgang mit hochaktiven o<strong>der</strong> sensibilisierenden<br />
Materialien.<br />
5.12 Während des gesamten Prozesses sind<br />
alle verwendeten Ausgangsmaterialien, Behältnisse<br />
mit Bulkware, wichtige Ausrüstungsteile<br />
und, soweit zutreffend, auch Räume zu kennzeichnen<br />
o<strong>der</strong> auf an<strong>der</strong>e Weise mit einem Hinweis<br />
auf das herzustellende Produkt o<strong>der</strong> Material,<br />
seine Chargenkennzeichnung und gegebenenfalls<br />
seine Stärke zu versehen. Soweit zutreffend,<br />
ist bei diesen Hinweisen auch die Produktionsstufe<br />
zu vermerken.<br />
5.13 Etiketten o<strong>der</strong> Hinweise an Behältnissen,<br />
Ausrüstung o<strong>der</strong> Räumen sind klar und eindeutig<br />
zu gestalten und haben <strong>der</strong> firmenintern festgelegten<br />
Aufmachung zu entsprechen. Es ist oft<br />
hilfreich, den Status (z. B. in Quarantäne, freigegeben,<br />
<strong>zur</strong>ückgewiesen, sauber usw.) außer in<br />
Übersetzung durch die Mitglie<strong>der</strong> <strong>der</strong> AG 42 des QS-Forums Schleswig-Holstein / Hamburg
5.14 Checks should be carried out to ensure<br />
that pipelines and other pieces of equipment<br />
used for the transportation of products from one<br />
area to another are connected in a correct manner.<br />
5.15 Any deviation from instructions or procedures<br />
should be avoided as far as possible. If a<br />
deviation occurs, it should be approved in<br />
writing by a competent person, with the<br />
involvement of the Quality Control Department<br />
when appropriate.<br />
5.16 Access to production premises should be<br />
restricted to authorised personnel.<br />
5.17 Normally, the production of non-medicinal<br />
products should be avoided in areas and with<br />
the equipment destined for the production of<br />
medicinal products.<br />
Worten auch mit unterschiedlichen Farben anzuzeigen.<br />
5.14 Durch Überprüfungen ist sicherzustellen,<br />
dass Rohrleitungen und an<strong>der</strong>e Ausrüstungsteile,<br />
die <strong>für</strong> den Transport eines Produkts von<br />
einem Bereich in einen an<strong>der</strong>en verwendet werden,<br />
ordnungsgemäß miteinan<strong>der</strong> verbunden<br />
sind.<br />
5.15 Jede Abweichung von Anweisungen o<strong>der</strong><br />
Verfahren ist, soweit irgend möglich, zu vermeiden.<br />
Kommt dennoch eine Abweichung vor, ist<br />
sie schriftlich von einer da<strong>für</strong> zuständigen Person,<br />
soweit erfor<strong>der</strong>lich unter Beteiligung <strong>der</strong><br />
Qualitätskontrollabteilung, zu genehmigen.<br />
5.16 Der Zutritt zu den Produktionsbereichen<br />
ist auf Befugte zu begrenzen.<br />
5.17 In <strong>der</strong> Regel sind Erzeugnisse, die keine<br />
<strong>Arzneimittel</strong> sind, nicht in Bereichen und mit<br />
Ausrüstungsteilen zu produzieren, die <strong>für</strong> die<br />
Produktion von <strong>Arzneimittel</strong>n bestimmt sind.<br />
Prevention of cross-contamination in production Vermeidung einer Kreuzkontamination bei <strong>der</strong><br />
Produktion<br />
5.18 Contamination of a starting material or of<br />
a product by another material or product must<br />
be avoided. This risk of accidental crosscontamination<br />
arises from the uncontrolled<br />
release of dust, gases, vapours, sprays or<br />
organisms from materials and products in<br />
process, from residues on equipment, and from<br />
operators’ clothing. The significance of this risk<br />
varies with the type of contaminant and of<br />
product being contaminated. Amongst the most<br />
hazardous contaminants are highly sensitising<br />
materials, biological preparations containing<br />
living organisms, certain hormones, cytotoxics,<br />
and other highly active materials. Products in<br />
which contamination is likely to be most<br />
significant are those administered by injection,<br />
those given in large doses and/or over a long<br />
time.<br />
5.19 Cross-contamination should be avoided<br />
by appropriate technical or organisational measures,<br />
for example:<br />
a) production in segregated areas (required for<br />
products such as penicillins, live vaccines, live<br />
bacterial preparations and some other biologicals),<br />
or by campaign (separation in time) followed<br />
by appropriate cleaning;<br />
5.18 Die Kontamination eines Ausgangsstoffs<br />
o<strong>der</strong> eines Produkts mit einem an<strong>der</strong>en Material<br />
o<strong>der</strong> Produkt muss vermieden werden. Die Gefahr<br />
einer unbeabsichtigten Kreuzkontamination<br />
entsteht bei <strong>der</strong> unkontrollierten Freisetzung von<br />
Staub, Gasen, Dämpfen, Aerosolen o<strong>der</strong> Organismen<br />
von in <strong>der</strong> Verarbeitung befindlichen Materialien<br />
und Produkten, aus Rückständen aus<br />
<strong>der</strong> Ausrüstung o<strong>der</strong> von <strong>der</strong> Arbeitskleidung <strong>der</strong><br />
Beschäftigten. Die Höhe des Risikos ist abhängig<br />
von <strong>der</strong> Art <strong>der</strong> Verunreinigung und dem betreffenden<br />
Produkt. Als beson<strong>der</strong>s gefährliche<br />
Verunreinigungen sind zu nennen: stark sensibilisierende<br />
Stoffe; biologische Zubereitungen, die<br />
lebende Organismen enthalten; bestimmte Hormone;<br />
Zytostatika und an<strong>der</strong>e hochwirksame<br />
Stoffe. Das Risiko ist beson<strong>der</strong>s hoch bei Parenteralia<br />
und solchen Produkten, die in hoher Dosierung<br />
und/o<strong>der</strong> über einen langen Zeitraum<br />
angewendet werden.<br />
5.19 Eine Kreuzkontamination ist durch geeignete<br />
technische o<strong>der</strong> organisatorische Maßnahmen<br />
zu verhin<strong>der</strong>n, beispielsweise durch:<br />
a) Produktion in räumlich getrennten Bereichen<br />
(erfor<strong>der</strong>lich <strong>für</strong> Produkte wie Penicilline,<br />
Lebendimpfstoffe, Zubereitungen, die lebende<br />
Bakterien enthalten, und an<strong>der</strong>e biologische<br />
Präparate) o<strong>der</strong> in Kampagnen (zeitlich getrennt)<br />
mit nachfolgen<strong>der</strong> angemessener Reini-<br />
Übersetzung durch die Mitglie<strong>der</strong> <strong>der</strong> AG 42 des QS-Forums Schleswig-Holstein / Hamburg
) providing appropriate air-locks and air extraction;<br />
c) minimising the risk of contamination caused<br />
by recirculation or re-entry of untreated or insufficiently<br />
treated air;<br />
d) keeping protective clothing inside areas<br />
where products with special risk of<br />
crosscontamination are processed;<br />
e) using cleaning and decontamination procedures<br />
of known effectiveness, as ineffective<br />
cleaning of equipment is a common source of<br />
cross-contamination;<br />
f) using “closed systems” of production;<br />
g) testing for residues and use of cleaning status<br />
labels on equipment.<br />
5.20 Measures to prevent cross-contamination<br />
and their effectiveness should be checked periodically<br />
according to set procedures<br />
Validation Validierung<br />
5.21 Validation studies should reinforce Good<br />
Manufacturing Practice and be conducted in accordance<br />
with defined procedures. Results and<br />
conclusions should be recorded.<br />
5.22 When any new manufacturing formula or<br />
method of preparation is adopted, steps should<br />
be taken to demonstrate its suitability for routine<br />
processing. The defined process, using the materials<br />
and equipment specified, should be<br />
shown to yield a product consistently of the required<br />
quality.<br />
5.23 Significant amendments to the manufacturing<br />
process, including any change in equipment<br />
or materials, which may affect product<br />
quality and/or the reproducibility of the process<br />
should be validated.<br />
5.24 Processes and procedures should un<strong>der</strong>go<br />
periodic critical re-validation to ensure<br />
that they remain capable of achieving the<br />
intended results.<br />
gung<br />
b) geeignete Schleusen und Luftabsaugung<br />
c) Minimierung des Risikos einer Kontamination,<br />
verursacht durch Umluft o<strong>der</strong> Rückführung von<br />
unbehandelter o<strong>der</strong> ungenügend behandelter<br />
Luft<br />
d) Belassen <strong>der</strong> Schutzkleidung innerhalb <strong>der</strong><br />
Bereiche, in denen Produkte mit beson<strong>der</strong>s<br />
großem Risiko einer Kreuzkontamination hergestellt<br />
werden<br />
e) Verwendung von Reinigungs- und Dekontaminationsverfahren<br />
mit nachgewiesener Wirksamkeit,<br />
da eine ungenügende Reinigung <strong>der</strong><br />
Ausrüstung eine häufige Ursache <strong>der</strong> Kreuzkontamination<br />
ist<br />
f) Einsatz "geschlossener Systeme" bei <strong>der</strong> Produktion<br />
g) Prüfung auf Rückstände und Kennzeichnung<br />
des Reinigungsstatus <strong>der</strong> Ausrüstung.<br />
5.20 Die Maßnahmen <strong>zur</strong> Verhütung <strong>der</strong><br />
Kreuzkontamination und ihre Wirksamkeit sind<br />
regelmäßig nach festgelegten Verfahren zu<br />
überprüfen.<br />
5.21 Validierungen unterstützen die Maßnahmen<br />
<strong>der</strong> <strong>Guten</strong> <strong>Herstellungspraxis</strong>, sie sind in<br />
Übereinstimmung mit festgelegten Verfahren<br />
durchzuführen. Die Ergebnisse und Schlussfolgerungen<br />
sind zu dokumentieren.<br />
5.22 Wenn eine neue Rezeptur o<strong>der</strong> Verarbeitungsmethode<br />
eingeführt wird, ist <strong>der</strong>en Eignung<br />
<strong>für</strong> den Routinebetrieb zu belegen. Es ist aufzuzeigen,<br />
dass <strong>der</strong> festgelegte Prozess bei Einsatz<br />
<strong>der</strong> spezifizierten Ausgangsmaterialien und<br />
<strong>der</strong> vorgegebenen Ausrüstung ein Produkt ergibt,<br />
das gleichbleibend die angestrebte Qualität<br />
aufweist.<br />
5.23 Wesentliche Än<strong>der</strong>ungen des Herstellungsprozesses,<br />
einschließlich aller Verän<strong>der</strong>ungen<br />
bei <strong>der</strong> Ausrüstung o<strong>der</strong> den Ausgangsmaterialien,<br />
die die Produktqualität und/o<strong>der</strong> die<br />
Reproduzierbarkeit des Prozesses beeinflussen<br />
können, sind zu validieren.<br />
5.24 Alle Anweisungen und Verfahren sind regelmäßig<br />
einer kritischen Revalidierung zu unterziehen,<br />
um sicherzustellen, dass sie weiterhin<br />
imstande sind, die angestrebten Ergebnisse zu<br />
Übersetzung durch die Mitglie<strong>der</strong> <strong>der</strong> AG 42 des QS-Forums Schleswig-Holstein / Hamburg
erreichen.<br />
Starting materials Ausgangsstoffe<br />
5.25 The purchase of starting materials is an<br />
important operation which should involve staff<br />
who have a particular and thorough knowledge<br />
of the suppliers.<br />
5.26 Starting materials should only be purchased<br />
from approved suppliers named in the<br />
relevant specification and, where possible, directly<br />
from the producer. It is recommended that<br />
the specifications established by the manufacturer<br />
for the starting materials be discussed with<br />
the suppliers. It is of benefit that all aspects of<br />
the production and control of the starting<br />
material in question, including handling, labelling<br />
and packaging requirements, as well as<br />
complaints and rejection procedures are<br />
discussed with the manufacturer and the<br />
supplier.<br />
5.27 For each delivery, the containers should<br />
be checked for integrity of package and seal and<br />
for correspondence between the delivery note<br />
and the supplier’s labels.<br />
5.28 If one material delivery is made up of different<br />
batches, each batch must be consi<strong>der</strong>ed<br />
as separate for sampling, testing and release.<br />
5.29 Starting materials in the storage area<br />
should be appropriately labelled (see Chapter 5,<br />
item 13). Labels should bear at least the following<br />
information:<br />
— the designated name of the product and the<br />
internal code reference where applicable;<br />
— a batch number given at receipt;<br />
— where appropriate, the status of the contents<br />
(e.g. in quarantine, on test, released, rejected);<br />
— where appropriate, an expiry date or a date<br />
beyond which retesting is necessary.<br />
When fully computerised storage systems are<br />
5.25 Der Einkauf <strong>der</strong> Ausgangsstoffe ist ein<br />
wichtiger Vorgang, an dem Mitarbeiter zu beteiligen<br />
sind, die über die Lieferanten sehr genaue<br />
und gründliche Kenntnisse haben.<br />
5.26 Ausgangsstoffe sind nur von genehmigten<br />
Lieferanten, die in <strong>der</strong> entsprechenden Spezifikation<br />
aufgeführt werden, möglichst direkt vom<br />
Hersteller, zu beziehen. Es wird empfohlen, die<br />
vom <strong>Arzneimittel</strong>hersteller festgelegten Spezifikationen<br />
<strong>der</strong> Ausgangsstoffe mit den Lieferanten/Herstellern<br />
zu diskutieren. Es ist von Vorteil,<br />
wenn alle Gesichtspunkte <strong>der</strong> Produktion und<br />
Kontrolle des jeweiligen Ausgangsstoffs, einschließlich<br />
<strong>der</strong> Anfor<strong>der</strong>ungen an dessen Handhabung,<br />
Kennzeichnung und Verpackung sowie<br />
Beanstandungen und Zurückweisungsverfahren<br />
zwischen dem <strong>Arzneimittel</strong>hersteller und dem<br />
Lieferanten/Ausgangsstoffhersteller erörtert werden.<br />
17<br />
5.27 Bei je<strong>der</strong> Lieferung ist zu überprüfen, ob<br />
Verpackung und Verschluss <strong>der</strong> Behältnisse unversehrt<br />
sind und die Angaben auf dem Lieferschein<br />
und den Etiketten des Lieferanten übereinstimmen.<br />
5.28 Falls eine Materiallieferung aus verschiedenen<br />
Chargen besteht, ist jede Charge hinsichtlich<br />
Probenahme, Prüfung und Freigabe getrennt<br />
zu betrachten.<br />
5.29 Im Lagerbereich befindliche Ausgangsstoffe<br />
sind in geeigneter Weise zu kennzeichnen<br />
(siehe Nummer 5.13). Die Kennzeichnung hat<br />
mindestens folgende Informationen zu enthalten:<br />
— den festgelegten Namen des Materials und,<br />
soweit zutreffend, den internen Referenzcode<br />
— die beim Wareneingang zugewiesene Chargenkennzeichnung<br />
— soweit angezeigt, den Status des Inhalts<br />
(z. B. in Quarantäne, in <strong>der</strong> Prüfung, freigegeben,<br />
<strong>zur</strong>ückgewiesen)<br />
— soweit angebracht, ein Verfalldatum o<strong>der</strong> ein<br />
Datum, ab dem eine Nachprüfung erfor<strong>der</strong>lich<br />
ist.<br />
Im Fall von vollständig computergesteuerten La-<br />
17 Unter Zugrundelegung allgemeiner GMP-Aspekte sind die Autoren <strong>der</strong> Meinung, dass die im deutschen abweichende<br />
Übersetzung im Gegensatz zum englischsprachigen Original den eigentlich beabsichtigten Aspekt<br />
wi<strong>der</strong>spiegelt.<br />
Übersetzung durch die Mitglie<strong>der</strong> <strong>der</strong> AG 42 des QS-Forums Schleswig-Holstein / Hamburg
used, all the above information need not necessarily<br />
be in a legible form on the label.<br />
5.30 There should be appropriate procedures<br />
or measures to assure the identity of the<br />
contents of each container of starting material.<br />
Bulk containers from which samples have been<br />
drawn should be identified (see Chapter 6, item<br />
13).<br />
5.31 Only starting materials which have been<br />
released by the Quality Control Department and<br />
which are within their shelf life should be used.<br />
5.32 Starting materials should only be dispensed<br />
by designated persons, following a written<br />
procedure, to ensure that the correct materials<br />
are accurately weighed or measured into<br />
clean and properly labelled containers.<br />
5.33 Each dispensed material and its weight or<br />
volume should be independently checked and<br />
the check recorded.<br />
5.34 Materials dispensed for each batch should<br />
be kept together and conspicuously labelled as<br />
such.<br />
Processing operations: intermediate and bulk<br />
products<br />
5.35 Before any processing operation is<br />
started, steps should be taken to ensure that the<br />
work area and equipment are clean and free<br />
from any starting materials, products, product<br />
residues or documents not required for the<br />
current operation.<br />
5.36 Intermediate and bulk products should be<br />
kept un<strong>der</strong> appropriate conditions.<br />
5.37 Critical processes should be validated<br />
(see "VALIDATION" in this Chapter).<br />
5.38 Any necessary in-process controls and<br />
environmental controls should be carried out<br />
and recorded.<br />
5.39 Any significant deviation from the<br />
expected yield should be recorded and<br />
investigated.<br />
gersystemen müssen die obigen Informationen<br />
nicht unbedingt in lesbarer Form auf dem Etikett<br />
enthalten sein.<br />
5.30 Mit geeigneten Verfahren o<strong>der</strong> Maßnahmen<br />
ist die Identität des Inhalts eines jeden Behältnisses<br />
mit Ausgangsstoffen sicherzustellen.<br />
Behältnisse, aus denen Proben entnommen<br />
wurden, sind entsprechend zu kennzeichnen<br />
(siehe Nummer 6.13).<br />
5.31 Es sind nur Ausgangsstoffe zu verwenden,<br />
die von <strong>der</strong> Qualitätskontrolle freigegeben<br />
wurden und <strong>der</strong>en Laufzeit nicht überschritten<br />
ist.<br />
5.32 Ausgangsstoffe sind nur von hierzu beauftragten<br />
Personen nach schriftlich festgelegten<br />
Verfahren <strong>zur</strong> Verarbeitung bereitzustellen, um<br />
sicherzustellen, dass die richtigen Stoffe in saubere<br />
und ordnungsgemäß gekennzeichnete Behältnisse<br />
exakt eingewogen o<strong>der</strong> abgemessen<br />
werden.<br />
5.33 Jedes <strong>zur</strong> Verarbeitung bereitgestellte<br />
Material, seine Masse o<strong>der</strong> Volumen ist von einer<br />
weiteren Person zu überprüfen und zu protokollieren.<br />
5.34 Die <strong>für</strong> jede einzelne Charge benötigten<br />
Materialien sind gemeinsam bereitzustellen und<br />
deutlich entsprechend zu kennzeichnen.<br />
Herstellungsvorgänge: Zwischenprodukte und<br />
Bulkware<br />
5.35 Vor jedem Herstellungsvorgang ist sicherzustellen,<br />
dass Arbeitsbereich und Ausrüstung<br />
sauber und frei von allen <strong>für</strong> diesen Vorgang<br />
nicht benötigten Ausgangsmaterialien, Produkten<br />
o<strong>der</strong> Unterlagen sowie Produktrückständen<br />
sind.<br />
5.36 Zwischenprodukte und Bulkware sind<br />
unter geeigneten Bedingungen aufzubewahren.<br />
5.37 Kritische Vorgänge sind zu validieren (siehe<br />
"Validierung" in diesem Kapitel).<br />
5.38 Alle erfor<strong>der</strong>lichen Inprozess- und Umgebungskontrollen<br />
sind durchzuführen und zu protokollieren.<br />
5.39 Jede signifikante Abweichung von <strong>der</strong> erwarteten<br />
Ausbeute ist zu protokollieren und zu<br />
untersuchen.<br />
Übersetzung durch die Mitglie<strong>der</strong> <strong>der</strong> AG 42 des QS-Forums Schleswig-Holstein / Hamburg
Packaging materials Verpackungsmaterialien<br />
5.40 The purchase, handling and control of primary<br />
and printed packaging materials shall be<br />
accorded attention similar to that given to starting<br />
materials.<br />
5.41 Particular attention should be paid to<br />
printed materials. They should be stored in adequately<br />
secure conditions such as to exclude<br />
unauthorised access. Cut labels and other loose<br />
printed materials should be stored and transported<br />
in separate closed containers so as to<br />
avoid mix-ups. Packaging materials should be<br />
issued for use only by authorised personnel following<br />
an approved and documented procedure.<br />
5.42 Each delivery or batch of printed or primary<br />
packaging material should be given a specific<br />
reference number or identification mark.<br />
5.43 Outdated or obsolete primary packaging<br />
material or printed packaging material should be<br />
destroyed and this disposal recorded.<br />
5.40 Der Beschaffung, <strong>der</strong> Handhabung und<br />
<strong>der</strong> Kontrolle <strong>der</strong> primären und/o<strong>der</strong> bedruckten<br />
Verpackungsmaterialien ist ebenso viel Aufmerksamkeit<br />
zu widmen wie den Ausgangsstoffen.<br />
5.41 Beson<strong>der</strong>e Vorsicht ist bei bedruckten<br />
Materialien geboten. Sie sind unter ausreichend<br />
sicheren Bedingungen zu lagern, um unbefugten<br />
Zugriff auszuschließen. Einzeletiketten und<br />
an<strong>der</strong>e lose, bedruckte Materialien sind in separaten,<br />
geschlossenen Behältnissen aufzubewahren<br />
und zu transportieren, um Verwechslungen<br />
zu vermeiden. Verpackungsmaterialien sind nur<br />
nach einem genehmigten und dokumentierten<br />
Verfahren von dazu befugtem Personal <strong>für</strong> den<br />
Gebrauch auszugeben.<br />
5.42 Jede Lieferung o<strong>der</strong> Charge von bedrucktem<br />
o<strong>der</strong> primärem Verpackungsmaterial ist mit<br />
einer spezifischen Kennzahl o<strong>der</strong> Markierung zu<br />
versehen.<br />
5.43 Nicht mehr zu verwendendes primäres<br />
o<strong>der</strong> bedrucktes Verpackungsmaterial ist zu vernichten.<br />
Die Vernichtung ist zu protokollieren.<br />
Packaging operations Herstellungsvorgänge (Verpackung)<br />
5.44 When setting up a programme for the<br />
packaging operations, particular attention should<br />
be given to minimising the risk of cross-contamination,<br />
mix-ups or substitutions. Different<br />
products should not be packaged in close<br />
proximity unless there is physical segregation.<br />
5.45 Before packaging operations are begun,<br />
steps should be taken to ensure that the work<br />
area, packaging lines, printing machines and<br />
other equipment are clean and free from any<br />
products, materials or documents previously<br />
used, if these are not required for the current<br />
operation. The line-clearance should be performed<br />
according to an appropriate check-list.<br />
5.46 The name and batch number of the<br />
product being handled should be displayed at<br />
each packaging station or line.<br />
5.47 All products and packaging materials to<br />
be used should be checked on delivery to the<br />
packaging department for quantity, identity and<br />
conformity with the Packaging Instructions.<br />
5.44 Bei <strong>der</strong> Planung <strong>der</strong> Herstellungsvorgänge<br />
(Verpackung) ist beson<strong>der</strong>s auf eine Risikominimierung<br />
hinsichtlich Kreuzkontamination,<br />
Untermischungen o<strong>der</strong> Verwechslungen zu achten.<br />
Ohne körperliche Trennung dürfen unterschiedliche<br />
Produkte nicht in unmittelbarer Nähe<br />
zueinan<strong>der</strong> verpackt werden.<br />
5.45 Vor Beginn <strong>der</strong> Verpackungsvorgänge ist<br />
sicherzustellen, dass <strong>der</strong> Arbeitsbereich, die<br />
Verpackungslinien, die Druckmaschinen und die<br />
sonstige Ausrüstung sauber und frei von allen<br />
zuvor verwendeten Produkten, Materialien o<strong>der</strong><br />
Unterlagen sind, wenn sie <strong>für</strong> diesen Vorgang<br />
nicht benötigt werden. Dies hat anhand einer<br />
geeigneten Checkliste zu erfolgen („lineclearance“).<br />
5.46 Je<strong>der</strong> Verpackungsplatz o<strong>der</strong> jede Verpackungslinie<br />
ist mit dem Namen und <strong>der</strong> Chargenbezeichnung<br />
des jeweils zu verpackenden<br />
Produkts zu kennzeichnen.<br />
5.47 Alle einzusetzenden Produkte und Verpackungsmaterialien<br />
sind bei <strong>der</strong> Bereitstellung<br />
<strong>für</strong> die Verpackungsvorgänge hinsichtlich Menge,<br />
Identität und Übereinstimmung mit den Her-<br />
Übersetzung durch die Mitglie<strong>der</strong> <strong>der</strong> AG 42 des QS-Forums Schleswig-Holstein / Hamburg
5.48 Containers for filling should be clean before<br />
filling. Attention should be given to avoiding<br />
and removing any contaminants such as glass<br />
fragments and metal particles.<br />
5.49 Normally, filling and sealing should be followed<br />
as quickly as possible by labelling. If it is<br />
not the case, appropriate procedures should be<br />
applied to ensure that no mix-ups or mislabelling<br />
can occur.<br />
5.50 The correct performance of any printing<br />
operation (for example code numbers, expiry<br />
dates) to be done separately or in the course of<br />
the packaging should be checked and recorded.<br />
Attention should be paid to printing by hand<br />
which should be re-checked at regular intervals.<br />
5.51 Special care should be taken when using<br />
cut-labels and when over-printing is carried out<br />
off-line. Roll-feed labels are normally preferable<br />
to cut-labels, in helping to avoid mix-ups.<br />
5.52 Checks should be made to ensure that<br />
any electronic code rea<strong>der</strong>s, label counters or<br />
similar devices are operating correctly.<br />
5.53 Printed and embossed information on<br />
packaging materials should be distinct and<br />
resistant to fading or erasing.<br />
5.54 On-line control of the product during packaging<br />
should include at least checking the following:<br />
a) general appearance of the packages;<br />
b) whether the packages are complete;<br />
c) whether the correct products and packaging<br />
materials are used;<br />
d) whether any over-printing is correct;<br />
e) correct functioning of line monitors.<br />
Samples taken away from the packaging line<br />
should not be returned.<br />
5.55 Products which have been involved in an<br />
unusual event should only be reintroduced into<br />
the process after special inspection,<br />
investigation and approval by authorised<br />
personnel. Detailed record should be kept of this<br />
stellungsanweisungen (Verpackung) zu überprüfen.<br />
5.48 Zu füllende Behältnisse haben sauber zu<br />
sein. Es ist sorgfältig darauf zu achten, dass<br />
Verunreinigungen wie Glassplitter o<strong>der</strong> Metallteilchen<br />
vermieden bzw. entfernt werden.<br />
5.49 Normalerweise ist die Etikettierung so<br />
schnell wie möglich nach dem Abfüllen und Verschließen<br />
durchzuführen. Sollte dies nicht <strong>der</strong><br />
Fall sein, sind Verwechslungen o<strong>der</strong> Falschetikettierungen<br />
durch geeignete Verfahren<br />
sicher zu vermeiden.<br />
5.50 Die einwandfreie Ausführung jedes Druckvorganges<br />
(z. B. Aufdruck von Chargenbezeichnungen,<br />
Verfalldaten), <strong>der</strong> getrennt o<strong>der</strong> während<br />
des Verpackens erfolgt, ist zu überprüfen<br />
und zu protokollieren. Beson<strong>der</strong>e Aufmerksamkeit<br />
erfor<strong>der</strong>t das manuelle Kennzeichnen, welches<br />
in regelmäßigen Abständen zu überprüfen<br />
ist.<br />
5.51 Es bedarf beson<strong>der</strong>er Sorgfalt, wenn Einzeletiketten<br />
verwendet und Aufdrucke nicht auf<br />
<strong>der</strong> Verpackungsanlage selbst (off-line) angebracht<br />
werden. Etiketten auf Rollen sind Einzeletiketten<br />
grundsätzlich vorzuziehen, da sich Untermischungen<br />
so besser vermeiden lassen.<br />
5.52 Die Funktionsfähigkeit elektronischer<br />
Code-Lesegeräte, Etikettenzähler o<strong>der</strong> ähnlicher<br />
Geräte ist durch Überprüfungen sicherzustellen.<br />
5.53 Gedruckte o<strong>der</strong> geprägte Angaben auf<br />
Verpackungsmaterialien haben deutlich, lichtecht<br />
und abriebfest zu sein.<br />
5.54 Die kontinuierliche Überprüfung des Produkts<br />
während des Verpackens (on-line) hat<br />
mindestens folgende Punkte zu umfassen:<br />
a) das Aussehen <strong>der</strong> Packungen<br />
b) die Vollständigkeit <strong>der</strong> Packungen<br />
c) den Einsatz <strong>der</strong> richtigen Produkte und Verpackungsmaterialien<br />
d) die Richtigkeit <strong>der</strong> Aufdrucke<br />
e) die einwandfreie Funktion <strong>der</strong> Überwachungsvorrichtungen<br />
<strong>der</strong> Anlage.<br />
Von <strong>der</strong> Verpackungslinie entfernte Proben sind<br />
nicht wie<strong>der</strong> (in den Prozess) <strong>zur</strong>ückzuführen.<br />
5.55 Produkte, die von einem beson<strong>der</strong>en Vorkommnis<br />
betroffen sind, dürfen nur nach sorgfältiger<br />
Überprüfung, Untersuchung und Genehmigung<br />
durch dazu befugtes Personal wie<strong>der</strong> in<br />
den Prozess eingeschleust werden. Detaillierte<br />
Übersetzung durch die Mitglie<strong>der</strong> <strong>der</strong> AG 42 des QS-Forums Schleswig-Holstein / Hamburg
operation. Aufzeichnungen darüber sind aufzubewahren.<br />
5.56 Any significant or unusual discrepancy<br />
observed during reconciliation of the amount of<br />
bulk product and printed packaging materials<br />
and the number of units produced should be<br />
investigated and satisfactorily accounted for before<br />
release.<br />
5.57 Upon completion of a packaging<br />
operation, any unused batch-coded packaging<br />
materials should be destroyed and the<br />
destruction recorded. A documented procedure<br />
should be followed if uncoded printed materials<br />
are returned to stock.<br />
Finished products Fertigprodukte<br />
5.58 Finished products should be held in quarantine<br />
until their final release un<strong>der</strong> conditions<br />
established by the manufacturer.<br />
5.59 The evaluation of finished products and<br />
documentation which is necessary before release<br />
of product for sale are described in Chapter<br />
6 (Quality Control).<br />
5.60 After release, finished products should be<br />
stored as usable stock un<strong>der</strong> conditions established<br />
by the manufacturer.<br />
5.56 Jede bei einer Bilanzierung festgestellte<br />
auffällige o<strong>der</strong> ungewöhnliche Abweichung<br />
zwischen <strong>der</strong> Menge an Bulkware, den bedruckten<br />
Verpackungsmaterialien und <strong>der</strong> Anzahl <strong>der</strong><br />
Packungen des Fertigprodukts ist vor <strong>der</strong> Freigabe<br />
zu untersuchen und ausreichend zu begründen.<br />
5.57 Mit <strong>der</strong> Chargenbezeichnung versehene,<br />
nicht verbrauchte Verpackungsmaterialien sind<br />
nach Beendigung des Verpackungsvorgangs zu<br />
vernichten. Die Vernichtung ist zu dokumentieren.<br />
Nicht mit einer Chargenbezeichnung versehene<br />
bedruckte Materialien dürfen nur nach<br />
schriftlich festgelegtem Verfahren an das Lager<br />
<strong>zur</strong>ückgegeben werden.<br />
5.58 Fertigprodukte sind bis zu ihrer endgültigen<br />
Freigabe unter vom Hersteller festgelegten<br />
Bedingungen in Quarantäne zu halten.<br />
5.59 Die vor <strong>der</strong> Freigabe von Fertigprodukten<br />
zum Verkauf erfor<strong>der</strong>liche Bewertung des Fertigprodukts<br />
und <strong>der</strong> Dokumentation wird in Kapitel<br />
6 (Qualitätskontrolle) beschrieben.<br />
5.60 Nach <strong>der</strong> Freigabe sind Fertigprodukte als<br />
verfügbarer Bestand unter den vom Hersteller<br />
festgelegten Bedingungen zu lagern.<br />
Rejected, recovered and returned materials Zurückgewiesene, aufgearbeitete und <strong>zur</strong>ückgegebene<br />
Materialien<br />
5.61 Rejected materials and products should<br />
be clearly marked as such and stored separately<br />
in restricted areas. They should either be<br />
returned to the suppliers or, where appropriate,<br />
reprocessed or destroyed. Whatever action is<br />
taken should be approved and recorded by<br />
authorised personnel.<br />
5.62 The reprocessing of rejected products<br />
should be exceptional. It is only permitted if the<br />
quality of the final product is not affected, if the<br />
specifications are met and if it is done in accordance<br />
with a defined and authorised procedure<br />
after evaluation of the risks involved. Record<br />
should be kept of the reprocessing.<br />
5.63 The recovery of all or part of earlier<br />
batches which conform to the required quality by<br />
5.61 Zurückgewiesene Materialien und Produkte<br />
sind eindeutig als solche zu kennzeichnen<br />
und geson<strong>der</strong>t in nicht allgemein zugänglichen<br />
Bereichen zu lagern. Sie sind entwe<strong>der</strong> an den<br />
Lieferanten <strong>zur</strong>ückzugeben, zu vernichten o<strong>der</strong><br />
können, sofern möglich, aufgearbeitet werden.<br />
Die jeweils durchgeführte Maßnahme ist von dazu<br />
befugtem Personal zu genehmigen und aufzuzeichnen.<br />
5.62 Die Aufarbeitung von <strong>zur</strong>ückgewiesenen<br />
Produkten sollte die Ausnahme sein. Eine Aufarbeitung<br />
ist nur zulässig, wenn die Qualität des<br />
Endprodukts nicht beeinträchtigt wird, wenn die<br />
Spezifikationen eingehalten werden und wenn<br />
die Aufarbeitung in Übereinstimmung mit einem<br />
definierten und genehmigten Verfahren nach<br />
Abschätzung <strong>der</strong> dabei bestehenden Risiken<br />
durchgeführt wird. Die Aufarbeitung ist zu protokollieren.<br />
5.63 Das vollständige o<strong>der</strong> teilweise Einbringen<br />
früherer Chargen in eine Charge desselben<br />
Übersetzung durch die Mitglie<strong>der</strong> <strong>der</strong> AG 42 des QS-Forums Schleswig-Holstein / Hamburg
incorporation into a batch of the same product at<br />
a defined stage of manufacture should be<br />
authorised beforehand. This recovery should be<br />
carried out in accordance with a defined procedure<br />
after evaluation of the risks involved, including<br />
any possible effect on shelf life. The recovery<br />
should be recorded.<br />
5.64 The need for additional testing of any finished<br />
product which has been reprocessed, or<br />
into which a recovered product has been incorporated,<br />
should be consi<strong>der</strong>ed by the Quality<br />
Control Department.<br />
5.65 Products returned from the market and<br />
which have left the control of the manufacturer<br />
should be destroyed unless without doubt their<br />
quality is satisfactory; they may be consi<strong>der</strong>ed<br />
for re-sale, re-labelling or recovery in a subsequent<br />
batch only after they have been<br />
critically assessed by the Quality Control<br />
Department in accordance with a written<br />
procedure. The nature of the product, any<br />
special storage conditions it requires, its<br />
condition and history, and the time elapsed<br />
since it was issued should all be taken into<br />
account in this assessment. Where any doubt<br />
arises over the quality of the product, it should<br />
not be consi<strong>der</strong>ed suitable for re-issue or re-use,<br />
although basic chemical reprocessing to recover<br />
active ingredient may be possible. Any action<br />
taken should be appropriately recorded.<br />
Produkts auf einer bestimmten Herstellungsstufe<br />
ist nur zulässig, wenn die einzubringenden<br />
Chargen die erfor<strong>der</strong>liche Qualität aufweisen<br />
und das Verfahren zuvor genehmigt wurde. Diese<br />
Umarbeitung ist in Übereinstimmung mit einem<br />
festgelegten Verfahren nach Abschätzung<br />
<strong>der</strong> dabei bestehenden Risiken, einschließlich<br />
einer möglichen Auswirkung auf die Haltbarkeitsdauer,<br />
durchzuführen und zu protokollieren.<br />
5.64 Die Entscheidung über die Notwendigkeit<br />
zusätzlicher Prüfungen eines Fertigprodukts,<br />
das auf- o<strong>der</strong> umgearbeitet wurde, liegt in <strong>der</strong><br />
Verantwortung <strong>der</strong> Qualitätskontrollabteilung.<br />
5.65 Aus dem Handel <strong>zur</strong>ückgegebene, <strong>der</strong><br />
Kontrolle des Herstellers zwischenzeitlich entzogene<br />
Produkte sind zu vernichten, es sei denn,<br />
sie weisen zweifelsfrei die erfor<strong>der</strong>liche Qualität<br />
auf. Für erneuten Verkauf, Umetikettierung o<strong>der</strong><br />
<strong>für</strong> ein Einbringen in eine spätere Charge können<br />
sie nur in Betracht kommen, wenn sie von<br />
<strong>der</strong> Qualitätskontrollabteilung nach einem<br />
schriftlich festgelegten Verfahren kritisch bewertet<br />
wurden. Dabei sind die Art des Produkts,<br />
evtl. erfor<strong>der</strong>liche beson<strong>der</strong>e Lagerungsbedingungen,<br />
<strong>der</strong> Zustand, die Historie sowie die Zeitspanne<br />
seit Auslieferung zu berücksichtigen.<br />
Sollte irgendein Zweifel an <strong>der</strong> Qualität des Produkts<br />
aufkommen, ist eine erneute Auslieferung<br />
o<strong>der</strong> erneute Verwendung nicht in Erwägung zu<br />
ziehen. Eine Rückgewinnung des Wirkstoffs<br />
kann jedoch möglich sein. Jede durchgeführte<br />
Maßnahme ist in angemessener Weise zu protokollieren.<br />
CHAPTER 6 QUALITY CONTROL KAPITEL 6 QUALITÄTSKONTROLLE<br />
Principle Grundsätze<br />
Quality Control is concerned with sampling,<br />
specifications and testing as well as the organisation,<br />
documentation and release procedures<br />
which ensure that the necessary and relevant<br />
tests are carried out, and that materials are not<br />
released for use, nor products released for sale<br />
or supply, until their quality has been judged<br />
satisfactory. Quality Control is not confined to<br />
laboratory operations, but must be involved in all<br />
decisions which may concern the quality of the<br />
product. The independence of Quality Control<br />
from Production is consi<strong>der</strong>ed fundamental to<br />
the satisfactory operation of Quality Control.<br />
(see also Chapter 1).<br />
Die Qualitätskontrolle befasst sich mit Probenahme,<br />
Spezifikationen und Prüfung sowie Organisation,<br />
Dokumentation und Freigabeverfahren.<br />
Sie hat sicherzustellen, dass die jeweils<br />
notwendigen und relevanten Prüfungen durchgeführt<br />
und we<strong>der</strong> Materialien <strong>für</strong> den Einsatz<br />
noch Produkte <strong>für</strong> den Verkauf o<strong>der</strong> die Auslieferung<br />
freigegeben werden, bevor ihre Qualität<br />
als <strong>der</strong> Vorgabe entsprechend beurteilt wurde.<br />
Die Qualitätskontrolle ist nicht auf Laborarbeiten<br />
beschränkt, son<strong>der</strong>n muss bei allen die<br />
Produktqualität betreffenden Entscheidungen<br />
beteiligt sein. Die Unabhängigkeit von <strong>der</strong> Produktion<br />
ist <strong>für</strong> das ordnungsgemäße Arbeiten<br />
<strong>der</strong> Qualitätskontrolle von grundlegen<strong>der</strong> Bedeutung<br />
(siehe auch Kapitel 1).<br />
Übersetzung durch die Mitglie<strong>der</strong> <strong>der</strong> AG 42 des QS-Forums Schleswig-Holstein / Hamburg
General Allgemeine Anfor<strong>der</strong>ungen<br />
6.1 Each hol<strong>der</strong> of a manufacturing authorisation<br />
should have a Quality Control Department.<br />
This department should be independent from<br />
other departments, and un<strong>der</strong> the authority of a<br />
person with appropriate qualifications and experience,<br />
who has one or several control laboratories<br />
at his disposal. Adequate resources<br />
must be available to ensure that all the Quality<br />
Control arrangements are effectively and reliably<br />
carried out.<br />
6.2 The principal duties of the head of Quality<br />
Control are summarised in Chapter 2. The Quality<br />
Control Department as a whole will also have<br />
other duties, such as to establish, validate and<br />
implement all quality control procedures, keep<br />
the reference samples of materials and<br />
products, ensure the correct labelling of<br />
containers of materials and products, ensure the<br />
monitoring of the stability of the products,<br />
participate in the investigation of complaints<br />
related to the quality of the product, etc. All<br />
these operations should be carried out in<br />
accordance with written procedures and, where<br />
necessary, recorded.<br />
6.3 Finished product assessment should embrace<br />
all relevant factors, including production<br />
conditions, results of in-process testing, a review<br />
of manufacturing (including packaging) documentation,<br />
compliance with Finished Product<br />
Specification and examination of the final finished<br />
pack.<br />
6.4 Quality Control personnel should have<br />
access to production areas for sampling and<br />
investigation as appropriate.<br />
6.1 Je<strong>der</strong> Inhaber einer Herstellungserlaubnis<br />
muss über eine Qualitätskontrollabteilung verfügen.<br />
Diese Abteilung hat von an<strong>der</strong>en Abteilungen<br />
unabhängig zu sein und unter <strong>der</strong> Leitung<br />
einer Person mit angemessener Qualifikation<br />
und Erfahrung zu stehen, die ein o<strong>der</strong> mehrere<br />
Kontrolllaboratorien <strong>zur</strong> Verfügung hat. Es<br />
müssen ausreichende Ressourcen verfügbar<br />
sein, damit alle Aufgaben <strong>der</strong> Qualitätskontrolle<br />
effektiv und zuverlässig ausgeführt werden können.<br />
6.2 Die Hauptaufgaben des Leiters <strong>der</strong> Qualitätskontrolle<br />
sind in Kapitel 2 zusammengefasst.<br />
Die Qualitätskontrollabteilung insgesamt hat<br />
u. a. noch weitere Aufgaben, wie die Festlegung,<br />
Validierung und In-Kraft-Setzung aller<br />
Qualitätskontrollverfahren, Aufbewahrung von<br />
Referenzmustern 18 von Ausgangsmaterialien<br />
und Produkten sowie die Sicherstellung <strong>der</strong> ordnungsgemäßen<br />
Kennzeichnung <strong>der</strong> Behältnisse,<br />
die diese Materialien und Produkte enthalten,<br />
Sicherstellung <strong>der</strong> fortlaufenden Stabilitätsüberprüfungen,<br />
Mitwirkung bei <strong>der</strong> Untersuchung<br />
von Beanstandungen hinsichtlich <strong>der</strong><br />
Produktqualität. Alle diese Vorgänge sind gemäß<br />
schriftlich festgelegter Verfahren durchzuführen<br />
und, falls erfor<strong>der</strong>lich, aufzuzeichnen.<br />
6.3 Die Bewertung des Fertigprodukts hat alle<br />
relevanten Gesichtspunkte zu berücksichtigen,<br />
einschließlich <strong>der</strong> Produktionsbedingungen, <strong>der</strong><br />
Ergebnisse von Inprozesskontrollen, <strong>der</strong> Überprüfung<br />
<strong>der</strong> Herstellungs- (einschließlich Verpackungs-)dokumentation,<br />
<strong>der</strong> Übereinstimmung<br />
mit <strong>der</strong> Spezifikation des Fertigprodukts<br />
und <strong>der</strong> Überprüfung <strong>der</strong> Fertigpackung.<br />
6.4 Das Personal <strong>der</strong> Qualitätskontrolle hat,<br />
soweit erfor<strong>der</strong>lich, Zugang zu den Produktionsbereichen<br />
zu haben, um Proben zu nehmen und<br />
Untersuchungen durchzuführen.<br />
Good Quality Control Laboratory Practice Gute Kontrolllabor-Praxis<br />
6.5 Control laboratory premises and equipment<br />
should meet the general and specific requirements<br />
for Quality Control areas given in<br />
Chapter 3.<br />
6.6 The personnel, premises, and equipment<br />
in the laboratories should be appropriate to the<br />
6.5 Räumlichkeiten und Ausrüstung von Kontrolllaboratorien<br />
haben den in Kapitel 3 beschriebenen<br />
allgemeinen und beson<strong>der</strong>en Anfor<strong>der</strong>ungen<br />
an Qualitätskontrollbereiche zu entsprechen.<br />
6.6 Das Personal, die Räumlichkeiten und die<br />
Ausrüstung in den Laboratorien haben den Auf-<br />
18<br />
Abweichende Regelungen in <strong>der</strong> AMWHV bezüglich <strong>der</strong> Verantwortung <strong>für</strong> und Lagerhaltung von Rückstellmustern.<br />
Übersetzung durch die Mitglie<strong>der</strong> <strong>der</strong> AG 42 des QS-Forums Schleswig-Holstein / Hamburg
tasks imposed by the nature and the scale of the<br />
manufacturing operations. The use of outside<br />
laboratories, in conformity with the principles<br />
detailed in Chapter 7, Contract Analysis, can be<br />
accepted for particular reasons, but this should<br />
be stated in the Quality Control records.<br />
Documentation Dokumentation<br />
6.7 Laboratory documentation should follow<br />
the principles given in Chapter 4. An important<br />
part of this documentation deals with Quality<br />
Control and the following details should be readily<br />
available to the Quality Control Department:<br />
• specifications;<br />
• sampling procedures;<br />
• testing procedures and records (including analytical<br />
worksheets and/or laboratory notebooks);<br />
• analytical reports and/or certificates;<br />
• data from environmental monitoring, where<br />
required;<br />
• validation records of test methods, where<br />
applicable;<br />
• procedures for and records of the calibration of<br />
instruments and maintenance of equipment<br />
6.8 Any Quality Control documentation<br />
relating to a batch record should be retained for<br />
one year after the expiry date of the batch and<br />
at least 5 years after the certification referred to<br />
in Article 51(3) of Directive 2001/83/EC.<br />
6.9 For some kinds of data (e.g. analytical<br />
tests results, yields, environmental controls) it is<br />
recommended that records are kept in a manner<br />
permitting trend evaluation.<br />
6.10 In addition to the information which is part<br />
of the batch record, other original data such as<br />
laboratory notebooks and/or records should be<br />
retained and readily available.<br />
gaben zu entsprechen, die sich aus Art und Umfang<br />
<strong>der</strong> Herstellungstätigkeiten ergeben. Der<br />
Einsatz externer Laboratorien kann in Übereinstimmung<br />
mit den in Kapitel 7 "Prüfung im Auftrag"<br />
beschriebenen Grundsätzen in bestimmten<br />
Fällen akzeptiert werden. Dies ist jedoch in den<br />
Protokollen <strong>der</strong> Qualitätskontrolle auszuweisen.<br />
6.7 Die Dokumentation im Labor hat den im<br />
Kapitel 4 genannten Anfor<strong>der</strong>ungen zu entsprechen.<br />
Ein wesentlicher Teil <strong>der</strong> dort genannten<br />
Dokumentation betrifft die Qualitätskontrolle. Die<br />
folgenden Unterlagen haben in <strong>der</strong> Qualitätskontrollabteilung<br />
je<strong>der</strong>zeit verfügbar zu sein:<br />
— Spezifikationen<br />
— Verfahren <strong>zur</strong> Probenahme<br />
— Prüfverfahren und Protokolle (einschließlich<br />
analytischer Arbeitsblätter und/o<strong>der</strong> Laborjournalen)<br />
— Analysenberichte und/o<strong>der</strong> -zertifikate<br />
— soweit erfor<strong>der</strong>lich, Daten aus <strong>der</strong> Überwachung<br />
<strong>der</strong> Umgebungsbedingungen<br />
— soweit zutreffend, Nachweise über die Validierung<br />
<strong>der</strong> Prüfverfahren<br />
— Verfahrensbeschreibungen <strong>für</strong> und Nachweise<br />
über die Kalibrierung von Geräten und die<br />
Wartung <strong>der</strong> Ausrüstung.<br />
6.8 Sämtliche Dokumente <strong>der</strong> Qualitätskontrolle,<br />
die ein bestimmtes Chargenprotokoll betreffen,<br />
sind ein Jahr über die Laufzeit <strong>der</strong> Charge<br />
und mindestens fünf Jahre über das Datum<br />
<strong>der</strong> Zertifizierung gemäß Artikel 51 Abs. 3 <strong>der</strong><br />
Direktive 2001/83/<strong>EG</strong> 19 hinaus aufzubewahren.<br />
6.9 Es wird empfohlen, einige Daten (z. B. Ergebnisse<br />
analytischer Prüfungen, Ausbeuten,<br />
Umgebungskontrollen usw.) so aufzubewahren,<br />
dass Trends ermittelt werden können.<br />
6.10 Zusätzlich zu den zu einem Chargenprotokoll<br />
gehörenden Informationen sind an<strong>der</strong>e<br />
Originalunterlagen wie Laborjournale und/o<strong>der</strong><br />
–aufzeichnungen aufzubewahren und schnell<br />
verfügbar zu halten.<br />
Sampling Probenahme (Musterzug)<br />
6.11 The sample taking should be done in accordance<br />
with approved written procedures that<br />
describe:<br />
19 Beziehungsweise Artikel 55 Abs. 3 <strong>der</strong> Direktive 2001/82/<strong>EG</strong><br />
6.11 Die Probenahme hat nach genehmigten,<br />
schriftlich festgelegten Verfahren zu erfolgen,<br />
die festlegen:<br />
Übersetzung durch die Mitglie<strong>der</strong> <strong>der</strong> AG 42 des QS-Forums Schleswig-Holstein / Hamburg
• the method of sampling;<br />
• the equipment to be used;<br />
• the amount of the sample to be taken;<br />
• instructions for any required sub-division of the<br />
sample;<br />
• the type and condition of the sample container<br />
to be used;<br />
• the identification of containers sampled;<br />
• any special precautions to be observed, especially<br />
with regard to the sampling of sterile or<br />
noxious materials;<br />
• the storage conditions;<br />
• instructions for the cleaning and storage of<br />
sampling equipment.<br />
6.12 Reference samples should be representative<br />
of the batch of materials or products from<br />
which they are taken. Other samples may also<br />
be taken to monitor the most stressed part of a<br />
process (e.g. beginning or end of a process).<br />
6.13 Sample containers should bear a label<br />
indicating the contents, with the batch number,<br />
the date of sampling and the containers from<br />
which samples have been drawn.<br />
6.14 Further guidance on reference and retention<br />
samples is given in Annex 19.<br />
Testing Prüfung<br />
6.15 Analytical methods should be validated.<br />
All testing operations described in the marketing<br />
authorisation should be carried out according to<br />
the approved methods.<br />
6.16 The results obtained should be recorded<br />
and checked to make sure that they are consistent<br />
with each other. Any calculations should<br />
be critically examined.<br />
6.17 The tests performed should be recorded<br />
and the records should include at least the following<br />
data:<br />
a) name of the material or product and, where<br />
applicable, dosage form;<br />
b) batch number and, where appropriate, the<br />
manufacturer and/or supplier;<br />
— das Verfahren <strong>der</strong> Probenahme<br />
— die zu verwendende Ausrüstung<br />
— die zu entnehmende Probemenge<br />
— Hinweise, falls die Probe aufzuteilen ist;<br />
— Art und Zustand des zu verwendenden Probenbehältnisses<br />
— die Kennzeichnung <strong>der</strong> beprobten Behältnisse<br />
— alle beson<strong>der</strong>en Vorsichtsmaßnahmen, insbeson<strong>der</strong>e<br />
im Hinblick auf die Probenahme<br />
steriler o<strong>der</strong> gesundheitsschädlicher Materialien<br />
— die Lagerungsbedingungen<br />
— Vorgaben <strong>für</strong> die Reinigung und Aufbewahrung<br />
<strong>der</strong> Ausrüstung <strong>für</strong> die Probenahme.<br />
6.12 Referenzmuster haben repräsentativ zu<br />
sein <strong>für</strong> die Charge des Materials o<strong>der</strong> Produktes,<br />
<strong>der</strong> sie entstammen. Zusätzliche Proben<br />
können entnommen werden, um kritische Prozessschritte<br />
zu überwachen, wie z. B. den Beginn<br />
o<strong>der</strong> das Ende eines Prozesses.<br />
6.13 Die Probenbehältnisse sind mit einem Etikett<br />
zu versehen, aus dessen Beschriftung sich<br />
<strong>der</strong> Inhalt des Behältnisses, die Chargenkennzeichnung,<br />
das Datum <strong>der</strong> Probenahme sowie<br />
ein Hinweis auf die Behältnisse, aus denen die<br />
Muster gezogen wurden, ergeben muss.<br />
6.14 Weiteres zu Referenz- und Rückstellmustern<br />
siehe Annex 19.<br />
6.15 Analytische Verfahren sind zu validieren.<br />
Alle in den Zulassungsunterlagen festgeschriebenen<br />
Prüfungen sind in Übereinstimmung mit<br />
den dort genehmigten Verfahren durchzuführen.<br />
6.16 Die erhaltenen Ergebnisse sind zu protokollieren<br />
und auf Plausibilität zu überprüfen. Alle<br />
Berechnungen sind kritisch nachzuprüfen.<br />
6.17 Die durchgeführten Prüfungen sind zu<br />
protokollieren. Die Protokolle haben mindestens<br />
die folgenden Angaben zu enthalten:<br />
a) den Namen des Materials o<strong>der</strong> Produktes,<br />
soweit zutreffend, auch dessen Darreichungsform<br />
b) die Chargenbezeichnung und, soweit zweckdienlich,<br />
den Hersteller und/o<strong>der</strong> Lieferanten<br />
Übersetzung durch die Mitglie<strong>der</strong> <strong>der</strong> AG 42 des QS-Forums Schleswig-Holstein / Hamburg
c) references to the relevant specifications and<br />
testing procedures;<br />
d) test results, including observations and calculations,<br />
and reference to any certificates of<br />
analysis;<br />
e) dates of testing;<br />
f) initials of the persons who performed the testing;<br />
g) initials of the persons who verified the testing<br />
and the calculations, where appropriate;<br />
h) a clear statement of release or rejection (or<br />
other status decision) and the dated signature of<br />
the designated responsible person.<br />
6.18 All the in-process controls, including those<br />
made in the production area by production personnel,<br />
should be performed according to<br />
methods approved by Quality Control and the<br />
results recorded.<br />
6.19 Special attention should be given to the<br />
quality of laboratory reagents, volumetric glassware<br />
and solutions, reference standards and<br />
culture media. They should be prepared in accordance<br />
with written procedures.<br />
6.20 Laboratory reagents intended for prolonged<br />
use should be marked with the preparation<br />
date and the signature of the person who<br />
prepared them. The expiry date of unstable reagents<br />
and culture media should be indicated on<br />
the label, together with specific storage conditions.<br />
In addition, for volumetric solutions, the<br />
last date of standardisation and the last current<br />
factor should be indicated.<br />
6.21 Where necessary, the date of receipt of<br />
any substance used for testing operations (e.g.<br />
reagents and reference standards) should be<br />
indicated on the container. Instructions for use<br />
and storage should be followed. In certain cases<br />
it may be necessary to carry out an identification<br />
test and/or other testing of reagent materials<br />
upon receipt or before use.<br />
6.22 Animals used for testing components,<br />
materials or products, should, where<br />
appropriate, be quarantined before use. They<br />
should be maintained and controlled in a<br />
manner that assures their suitability for the<br />
intended use. They should be identified, and<br />
adequate records should be maintained,<br />
c) eine Bezugnahme auf die betreffenden Spezifikationen<br />
und Prüfverfahren<br />
d) die Prüfergebnisse, einschließlich <strong>der</strong> Beobachtungen<br />
und Berechnungen sowie einen<br />
Bezug auf Analysenzertifikate<br />
e) die Datumsangaben <strong>der</strong> Prüfungen<br />
f) die Namenszeichen <strong>der</strong> Personen, die die<br />
Prüfungen durchgeführt haben<br />
g) soweit vorgesehen, die Namenszeichen <strong>der</strong><br />
Personen, die solche Prüfungen und Berechnungen<br />
kontrolliert haben<br />
h) eine eindeutige Aussage <strong>zur</strong> Freigabe o<strong>der</strong><br />
Zurückweisung (o<strong>der</strong> eine an<strong>der</strong>e Entscheidung<br />
über den Status) mit Datum und Unterschrift <strong>der</strong><br />
hier<strong>für</strong> verantwortlichen Person.<br />
6.18 Alle Inprozesskontrollen, auch die im Produktionsbereich<br />
vom dortigen Personal durchgeführten,<br />
haben nach Verfahren zu erfolgen, die<br />
von <strong>der</strong> Qualitätskontrolle genehmigt sind. Die<br />
Ergebnisse sind zu protokollieren.<br />
6.19 Auf die Eignung von Laborreagenzien,<br />
Volumenmessgeräten, volumetrischen Lösungen,<br />
Referenzsubstanzen und Kulturmedien ist<br />
beson<strong>der</strong>s zu achten. Ihre Zubereitung hat nach<br />
schriftlich festgelegten Verfahren zu erfolgen.<br />
6.20 Laborreagenzien, die <strong>für</strong> einen längeren<br />
Gebrauch vorgesehen sind, sind mit dem Datum<br />
ihres Ansatzes und dem Namenszeichen <strong>der</strong><br />
Person zu versehen, die sie hergestellt hat. Bei<br />
instabilen Reagenzien und Kulturmedien sind<br />
das Verfalldatum sowie beson<strong>der</strong>e Lagerungsbedingungen<br />
auf dem Etikett anzugeben. Zusätzlich<br />
sind bei volumetrischen Lösungen das<br />
Datum <strong>der</strong> letzten Einstellung und <strong>der</strong> ermittelte<br />
Faktor zu vermerken.<br />
6.21 Falls erfor<strong>der</strong>lich, ist bei den <strong>für</strong> die Prüfungen<br />
verwendeten Substanzen (z. B. Reagenzien<br />
und Referenzsubstanzen) auf dem Behältnis<br />
das Eingangsdatum zu vermerken. Hinweise<br />
zu Gebrauch und Aufbewahrung sind zu befolgen.<br />
In bestimmten Fällen kann es erfor<strong>der</strong>lich<br />
sein, eine Identitätsprüfung und/o<strong>der</strong> einen<br />
Eignungstest <strong>der</strong> Reagenzien beim Wareneingang<br />
o<strong>der</strong> vor Gebrauch durchzuführen.<br />
6.22 Tiere, die bei <strong>der</strong> Prüfung von Bestandteilen,<br />
Materialien o<strong>der</strong> Produkten eingesetzt<br />
werden, sind, soweit erfor<strong>der</strong>lich, vor ihrer Verwendung<br />
in Quarantäne zu halten. Sie sind so<br />
zu halten und zu überwachen, dass sie <strong>für</strong> die<br />
beabsichtigte Prüfung geeignet sind. Sie sind zu<br />
identifizieren, die Historie ihrer Verwendung ist<br />
Übersetzung durch die Mitglie<strong>der</strong> <strong>der</strong> AG 42 des QS-Forums Schleswig-Holstein / Hamburg
showing the history of their use. in Protokollen umfassend zu dokumentieren.<br />
On-going stability programme Fortlaufendes Stabilitätsprogramm<br />
6.23 After marketing, the stability of the medicinal<br />
product should be monitored according to a<br />
continuous appropriate programme that will permit<br />
the detection of any stability issue (e.g.<br />
changes in levels of impurities or dissolution<br />
profile) associated with the formulation in the<br />
marketed package.<br />
6.24 The purpose of the on-going stability programme<br />
is to monitor the product over its shelf<br />
life and to determine that the product remains,<br />
and can be expected to remain, within specifications<br />
un<strong>der</strong> the labelled storage conditions.<br />
6.25 This mainly applies to the medicinal product<br />
in the package in which it is sold, but consi<strong>der</strong>ation<br />
should also be given to the inclusion<br />
in the programme of bulk product. For example,<br />
when the bulk product is stored for a long period<br />
before being packaged and/or shipped from a<br />
manufacturing site to a packaging site, the impact<br />
on the stability of the packaged product<br />
should be evaluated and studied un<strong>der</strong> ambient<br />
conditions. In addition, consi<strong>der</strong>ation should be<br />
given to intermediates that are stored and used<br />
over prolonged periods. Stability studies on reconstituted<br />
product are performed during<br />
product development and need not be<br />
monitored on an on-going basis. However, when<br />
relevant, the stability of reconstituted product<br />
can also be monitored.<br />
6.26 The on-going stability programme should<br />
be described in a written protocol following the<br />
general rules of Chapter 4 and results<br />
formalised as a report. The equipment used for<br />
the on-going stability programme (stability<br />
chambers among others) should be qualified<br />
and maintained following the general rules of<br />
Chapter 3 and annex 15.<br />
6.27 The protocol for an on-going stability programme<br />
should extend to the end of the shelf<br />
life period and should include, but not be limited<br />
to, the following parameters:<br />
• number of batch(es) per strength and different<br />
batch sizes, if applicable<br />
• relevant physical, chemical, microbiological<br />
and biological test methods<br />
6.23 Nach <strong>der</strong> Markteinführung ist die Stabilität<br />
eines <strong>Arzneimittel</strong>s fortlaufend mit einem geeigneten<br />
Verfahren zu überwachen, welches es erlaubt,<br />
mögliche stabilitätsbezogene Probleme<br />
beim Fertigprodukt (z. B. Verän<strong>der</strong>ungen hinsichtlich<br />
des Gehaltes an Verunreinigungen<br />
o<strong>der</strong> des Freisetzungsverhaltens) aufzudecken.<br />
6.24 Zweck des fortlaufenden Stabilitätsprogramms<br />
ist es, das Fertigprodukt während seiner<br />
Laufzeit dahingehend zu überwachen, ob es<br />
unter den angegebenen Lagerungsbedingungen<br />
die (festgelegten) Spezifikationen erfüllt o<strong>der</strong><br />
voraussichtlich erfüllen wird.<br />
6.25 Dies gilt hauptsächlich <strong>für</strong> das <strong>Arzneimittel</strong><br />
als Fertigprodukt, doch ist auch die Einbeziehung<br />
<strong>der</strong> Bulkware in das Programm zu erwägen.<br />
Wenn beispielsweise eine Bulkware über<br />
einen längeren Zeitraum vor ihrer Verpackung<br />
und/o<strong>der</strong> ihrem Versand vom Produktionsbetrieb<br />
zum Verpackungsbetrieb gelagert wird, ist <strong>der</strong><br />
Einfluss <strong>der</strong> Lagerung auf die Stabilität des Fertigproduktes<br />
zu überprüfen und zu bewerten.<br />
Außerdem sind die Zwischenprodukte zu berücksichtigen,<br />
die über längere Zeiträume gelagert<br />
und eingesetzt werden. Für vor Gebrauch<br />
rekonstituierte <strong>Arzneimittel</strong> sind Stabilitätsprüfungen<br />
Bestandteil <strong>der</strong> Produktentwicklung,<br />
es bedarf hierzu (prinzipiell) keines fortlaufenden<br />
Stabilitätsprogramms. Jedoch kann erfor<strong>der</strong>lichenfalls<br />
auch die Stabilität eines rekonstituierten<br />
Produkts überwacht werden.<br />
6.26 Das fortlaufende Stabilitätsprogramm ist<br />
schriftlich festzulegen, dabei sind die allgemeinen<br />
Regeln des Kapitels 4 zu beachten. Die Ergebnisse<br />
sind in Form eines Berichtes darzustellen.<br />
Die im Rahmen des fortlaufenden Stabilitätsprogramms<br />
verwendete Ausrüstung (u. a.<br />
Klimakammern) sind entsprechend <strong>der</strong> allgemeinen<br />
Regeln des Kapitels 3 sowie des Annexes<br />
15 zu qualifizieren und zu warten.<br />
6.27 Das fortlaufende Stabilitätsprogramm hat<br />
sich bis zum Ende <strong>der</strong> jeweiligen Laufzeit zu erstrecken;<br />
mindestens folgende Parameter sind<br />
vorzugeben:<br />
— die Anzahl <strong>der</strong> Chargen je Stärke und unterschiedlicher<br />
Chargengröße, soweit zutreffend<br />
— die relevanten physikalischen, chemischen,<br />
mikrobiologischen und biologischen Prüfverfahren<br />
Übersetzung durch die Mitglie<strong>der</strong> <strong>der</strong> AG 42 des QS-Forums Schleswig-Holstein / Hamburg
• acceptance criteria<br />
• reference to test methods<br />
• description of the container closure system(s)<br />
• testing intervals (time points)<br />
• description of the conditions of storage (standardised<br />
ICH conditions for long term testing,<br />
consistent with the product labelling, should be<br />
used)<br />
• other applicable parameters specific to the medicinal<br />
product.<br />
6.28 The protocol for the on-going stability programme<br />
can be different from that of the initial<br />
long-term stability study as submitted in the marketing<br />
authorisation dossier provided that this is<br />
justified and documented in the protocol (for example<br />
the frequency of testing, or when<br />
updating to ICH recommendations).<br />
6.29 The number of batches and frequency of<br />
testing should provide a sufficient amount of<br />
data to allow for trend analysis. Unless<br />
otherwise justified, at least one batch per year of<br />
product manufactured in every strength and<br />
every primary packaging type, if relevant, should<br />
be included in the stability programme (unless<br />
none are produced during that year). For<br />
products where on-going stability monitoring<br />
would normally require testing using animals<br />
and no appropriate alternative, validated<br />
techniques are available, the frequency of<br />
testing may take account of a risk-benefit<br />
approach. The principle of bracketing and<br />
matrixing designs may be applied if scientifically<br />
justified in the protocol.<br />
6.30 In certain situations, additional batches<br />
should be included in the on-going stability programme.<br />
For example, an on-going stability<br />
study should be conducted after any significant<br />
change or significant deviation to the process or<br />
package. Any reworking, reprocessing or recovery<br />
operation should also be consi<strong>der</strong>ed for<br />
inclusion.<br />
6.31 Results of on-going stability studies<br />
should be made available to key personnel and,<br />
in particular, to the Qualified Person(s). Where<br />
on-going stability studies are carried out at a site<br />
— die Akzeptanzkriterien<br />
— eine Bezugnahme auf die Prüfverfahren<br />
— eine Beschreibung des o<strong>der</strong> <strong>der</strong> Verschlusssystems/e<br />
<strong>der</strong> Behältnisse<br />
— die Prüfintervalle (Zeitpunkte)<br />
— eine Beschreibung <strong>der</strong> Lagerungsbedingungen<br />
(es sind die standardisierten ICH-Bedingungen<br />
<strong>für</strong> Langzeitstudien zu verwenden, entsprechend<br />
den Angaben <strong>der</strong> Kennzeichnung)<br />
— sonstige <strong>für</strong> das <strong>Arzneimittel</strong> spezifische<br />
Parameter.<br />
6.28 Das fortlaufende Stabilitätsprogramm<br />
kann sich von dem <strong>der</strong> ursprünglichen Langzeitstabilitätsstudie<br />
in den Zulassungsunterlagen<br />
unterscheiden, vorausgesetzt, dass dies begründet<br />
und im Programm festgelegt wird (z. B.<br />
Häufigkeit <strong>der</strong> Prüfung o<strong>der</strong> Aktualisierungen<br />
aufgrund von ICH-Empfehlungen).<br />
6.29 Die Anzahl <strong>der</strong> geprüften Chargen und die<br />
Häufigkeit <strong>der</strong> Prüfung müssen eine ausreichende<br />
Datenmenge liefern, um Trendanalysen zu<br />
ermöglichen. Grundsätzlich ist von jedem hergestellten<br />
Produkt pro Jahr mindestens eine Charge<br />
je<strong>der</strong> Stärke und, soweit zutreffend, jedes<br />
Primärverpackungsmaterials in das Stabilitätsprogramm<br />
einzubeziehen (es sei denn, im entsprechenden<br />
Jahr wurde keine Charge hergestellt).<br />
Bei Produkten, <strong>der</strong>en fortlaufende Stabilitätsüberwachung<br />
normalerweise eine Prüfung<br />
unter Verwendung von Tieren erfor<strong>der</strong>t, kann<br />
bei <strong>der</strong> Entscheidung über die Häufigkeit <strong>der</strong><br />
Prüfung eine Nutzen-Risiko-Bewertung herangezogen<br />
werden, sofern keine geeigneten alternativen<br />
validierten Prüfverfahren <strong>zur</strong> Verfügung<br />
stehen. Das Prinzip des Bracketing- und<br />
Matrixing-Designs kann angewendet werden,<br />
wenn dies im Programm wissenschaftlich begründet<br />
wird.<br />
6.30 Unter Umständen sind zusätzliche Chargen<br />
in das fortlaufende Stabilitätsprogramm einzubeziehen,<br />
zum Beispiel bei je<strong>der</strong> signifikanten<br />
Än<strong>der</strong>ung o<strong>der</strong> Abweichung im (Produktions-)<br />
Prozess (inkl. Verpackung). Bei jedem Aufarbeitungs-,<br />
Umarbeitungs- o<strong>der</strong> Rückgewinnungsprozess<br />
ist die Aufnahme in das Programm zu<br />
erwägen.<br />
6.31 Die Ergebnisse von fortlaufenden Stabilitätsuntersuchungen<br />
sind dem Schlüsselpersonal,<br />
insbeson<strong>der</strong>e <strong>der</strong>/den Sachkundigen Person(en)<br />
<strong>zur</strong> Verfügung zu stellen. Werden die<br />
Übersetzung durch die Mitglie<strong>der</strong> <strong>der</strong> AG 42 des QS-Forums Schleswig-Holstein / Hamburg
other than the site of manufacture of the bulk or<br />
finished product, there should be a written<br />
agreement between the parties concerned. Results<br />
of on-going stability studies should be<br />
available at the site of manufacture for review by<br />
the competent authority.<br />
6.32 Out of specification or significant atypical<br />
trends should be investigated. Any confirmed<br />
out of specification result, or significant negative<br />
trend, should be reported to the relevant competent<br />
authorities. The possible impact on<br />
batches on the market should be consi<strong>der</strong>ed in<br />
accordance with chapter 8 of the GMP Guide<br />
and in consultation with the relevant competent<br />
authorities.<br />
6.33. A summary of all the data generated, including<br />
any interim conclusions on the programme,<br />
should be written and maintained. This<br />
summary should be subjected to periodic<br />
review.<br />
CHAPTER 7 CONTRACT MANUFACTURE<br />
AND ANALYSIS<br />
Principle Grundsätze<br />
Contract manufacture and analysis must be correctly<br />
defined, agreed and controlled in or<strong>der</strong> to<br />
avoid misun<strong>der</strong>standings which could result in a<br />
product or work of unsatisfactory quality. There<br />
must be a written contract between the Contract<br />
Giver and the Contract Acceptor which clearly<br />
establishes the duties of each party. The contract<br />
must clearly state the way in which the<br />
Qualified Person releasing each batch of<br />
product for sale exercises his full responsibility.<br />
fortlaufenden Stabilitätsstudien in einer an<strong>der</strong>en<br />
Betriebsstätte als <strong>der</strong> Herstellungsstätte <strong>der</strong><br />
Bulkware o<strong>der</strong> des Fertigprodukts durchgeführt,<br />
bedarf es einer schriftlichen Vereinbarung zwischen<br />
den beteiligten Parteien. Die Ergebnisse<br />
<strong>der</strong> fortlaufenden Stabilitätsstudien sind in <strong>der</strong><br />
Herstellungsstätte <strong>zur</strong> Überprüfung durch die<br />
zuständige Behörde <strong>zur</strong> Verfügung zu halten.<br />
6.32 Ergebnisse außerhalb <strong>der</strong> Spezifikation<br />
o<strong>der</strong> auffällige atypische Trends sind zu untersuchen.<br />
Jedes bestätigte OoS-Ergebnis o<strong>der</strong> ein<br />
signifikanter negativer Trend sind den jeweils<br />
zuständigen Behörden zu melden. Einer<br />
möglichen Auswirkung auf die im Markt befindliche<br />
Chargen ist in Übereinstimmung mit dem<br />
Kapitel 8 des <strong>EG</strong>-GMP-<strong>Leitfaden</strong>s und in Abstimmung<br />
mit <strong>der</strong> jeweiligen zuständigen Behörde<br />
nachzugehen.<br />
6.33 Eine Zusammenfassung aller erhaltenen<br />
Daten, einschließlich je<strong>der</strong> zwischenzeitlich getroffenen<br />
Schlussfolgerung hinsichtlich des Stabilitätsprogramms,<br />
ist schriftlich zu erstellen und<br />
aufzubewahren. Die Zusammenfassung ist regelmäßig<br />
zu aktualisieren.<br />
Kapitel 7 Herstellung und Prüfung im Auftrag<br />
Herstellung und Prüfung im Auftrag müssen genau<br />
definiert, vereinbart und überprüft werden,<br />
um Missverständnisse zu vermeiden, aus denen<br />
sich ein Produkt o<strong>der</strong> eine Dienstleistung von<br />
un<strong>zur</strong>eichen<strong>der</strong> Qualität ergeben könnte.<br />
Zwischen Auftraggeber und Auftragnehmer<br />
muss ein schriftlicher Vertrag bestehen, <strong>der</strong> die<br />
Verantwortlichkeiten je<strong>der</strong> Seite klar festlegt.<br />
Aus dem Vertrag muss eindeutig hervorgehen,<br />
auf welche Weise die Sachkundige Person, die<br />
jede Produktcharge zum Inverkehrbringen freigibt,<br />
ihrer Verantwortung voll gerecht wird.<br />
Note:<br />
Anmerkung:<br />
This Chapter deals with the responsibilities of Dieses Kapitel behandelt die Verantwortlichkei-<br />
manufacturers towards the Competent Authoriten <strong>der</strong> Hersteller gegenüber den zuständigen<br />
ties of the Member States with respect to the Behörden <strong>der</strong> Mitgliedstaaten im Hinblick auf die<br />
granting of marketing and manufacturing Erteilung von Zulassungen und Herstellungs-<br />
authorisations. It is not intended in any way to erlaubnissen. Seine Regelungen sollen keines-<br />
affect the respective liability of contract accepfalls die jeweilige Haftung des Auftraggebers<br />
tors and contract givers to consumers; this is bzw. Auftragnehmers gegenüber dem Ver-<br />
governed by other provisions of Community and braucher berühren; dies wird durch an<strong>der</strong>e Be-<br />
national law.<br />
stimmungen <strong>der</strong> Gemeinschaft und durch nationales<br />
Recht geregelt.<br />
Übersetzung durch die Mitglie<strong>der</strong> <strong>der</strong> AG 42 des QS-Forums Schleswig-Holstein / Hamburg
General Allgemeine Anfor<strong>der</strong>ungen<br />
7.1 There should be a written contract covering<br />
the manufacture and/or analysis arranged<br />
un<strong>der</strong> contract and any technical arrangements<br />
made in connection with it.<br />
7.2 All arrangements for contract manufacture<br />
and analysis including any proposed changes in<br />
technical or other arrangements should be in<br />
accordance with the marketing authorisation for<br />
the product concerned.<br />
The Contract Giver Der Auftraggeber<br />
7.3 The Contract Giver is responsible for assessing<br />
the competence of the Contract Acceptor<br />
to carry out successfully the work required<br />
and for ensuring by means of the contract that<br />
the principles and guidelines of GMP as interpreted<br />
in this Guide are followed.<br />
7.4 The Contract Giver should provide the<br />
Contract Acceptor with all the information necessary<br />
to carry out the contracted operations correctly<br />
in accordance with the marketing authorisation<br />
and any other legal requirements. The<br />
Contract Giver should ensure that the Contract<br />
Acceptor is fully aware of any problems associated<br />
with the product or the work which might<br />
pose a hazard to his premises, equipment, personnel,<br />
other materials or other products.<br />
7.5 The Contract Giver should ensure that all<br />
processed products and materials delivered to<br />
him by the Contract Acceptor comply with their<br />
specifications or that the products have been<br />
released by a Qualified Person.<br />
The Contract Acceptor Der Auftragnehmer<br />
7.6 The Contract Acceptor must have adequate<br />
premises and equipment, knowledge and<br />
experience, and competent personnel to carry<br />
out satisfactorily the work or<strong>der</strong>ed by the Contract<br />
Giver. Contract manufacture may be un<strong>der</strong>taken<br />
only by a manufacturer who is the<br />
hol<strong>der</strong> of a manufacturing authorisation.<br />
20 Vgl. die ausführlicheren Regelungen im Annex 16<br />
7.1 Es ist ein schriftlicher Vertrag abzuschließen,<br />
<strong>der</strong> die Herstellung und/o<strong>der</strong> Prüfung<br />
im Auftrag und alle damit in Zusammenhang<br />
stehenden technischen Vereinbarungen umfasst.<br />
7.2 Alle Vereinbarungen über die Herstellung<br />
und Prüfung im Auftrag, einschließlich aller vorgesehenen<br />
Än<strong>der</strong>ungen an technischen o<strong>der</strong><br />
an<strong>der</strong>en Vereinbarungen, haben mit den Zulassungsunterlagen<br />
des betreffenden Produkts<br />
übereinzustimmen.<br />
7.3 Der Auftraggeber ist verantwortlich <strong>für</strong> die<br />
Beurteilung, ob <strong>der</strong> Auftragnehmer in <strong>der</strong> Lage<br />
ist, die vorgesehenen Aufgaben ordnungsgemäß<br />
auszuführen. Er hat durch den Vertrag<br />
sicherzustellen, dass die in diesem <strong>Leitfaden</strong><br />
dargelegten Grundsätze und Leitlinien <strong>der</strong><br />
<strong>Guten</strong> <strong>Herstellungspraxis</strong> befolgt werden.<br />
7.4 Der Auftraggeber hat dem Auftragnehmer<br />
alle nötigen Informationen <strong>zur</strong> Verfügung zu<br />
stellen, damit dieser die in Auftrag gegebenen<br />
Aufgaben korrekt in Übereinstimmung mit den<br />
Zulassungsunterlagen und allen weiteren rechtlichen<br />
Vorgaben ausführen kann. Der Auftraggeber<br />
hat sicherzustellen, dass sich <strong>der</strong> Auftragnehmer<br />
aller Beson<strong>der</strong>heiten bewusst ist, die<br />
mit dem Produkt o<strong>der</strong> den Aufgaben in Zusammenhang<br />
stehen und die ein Risiko <strong>für</strong> seine<br />
Räumlichkeiten, die Ausrüstung, das Personal<br />
o<strong>der</strong> <strong>für</strong> an<strong>der</strong>e Materialien o<strong>der</strong> an<strong>der</strong>e Produkte<br />
darstellen könnten.<br />
7.5 Der Auftraggeber hat sicherzustellen,<br />
dass alle ihm vom Auftragnehmer gelieferten<br />
bearbeiteten Produkte und Materialien ihren<br />
Spezifikationen entsprechen o<strong>der</strong> diese Produkte<br />
durch eine Sachkundige Person freigegeben<br />
wurden. 20<br />
7.6 Der Auftragnehmer muss über geeignete<br />
Räumlichkeiten und Ausrüstung, Sachkenntnis<br />
und Erfahrung sowie über kompetentes Personal<br />
verfügen, um die vom Auftraggeber übertragenen<br />
Aufgaben zufriedenstellend ausführen zu<br />
können. Auftragsherstellung kann nur von einem<br />
Hersteller übernommen werden, <strong>der</strong> eine Herstellungserlaubnis<br />
besitzt.<br />
Übersetzung durch die Mitglie<strong>der</strong> <strong>der</strong> AG 42 des QS-Forums Schleswig-Holstein / Hamburg
7.7 The Contract Acceptor should ensure that<br />
all products or materials delivered to him are<br />
suitable for their intended purpose.<br />
7.8 The Contract Acceptor should not pass to<br />
a third party any of the work entrusted to him<br />
un<strong>der</strong> the contract without the Contract Giver’s<br />
prior evaluation and approval of the arrangements.<br />
Arrangements made between the Contract<br />
Acceptor and any third party should ensure<br />
that the manufacturing and analytical<br />
information is made available in the same way<br />
as between the original Contract Giver and<br />
Contract Acceptor.<br />
7.9 The Contract Acceptor should refrain from<br />
any activity which may adversely affect the quality<br />
of the product manufactured and/or analysed<br />
for the Contract Giver.<br />
The Contract Der Vertrag<br />
7.10 A contract should be drawn up between<br />
the Contract Giver and the Contract Acceptor<br />
which specifies their respective responsibilities<br />
relating to the manufacture and control of the<br />
product. Technical aspects of the contract<br />
should be drawn up by competent persons<br />
suitably knowledgeable in pharmaceutical<br />
technology, analysis and Good Manufacturing<br />
Practice. All arrangements for manufacture and<br />
analysis must be in accordance with the<br />
marketing authorisation and agreed by both<br />
parties.<br />
7.11 The contract should specify the way in<br />
which the Qualified Person releasing the batch<br />
for sale ensures that each batch has been<br />
manufactured and checked for compliance with<br />
the requirements of Marketing Authorisation.<br />
7.12 The contract should describe clearly who<br />
is responsible for purchasing materials, testing<br />
and releasing materials, un<strong>der</strong>taking production<br />
and quality controls, including in-process controls,<br />
and who has responsibility for sampling<br />
and analysis. In the case of contract analysis,<br />
the contract should state whether or not the<br />
Contract Acceptor should take samples at the<br />
premises of the manufacturer.<br />
7.13 Manufacturing, analytical and distribution<br />
records, and reference samples should be kept<br />
by, or be available to, the Contract Giver. Any<br />
records relevant to assessing the quality of a<br />
7.7 Der Auftragnehmer hat sicherzustellen,<br />
dass alle an ihn gelieferten Produkte o<strong>der</strong> Materialien<br />
<strong>für</strong> ihren vorgesehenen Zweck geeignet<br />
sind.<br />
7.8 Der Auftragnehmer darf ohne vorherige<br />
Bewertung und Genehmigung <strong>der</strong> Vereinbarungen<br />
durch den Auftraggeber keine ihm vertraglich<br />
übertragene Aufgabe an einen Dritten weitergeben.<br />
Bei Vereinbarungen zwischen Auftragnehmer<br />
und Dritten ist sicherzustellen, dass<br />
die Informationen über Herstellung und Prüfung<br />
in gleicher Weise <strong>zur</strong> Verfügung stehen wie<br />
zwischen dem ursprünglichen Auftraggeber und<br />
Auftragnehmer.<br />
7.9 Der Auftragnehmer hat alles zu unterlassen,<br />
was die Qualität des <strong>für</strong> den Auftraggeber<br />
hergestellten und/o<strong>der</strong> geprüften Produkts nachteilig<br />
beeinflussen könnte.<br />
7.10 Zwischen Auftraggeber und Auftragnehmer<br />
ist ein Vertrag abzuschließen, <strong>der</strong> ihre jeweiligen<br />
Verantwortlichkeiten hinsichtlich Herstellung<br />
und Prüfung des Produkts festlegt.<br />
Technische Aspekte des Vertrags sind von kompetenten<br />
Personen abzufassen, die über geeignete<br />
Kenntnisse in pharmazeutischer Technologie,<br />
Analytik und <strong>der</strong> <strong>Guten</strong> <strong>Herstellungspraxis</strong><br />
verfügen. Alle Vereinbarungen über Herstellung<br />
und Prüfung müssen mit den For<strong>der</strong>ungen <strong>der</strong><br />
Zulassung übereinstimmen und von beiden<br />
Parteien genehmigt sein.<br />
7.11 In dem Vertrag ist festzulegen, auf welche<br />
Weise die Sachkundige Person, die die Chargen<br />
zum Inverkehrbringen freigibt 15 , sicherstellt,<br />
dass jede Charge in Übereinstimmung mit den<br />
Vorgaben <strong>der</strong> Zulassungsunterlagen hergestellt<br />
und geprüft wurde.<br />
7.12 Im Vertrag ist klar zu beschreiben, wer <strong>für</strong><br />
den Materialeinkauf, <strong>für</strong> die Prüfung und Freigabe<br />
von Materialien, <strong>für</strong> die Durchführung <strong>der</strong><br />
Produktion und Qualitätskontrollen, einschließlich<br />
Inprozesskontrollen, verantwortlich ist und in<br />
wessen Verantwortungsbereich Probenahme<br />
und Prüfung fallen. Im Falle <strong>der</strong> Auftragsprüfung<br />
hat aus dem Vertrag hervorzugehen, ob Probenahmen<br />
durch den Auftragnehmer in den Räumlichkeiten<br />
des Herstellers durchzuführen sind<br />
o<strong>der</strong> nicht.<br />
7.13 Herstellungs-, Prüf- und Vertriebsprotokolle<br />
sowie Referenzmuster sind vom Auftraggeber<br />
aufzubewahren o<strong>der</strong> müssen ihm bei Bedarf <strong>zur</strong><br />
Verfügung stehen. Alle <strong>für</strong> die Qualitätsbewer-<br />
Übersetzung durch die Mitglie<strong>der</strong> <strong>der</strong> AG 42 des QS-Forums Schleswig-Holstein / Hamburg
product in the event of complaints or a suspected<br />
defect must be accessible and specified<br />
in the defect/recall procedures of the Contract<br />
Giver.<br />
7.14 The contract should permit the Contract<br />
Giver to visit the facilities of the Contract Acceptor.<br />
7.15 In the case of contract analysis, the Contract<br />
Acceptor should un<strong>der</strong>stand that he is subject<br />
to Inspection by the competent Authorities.<br />
CHAPTER 8 COMPLAINTS AND PRODUCT<br />
RECALL<br />
Principle Grundsätze<br />
All complaints and other information concerning<br />
potentially defective products must be reviewed<br />
carefully according to written procedures. In<br />
or<strong>der</strong> to provide for all contingencies, and in accordance<br />
with Article 28 of Directive<br />
75/319/EEC, a system should be designed to<br />
recall, if necessary, promptly and effectively<br />
products known or suspected to be defective<br />
from the market.<br />
Complaints Beanstandungen<br />
8.1 A person should be designated responsible<br />
for handling the complaints and deciding the<br />
measures to be taken together with sufficient<br />
supporting staff to assist him. If this person is<br />
not the Qualified Person, the latter should be<br />
made aware of any complaint, investigation or<br />
recall.<br />
8.2 There should be written procedures<br />
describing the action to be taken, including the<br />
need to consi<strong>der</strong> a recall, in the case of a complaint<br />
concerning a possible product defect.<br />
8.3 Any complaint concerning a product<br />
defect should be recorded with all the original<br />
details and thoroughly investigated. The person<br />
responsible for Quality Control should normally<br />
be involved in the study of such problems.<br />
8.4 If a product defect is discovered or suspected<br />
in a batch, consi<strong>der</strong>ation should be given<br />
to checking other batches in or<strong>der</strong> to determine<br />
tung eines Produkts relevanten Aufzeichnungen<br />
müssen im Falle einer Beanstandung o<strong>der</strong> eines<br />
vermuteten Fehlers zugänglich und in den entsprechenden<br />
Verfahrensanweisungen des Auftraggebers<br />
zum Umgang mit Fehlern sowie <strong>für</strong><br />
Produktrückrufe vorgegeben sein.<br />
7.14 Im Vertrag ist dem Auftraggeber das<br />
Recht ein<strong>zur</strong>äumen, die Einrichtungen des Auftragnehmers<br />
zu besichtigen.<br />
7.15 Im Falle von Auftragsanalytik muss sich<br />
<strong>der</strong> Auftragnehmer bewusst sein, dass er <strong>der</strong> Inspektion<br />
durch die zuständigen Behörden unterworfen<br />
ist.<br />
Kapitel 8 Beanstandungen und Produktrückruf<br />
Alle Beanstandungen und an<strong>der</strong>en Informationen<br />
über möglicherweise fehlerhafte Produkte<br />
müssen nach schriftlich festgelegten Verfahren<br />
sorgfältig überprüft werden. In Übereinstimmung<br />
mit Artikel 28 <strong>der</strong> Direktive 75/319/EWG<br />
(entspricht Artikel 117 <strong>der</strong> Direktive 2001/83/<strong>EG</strong><br />
bzw. Artikel 84 <strong>der</strong> Direktive 2001/82/<strong>EG</strong>) sind<br />
systematische und umfassende Vorkehrungen<br />
zu treffen, um erfor<strong>der</strong>lichenfalls Produkte mit<br />
erwiesenen o<strong>der</strong> vermuteten Mängeln schnell<br />
und wirkungsvoll vom Markt <strong>zur</strong>ückrufen zu<br />
können.<br />
8.1 Es ist eine verantwortliche Person zu benennen,<br />
die die Beanstandungen bearbeitet und<br />
die erfor<strong>der</strong>lichen Maßnahmen festlegt. Ihr ist<br />
genügend Hilfspersonal <strong>zur</strong> Verfügung zu stellen.<br />
Ist diese Person nicht mit <strong>der</strong> Sachkundigen<br />
Person identisch, ist letztere über jede Beanstandung,<br />
Überprüfung und jeden Rückruf zu informieren.<br />
8.2 Es bedarf schriftlicher Verfahrensanweisungen<br />
<strong>für</strong> das Vorgehen bei Beanstandungen<br />
eines möglicherweise fehlerhaften Produktes bis<br />
hin zu dem Fall eines möglichen Rückrufs.<br />
8.3 Jede Produktbeanstandung ist mit allen<br />
Originalinformationen aufzuzeichnen und gründlich<br />
zu untersuchen. Die <strong>für</strong> die Qualitätskontrolle<br />
verantwortliche Person ist in <strong>der</strong> Regel an dieser<br />
Untersuchung zu beteiligen.<br />
8.4 Wenn ein Fehler an einer Charge festgestellt<br />
o<strong>der</strong> vermutet wird, ist auch die Überprüfung<br />
an<strong>der</strong>er möglicherweise betroffener Char-<br />
Übersetzung durch die Mitglie<strong>der</strong> <strong>der</strong> AG 42 des QS-Forums Schleswig-Holstein / Hamburg
whether they are also affected. In particular,<br />
other batches which may contain reworks of the<br />
defective batch should be investigated.<br />
8.5 All the decisions and measures taken as a<br />
result of a complaint should be recorded and<br />
referenced to the corresponding batch records.<br />
8.6 Complaints records should be reviewed<br />
regularly for any indication of specific or recurring<br />
problems requiring attention and<br />
possibly the recall of marketed products.<br />
8.7 Special attention should be given to<br />
establishing whether a complaint was caused by<br />
counterfeiting.<br />
8.8 The Competent Authorities should be informed<br />
if a manufacturer is consi<strong>der</strong>ing action<br />
following possibly faulty manufacture, product<br />
deterioration, detection of counterfeiting or any<br />
other serious quality problems with a product.<br />
Recalls Rückrufe<br />
8.8 A person should be designated as<br />
responsible for execution and co-ordination of<br />
recalls and should be supported by sufficient<br />
staff to handle all the aspects of the recalls with<br />
the appropriate degree of urgency. This<br />
responsible person should normally be<br />
independent of the sales and marketing<br />
organisation. If this person is not the Qualified<br />
Person, the latter should be made aware of any<br />
recall operation.<br />
8.9 There should be established written procedures,<br />
regularly checked and updated when<br />
necessary, in or<strong>der</strong> to organise any recall activity.<br />
8.10 Recall operations should be capable of<br />
being initiated promptly and at any time.<br />
8.11 All Competent Authorities of all countries<br />
to which products may have been distributed<br />
should be informed promptly if products are intended<br />
to be recalled because they are, or are<br />
suspected of being defective.<br />
21 Entspricht dem Stufenplanbeauftragten nach § 63a AMG<br />
gen in Betracht zu ziehen. Es sind insbeson<strong>der</strong>e<br />
die Chargen zu überprüfen, die aufbereitetes<br />
Material aus <strong>der</strong> fehlerhaften Charge enthalten<br />
können.<br />
8.5 Alle aufgrund einer Beanstandung getroffenen<br />
Entscheidungen und Maßnahmen sind mit<br />
Querverweis auf die entsprechenden Chargenprotokolle<br />
zu dokumentieren.<br />
8.6 Die Aufzeichnungen über Beanstandungen<br />
sind regelmäßig daraufhin zu überprüfen,<br />
ob sich Hinweise auf spezifische o<strong>der</strong> sich wie<strong>der</strong>holende<br />
Probleme ergeben, die eine beson<strong>der</strong>e<br />
Aufmerksamkeit und möglicherweise einen<br />
Rückruf erfor<strong>der</strong>n.<br />
8.7 Mit beson<strong>der</strong>er Aufmerksamkeit ist zu<br />
überprüfen, ob eine Beanstandung auf eine <strong>Arzneimittel</strong>fälschung<br />
<strong>zur</strong>ückzuführen ist.<br />
8.8 Sollte <strong>der</strong> Hersteller Maßnahmen in Erwägung<br />
ziehen, die auf einer möglicherweise fehlerhaften<br />
Herstellung, einer Zersetzung des Produktes,<br />
<strong>der</strong> Aufdeckung einer <strong>Arzneimittel</strong>fälschung<br />
o<strong>der</strong> irgendeinem an<strong>der</strong>en ernsthaften<br />
Qualitätsproblem beruhen, sind die zuständigen<br />
Behörden zu informieren.<br />
8.9 Es ist eine Person zu benennen, die <strong>für</strong><br />
die Durchführung und Koordination von Rückrufen<br />
verantwortlich ist. Ihr ist genügend Personal<br />
<strong>zur</strong> Verfügung zu stellen, damit alle Aspekte eines<br />
Rückrufs mit <strong>der</strong> erfor<strong>der</strong>lichen Dringlichkeit<br />
behandelt werden können. Diese verantwortliche<br />
Person hat üblicherweise unabhängig von<br />
Vertrieb und Marketing zu sein. 21 Falls diese<br />
Person nicht mit <strong>der</strong> Sachkundigen Person identisch<br />
ist, ist letztere über jeden Rückruf zu informieren.<br />
8.10 Es bedarf schriftlicher, regelmäßig überprüfter<br />
und, wenn nötig, aktualisierter Anweisungen,<br />
um bei Rückrufen vorbereitet zu sein.<br />
8.11 Die Maßnahmen <strong>für</strong> einen Rückruf müssen<br />
je<strong>der</strong>zeit unverzüglich in Gang gesetzt werden<br />
können.<br />
8.12 Wenn beabsichtigt ist, Produkte wegen eines<br />
erwiesenen o<strong>der</strong> vermuteten Mangels <strong>zur</strong>ück<strong>zur</strong>ufen,<br />
sind die zuständigen Behörden aller<br />
Län<strong>der</strong>, in die die Produkte möglicherweise<br />
geliefert wurden, unverzüglich zu benachrichtigen.<br />
Übersetzung durch die Mitglie<strong>der</strong> <strong>der</strong> AG 42 des QS-Forums Schleswig-Holstein / Hamburg
8.12 The distribution records should be readily<br />
available to the person(s) responsible for recalls,<br />
and should contain sufficient information on<br />
wholesalers and directly supplied customers<br />
(with addresses, phone and/or fax numbers inside<br />
and outside working hours, batches and<br />
amounts delivered), including those for exported<br />
products and medical samples.<br />
8.13 Recalled products should be identified<br />
and stored separately in a secure area while<br />
awaiting a decision on their fate.<br />
8.14 The progress of the recall process should<br />
be recorded and a final report issued, including<br />
a reconciliation between the delivered and recovered<br />
quantities of the products.<br />
8.15 The effectiveness of the arrangements for<br />
recalls should be evaluated from time to time.<br />
8.13 Unterlagen über den Vertrieb sind <strong>der</strong><br />
(den) <strong>für</strong> Rückrufe verantwortlichen Person(en)<br />
prompt <strong>zur</strong> Verfügung zu stellen. Daraus haben<br />
sich ausreichende Informationen über Großhändler<br />
und direkt belieferte Kunden (einschließlich<br />
Adressen, Telefon- und/o<strong>der</strong> Faxnummern<br />
während und außerhalb <strong>der</strong> Arbeitszeit,<br />
sowie <strong>der</strong> gelieferten Chargen und Mengen)<br />
zu ergeben. Das Gleiche gilt auch <strong>für</strong> exportierte<br />
Produkte und unverkäufliche Muster.<br />
8.14 Zurückgerufene Produkte sind als solche<br />
zu kennzeichnen sowie getrennt und in einem<br />
gesicherten Bereich zu lagern, solange eine<br />
Entscheidung über ihre weitere Bestimmung<br />
aussteht.<br />
8.15 Der Ablauf <strong>der</strong> Rückrufaktion ist zu dokumentieren.<br />
Es ist ein Abschlussbericht mit einer<br />
Bilanzierung <strong>der</strong> ausgelieferten und <strong>der</strong> <strong>zur</strong>ückerhaltenen<br />
Produktmengen zu erstellen.<br />
8.16 Die Eignung <strong>der</strong> Rückrufverfahren ist regelmäßig<br />
zu bewerten.<br />
CHAPTER 9 SELF INSPECTION KAPITEL 9 SELBSTINSPEKTION<br />
Principle Grundsätze<br />
Self inspections should be conducted in or<strong>der</strong> to<br />
monitor the implementation and compliance with<br />
Good Manufacturing Practice principles and to<br />
propose necessary corrective measures.<br />
9.1 Personnel matters, premises, equipment,<br />
documentation, production, quality control, distribution<br />
of the medicinal products, arrangements<br />
for dealing with complaints and recalls, and self<br />
inspection, should be examined at intervals following<br />
a pre-arranged programme in or<strong>der</strong> to<br />
verify their conformity with the principles of<br />
Quality Assurance.<br />
9.2 Self inspections should be conducted in<br />
an independent and detailed way by designated<br />
competent person(s) from the company. Independent<br />
audits by external experts may also be<br />
useful.<br />
Selbstinspektionen sind durchzuführen, um die<br />
Anwendung und Beachtung <strong>der</strong> Regeln <strong>der</strong><br />
<strong>Guten</strong> <strong>Herstellungspraxis</strong> zu überwachen und<br />
Vorschläge <strong>für</strong> notwendige Korrekturmaßnahmen<br />
zu machen.<br />
9.1 Personalbezogene Belange, Räumlichkeiten,<br />
Ausrüstung, Dokumentation, Produktion,<br />
Qualitätskontrolle, Vertrieb von <strong>Arzneimittel</strong>n,<br />
Vorkehrungen <strong>zur</strong> Behandlung von Beanstandungen<br />
und Abwicklung von Rückrufen sowie<br />
die Durchführung von Selbstinspektionen sind in<br />
regelmäßigen Abständen nach einem im voraus<br />
festgelegten Programm zu überprüfen, um ihre<br />
Übereinstimmung mit den Grundsätzen <strong>der</strong><br />
Qualitätssicherung festzustellen.<br />
9.2 Selbstinspektionen sind von einem o<strong>der</strong><br />
mehreren beauftragten, vom auditierten Bereich<br />
unabhängigen Experten des Unternehmens bis<br />
auf Detailebene durchzuführen. Audits durch externe<br />
Sachverständige können ebenfalls hilfreich<br />
sein.<br />
Übersetzung durch die Mitglie<strong>der</strong> <strong>der</strong> AG 42 des QS-Forums Schleswig-Holstein / Hamburg
9.3 All self inspections should be recorded.<br />
Reports should contain all the observations<br />
made during the inspections and, where applicable,<br />
proposals for corrective measures. Statements<br />
on the actions subsequently taken should<br />
also be recorded.<br />
9.3 Alle Selbstinspektionen sind zu dokumentieren.<br />
Die Berichte haben alle während <strong>der</strong> Inspektion<br />
gemachten Beobachtungen und, soweit<br />
zutreffend, Vorschläge <strong>für</strong> Korrekturmaßnahmen<br />
zu enthalten. Über die anschließend ergriffenen<br />
Maßnahmen sind ebenfalls Aufzeichnungen<br />
zu führen.<br />
Übersetzung durch die Mitglie<strong>der</strong> <strong>der</strong> AG 42 des QS-Forums Schleswig-Holstein / Hamburg